
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUntangling the catalytic importance of the Se oxidation state in organoselenium-mediated oxygen-transfer reactions: the conversion of aniline to nitrobenzene†
Andrea
Madabeni
 a,
Damiano
Tanini
b,
Antonella
Capperucci
b and
Laura
Orian
*a
a,
Damiano
Tanini
b,
Antonella
Capperucci
b and
Laura
Orian
*a
aDipartimento di Scienze Chimiche Università Degli Studi di Padova, Via Marzolo 1, 35131 Padova, Italy. E-mail: laura.orian@unipd.it
bDipartimento di Chimica ‘Ugo Schiff’ Università Degli Studi di Firenze, Via Della Lastruccia 3-13, Sesto Fiorentino, Firenze, Italy
First published on 27th June 2024
Abstract
Seleninic acids and their precursors are well-known oxygen-transfer agents that can catalyze several oxidations with H2O2 as the final oxidant. Until very recently, the Se(IV) “peroxyseleninic” acid species has been considered the only plausible catalytic oxidant. Conversely, in 2020, the involvement of Se(VI) “peroxyselenonic” acid has been proposed for the selenium mediated epoxidation of alkenes. In this work, we theoretically probe different mechanisms of H2O2 activation and of Se(IV) to Se(VI) interconversion. In addition, we investigate through a combined theoretical (DFT) and experimental approach the mechanistic steps leading to the oxidation of aniline to nitrobenzene, when Se(IV) seleninic acid or Se(VI) selenonic acids are used as catalysts and H2O2 as the oxidant. This process encompasses three subsequent organoselenium mediated oxidations by H2O2. These results provide a mechanistic explanation of the advantages and disadvantages of both oxidation states (IV and VI) in the different stages of catalytic oxygen-transfer reactions: hydrogen peroxide activation and actual substrate oxidation. While the Se(VI) “peroxyselenonic” acid is found to be a better oxidant, the privileged role of “peroxyseleninic” acid as the main active species is assessed and its origin is identified in the lower catalyst-distortion that seleninic acid undergoes when activating H2O2. Conversely, the higher catalyst-distortion that characterizes the reaction of selenonic acid with H2O2 supports an inactivating role of Se(IV) to Se(VI) interconversion.
Introduction
Oxidations are an important class of reactions allowing the manipulation of a variety of functional groups.1 While many effective oxidants are available, the employment of green oxidants such as hydrogen peroxide for the practical oxidation of organic substrates usually requires the use of a catalyst to activate hydrogen peroxide into an oxygen-transfer species which is kinetically more reactive.2,3Organoselenides are recognized as effective oxygen-transfer catalysts1 in epoxidation/dihydroxylation reactions,4–7 sulfoxidation,8 aldehyde9 and amine oxidations,10,11 with H2O2 as the final oxidant. For reactions catalyzed by phenyl seleninic acid 1 or its respective precursor (e.g., diphenyl diselenide 2), until recently, phenyl peroxyseleninic acid 3 was deemed the only plausible active catalytic intermediate (Scheme 1).
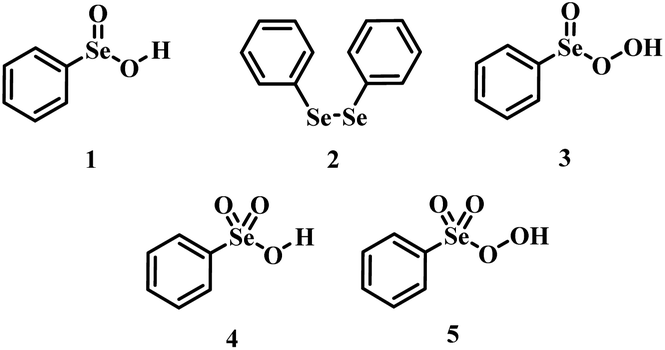 | ||
| Scheme 1 Organoselenides employed as catalysts or pre-catalysts for selenium mediated oxygen-transfer reactions and postulated plausible active oxidants. | ||
The first hypothetical participation of peroxyseleninic acids in oxidation reactions dates back to the early days of organoselenium chemistry, when the oxidation of seleninic to peroxyseleninic acid was postulated to rationalize the autocatalytic behavior of selenide oxidation.12 Later on, their participation in various organic substrate oxidations was proposed4,8,13 and it consolidated in the past fifty years.1,14 In 2020, Back and coworkers6 reported for the first time evidence of the involvement of the high oxidation state intermediates selenonic 4 and peroxyselenonic acid 5 in the selenium-mediated epoxidation of cyclooctene. This observation, while thought-provoking, was suggested to be not completely general. Indeed, in 2021, evidence was reported for the involvement of the “conventional” peroxyseleninic acid 3 in another selenium catalyzed oxygen-transfer process, i.e., the on-water oxidation of aniline to nitrobenzene11 (Scheme 2).
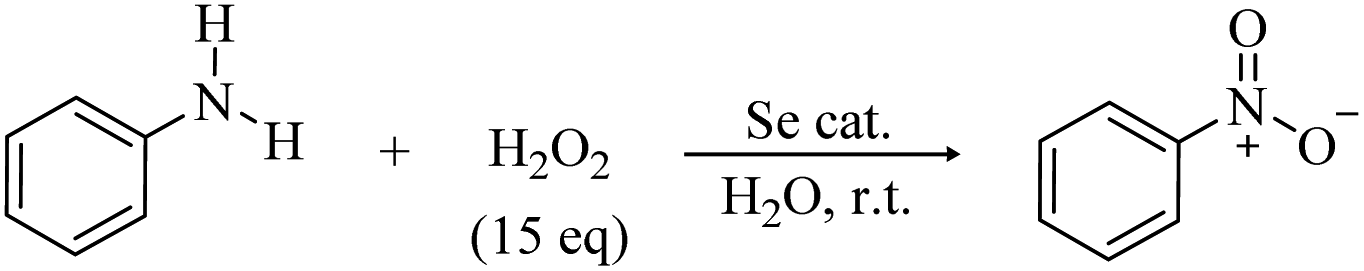 | ||
| Scheme 2 On-water selenium catalyzed oxidation of aniline to nitrobenzene at room temperature (r.t). | ||
In the work by Tanini et al.,11 Se(VI) selenonic acid 4 was recovered in the water after complete oxidation of the substrate but was found inactive towards further catalytic activity if not reduced back to Se(IV) seleninic acid 1. Conversely, in the selenium-mediated epoxidation of alkenes, selenonic acid was found to be more active than seleninic acid itself.6 Puzzled by these apparently conflicting results, we chose to investigate accurately the mechanistic details of Se-promoted H2O2 activation, which should be a key reactive step common to all reactions catalyzed by 1 or 2.
Despite the long-standing experimental experience in the field, to the best of our knowledge, no detailed theoretical mechanistic investigation has ever been carried out on catalytic oxygen-transfer reactions mediated by organoselenides. Only a previous report discussed peroxyseleninic acid 3 formation starting from the parent diphenyl diselenide 2, addressing the autocatalytic decomposition of H2O2.15 Indeed, while much theoretical mechanistic effort was devoted in the past twenty years to the elucidation of glutathione peroxidase-like catalytic potential of organoselenides,16,17 other catalytic reactions remained somewhat unexplored in silico. Among the different selenium catalyzed oxygen transfer processes,1 we chose to focus on the aniline oxidation to nitrobenzene, which represents an intriguing case involving three consecutive catalytic oxygen-transfers. Indeed, according to previous studies,10,18 the oxidation of aniline to nitrobenzene goes through hydroxyanilines and nitrosobenzene as intermediates, which are progressively oxidized (Scheme 3).
 | ||
| Scheme 3 Oxidation of aniline to nitrobenzene, with the three oxygen-transfer steps highlighted. | ||
As previously mentioned, no theoretical mechanistic investigation has to date been reported for these and similar catalytic reactions.11 Additionally, while experimental evidence supports a Se(IV) only catalysis, no theoretical explanation as to why only one oxidation state of Se is active in the reaction is available until now. Thus, with the inactive nature of the Se(VI) selenonic acid as a catalyst in mind, we aim to provide a detailed mechanistic picture of the title reaction to understand the different properties of Se(IV) and Se(VI) acids in the three different progressive oxygen-transfers and the hydrogen peroxide activation step.
Results and discussion
Previous experimental evidence showed that seleninic acid and selenonic acid alone do not oxidize aniline.10,11 Thus, the catalyst must first react with hydrogen peroxide to produce the actual oxygen-transfer species. In oxygen-transfer processes catalyzed by Se(IV) or Se(VI), the activation of H2O2 is thought to occur via the formation of the corresponding organoselenium peroxyacid, and this oxygen-transfer species is believed to be fundamental also for other organoselenium catalyzed oxidations.1,14 Since this step is required to produce the active oxidant in the reaction, we first discuss the formation of these species starting from the corresponding acids and hydrogen peroxide. Different mechanisms for peroxyacid formation were investigated, both concerted and stepwise, and the most favorable energetics were obtained for a mechanistic hypothesis that closely matches the one made by Back et al.,6 in which the H2O2 activation step is independent of the nature of the substrate and proceeds in two steps. This stepwise process proceeds through the intermediate formation of a peroxy selenurane (I), which can then undergo a non-redox dehydration to the corresponding peroxyacid (Pox) (Scheme 4).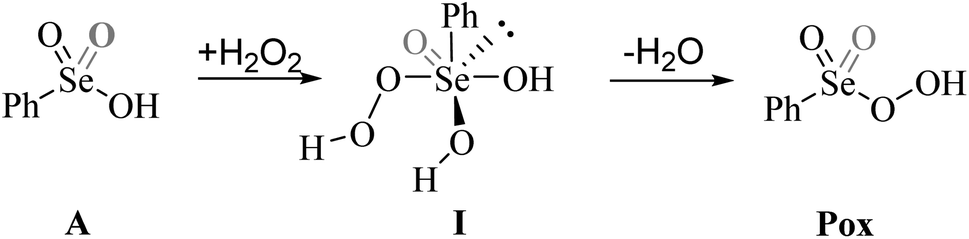 | ||
| Scheme 4 Hydrogen peroxide activation by organoselenium acids (A) in the form of peroxyacids (Pox). The grey double bond represents the additional bond present only in the Se(VI) selenonic acid and derivatives. | ||
In the H2O2 activation process (from A to Pox), Se(IV) seleninic acid 1 appears to be the best reactant. Indeed, not only the formation of the peroxyseleninic acid 3 is slightly more favored thermodynamically as compared to the same process for Se(VI), i.e. the conversion of 4 to 5, (ΔGr of −4.7 and −1.7 kcal mol−1 respectively), but the overall process occurs on a much lower energy surface. While the formation of the peroxy selenurane (I) is endergonic for both Se oxidation states, the dehydration then occurs with a relatively low activation energy. Both H2O2 addition and the peroxy selenurane (I) dehydration are located much lower on the PES when seleninic acid is involved, as compared to selenonic acid. Particularly, the selenurane (I) itself is much more stable for Se(IV) than for Se(VI), being located on the PES respectively at 13.9 and 23.8 kcal mol−1. This energy difference is roughly conserved also when comparing TS1 and TS2, i.e., in the transition states for the H2O2 addition (from A to I) and dehydration processes (from I to Pox). Thus, overall, the peroxyacid Pox formation occurs with an activation energy 10 kcal mol−1 lower for seleninic acid than for selenonic acid, making the former much more privileged energetically in H2O2 activation (ΔG‡ of 22.1 and 31.7 kcal mol−1 for H2O2 addition, TS1, and of 24.0 and 33.9 kcal mol−1 for the dehydration, TS2, respectively). The choice of the solvent did not affect these outcomes, even if highly non-polar solvents (e.g., benzene) were found to moderately decrease the height of all the barriers as compared to polar solvents. (Table S1†) Along the manuscript, only data in water will be discussed for consistency with the previous experimental work by Tanini et al.,11 which was carried out under on-water conditions (see also Computational details).
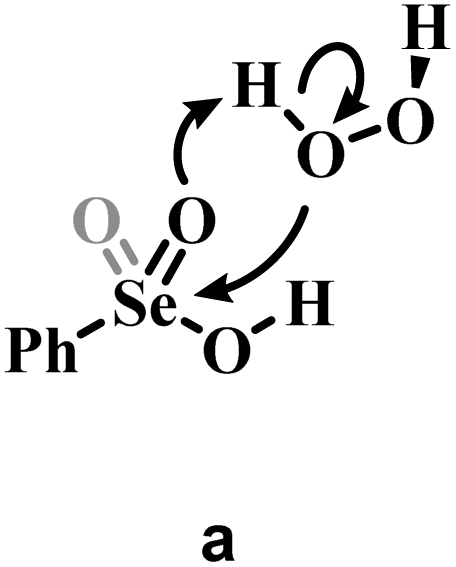
Conversely, two other possible H2O2 activation mechanisms (all independent of the nature of the substrate) were found to be kinetically less favored by more than 10 kcal mol−1. Both investigated concerted processes (Table 1b and c) have a higher activation energy than the corresponding H2O2 addition step (Table 1a) and higher than the overall stepwise H2O2 activation process. Particularly, the previously proposed concerted mechanism,15 in which one proton of H2O2 is transferred to the –OH group of the acids 1 or 4 (Table 1b) has an activation energy already more than 10 kcal mol−1 higher than H2O2 addition, in which the proton of H2O2 is transferred to the corresponding Se–O double bond moiety (Table 1a). A concerted mechanism corresponding to an “O insertion” within the Se–OH bond of seleninic and selenonic acids, thus turning the –OH function into the –OOH function (Table 1c), displays an even higher, unfeasible activation energy. These results suggest that the formation of the peroxy selenurane intermediate (I) is pivotal in the actual conversion between the acid and the peroxyacid species. Interestingly, also for the two concerted (unlikely) mechanisms, Se(IV) appears to be consistently more reactive, a result further corroborating our previous conclusions about H2O2 activation by seleninic and selenonic acids, and suggesting that the relative inertia of Se(VI) in activating H2O2 might be intrinsic in its high oxidation state.
| ΔG‡ | ||
|---|---|---|
| Se(IV) | Se(VI) | |
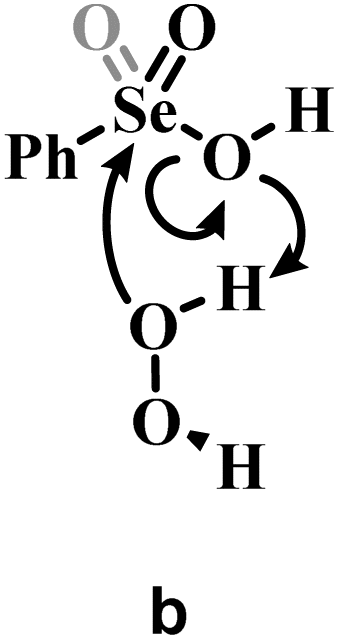
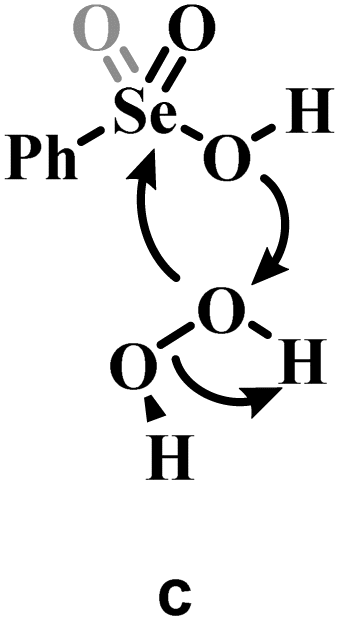
a Grey portions of the Lewis structures represents the additional Se![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond present in Se(VI) structures only.
b The H2O2 addition process sketched in a is represented for comparison with b and c, with its relative activation energy (TS1). The overall barrier towards peroxyseleninic/peroxyselenonic acid is slightly higher (TS2), corresponding to dehydration of the selenurane (I). However, it remains much lower than both processes b and c. Level of theory: COSMO-M06//OPBE. O bond present in Se(VI) structures only.
b The H2O2 addition process sketched in a is represented for comparison with b and c, with its relative activation energy (TS1). The overall barrier towards peroxyseleninic/peroxyselenonic acid is slightly higher (TS2), corresponding to dehydration of the selenurane (I). However, it remains much lower than both processes b and c. Level of theory: COSMO-M06//OPBE.
|
||
| a | 22.1b | 31.7b |
| b | 36.9 | 48.2 |
| c | 61.9 | 74.0 |
|
|
|
To provide a quantitative discussion on the effect of the oxidation state on the H2O2 activation, ASA and EDA were performed. (see Computational methods). Particularly, the addition of H2O2 to seleninic and selenonic acid was compared to provide a detailed insight into the chalcogen different oxidation state effect in the most favored pathway, i.e., the stepwise activation (Scheme 3).
The ASA for the H2O2 addition step is represented in Fig. 1a. All energies were plotted at consistent values of O–H bond breaking. This reaction coordinate (r.c.) undergoes a well-defined variation along the reaction since the proton is transferred from H2O2 to the SeO bond of seleninic and selenonic acid. Analogous conclusions can be drawn by analysis of the complementary r.c. i.e., the H–O bond formation in the peroxy selenurane (I). (Fig. S1†) It can be seen how, from the beginning of the reaction to the TS, which occurs roughly at the same value of r.c. for the two reactions (at ca. 0.35 Å) the ΔEintfor both OS is superimposed, suggesting that both seleninic and selenonic acids interact similarly with H2O2. Conversely, seleninic acid has a consistently lower ΔEstr, which appears to be the main player in its reduced activation energy in H2O2 addition.
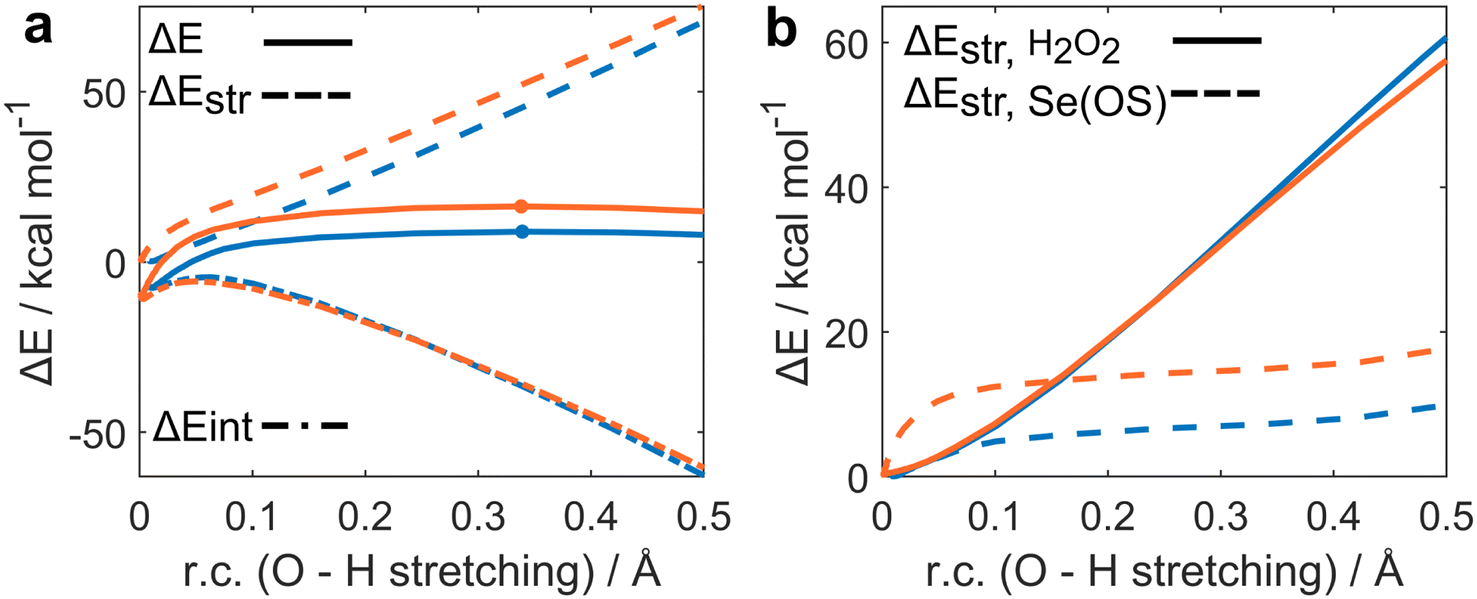 | ||
| Fig. 1 Activation strain (a) and strain decomposition (b) analysis of H2O2 addition to seleninic (blue) and selenonic (orange) acids. Level of theory: M06//OPBE. | ||
The strain energy was then decomposed into the contribution of each fragment i.e., a ΔEstr,H2O2 accounting for H2O2 deformation only, and ΔEstr,Se(OS) associated with seleninic/selenonic acid deformation alone (Fig. 1b). What can be inferred is that the higher strain, and thus the higher activation energy predicted for H2O2 addition to Se(VI), is due exclusively to the higher ΔEstr,Se(VI) when compared to ΔEstr,Se(IV). Indeed, seleninic acid has a lower ΔEstr,Se(OS) along the whole r.c., when compared to selenonic acid. We interpreted this result as an effect of the different environment around the Se nucleus in the two OSs. Indeed, selenonic acid has four groups in the surrounding of Se (a phenyl group, two oxygens and one hydroxyl group), while seleninic has only three groups (one SeO less than selenonic acid): thus, the distortion required to collocate the new OOH group around Se is intuitively larger for the former than for the latter, as reflected by its higher ΔEstr.
The oxidation of aniline to nitrobenzene
In the previous section, we showed that Se(VI) activates hydrogen peroxide in the form of a peroxyacid less efficiently than Se(IV). Nevertheless, in the context of a catalytic mechanism, to properly assess the performances of each oxidation state in aniline conversion, the full mechanism must be investigated. We want to stress that apart from the specific interest in the reaction mechanism of aniline oxidation, this catalytic mechanism also provides an interesting archetypal case study since it represents a case of three consecutive oxygen-transfers in which the nucleophile is progressively more and more oxidized in nature (Scheme 5).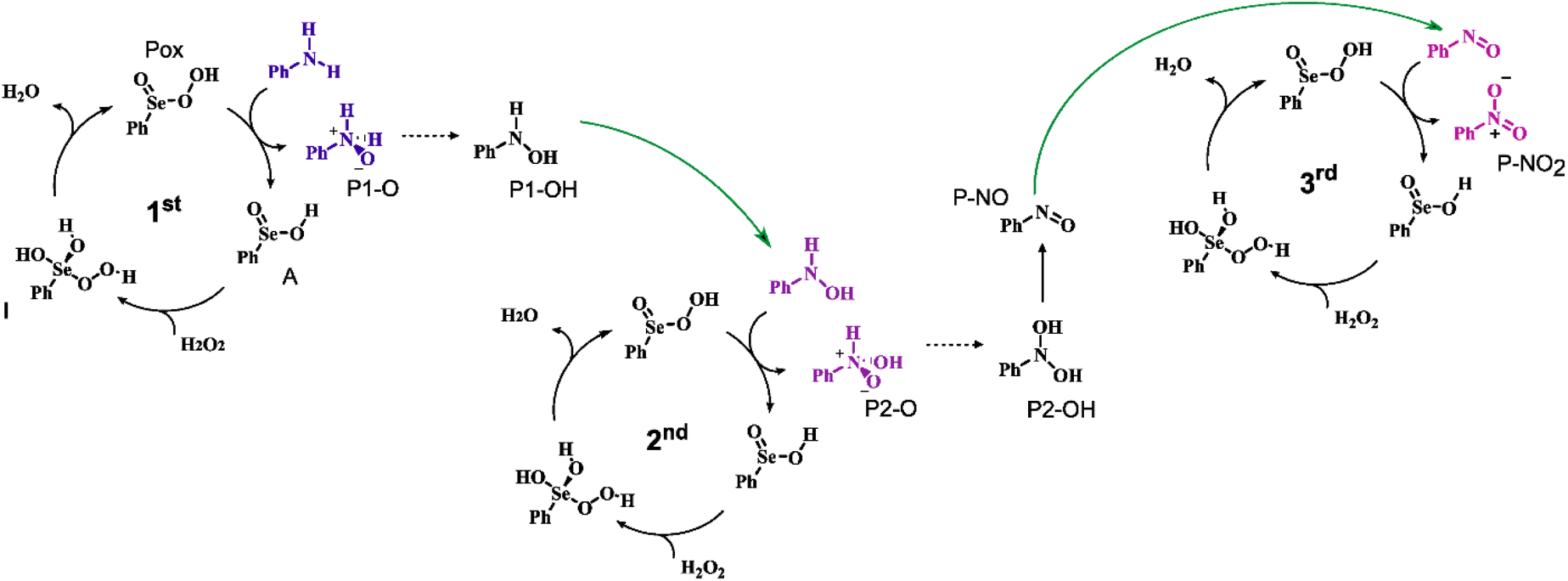 | ||
| Scheme 5 Catalytic cycles for the description of the overall oxidation to nitrobenzene. Only Se(IV) species are represented. Analogous Se(VI) species have one additional Se–O formal double bond. Dashed steps occur with a low to non-appreciable activation energy and corresponds to dashed lines in Fig. 2a. Green arrows represent the products which exit from one cycle to enter the next one. The substrate oxidation step is represented in purple. | ||
Bearing in mind the inactive nature of selenonic acid 4 in the selenium mediated oxidation of aniline to nitrobenzene, when compared to seleninic acid 1 – further highlighted by the experiments reported in Scheme 6 – we then screened through DFT calculations the potential energy surface (PES) for the complete catalytic mechanism from the fully reduced (aniline) to the fully oxidized substrate (nitrobenzene) (Fig. 2a). Both seleninic and selenonic acids were theoretically investigated as possible catalysts, for a straight comparison of two PESs and to gain insight into the role of the Se oxidation state in the catalytic mechanism. As previously hinted, this process can be described by three sequential catalytic cycles, each involving the activation of H2O2. The most stable Gibbs free energy profile (see Computational methods for a full description of Gibbs free energy calculations) is represented in Fig. 2a. The key mechanistic features of the process will be described, as deduced by DFT calculations.
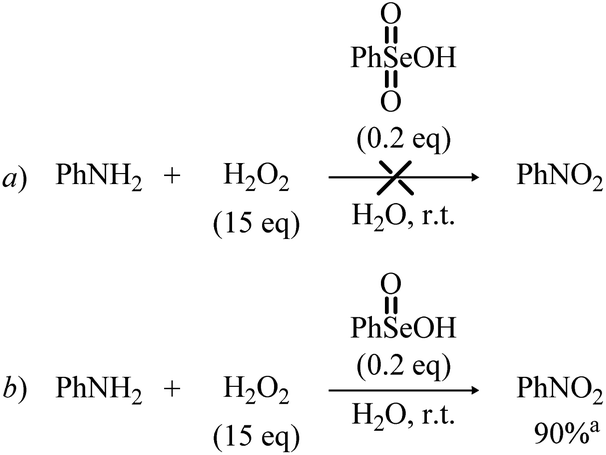 | ||
| Scheme 6 Reaction of aniline with H2O2 in the presence of Se(VI) and Se(IV) species. aIsolated yield is given. | ||
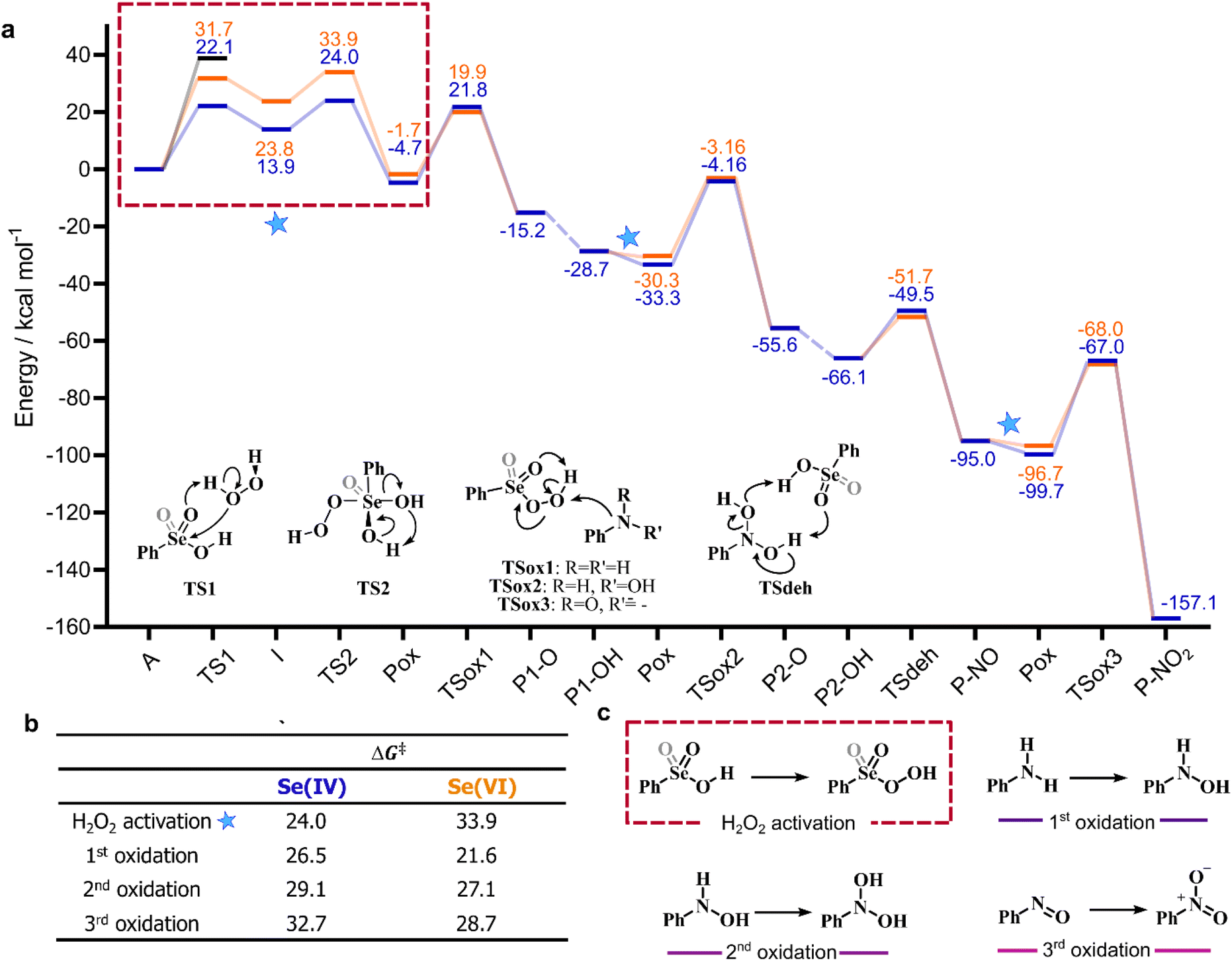 | ||
| Fig. 2 (a) Gibbs free energy profile (kcal mol−1) for the oxidation of aniline to nitrobenzene: Se(IV) mediated process (blue); Se(VI) mediated process (orange). All energies are relative to the free seleninic/selenonic acid and H2O2. The black line represents the activation energy for direct aniline oxidation by H2O2 (i.e., the uncatalyzed process). Level of theory: COSMO-M06//OPBE. Lines are intended as a guide to the eye only. Only the first H2O2 activation step is represented (within a dashed red box); the subsequent analogous steps are omitted, and their position is indicated by a star. Dashed lines are acid/base processes occurring without or with very low activation energies. (b) Activation energies (kcal mol−1) for the key steps of the mechanism. (c) Schematic representation of the H2O2 activation process and of the three consecutive oxidative steps. | ||
Analogous pathways for the Se catalyzed conversion of aniline to nitrobenzene were found regardless of the oxidation state of the Se nucleus, i.e., for both seleninic and selenonic acid. In both cases, the acid (A) reacts first with H2O2 to produce the corresponding peroxyacid (Pox). (vide supra). Three consecutive catalytic cycles progressively lead to the oxidation of aniline to the corresponding mono hydroxylamine (P1-OH), dihydroxylamine (P2-OH) and lastly to nitrobenzene (P-NO2). In each cycle, the peracid generated by H2O2 activation (Fig. 2c) reacts with the substrate in a SN2 fashion, producing the N-oxide products (P1-O and P2-O) and the corresponding seleninic or selenonic acid. Under the acidic conditions in which the reaction takes place, the primary N-oxide is easily protonated by either 1 or 4 on the O position, and then easily deprotonated on the N position, leading to the mono hydroxyl amine and dihydroxylamine (P1-OH and P2-OH). For aniline N-oxide (P1-O), the process was found to proceed without any appreciable activation energy, while, for aniline hydroxylamine N-oxide (P2-O), the process occurs with a low activation energy of less than 2 kcal mol−1 (Table S2†).
The involvement of N-phenylhydroxylamine in the mechanism of the Se-catalyzed oxidation of anilines was also confirmed through the control experiments summarized in Scheme 7. In the presence of catalytic amounts of benzeneseleninic acid, N-phenylhydroxylamine reacted with H2O2 to afford nitrobenzene, although significant amounts of the related azoxybenzene (diphenyldiazene oxide) was formed (Scheme 7, reaction a). Remarkably, the reaction of N-phenylhydroxylamine with H2O2 in the presence of benzeneselenonic acid (Scheme 7, reaction b) as well as the control experiment conducted under Se-free conditions (Scheme 7, reaction c) did not afford nitrobenzene and only diphenyldiazene oxide was detected.
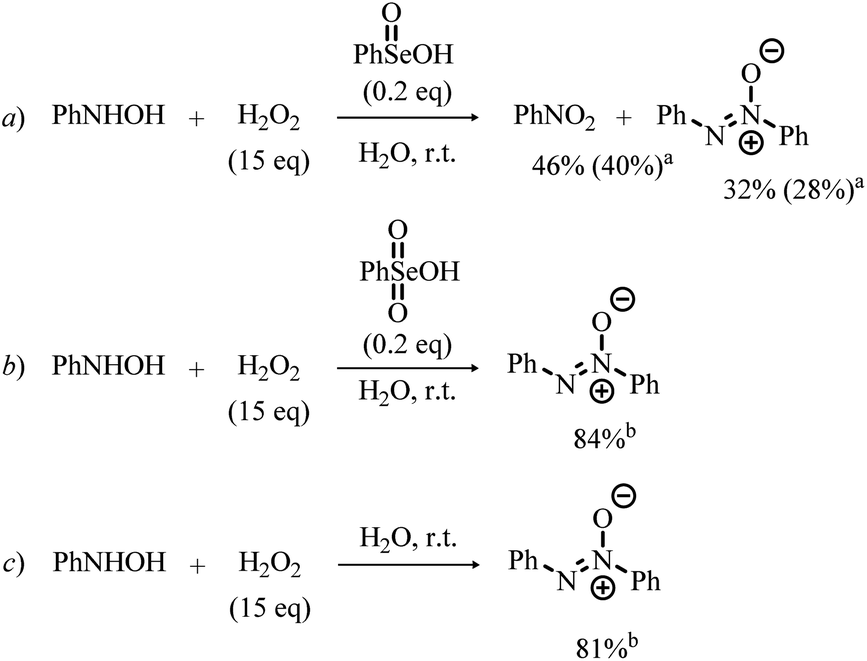 | ||
| Scheme 7 Reaction of N-phenylhydroxylamine with H2O2 in the presence of benzeneseleninic acid (reaction a), benzeneselenonic acid (reaction b) and Se-free control experiment (reaction c). aConversion is reported (isolated yield is given in parentheses). bIsolated yield is given. | ||
Before the 3rd catalytic cycle, P2-OH undergoes an acid catalyzed dehydration reaction to nitrosobenzene (P-NO), in which either 1 or 4 acts as amphoteric acid/base catalyst with an overall activation energy of 16.6 and 14.4 kcal mol−1, respectively, and a reaction energy of −28.9 kcal mol−1. Lastly, in the final oxidative cycle, P-NO is oxidized to the final species, nitrobenzene (P-NO2). The potentially competitive direct oxidation of P2-OH to the corresponding N-oxide followed by dehydration to P-NO2 was found to be less favored kinetically than the dehydration to P-NO, even if it has a lower activation energy than the oxidation of P-NO to P-NO2. (Table S3†) Thus, Se-catalyzed dehydration to P-NO was deemed more likely. Additionally, the formation of P-NO was confirmed in previous Se-catalyzed oxidations of aniline, and it is likely the key intermediate in the formation of azoxy benzene, one of the side products of the reaction.11 Direct evidence of the role of nitrosobenzene as a reaction intermediate in the Se-catalyzed process was provided by the control experiments reported in Scheme 8. Indeed, nitrobenzene was directly achieved in good yield upon treatment of nitrosobenzene with H2O2 in the presence of a catalytic amount of benzeneseleninic acid (Scheme 8, reaction a). On the other hand, the reactions of Scheme 8 further highlighted the key role of Se(IV) species in the oxidation of aniline. The plausible nitroso intermediate was recovered unreacted after treatment with H2O2 in the presence of Se(VI) catalysts such as benzeneselenonic acid (Scheme 8, reaction b). As expected, no oxidation occurred in the absence of selenium-containing species (Scheme 8, reaction c).
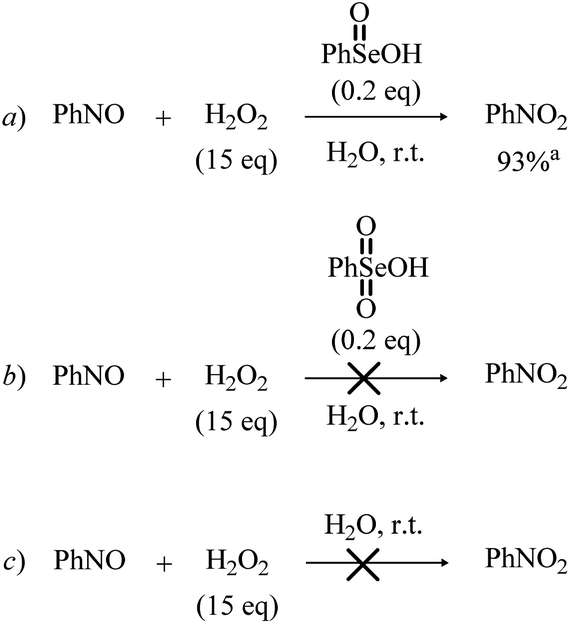 | ||
| Scheme 8 Reaction of nitrosobenzene with H2O2 in the presence of benzeneseleninic acid (reaction a), benzeneselenonic acid (reaction b) and the Se-free control experiment (reaction c). aIsolated yield is given. | ||
The direct participation of this peroxy selenurane in the oxidation of the substrate was excluded on the basis of the activation energy of the process, e.g., for Se(IV), aniline oxidation to P1-O occurs with an activation energy of ca. 45 kcal mol−1 and of ca. 26 kcal mol−1 when the peroxy selenurane (I), and the peroxyacid (Pox) acts as the oxidant, respectively (Table S4†).
Following H2O2 activation, the three oxidations occur with increasing activation energies, regardless of the oxidation state of the catalyst. That is, oxidation of aniline has the lowest activation energy (Fig. 2b, 1st oxidation) and oxidation of P-NO the highest one (Fig. 2b, 3rd oxidation). Intuitively, since the amine acts as the nucleophile in the reaction, each increment of the N oxidation state reduces the nucleophilicity of the substrate due to the electron-withdrawing nature of each new binding oxygen group. Indeed, the Hirshfeld partial charge19,20 on N increases from −0.212 to −0.088 and to −0.003 going from aniline to P1-OH to P-NO, thus highlighting a reduced nucleophilicity of the oxidized intermediates. Similarly, the barrier for the three acid-catalyzed processes increases, i.e., while the isomerization of P1-O proceeds without any appreciable activation energy, and the conversion of P2-O to P2-OH has a low activation energy of less than 2 kcal mol−1 when seleninic acid 1 act as the acid catalyst, the dehydration of P2-OH to P-NO has an activation energy of 14–16 kcal mol−1. Thus, excluding the two previous almost barrierless acid catalyzed processes, this step has the lowest barrier of the overall mechanism by 10 kcal mol−1 or more for both oxidation states.
Overall, the whole process from aniline to nitrobenzene is strongly exergonic by more than 150 kcal mol−1. All the reaction steps of the mechanism are exergonic or almost isergonic, apart from the H2O2 addition to 1 or 4, leading to the formation of the intermediate peroxy selenurane (I) which is destabilized with respect to the corresponding acids. In contrast, the reaction energy of each sequential oxidation becomes more and more negative along the pathway, starting from a ΔGr of −28.6 kcal mol−1 for the oxidation of aniline to P1-OH and reaching a ΔGr of −62.1 for the final oxidation of P-NO to P-NO2.
Comparison between Se(IV) and Se(VI) cycles
The overall representation of the energy profile leading to the conversion of aniline to P-NO2, in which a clear comparison between the performance of Se(IV) and Se(VI) species can be made, is shown in Fig. 2a. As initially highlighted, Se(IV) outperforms Se(VI) in the hydrogen peroxide activation step, leading to a more stable peroxyacid with a lower activation energy (Fig. 2b). Conversely, Se(VI) appears to perform consistently better in each of the following reaction steps, i.e., the three substrate oxidations and the Se-catalyzed dehydration of P2-OH to P-NO. Focusing on the three former reactions, activation energies for Se(VI) mediated processes are 2–5 kcal mol−1 lower than those for the corresponding Se(IV) mediated processes. Thus, peroxyselenonic acid 5 is a better oxidant from the kinetic point of view than peroxyseleninic acid 3. Part of this activation energy lowering is clearly due to the lower stability of peroxyselenonic than selenonic acid (as compared to the couple peroxyseleninic/seleninic acid) assessed on the basis of their ΔGr, as previously mentioned. Thus, while Se(VI) appears to be the best oxidant for the reactions under investigation, Se(IV) appears to be the best species in the H2O2 activation process, with a much higher energy gap between the two PESs in this latter case.Theoretical comparison of the catalytic performance
While the analysis of the PESs already provides valuable insight into the catalytic potential of Se(IV) and Se(VI) species, a much more quantitative index of their catalytic potential is represented by the calculated turnover frequency (TOF) of each catalytic cycle, when the two different catalysts, i.e. seleninic and selenonic acid, are employed. This quantity is directly related to an experimentally accessible parameter, and accounts for all steps of each cycle at once in the evaluation of the performance of the catalyst. Within the energetic span model proposed by the Kozuch and Shaik, who elaborated the idea of Amatore and Jutand,21 it is possible, starting from the energy landscape constructed with quantum mechanics calculations, to obtain a well-defined TOF value.22–24 Additionally, this procedure allows to gain insight into the states which mostly (or totally) determine the value of the TOF, i.e. the TOF determining transition state (TDTS) and the TOF determining intermediate (TDI) (see additional Computational details in the ESI† for further information).25In Fig. 3, a representative close-up of the first catalytic cycle is illustrated; the TDI and the TDTS are shown for both Se(IV) and Se(VI) catalyzed processes. The two downhill cycles bear close similarity until Pox, differing only in the substrate oxidations. Notably, the TDI and TDTS positions do not change in the other two cycles. For the Se(VI) catalyzed processes, the TDI/TDTS are always identified within the H2O2 activation step corresponding respectively to the selenonic acid and to the Se(VI) peroxyseleurane dehydration (A and TS2 in Fig. 3). For all three Se(VI) catalyzed oxidations, this step has the overall largest transition state – minimum energy difference, i.e. the energetic span of the cycle.
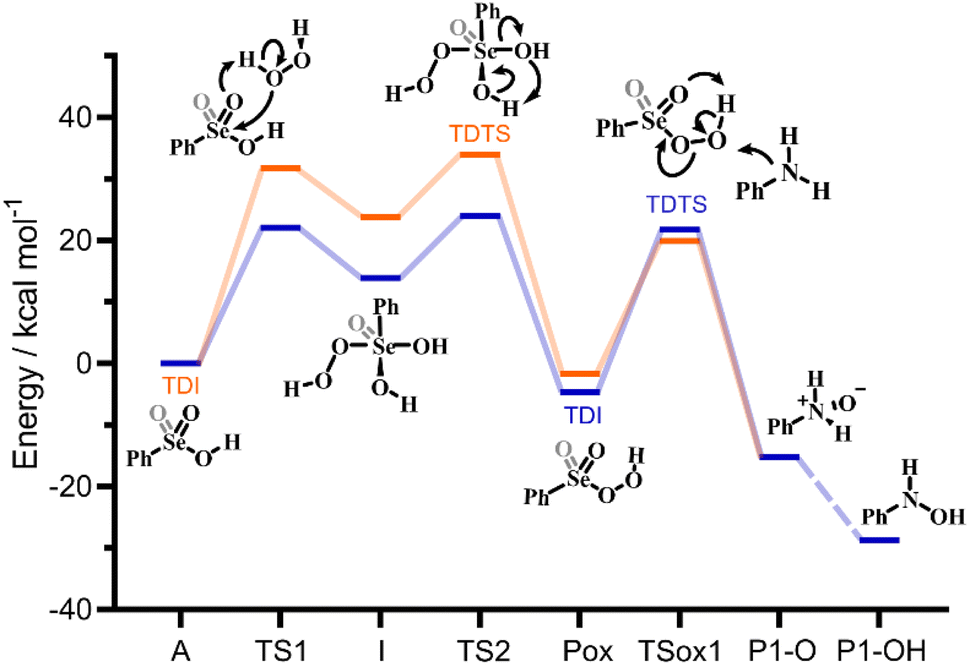 | ||
| Fig. 3 Gibbs free energy profile, intermediates, transition states, and calculated TDI and TDTS for the 1st catalytic cycle: Se(IV) mediated process (blue); Se(VI) mediated process (orange). Grey portions on the Lewis structures represent the additional Se–O bond which is present only in Se(VI) structures. Level of theory: COSMO-M06//OPBE. | ||
Conversely, for the Se(IV) catalyzed processes, the TDI/TDTS are always identified within the substrate oxidation step, corresponding to the peroxyacid and to the SN2 oxygen-transfer transition state (Pox and TSox in Fig. 3), respectively. In the two downhill cycles, as previously outlined, this couple remains the TDI/TDTS, but since the three oxidations appear to have progressively increasing activation energies (Fig. 2b) the energetic span of Se(IV) cycles increases along the overall mechanism, with the first cycle having the lowest and the last having the largest span, always corresponding to the substrate oxidation activation energy.
To verify how these mechanistic differences affect the overall process, the six TOFs for the three Se(IV)/Se(VI) catalyzed oxidations were computed, and their ratio was evaluated (Table 2) to quantify the different performance of Se(VI) and Se(IV) catalysis.
|
|
|
|---|---|
| 1st cycle | 3·10−6 |
| 2nd cycle | 3·10−4 |
| 3rd cycle | 1·10−1 |
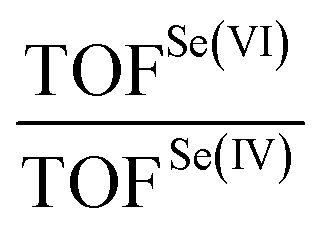
From these results, it emerges that the differences between Se(VI) and Se(IV) as catalysts go attenuating along the overall mechanism, with the greatest differences in the first cycle (highest TOF ratio) and the smallest differences in the last cycle (lowest TOF ratio). This result comes from the different nature of the TDI/TDTS couples when Se(VI) selenonic and Se(IV) seleninic acids are the catalysts, as above described. Indeed, since for Se(VI) the TDI and the TDTS remain the same in the three cycles, and correspond to the same energetic span, all three Se(VI) catalyzed processes have the same TOF. Conversely, since the activation energy of the three substrate oxidations increases along the mechanism, and this activation energy matches to the energetic span of Se(IV) catalyzed processes, their TOF becomes lower along the overall conversion from aniline to nitrobenzene, thus narrowing down the TOF ratio from ca. 10−6 to ca. 10−1. Most importantly, the data reported in Table 2 clearly reveal that Se(IV) is always a better catalyst for aniline oxidation than the corresponding Se(VI) species, since in all cases the TOF ratio is smaller than 1 by at least one order of magnitude. Similar conclusions hold true at different levels of theory (Fig. S2, Tables S5, S6, and relative discussion†). These results further corroborate the privileged role of seleninic acid 1 in the organoselenium catalyzed oxidation of aniline to nitrobenzene, that is, peroxyseleninic acid 3 appears to be a much better oxygen-transfer catalyst than peroxyselenonic acid 5, even if the latter is in principle a better oxidizing agent (Fig. 2b). The central role of Se(IV) catalytic species in the oxidation of plausible reaction intermediates is further highlighted by the control experiments reported in Schemes 7 and 8, as previously highlighted.
Insight into Se(IV) to Se(VI) interconversion
While the data above discussed highlight the privileged role of the Se(IV) oxidation state in the reaction under investigation, one could argue that Se(IV) seleninic acid cannot catalyze the three reactions simply because it is rapidly oxidized to Se(VI) selenonic acid under the conditions in which the reaction takes place.According to Table 2, this conversion would result in a catalyst inactivation. However, we followed this hypothesis, since Back and coworkers reported the fast formation of Se(VI) species when diphenyl diselenide is treated with H2O2,6 and Tanini and coworkers observed the formation of Se(VI) selenonic acid in the water recovered after the complete oxidation of aniline to nitrobenzene.11
Guided by the inspiring work of Back et al., we first probed the mechanistic hypothesis that the peroxy selenurane (I) on the Se(IV) PES acts as the key intermediate in the interconversion. Indeed, it was previously postulated that while a non-redox dehydration would lead to peroxyacid, as previously described, an alternative redox dehydration should lead to selenonic acid (Scheme 9a). Unfortunately, while such interconversion is strongly favored thermodynamically, it proved to be quite troublesome kinetically (Fig. 4).
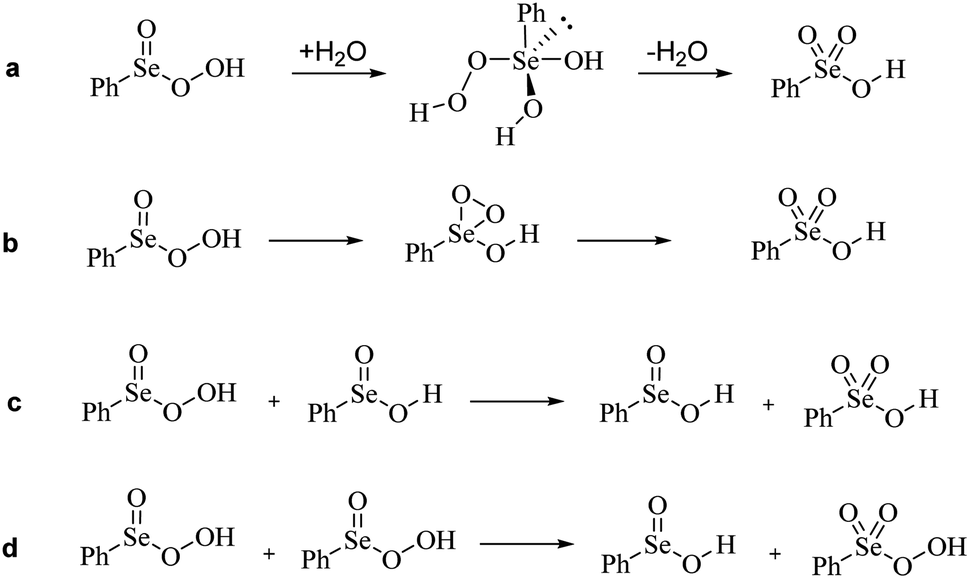 | ||
| Scheme 9 Plausible pathways for Se(IV) to Se(VI) interconversion. (a) Redox dehydration of peroxy selenurane. (b) Syper hypothesis, through a peroxo transition state/intermediate. (c) Direct oxidation of seleninic acid by peroxyseleninic acid. (d) Dismutation of peroxyseleninic acid to seleninic and peroxyselenonic acid. | ||
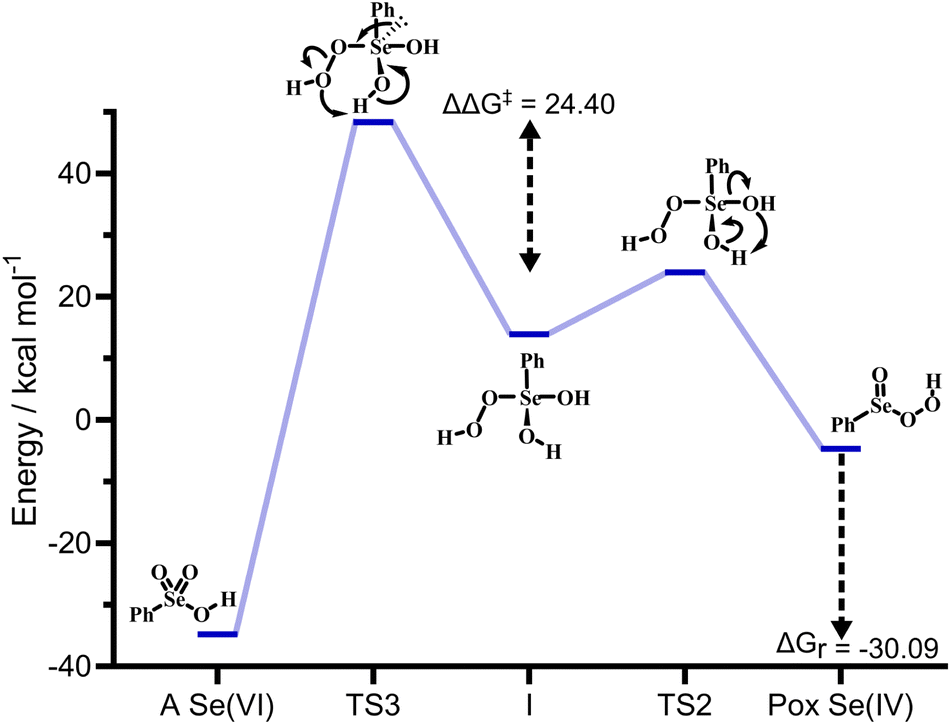 | ||
| Fig. 4 Gibbs free energy profile (kcal mol−1) and relative structures for the interconversion of peroxyseleninic acid (Pox) to selenonic acid (A). All energies are relative to seleninic acid and H2O2. Level of theory: COSMO-M06//OPBE. | ||
Particularly, with respect to peroxyseleninic acid (Pox in Fig. 4), the formation of selenonic acid (A in Fig. 4) is exergonic by ca. 30 kcal mol−1. As previously mentioned, the peroxy selenurane (I) is destabilized by ca. 13 kcal mol−1 with respect to seleninic acid and its dehydration to peroxyseleninic acids occurs thorough a low-lying transition state (TS2). Conversely, its dehydration to selenonic acid goes through a transition state TS3 located way higher on the PES, such that the process is kinetically disfavored by ca. 24.40 kcal mol−1. Overall, with respect to the Se(IV) peroxyseleninic acid Pox, TS3 is located 53.1 kcal mol−1 higher in energy. Thus, while the peroxyselenurane is a key intermediate in H2O2 activation, it does not seem to be involved in the interconversion between Se(IV) and Se(VI). Intrigued by this result, we explored a couple of alternative plausible mechanisms. To the best of our knowledge, the question of the formation of selenonic acid was first tentatively addressed by Syper and coworkers in 1987,26 who invoked a peroxo transition state/intermediate already envisioned by Sharpless and Hori in 1978 (Scheme 9b).4
A transition state for the formation of such peroxo intermediate was found to have an activation energy of 52.5 kcal mol−1 with respect to peroxyseleninic acid, not much lower than the redox-dehydration, which has an overall activation energy of 53.1 kcal mol−1 with respect to peroxyseleninic acid. Thus, these two processes were deemed equally unlikely to occur. Further evolution of the peroxo intermediate to selenonic acid was not investigated, because the barrier for its formation was prohibitively high.
Since the active role of peroxyseleninic acid in the autocatalytic oxidation of organoselenides was observed in the past,15 we checked whether a similar “autocatalytic” oxidation might operate also in this case (Scheme 9c). Such process, in which one equivalent of peroxyseleninic acid 3 oxidizes some residual seleninic acid 1 to selenonic acid 4, while being reduced back to seleninic acid, occurs with an activation energy of 37.5 kcal mol−1 at our level of theory, way more favorable than the two previous proposed mechanisms, but still higher than the H2O2 activation process of seleninic acid, which has an overall barrier of 23.98 kcal mol−1 (Fig. 2b). Thus, such a process does not appear to be autocatalytic in nature. Most importantly, since all mechanistic proposals in Scheme 9a–c have an activation energy higher than each of the three substrate oxidations by peroxyseleninic acid (Fig. 2b), we conclude that no conversion to selenonic acid should occur, as long as there is some substrate left to undergo catalytic oxidation. This is consistent with the hypothesis by Tanini et al. that selenonic acid forms by overoxidation of the catalyst after all aniline and derivatives have been fully oxidized to the final product.11
Lastly, we checked whether peroxyseleninic acid 3 might undergo a self-oxidation reaction directly to peroxyselenonic acid 5, thus bypassing the high activation energy required by selenonic acid in H2O2 activation (Scheme 9d). Selenonic acid would be finally produced from the oxidation of the organic substrate, which is a favored process (Fig. 2b). However, also this self-oxidation occurs with a rather high activation energy of 39.8 kcal mol−1 at our level of theory, higher than that of the previously described oxidation to selenonic acid. Thus, also this process is not expected to take place in the presence of aniline and its intermediates towards nitrobenzene.
Activation strain and energy decomposition analysis of substrate oxidation
While we previously discussed the privileged nature of Se(IV) as both an hydrogen peroxide activator and an oxygen-transfer catalyst, our calculations also highlight its worst performance in the effective organic substrate oxidation. To fully untangle the role of the oxidation state in organoselenium catalyzed oxygen-transfers, a clear molecular picture of the peroxyseleninic and peroxyselenonic acid performances in the actual oxygen-transfer step is desirable. To provide a quantitative discussion on the effect of the oxidation state on the SN2-like oxidation of aniline, ASA and EDA were performed (see Computational methods). Particularly, the oxidation of aniline to aniline-N-oxide by peroxyseleninic and peroxyselenonic acid was compared to provide a detailed insight into the different oxidation state effect in the 1st catalytic cycle. The oxidative potential of the two peroxyacids was analyzed (Fig. 5a–c).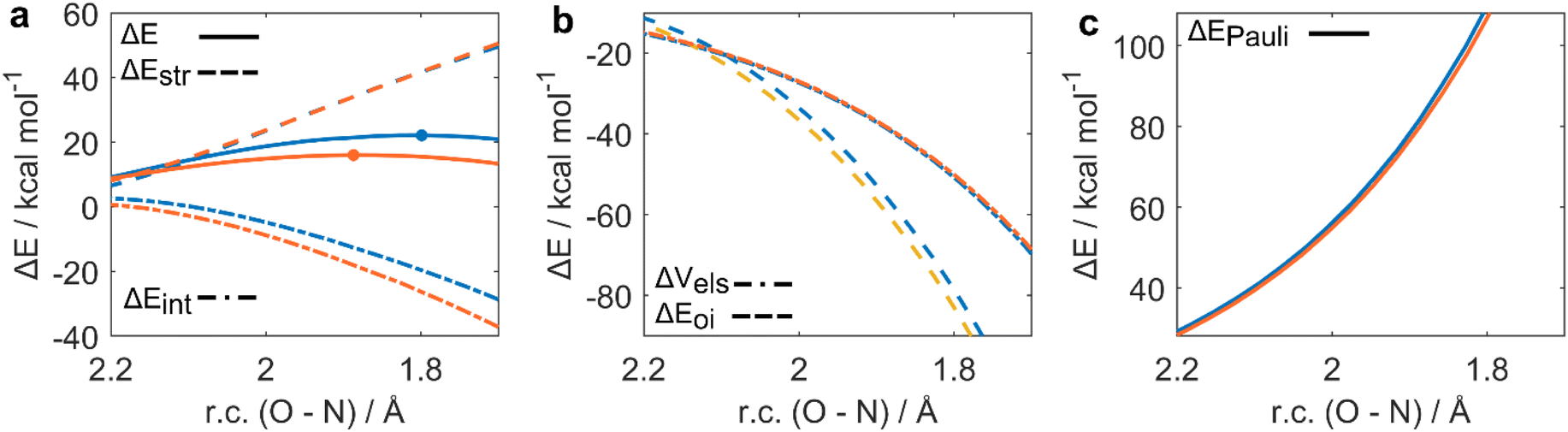 | ||
| Fig. 5 Activation strain (a) and energy decomposition analysis (b and c) of aniline oxidation to aniline N-oxide (P1-O) by peroxyseleninic (blue) and peroxyselenonic (orange) acids. Level of theory: M06//OPBE. | ||
In the actual oxygen-transfer step, peroxyselenonic acid acts as the best oxidant. To analyze this step, ASA was plotted along the O–N distance, which undergoes a well-defined change along the reaction. Analogous conclusions can be drawn by investigating the complementary reaction coordinate i.e., O–O bond breaking (Fig. S1†). ASA (Fig. 5a) reveals how for the two reactions, similar values of ΔEstr occur at consistent points of r.c. Conversely, the ΔEint value alone accounts for the trend in activation energy, being systematically more stabilizing for Se(VI) than for Se(IV), and thus correlating with the lower ΔE of the former. To understand what factors are responsible for this effect, ΔEint was partitioned according to EDA (Fig. 5b and c). The main factor responsible for the lower ΔEint of Se(VI) is ΔEOI, which is consistently more stabilizing for peroxyselenonic acid.
An EDA-NOCV calculation was performed to characterize the orbital interaction between the two reactants. Within the EDA-NOCV approach, ΔEOI can be further decomposed into contributions associated with NOCV. For each NOCV pair, a deformation density can be visualized, associated with a certain charge transfer degree from one fragment to the other (see Computational methods).
The deformation density associated with the NOCV with the greater eigenvalue (i.e., to the greatest charge transfer between the two fragments), is represented in Fig. 6a. It appears that for both Se(IV) and Se(VI) the strongest orbital interaction leads to a charge depletion from the nitrogen lone pair, and to charge accumulation on the oxygen atom of the peroxy bond directly bonded to Se, and in the O–N internuclear region, due to the new bond formation. This representation is consistent with the SN2-like nature of the reaction, in which aniline acts as the nucleophile and the peroxyacid as the electrophile. Additionally, the absolute value of the eigenvalue associated with the NOCV of the Se(VI) reaction is greater than that for Se(IV), being equal to 0.83 and 0.80, respectively, thus unveiling a higher charge-transfer character for the former. Since the nucleophile is the same, this can be associated with the better electron-accepting properties of the peroxyacid in the highest OS. In fact, the couple of canonical Kohn–Sham molecular orbitals (KS-MO) mainly associated with this charge-transfer interaction can be identified with the HOMO–LUMO couple, in which the HOMO of the nucleophile (mostly the nitrogen lone pair) interacts with the LUMO of the electrophile (mostly localized on the peroxide bond). The peroxyacid of Se(VI) has a lower LUMO that better matches to the energy of aniline's HOMO (Fig. 6b and c). Overall, this is the main factor responsible for the stronger Se(VI) orbital interaction, rationalizing the better performance of peroxyselenonic acid in the substrate oxidation step, if it is indeed produced in the reaction mixture.
 | ||
| Fig. 6 Analysis at consistent geometries, r.c. (O–N) = 1.92 A. (a) Deformation density associated with the NOCVs with the largest eigenvalue (νk). Red areas are associated with charge depletion, while blue lobes are associated with charge accumulation. (b) Schematic representation of the HOMO (aniline)–LUMO(peroxyacid) orbital interaction and KS-MO for Se(VI), isodensity value: (0.03). c. ΔEOI, HOMO(aniline)–LUMO(peroxyacid) energy gap (ΔεHOMO–LUMO) and HOMO(aniline)–LUMO(peroxyacid) orbital overlap (〈HOMO|LUMO〉). Level of theory: M06//OPBE. | ||
Conclusions
In this work, an extensive theoretical mechanistic investigation has been carried out on the on-water organoselenium catalyzed oxidation of aniline to nitrobenzene using H2O2 as the final oxidant, with particular attention to the H2O2 activation step. A careful analysis of the PES, carried out in the framework of the energetic span model, showed how the Se(VI) selenonic acid catalyst has a poorer catalytic performance (evaluated on the basis of calculated TOFs) in all three catalytic cycles progressively oxidizing aniline to nitrobenzene, highlighting the privileged role of Se(IV) seleninic acid species in the title catalytic oxygen-transfer reaction.Additionally, our analysis pinpointed how peroxyselenonic acid 5 is indeed a stronger oxidant (at least from the kinetic point of view) than peroxyseleninic acid 3, since all three substrate oxidations occur with a lower activation energy. Thus, if peroxyselenonic acid 5 is indeed produced, a faster reactivity is envisioned. Conversely, selenonic acid 4 performs worse than seleninic acid 1 in H2O2 activation, thus rationalizing its poor catalytic performance. Through ASA, we interpreted the sluggish H2O2 activation by selenonic acid as an effect of the higher distortion associated with this step when compared to the analogous one for seleninic acid. Conversely, the enhanced electrophilicity of peroxyselenonic acid was associated with its reinforced orbital interaction, mainly granted by its low-lying LUMO. Nevertheless, it is important to stress once again that these two opposite effects (i.e., Se(IV) has a lower ΔEstr in the H2O2 activation step, and Se(VI) has a stronger ΔEOI in the substrate oxidation step) lead to a TOF which remains consistently in favor of the Se(IV) oxidation state. Thus, in the context of a catalytic mechanism, the less favorable orbital matching of peroxyseleninic acid with the organic substrate is favorably counterbalanced by the lower distortion associated with its H2O2 activation step, which makes the Se(IV) reaction pathway overall more productive.
Lastly, the conversion of seleninic to selenonic acid under oxidizing conditions was found to proceed with a higher activation energy than the three substrate oxidations by peroxyseleninic acid. Thus, the formation of the Se(VI) species is expected only when the substrate is fully converted to nitrobenzene.
This work provides a detailed mechanistic picture on the catalytic oxygen-transfer behavior of the popular organoselenium catalysts derived from the commercial diphenyl diselenide, shedding light on the physico–chemical role of the oxidation state in their catalytic performance, spanning from the H2O2 activation to their oxidative potential toward the organic substrate. Additionally, to the best of our knowledge, this is the first comprehensive theoretical attempt to rationalize the oxygen-transfer catalytic cycles of seleninic acid and derivatives, providing a theoretical complementary basis to the existing experimental knowledge and tackling the general problem of identifying the active and inactive intermediates in organoselenium catalyzed reactions. Based on these results, we can hypothesize that the way in which the specific organic substrate and the reaction conditions (e.g., choice of the solvent, homogeneous vs. heterogeneous conditions) affects the balance between the hydrogen peroxide activation step and the actual substrate oxidation step might shift the catalysis either toward Se(IV) or Se(VI) based catalytic cycles. Indeed, if Se(VI) can be formed competitively with the substrate oxidation, and the environment somewhat facilitates Se(VI) hydrogen peroxide activation, both oxidation states might contribute to the catalysis. How this balance is modified in other organoselenium catalyzed oxygen transfer is currently under investigation.
Experimental section
Synthetic procedures
Aniline, nitrosobenzene, and benzeneseleninic acid were purchased and used as received, without further purification. Benzeneselenonic acid was prepared according to the literature.6,11,26N-Phenylhydroxylamine was synthesized upon reduction of nitrobenzene.27 Experimental details are reported in the ESI.†Computational details
DFT calculations were carried out with the ADF2019.307 software.28,29 All geometries were optimized employing OPBE density functional30 combined with a small-core TZ2P basis set. Scalar relativistic corrections have been included in all calculations at the zeroth-order regular-approximation (ZORA) as implemented in ADF.31 Thus, all optimizations are at the ZORA-OPBE/TZ2P level of theory. This level of theory was found to properly reproduce geometrical features of organochalcogenides32 and is recognized as one of the best GGAs to tackle SN2 reactions,30,33 such as the oxidations studied in this work. Vibrational analysis was performed to confirm the nature of each optimized geometry: minima displayed all positive frequencies, while transition states displayed one imaginary frequency associated with the motion of the atoms along the reaction. Accurate single points energies were then computed with two different functionals, the hybrid OPBE0 and the meta-hybrid M06,34,35 combined with an all-electron TZ2P basis set. Both functionals were previously benchmarked on organochalcogen reactions in different oxidation states and provided results within a couple of kcal mol−1 when compared against highly correlated reference calculations.36 OPBE, OPBE0 and M06 provide a consistent, qualitatively analogous, description of the PES (Tables S5, S6 and Fig. S2†). Along the manuscript, only M06 energies are discussed. Solvation effects were included employing the conductor-like screening model (COSMO) as implemented in ADF,37,38 using the standard parameters (dielectric constant and effective radius) for water. The radii of the atoms were taken to be the MM3 van der Waals radii divided by 1.2, as per default in ADF. Thermodynamics corrections were evaluated at the ZORA-OPBE/TZ2P level of theory under perfect gas approximation using standard statistical thermodynamic relationships and added to the electronic energy calculated at the COSMO-ZORA-M06/TZ2P-ae level of theory. This level of theory is labelled M06//OPBE.DLPNO-CCSD(T) calculations with the cc-pVTZ-DK basis set and Douglas–Kroll–Hess (DKH) scalar relativistic correction39,40 have been performed on critical steps of the reaction mechanism to corroborate the DFT based discussion.41 The electronic energy was obtained by DLPNO-CCSD(T) single point calculations with TightPNO criteria, using ZORA-OPBE/TZ2P optimized geometries. Orca 4.2 was used for all DLPNO-CCSD(T) calculations.42,43 The thermodynamic correction and solvation energy were estimated at the ZORA-OPBE/TZ2P level of theory, and COSMO-ZORA-OPBE/TZ2P level of theory respectively. We label this level of theory DLPNO-CCSD(T)//OPBE. The results are comparable to those obtained at the DFT level of theory employed in the manuscript and in the ESI.† (Table S7 and S8†). DLPNO-CCSD(T)//OPBE calculations were also used to probe the effect of one water molecule in the reaction mechanism, which was found to be modest (Table S9†).
Activation strain analysis (ASA, eqn (1)) and energy decomposition analysis (EDA, eqn (2)) were applied along a suitable reaction coordinate (r.c.) ζ to gain further insight into the factors at the origin of the investigated reactivity.44–48
| ΔE(ζ) = ΔEstrain(ζ) + ΔEint(ζ) | (1) |
| ΔEint(ζ) = ΔVelstat(ζ) + ΔEPauli(ζ) + ΔEOI(ζ) | (2) |
Along the manuscript, only M06//OPBE results are discussed, since the EDA partitioning scheme here employed is not compatible with DLPNO-CCSD(T) calculations. The absolute value of the TOF at M06//OPBE is reported in the ESI (Table S10†), while in the main text only TOF ratios are discussed.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization: all the authors; investigation: A. M. (theory and computations) and D. T. (experiments); data curation and formal analysis: A. M., methodology: A. M. and D. T.; writing – original draft: A. M. and D. T.; writing – review & editing: all the authors; fundings and supervision: L. O.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Università degli Studi di Padova has supported this research. CNAF (https://www.cnaf.infn.it/) is acknowledged for the generous allocation of computational resources. Part of the calculations were performed on adacloud@CINECA, thanks to the Iscra C DIRAC-1 (PI: Laura Orian). This research is part of the scientific activity of the international multidisciplinary network SeS redox and Catalysis. The financial support provided by the MUR–Dipartimenti di Eccellenza 2023–2027 (DICUS 2.0) to the Department of Chemistry “Ugo Schiff” of the University of Florence is acknowledged.References
- A. J. Pacuła-Miszewska, L. Sancineto, Oxygen-Transfer Reactions Catalyzed by Organoselenium Compounds, Organochalcogen Compounds, Advances in Green and Sustainable Chemistry, 2022, pp. 219–250 Search PubMed.
- J. Piera and J. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- E. T. Poursaitidis, P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem. Sci., 2023, 15, 1177–1203 RSC.
- T. Hori and K. B. Sharpless, J. Org. Chem., 1978, 43, 1689–1697 CrossRef CAS.
- S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881–2884 CrossRef CAS.
- K. N. Sands, E. Mendoza Rengifo, G. N. George, I. J. Pickering, B. S. Gelfand and T. G. Back, Angew. Chem., Int. Ed., 2020, 59, 4283–4287 CrossRef CAS PubMed.
- G. J. Ten Brink, B. C. M. Fernandes, M. C. A. Van Vliet, I. W. C. E. Arends and R. A. Sheldon, J. Chem. Soc., Perkin Trans. 1, 2001, 224–228 RSC.
- H. J. Reich, F. Chow and S. L. Peake, Synthesis, 1978, 1978, 299–301 CrossRef.
- K. H. Tan, W. Xu, S. Stefka, D. E. Demco, T. Kharandiuk, V. Ivasiv, R. Nebesnyi, V. S. Petrovskii, I. I. Potemkin and A. Pich, Angew. Chem., Int. Ed., 2019, 58, 9791–9796 CrossRef CAS PubMed.
- D. Zhao, M. Johansson and J. E. Bäckvall, Eur. J. Org. Chem., 2007, 4431–4436 CrossRef CAS.
- D. Tanini, C. Dalia and A. Capperucci, Green Chem., 2021, 23, 5680–5686 RSC.
- H. J. Reich, I. L. Reich and J. M. Renga, J. Am. Chem. Soc., 1973, 95, 5813–5815 CrossRef CAS.
- P. A. Grieco, Y. Yokoyama, S. Gilman and M. Nishizawa, J. Org. Chem., 1977, 42, 2034–2036 CrossRef CAS.
- D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed.
- G. Ribaudo, M. Bellanda, I. Menegazzo, L. P. Wolters, M. Bortoli, G. Ferrer-Sueta, G. Zagotto and L. Orian, Chem.–Eur. J., 2017, 23, 2405–2422 CrossRef CAS PubMed.
- C. A. Bayse and K. N. Ortwine, Eur. J. Inorg. Chem., 2013, 3680–3688 CrossRef CAS.
- L. Orian and L. Flohé, Antioxidants, 2021, 10, 1–22 CrossRef PubMed.
- K. Ravi, B. D. Bankar, S. Jindani and A. V. Biradar, ACS Omega, 2019, 4, 9453–9457 CrossRef CAS PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- C. Amatore and A. Jutand, J. Organomet. Chem., 1999, 576, 254–278 CrossRef CAS.
- S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355–3365 CrossRef CAS PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- D. Garay-Ruiz and C. Bo, ACS Catal., 2020, 10, 12627–12635 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, ChemPhysChem, 2011, 12, 1413–1418 CrossRef CAS PubMed.
- L. Syper and J. Młochowski, Tetrahedron, 1987, 43, 207–213 CrossRef CAS.
- A. Capperucci, M. Clemente, A. Cenni and D. Tanini, ChemSusChem, 2023, 16, e202300086 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- ADF2019, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com Search PubMed.
- M. Swart, A. W. Ehlers and K. Lammertsma, Mol. Phys., 2004, 102, 2467–2474 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- F. Zaccaria, L. P. Wolters, C. Fonseca Guerra and L. Orian, J. Comput. Chem., 2016, 37, 1672–1680 CrossRef CAS PubMed.
- M. Swart, M. Solà and F. M. Bickelhaupt, J. Comput. Chem., 2007, 28, 1551–1560 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Madabeni, S. Zucchelli, P. A. Nogara, J. B. T. Rocha and L. Orian, J. Org. Chem., 2022, 87, 11766–11775 CrossRef CAS PubMed.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Neese, A. Wolf, T. Fleig, M. Reiher and B. A. Hess, J. Chem. Phys., 2005, 122, 204107 CrossRef PubMed.
- D. A. Pantazis and F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 363–374 CAS.
- D. G. Liakos, Y. Guo and F. Neese, J. Phys. Chem. A, 2020, 124, 90–100 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–6 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- L. P. Wolters and F. M. Bickelhaupt, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 324–343 CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- F. M. Bickelhaupt and E. J. Baerends, Rev. Comput. Chem., 2007, 1–86 Search PubMed.
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed.
- I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC.
- L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, Nat. Rev. Chem, 2019, 3, 48–63 CrossRef CAS.
- A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS PubMed.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details, energetics and TOF calculations carried out with OPBE and OPBE0 density functionals, additional activation and reaction energies, Cartesian coordinates and electronic energies of all structures discussed in the article and DLPNO-CCSD(T) calculations. Synthetic procedures and experimental details on control experiments are also reported. See DOI: https://doi.org/10.1039/d4sc03329a |
| This journal is © The Royal Society of Chemistry 2024 |