
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing local K+ adsorption by high-density cube corners for efficient electroreduction of CO2 to C2+ products†
Hu
Zang
a,
Changjiang
Liu
a,
Qinyuan
Ji
a,
Jiahao
Wang
a,
Haiyan
Lu
a,
Nan
Yu
a and
Baoyou
Geng
 *ab
*ab
aCollege of Chemistry and Materials Science, The Key Laboratory of Functional Molecular Solids, Ministry of Education, The Key Laboratory of Electrochemical Clean Energy of Anhui Higher Education Institutes, Anhui Provincial Engineering Laboratory for New-Energy Vehicle Battery Energy-Storage Materials, Anhui Normal University, Jiuhua Road 189, Wuhu, 241002, China. E-mail: bygeng@mail.ahnu.edu.cn
bInstitute of Energy, Hefei Comprehensive National Science Center, Hefei, 230031, China
First published on 10th June 2024
Abstract
Reducing carbon dioxide (CO2) to high value-added chemicals using renewable electricity is a promising approach to reducing CO2 levels in the air and mitigating the greenhouse effect, which depends on high-efficiency electrocatalysts. Copper-based catalysts can be used for electroreduction of CO2 to produce C2+ products with high added value, but suffer from poor stability and low selectivity. Herein, we propose a strategy to enhance the field effect by varying the cubic corner density on the surface of Cu2O microspheres for improving the electrocatalytic performance of CO2 reduction to C2+ products. Finite element method (FEM) simulation results show that the high density of cubic corners helps to enhance the local electric field, which increases the K+ concentration on the catalyst surface. The results of CO2 electroreduction tests show that the FEC2+ of the Cu2O catalyst with high-density cubic corners is 71% at a partial current density of 497 mA cm−2. Density functional theory (DFT) calculations reveal that Cu2O (111) and Cu2O (110) can effectively reduce the energy barrier of C–C coupling and improve the FEC2+ at high K+ concentrations relative to Cu2O (100). This study provides a new perspective for the design and development of efficient CO2RR catalysts.
Introduction
The massive extraction and utilization of fossil fuels has brought about a dramatic increase in the amount of CO2 in the atmosphere, leading to global warming and climate change, which poses serious environmental problems.1 The efficient capture and recycling of CO2 is widely recognized as the key to mitigating environmental problems related to the greenhouse effect and sea level rise, and promoting sustainable development.2–4 In recent years, it has been found that the electrochemical conversion of CO2 into C2+ products with high application value, using renewable electricity, is one of the most promising technologies for realizing the carbon cycle.5–12 Currently, Cu-based catalysts are attracting attention as the only catalysts that can realize C–C coupling and electroreduction of CO2 to C2+ products.13–21Nevertheless, the thermodynamics and kinetics of C–C coupling are relatively slow in the CO2RR.8,22–26 Therefore, modification of Cu-based catalysts, such as crystal surface modulation,27–29 surface restructuring,30–33 and oxidation state optimization,16,34–36 to improve the efficiency of C2+ products remains a hot topic and challenge. The adjustment of the crystal surface or surface structure has a significant effect on improving the performance of Cu-based catalysts.37 On the one hand, the exposed faces of Cu-based catalysts have a great influence on the catalytic activity and selectivity in the CO2RR process.38,39 On the other hand, the field effect induced by the tip structure of Cu-based catalysts plays an important role in the CO2RR process.40–42 Numerous studies have found that the active sites of the CO2RR are usually located on the top corners and ridges of the catalysts.43–45 Previous reports have focused on the effects of vertex curvature or array orientation on the field effect; however, the vertices and ridges on the exposed surface are particularly critical to catalytic activity and selectivity, while the effects of vertex density or tip-exposed crystal faces on the catalyst surface for the catalytic performance have been rarely reported, and also their effects on the CO2RR are unknown.
Herein, we investigated the effect of the density of cubic corners on the field effect with FEM simulations, and the effect of the vertices and ridges on the cubic corners on the catalytic performance was studied by DFT calculations. The FEM simulation results found that the higher density of the cube corners had a stronger influence on the field effect, which is favorable for the aggregation and adsorption of K+. In the experiment, we prepare a Cu2O microsphere with cubic corners on the surface,46 which is the same as the results of FEM simulation. DFT calculations demonstrate that, at stronger electric fields and higher K+ concentrations, the Cu2O (111) and (110) faces on the cube corner contribute to the adsorption and C–C coupling of *CO intermediates, as well as to the desorption of C2+ products. This work fills the gap in the application of the localized electric field induced by different density nano-arrays in CO2 electroreduction, and also illustrates the roles of vertices and ridges on the exposed crystal in CO2 electroreduction, which provides new perspectives for the design of efficient CO2RR catalysts and contributes to the mitigation of environmental problems caused by greenhouse gases.
Results and discussion
FEM simulation
We used COMSOL Multiphysics field simulations to investigate the effect of the density of cubic corners on the local electric field and K+ concentration. Five models with different cube corner densities (Fig. S1†) were developed using FEM, and the localized electric field and K+ concentration on their cube corners were also investigated.As shown in Fig. 1a–c and S2–S4,† significant local electric field aggregation effects can be observed for a single cube corner. According to the principle of electric field superposition, as the number of cube corners increases, the electric field strength of the cube angle shows an increasing trend. M3 is a microsphere composed of cubic inflections (Fig. 1c), which has the highest density of cubic corners and the maximum localized electric field strength of 565 V m−1. We simulated the K+ concentration on the cubic corner surface using the transport of diluted species (tds) physics modules in COMSOL Multiphysics. From Fig. 1a and b and S5–S7,† it is found that the distribution of K+ concentration is positively correlated with the distribution of local electric field strength. As the local electric field intensity increases, the K+ concentration on the cube corner surface gradually increases, with M3 having the maximum K+ concentration of 3.39 M. The localized electric field facilitates the adsorption and aggregation of K+, which is mainly caused by the field effect induced reagent concentration, because positively charged K+ will be aggregated on the corner surfaces of the cube possessing a strong negative electric field under electrostatic action. The high concentrations of K+ facilitate the adsorption of CO2 and the generation of C2+ products (Fig. 1d).
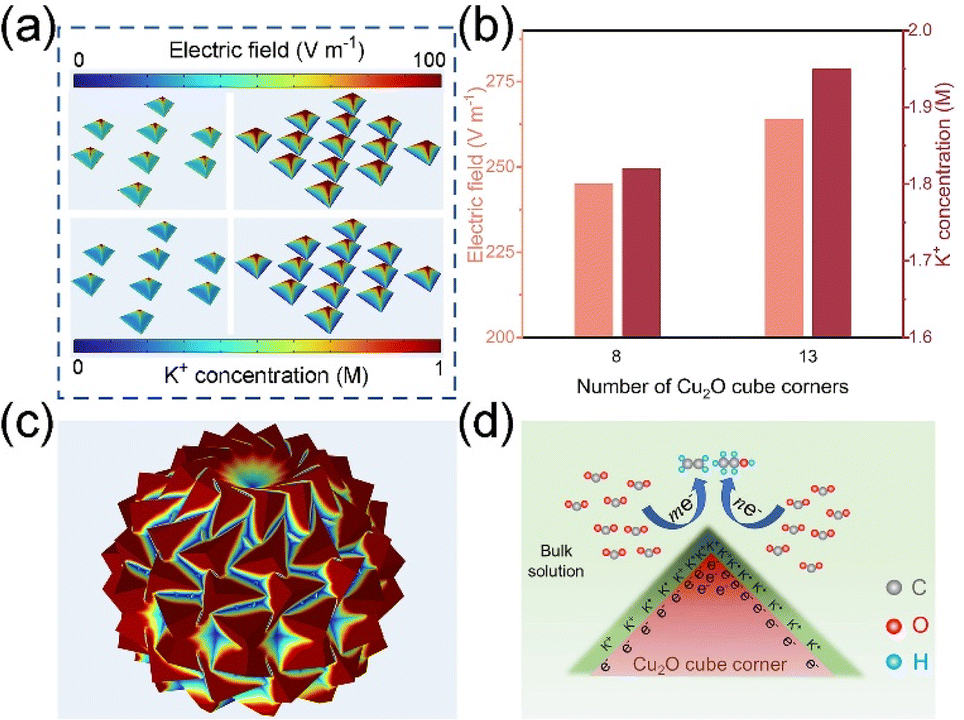 | ||
| Fig. 1 (a) The electric field (upper half) and K+ concentration (lower half) distribution on M-4 and M-5 through COMSOL Multiphysics simulations. (b) The electric field and K+ concentration at the tips of M-4 and M-5. (c) The electric field distribution on M-3. (d) A schematic illustration of the local electric field and K+ ions on the Cu2O cube corner for promoting C2 formation. | ||
Catalyst synthesis and characterization
Cu2O-X microspheres with cube corners were synthesized using a solvothermal method (X is the volume of water used, which is 0, 0.5, 1, 2, 4, and 10 mL respectively, and the corresponding samples are named Cu2O-0, Cu2O-0.5, Cu2O-1, Cu2O-2, Cu2O-4, and Cu2O-10, respectively). The synthesis mechanism and synthesis process of Cu2O-0 are shown in Fig. 2a and S8.† In order to better observe the growth mechanism of the Cu2O-0 catalyst, its liquid color and product morphology were investigated over time during the synthesis process (Fig. S9†). The scanning electron microscopy (SEM) images of Cu2O-0 are shown in Fig. 2b and c, and it can be seen that the surface of Cu2O-0 microspheres has a large number of cube corners. The transmission electron microscopy (TEM) image (Fig. 2d) shows more visually that Cu2O-0 consists of microspheres with numerous vertices and ridges on the surface. The angles of the vertices are mostly 90°, which better indicates that the tips of the surface on the Cu2O-0 microspheres are cubic inflection corners. From the inset in Fig. 2b, the air/water contact angle of Cu2O-0 is 141°, which indicates that it has good hydrophobicity. Fig. S10† shows the air/water contact angle of Cu2O-X. Fig. 2e–g show the high-resolution TEM (HRTEM) images of Cu2O-0. Fig. 2e shows the Cu2O crystal planes at the corners of different cubes (as shown in the inset). HRTEM images (Fig. 2f and g) and FFT patterns (inset) show that the lattice spaces are 0.301, 0.246 and 0.213 nm respectively, which can be well matched with the (110), (111) and (200) crystal planes of cubic Cu2O. The SEM and HRTEM analyses showed that the morphology of Cu2O-X did not change significantly with increasing water content (Fig. S11†). The peaks located at 29.6°, 36.4°, and 42.3° correspond to the (110), (111), and (200) crystal planes of Cu2O (JCPDS: PDF#99-0041), respectively, which indicates that the synthesized material is predominantly Cu2O and has a good crystallinity (Fig. 2h). Fig. S12† shows that the Cu2O-X catalysts with different water contents have the same Cu2O phase. Fig. 2i shows that Cu is predominantly +1-valent, some of which is +2-valent; the Cu2+ in the XPS is most likely caused by the oxidation of the Cu2O when it is exposed in the air. In summary, the synthesized catalysts are consistent with the FEM model.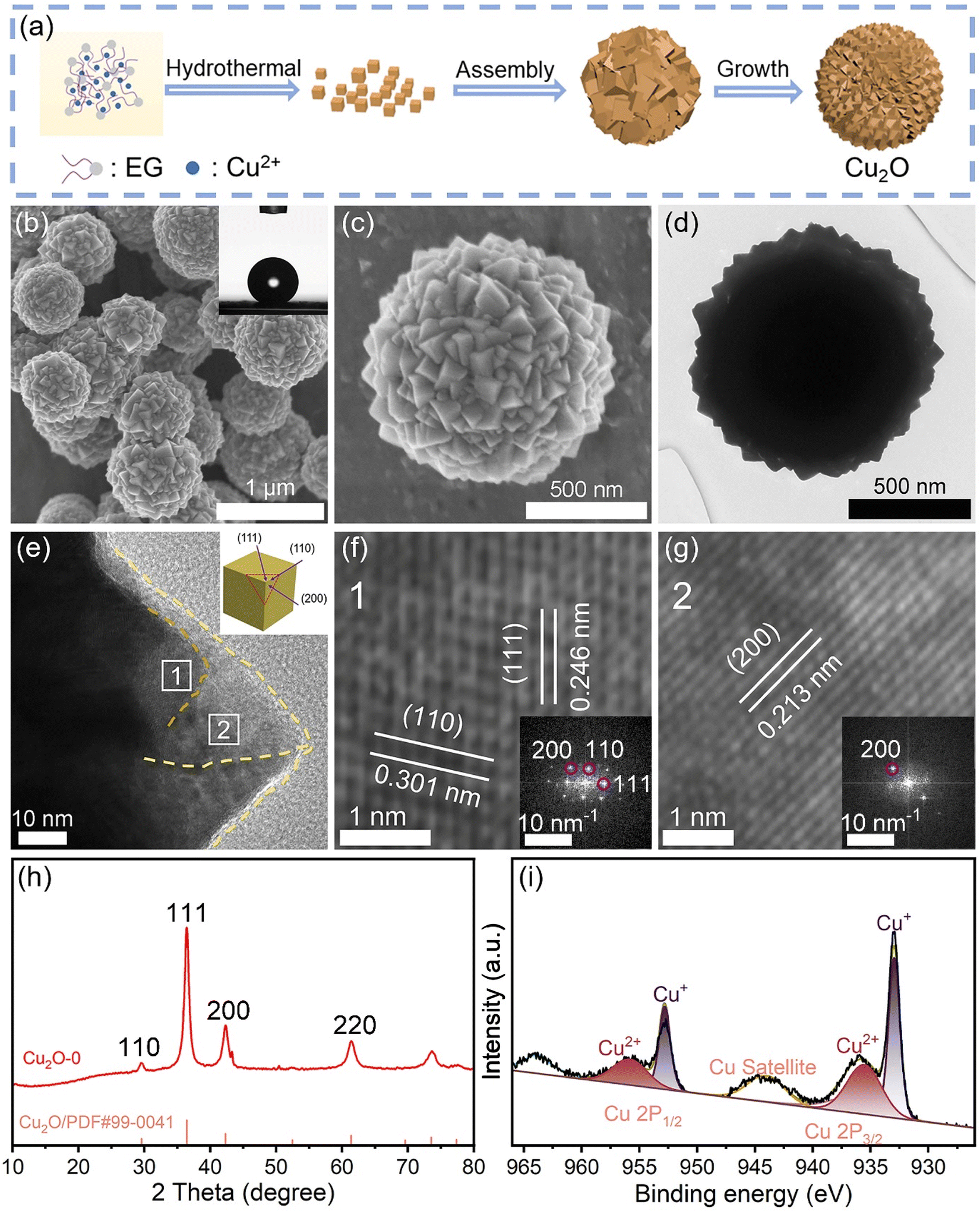 | ||
| Fig. 2 (a) Schematic formation mechanism of the Cu2O-0 catalyst. (b and c) SEM images of the Cu2O-0 catalyst. (d) TEM image of the Cu2O-0 catalyst. (e–g) HRTEM images of the Cu2O-0 catalyst. (h) The XRD pattern of the Cu2O-0 catalyst. (i) The XPS spectrum of the Cu2O-0 catalyst. | ||
Electrochemical CO2RR performance evaluation
Tests of the CO2 electroreduction properties of the catalysts were performed in a flow cell (Fig. S13†). The linear sweep voltammetry (LSV) test results (Fig. 3a and S14†) show that Cu2O-0 has greater positive onset potential and greater negative current density, indicating that Cu2O-0 has higher CO2RR activity. The liquid and gas products were examined by 1H NMR spectroscopy (Fig. S15†) and gas chromatography (GC) (Fig. S16 and S17†), respectively. In terms of product distribution, the reduction products of all Cu2O-X catalysts had the same trend with increasing applied current, with the faradaic efficiency (FE) of C2+ increasing and then decreasing, and the FE of H2 decreasing and then increasing (Fig. 3b and S18†). From Fig. 3c and d, it was found that the Cu2O-0 catalyst possessed the largest FEC2+ and the smallest FEH2. The FEC2+ and FEH2 of the Cu2O-0 catalyst were 71% and 12% at a partial current density of 497 mA cm−2, respectively. Moreover, the FEC2+ of the Cu2O-0 catalyst was 1.8 times higher than the FEC2+ of the Cu2O-10 catalyst.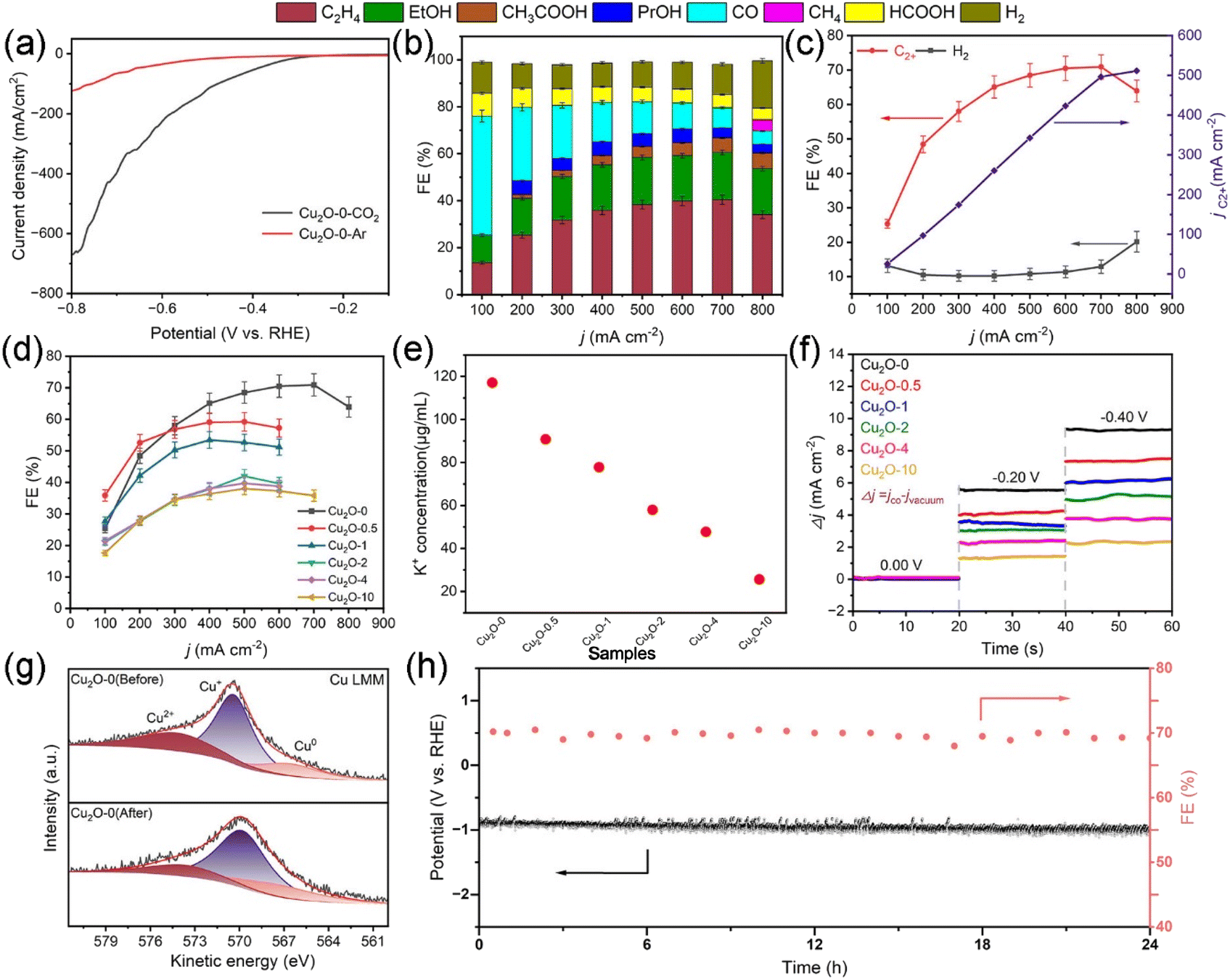 | ||
| Fig. 3 (a) The LSV curves of the Cu2O-0 catalyst under an N2 atmosphere and CO2 atmosphere. (b) FEs of various products on the Cu2O-0 catalyst during the CO2RR at different current densities. (c) C2+ and H2 FEs and current densities vs. applied potential of the Cu2O-0 catalyst. (d) C2+ FEs of Cu2O-X catalysts. (e) Comparison of the adsorbed-K+ concentration on Cu2O-X catalysts. (f) Results of CO adsorption responses on Cu2O-X catalysts under different applied voltages. (g) Deconvoluted Cu Auger LMM spectra of the Cu2O-0 catalyst before and after the CO2RR. (h) CO2RR stability on the Cu2O-0 catalyst at 700 mA cm−2. | ||
We have conducted multiple studies to better understand the differences in catalytic performance of different Cu2O-X catalysts. By recording the cyclic voltammetry curves (Fig. S19†) of Cu2O-X catalysts at different scan rates in the non-Faraday zone, their double-layer capacitance (Cdl) (Fig. S20†) was calculated, which was used to estimate their surface roughness coefficient (Rf) and electrochemically active surface area (ECSA) (Fig. S21 and Table S1†). The results showed that the Cu2O-0 catalyst had the largest Cdl (42.8 mF cm−2), Rf (713.3), and ECSA (713.3 cm2), thus indicating that the Cu2O-0 catalyst can produce more active sites than other Cu2O-X catalysts, which is more favorable for the CO2RR. Meanwhile, Fig. S22† shows that the Cu2O-0 catalyst has the smallest Tafel slope, which also indicates that the Cu2O-0 catalyst has the best CO2RR performance.
According to the FEM simulation results, it is illustrated that the high density of cubic corners contributes to the adsorption of K+ and CO. To validate the FEM simulation results and to demonstrate the advantages of high-density cubic corners, we performed K+ and CO adsorption experiments (schematic shown in Fig. S23 and S24†). Fig. 3e and f and Table S2† show that the adsorption capacity of K+ and CO on the Cu2O-X catalysts increases gradually with the increase of the cube corner density, and the Cu2O-0 catalyst has the largest cube corner density and the strongest K+ and CO adsorption capacity.
As shown in Fig. 3h, the Cu2O-0 catalyst also exhibited excellent long-term stability, operating at a constant current density of 700 mA cm−2 for 24 h without a significant change in the applied voltage or FEC2+, with the FEC2+ remaining around 70%. In order to better understand the long-term stability and selectivity of the Cu2O-0 catalyst, SEM, XRD, and XPS were used to characterize the Cu2O-X catalyst before and after the CO2RR. From Fig. S25,† the morphology of the Cu2O-0 catalyst was almost unchanged before and after the CO2RR. XRD (Fig. S26 and S27†) patterns showed that the physical phase of the Cu2O-0 catalyst was maintained before and after the reaction. The Cu Auger LMM spectra (Fig. 3g and S28†) were recorded and deconvoluted to distinguish Cu+ and Cu0 before and after the CO2RR.47 The ratio of the Cu0 fraction increased from 17.14% to 20.23%, and the ratio of the Cu+ fraction decreased from 68.72% to 65.34% in the Cu2O-0 catalyst. The Cu Auger LMM spectra illustrated that Cu+ in the Cu2O-0 catalyst was well maintained during the CO2RR process. The reason for Cu+ being maintained in the Cu2O-0 catalyst may be due to the high density of cubic corners on the Cu2O-0 catalyst microspheres, which gives the surface a strong localized electric field and high K+ concentration, leading to accelerated electron transport on the cubic corner surface and the inhibition of the redox reaction of Cu+/Cu0.47 These lead to the excellent selectivity and long-term stability of the Cu2O-0 catalyst.
In situ Raman and in situ FT-IR measurements
To further understand the reaction mechanism of the CO2RR, we performed in situ Raman and in situ FT-IR tests on the Cu2O-0 and Cu2O-4 during the CO2RR. The in situ Raman testing equipment is shown in Fig. S29.† As shown in Fig. 4a and S30a,† Cu2O-0 and Cu2O-4 show two peaks around ∼280 and ∼365 cm−1, which are caused by the rotational and stretching vibrations of *CO on the Cu active site after applying the voltage.48–50 In addition, the Cu2O-0 and Cu2O-4 catalysts have three peaks associated with Cuδ+ near ∼415, ∼520, and ∼620 cm−1 in the low-energy region (Fig. 4b and S30b†), corresponding to the modes resulting from a multi-phonon process – the T2g vibrational mode and the T1u mode of Cu2O.47,49,51–55 With the increase of applied potential, the Cu+ characteristic peaks (∼520 cm−1) of Cu2O-0 catalysts are well preserved, and this is consistent with the XRD (Fig. S26†) and XPS (Fig. 3g) results measured before and after the CO2RR, fully indicating that the Cu valence state in the Cu2O-0 catalyst is stable during the CO2RR process. In the high-energy region, the signals near ∼1015 and ∼1066 cm−1 are attributed to the adsorption of HCO3− and CO32− (Fig. 4c and S30c†),50,56 respectively, which are formed on the surface of the catalyst after CO2 is dissolved in the electrolyte. The local hydrogen proton concentration and local microenvironment around the catalysts during the CO2RR were evaluated by monitoring the ratio of HCO3− and CO32− adsorbed on the catalyst surface (Fig. S31†).56,57 The results show that the local hydrogen proton depletion on the surface of the Cu2O-0 catalyst is more stable than that of the Cu2O-4 catalyst, thus maintaining the stability of the local microenvironment and the durability of the C2+ products on the catalyst.50 In addition, the signals generated in the regions around ∼1840 cm−1 and ∼2060 cm−1 belong to the bridge-boundary *CO (*CObridge) and top-boundary *CO (*COatop) conformations,48,58 respectively. As the cathode applied voltage became more negative, the *CObridge signal on the Cu2O-0 catalyst changed weakly (Fig. 4d), and the *CObridge signal on the Cu2O-4 catalyst basically disappeared (Fig. S30d†), suggesting that most of the *CO intermediates were bound at the top and a small portion of them were bound at the bridge sites in the Cu2O-0 catalyst, whereas the *CO intermediates were mainly from the top of the Cu2O-4 catalyst. The mixing of *CObridge and *COatop is more favorable for the C–C coupling.59,60 Therefore, the *CO intermediates on the Cu2O-0 catalyst are more favorable for C–C coupling and the production of C2+ products.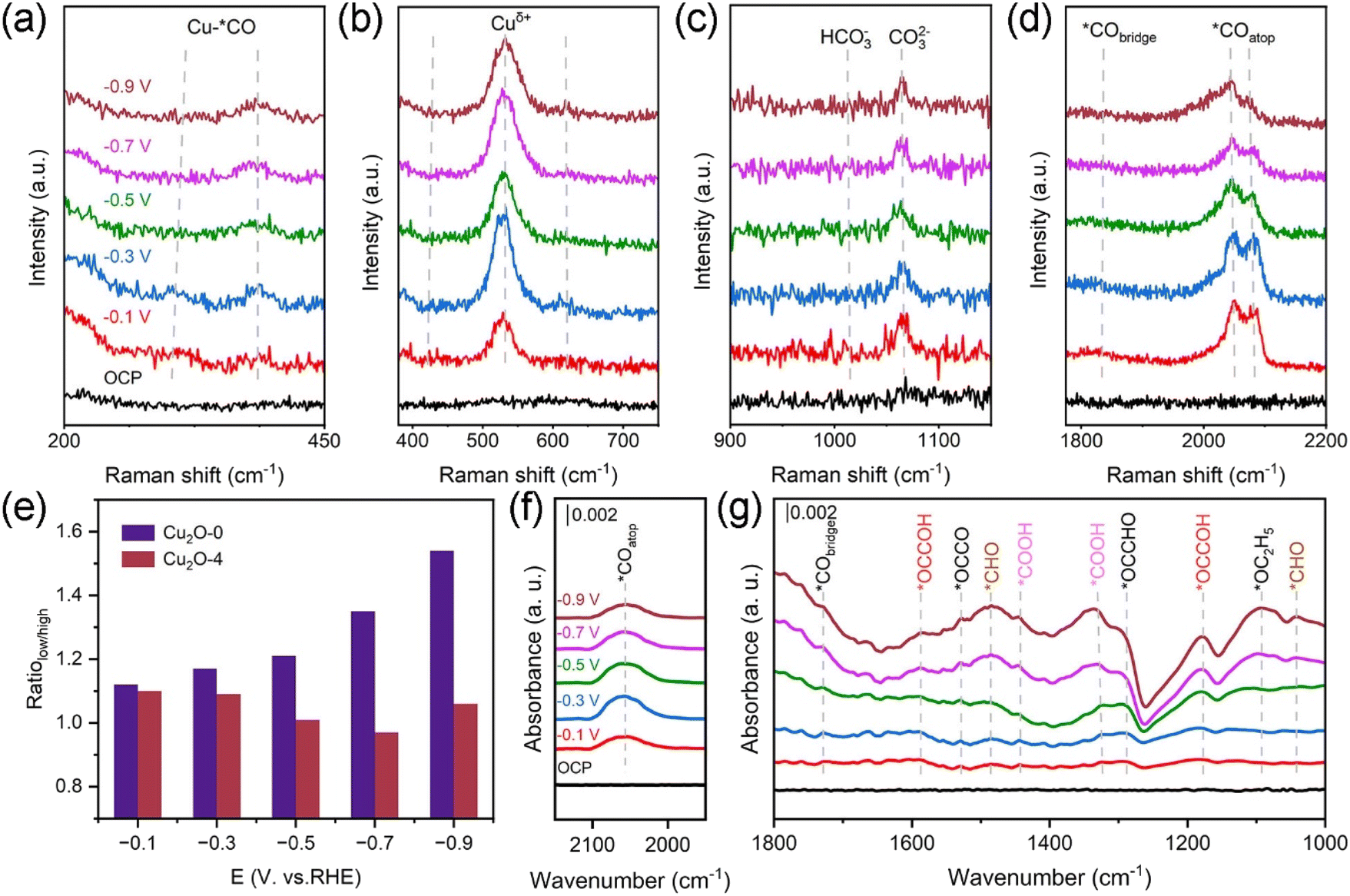 | ||
| Fig. 4 In situ Raman spectra of the Cu2O-0 catalyst in the range 200–450 cm−1 (a), 400–750 cm−1 (b), 900–1150 cm−1 (c) and 1800–2200 cm−1 (d) at various applied potentials (vs. RHE). (e) Ratio of the low-frequency and high-frequency bands in the range 2000–2200 cm−1 of Cu2O-0 and Cu2O-4 catalysts at various applied potentials. (f and g) In situ FT-IR spectra of the Cu2O-0 catalyst at various applied potentials (vs. RHE). | ||
The *COatop intermediates on the catalyst can be divided into low and high frequency bands (LFB and HFB). According to previous studies, the signal in the LFB belongs to a dynamic *CO intermediate, which contributes to subsequent C–C coupling. The HFB stretching band is attributed to the separated static *CO intermediate, which makes it difficult to perform C–C coupling.48,60 As seen in Fig. 4e, we compared the LFB and HFB band intensities. The low/high value of the Cu2O-0 catalyst is larger than that of the Cu2O-4 catalyst, indicating that the Cu2O-0 catalyst is more favorable for C–C coupling and C2+ product generation.50 Through observing the changes in the intensity of the *COatop peaks, it is found that as the external applied voltage becomes more negative, the *CO intermediates first accumulate, then C–C coupling generates C2+ products on the Cu2O-0 catalyst. Due to the involvement of more *CO intermediates in C–C coupling, the *CO peak intensity shows a decreasing trend on the Cu2O-0 catalyst (Fig. 4d),61 while the relative intensity of the HFB (Fig. S30d†) tends to increase at higher potentials on the Cu2O-4 catalyst, indicating an increase in static *CO intermediates and a decrease in C–C coupling ability on the Cu2O-4 catalyst.48 This is consistent with the electrochemical CO2RR results.
In situ FT-IR tests was performed to investigate the reaction pathways of the C2+ products (Fig. S32†). In Fig. 4f and g and S33,† the peaks at ∼1720 and ∼2050 cm−1 are the characteristic peaks of *CObridge and *COatop,62,63 respectively. This is the same species of *CO intermediates observed in in situ Raman spectra. The characteristic peaks at ∼1330 and ∼1440 cm−1 belong to the C–OH and C–O stretching modes of *COOH intermediates, respectively, which help to generate *CO intermediates on the catalysts.64,65 Compared with Cu2O-4, the *COOH peak intensity on Cu2O-0 is significantly stronger, but its *COatop peak intensity is weak, indicating that the *CO intermediates produced by *COOH on Cu2O-0 are more involved in C–C coupling and C2+ products.66 The absorption peaks near ∼1090, ∼1180 and ∼1590 cm−1 are attributed to *OC2H5 and *OCCOH intermediates, respectively, which are key intermediates of C2+ products.56,63,67 The *OCCO intermediates appear at ∼1530 cm−1, indicating that the C–C coupling mode on the catalyst is likely due to dimerization of the *CO intermediate, leading to further hydrogenation into *OCCOH.68–70 As the applied voltage becomes more negative, the accumulation of *CHO (∼1041 and ∼1480 cm−1) and the weakening of *OCCHO (∼1290 cm−1) peak intensity on the Cu2O-0 catalyst indicate that it is difficult to achieve C–C coupling of *CO + *CHO, which also indirectly confirms that the C–C coupling mode is *CO + *CO.62,71
The performance differences between Cu20-0 and Cu20-4 were understood by in-depth analysis of *CO intermediates. Research findings suggest that the intermixing of *CObridge and *COatop was more favorable for the production of the C2+ products, which may be due to the ease of *CObridge in promoting *CO dimerization.72 With the increase of negative voltage, the intensity of *CObridge on Cu2O-0 does not change obviously, but the intensity of the *COatop first enhances and then decreases, indicating that *CO intermediates accumulate first, and then more *CO intermediates participate in the C–C coupling process on Cu2O-0.63 The change trend of *COatop on the Cu2O-0 catalyst in the in situ FT-IR spectrum is consistent with its in situ Raman spectrum (Fig. 4d). However, as the applied voltage increases, the *CObridge peak (∼1720 cm−1) of the Cu2O-4 catalyst decreases and almost disappears. At the same time, the intensity of the *COatop peak (∼2050 cm−1) gradually increases (Fig. S33†), indicating that C–C coupling occurs with difficulty on the Cu2O-4 catalyst.63 The results of in situ FT-IR are consistent with those of in situ Raman and electrochemical CO2RR.
DFT theoretical calculations
DFT theoretical calculations were used to gain a deeper understanding of the mechanism of Cu2O-X catalysts promoting the electro-reduction of CO2 to C2+ products. We performed DFT calculations to investigate the adsorption capacity of the reaction intermediate *CO and the product C2H4 at the Cu2O (100), (111) and (110) interface at different K+ concentrations as well as the Gibbs free energy of the C–C coupling. We developed a DFT model of the same Cu2O crystal surface as the catalyst under study (Fig. S34†). As shown in Fig. 5a and b and S35–S40, and Tables S3 and S4,† *CO and *C2H4 adsorption on the Cu2O (100) crystallite were stronger than that on Cu2O (111) and (110) crystallites. It was very difficult for C2H4 to overflow from the Cu2O (100) crystal surface, so the amount of C2+ product produced by the Cu2O (100) crystal surface was low. Unlike the Cu2O (100) crystal faces, the adsorption of *CO was weaker on the Cu2O (111) and (110) crystal faces, but the introduction of K+ led to the enhancement of *CO adsorption; at the same time, the adsorption of *C2H4 on the Cu2O (111) and (110) crystal faces was weaker, and the C2H4 produced was readily desorbed out from the Cu2O (111) and (110) crystal faces, so the amount of C2+ product produced by the Cu2O (111) and (110) crystal faces was higher.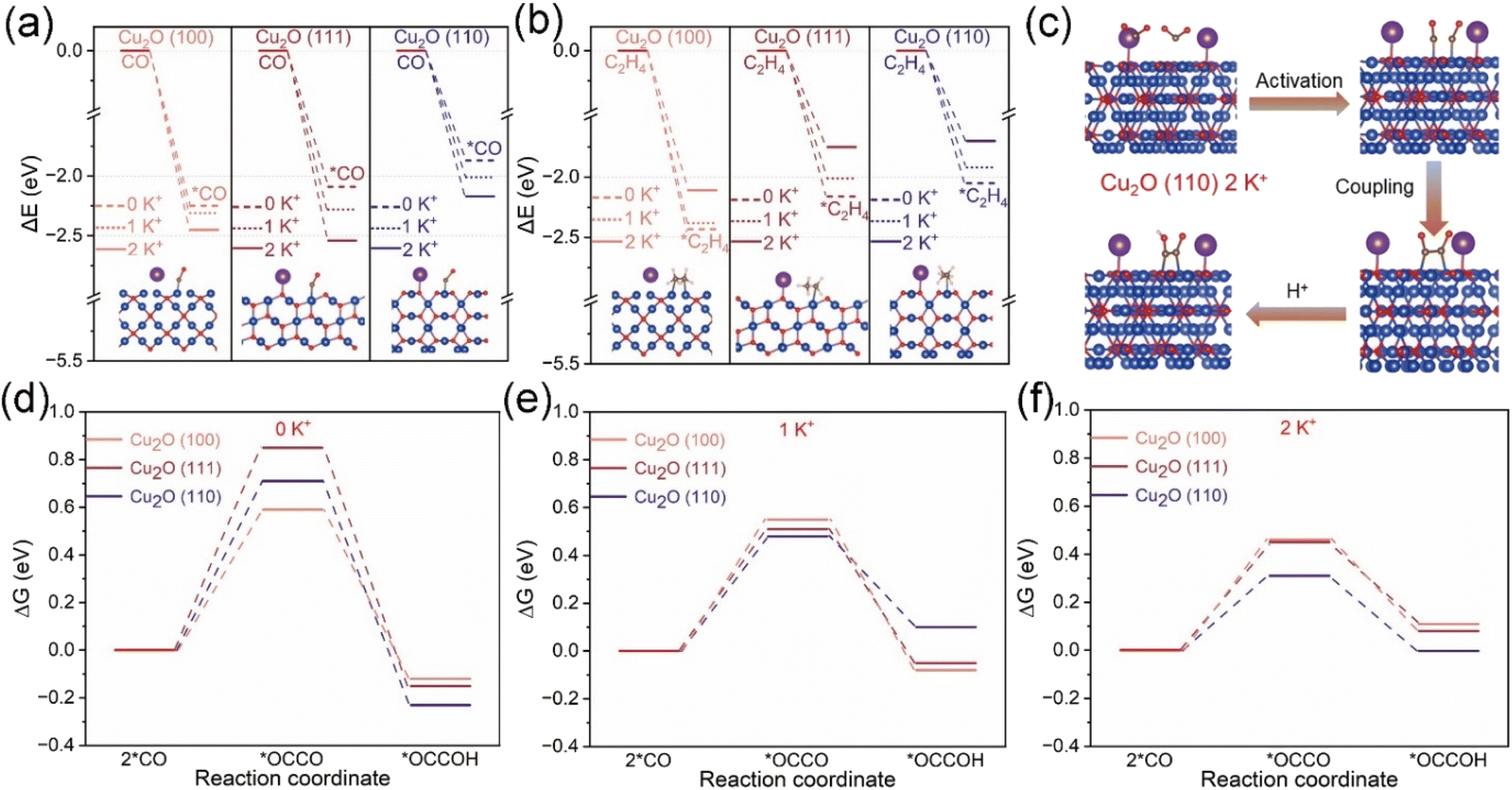 | ||
| Fig. 5 Adsorption energies of (a) CO and (b) C2H4 on three crystalline surfaces of Cu2O (100), (111) and (110) at three K+ concentrations of 0 K+, 1 K+ and 2 K+, respectively. (c) Adsorption configurations of reaction intermediates on the Cu2O (110) structure at the K+ concentrations of 2 K+. Gibbs free energy pathways for C–C coupling on three crystalline surfaces of Cu2O (100), (111) and (110) at three K+ concentrations of 0 K+ (d), 1 K+ (e) and 2 K+ (f), respectively. | ||
From the in situ characterization and DFT calculations (Fig. S41†), the pathway diagram of the Cu2O-X catalyst to generate C2+ products was constructed and is shown in Fig. S42.†61 The critical step to generate C2+ products is the coupling of two *CO intermediates into *OCCO intermediates. Fig. 5c shows the schematic diagram of the CO2RR on the Cu2O (110) crystal surface under 2 K+ concentration. Fig. S43–S45† are schematic diagrams of the modeling of C–C coupling as well as hydrogenation. From Fig. 5d–f and S46–S49, and Table S5,† it is found that the Gibbs free energies of C–C coupling on all three crystalline surfaces of Cu2O (100), (111) and (110) are decreasing with the increase of K+ concentration. The Gibbs free energy of C–C coupling on the Cu2O (110) (0.31 eV) and (111) (0.45 eV) crystal surfaces is smaller than that on the Cu2O (100) (0.46 eV) crystal surface at 2 K+ concentration, which suggests that the Cu2O (110) and (111) crystal surfaces are more favorable for C–C coupling at high K+ concentration. At the same time, experimental support was provided for the DFT calculation results (Fig. S50†). In the experiment, Cu2O catalysts with different crystal planes were synthesized and the electrocatalytic CO2 reduction performance was studied. It was found that the experimental results were consistent with the DFT calculation results.
Conclusions
In summary, we propose a strategy to enhance the field effect and thus improve the catalyst's performance for the electroreduction of CO2 by changing the density of cubic corner arrays on the catalyst surface. CO2RR experimental results indicated that the FEC2+ of the Cu2O catalyst with high-density cubic corners was 71% at a partial current density of 497 mA cm−2, and the FEC2+ of this Cu2O-0 catalyst was 1.8 times higher than the FEC2+ of the microcubic Cu2O catalyst. Moreover, the cubic corner arrays on the surface of Cu2O microspheres help to improve the hydrophobicity of the catalyst and reduce the occurrence of its competing HER. FEM simulations and DFT calculations demonstrated that the high density of cubic corners contributed to the enhancement of the local electric field, which increased the K+ concentration in the local microenvironment and enhanced the adsorption of *CO intermediates on the catalyst surface. The increase in the density of cubic corners leads to a higher number of vertices (Cu2O (111)) and ridges (Cu2O (110)). Relative to Cu2O (100), Cu2O (111) and Cu2O (110) can effectively reduce the energy barrier of C–C coupling and improve the Faraday efficiency of C2+ products at high K+ concentrations. This strategy can not only effectively improve the CO2RR performance of the catalysts, but also indicates that the surface structure of the catalysts plays an important role in their CO2RR performance, which provides a new idea for the design of efficient CO2RR catalysts.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
Hu Zang: conceptualization, data curation, formal analysis, investigation, methodology, software, writing – original draft, and writing – review & editing; Changjiang Liu, Qinyuan Ji, Jiahao Wang, and Haiyan Lu: data curation, formal analysis, investigation, and software; Nan Yu: investigation, methodology, software and validation; Baoyou Geng: conceptualization, formal analysis, investigation, methodology, resources, funding acquisition, supervision, project administration, and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the National Natural Science Foundation of China (22171005), the Anhui Province Outstanding Research and Innovation Team Project for Universities (2023AH010030), and the University Synergy Innovation Program of Anhui Province (GXXT-2020-005, GXXT-2021-012, GXXT-2021-013, and GXXT-2022-007) for supporting this work.References
- P. Falkowski, R. J. Scholes and E. Boyle, Science, 2000, 290, 291–296 CrossRef CAS PubMed.
- B. Obama, Science, 2017, 355, 126–129 CrossRef CAS PubMed.
- A. J. Welch, E. Dunn and J. S. Duchene, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- X. Tan, C. Yu and Y. Ren, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- L. P. De, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef PubMed.
- O. S. Bushuyev, L. P. De, C. T. Dinh, L. Tao, G. Saur, J. Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- D. F. Gao, R. M. Aran-ais and H. S. Jeon, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- A. D. Handoko, F. X. Wei, Jenndy, S. Y. Boon and W. S. Zhi, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- X. Mao, T. W. He, G. Kour, H. Q. Yin, C. Y. Ling, G. P. Gao, Y. G. Jin, Q. J. Liu, A. P. O'Mullane and A. J. Du, Chem. Sci., 2024, 15, 3330–3338 RSC.
- H. R. M. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- L. Zhu, Y. Lin, K. Liu, E. Cortes, H. Li, J. Hu, A. Yamaguchi, X. Liu, M. Miyauchi, J. Fu and M. Liu, Chin. J. Catal., 2021, 42, 1500–1508 CrossRef CAS.
- X. Zhi, Y. Jiao, Y. Zheng, K. Davey and S. Z. Qiao, J. Mater. Chem. A, 2021, 9, 6345–6351 RSC.
- T. Zhang, Z. Li, J. Zhang and J. Wu, J. Catal., 2020, 387, 163–169 CrossRef CAS.
- Y. C. Li, E. P. Delmo, G. Y. Hou, X. L. Cui, M. Zhao, Z. H. Tian, Y. Zhang and M. H. Shao, Angew. Chem., Int. Ed., 2023, e202313522 CAS.
- K. Yao, Y. Xia, J. Li, N. Wang, J. Han, C. Gao, M. Han, G. Shen, Y. Liu, A. Seifitokaldani, X. Sun and H. Liang, J. Mater. Chem. A, 2020, 8, 11117–11123 RSC.
- Y. D. Wang, R. Y. Zhao, Y. P. Liu, F. T. Zhang, Y. P. Wang, Z. H. Wu, B. X. Han and Z. M. Liu, Chem. Sci., 2024, 15, 4140–4145 RSC.
- Z. Gu, H. Shen, Z. Chen, Y. Yang, C. Yang, Y. Ji, Y. Wang, C. Zhu, J. Liu, J. Li, T.-K. Sham, X. Xu and G. Zheng, Joule, 2021, 5, 429–440 CrossRef CAS.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- X. Zhang, K. Liu, J. Fu, H. Li, H. Pan, J. Hu and M. Liu, Front. Phys., 2021, 16, 63500 CrossRef.
- B. Yang, K. Liu, H. Li, C. Liu, J. Fu, H. Li, J. E. Huang, P. Ou, T. Alkayyali and C. Cai, J. Am. Chem. Soc., 2022, 144, 3039–3049 CrossRef CAS PubMed.
- X. Zhang and Z. Zhou, J. Phys. Chem. C, 2022, 126, 3820–3829 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- M. B. Ross, P. De Luna, Y. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- X. Hu, S. Yao, L. T. Chen, X. Zhang, M. G. Jiao, Z. Y. Lu and Z. Zhou, J. Mater. Chem. A, 2021, 9, 23515–23521 RSC.
- P. S. Li, J. H. Bi, J. Y. Liu, Q. G. Zhu, C. J. Chen, X. F. Sun, J. L. Zhang, Z. M. Liu and B. X. Han, Chem. Sci., 2023, 14, 310–316 RSC.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- C. Tang, J. Shi, X. Bai, A. Hu, N. Xuan, Y. Yue, T. Ye, B. Liu, P. Li, P. Zhuang, J. Shen, Y. Liu and Z. Sun, ACS Catal., 2020, 10, 2026–2032 CrossRef CAS.
- Y. Wang, H. Shen, K. J. T. Livi, D. Raciti, H. Zong, J. Gregg, M. Onadeko, Y. Wan, A. Watson and C. Wang, Nano Lett., 2019, 19, 8461–8468 CrossRef CAS PubMed.
- Z. Sun, J. Dong, C. Chen, S. Zhang and Y. Zhu, J. Chem. Technol. Biotechnol., 2021, 96, 1161–1175 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- X. Tan, H. J. Zhu, C. He, Z. W. Zhuang, K. A. Sun, C. Zhang and C. Chen, Chem. Sci., 2024, 15, 4292–4312 RSC.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS.
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H. S. Oh, B. K. Min and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC.
- L. P. De, B. R. Quintero, C. T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorovic, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef.
- Y. Y. Xue, Y. B. Guo, H. J. Cui and Z. Zhou, Small Methods, 2021, 5, 2100736 CrossRef CAS PubMed.
- H. Zhang, C. H. He, S. M. Han, Z. Y. Du, L. Wang, Q. B. Yun, W. B. Cao, B. W. Zhang, Y.-H. Tian and Q. P. Lu, Chin. Chem. Lett., 2022, 33, 3641–3649 CrossRef CAS.
- Y. G. Gao, Q. Wu, X. Z. Liang, Z. Y. Wang, Z. K. Zheng, P. Wang, Y. Y. Liu, Y. Dai, M.-H. Whangbo and B. B. Huang, Adv. Sci., 2020, 7, 1902820 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- Y. J. Zhou, Y. Q. Liang, J. W. Fu, K. Liu, Q. Chen, X. Q. Wang, H. M. Li, L. Zhu, J. H. Hu, H. Pan, M. Miyauchi, L. X. Jiang, E. Cortés and M. Liu, Nano Lett., 2022, 22, 1963–1970 CrossRef CAS PubMed.
- Z. Z. Niu, F. Y. Gao, X. L. Zhang, P. P. Yang, R. Liu, L. P. Chi, Z. Z. Wu, S. Qin, X. X. Yu and M. R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed.
- W. L. Zhu, Y. J. Zhang, H. Y. Zhang, H. F. Lv, Q. Li and S. H. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- W. L. Zhu, R. Michalsky, H. F. Lv, X. L. Sun, A. A. Peterson and S. H. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- S. Back, M. S. Yeom and Y. S. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- D. S. Chen, W. B. Yu, Z. Deng, J. Liu, J. Y. Jin, M. Wu, L. H. Chen and B. L. Su, RSC Adv., 2015, 5, 55520–55526 RSC.
- T. T. Zhao, J. H. Li, J. D. Liu, F. M. Liu, K. Q. Xu, M. Yu, W. Xu and F. Y. Cheng, ACS Catal., 2023, 13, 4444–4453 CrossRef CAS.
- A. Xu, S. F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F. Y. Wu, Z. Y. Lin, H. J. Tsai, T. J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D. H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- S. Jiang, K. Klingan, C. Pasquini and H. Dau, J. Chem. Phys., 2019, 150, 041718 CrossRef PubMed.
- M. W. Fang, M. L. Wang, Z. W. Wang, Z. X. Zhang, H. C. Zhou, L. M. Dai, Y. Zhu and L. Jiang, J. Am. Chem. Soc., 2023, 145, 11323–11332 CrossRef CAS PubMed.
- X. Y. Chen, D. A. Henckel, U. O. Nwabara, Y. Y. Li, A. I. Frenkel, T. T. Fister, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2020, 10, 672–682 CrossRef CAS.
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- G.-Y. Duan, X.-Q. Li, G.-R. Ding, L.-J. Han, B.-H. Xu and S.-J. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202110657 CrossRef CAS PubMed.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- A. Singhal, M. R. Pai, R. Rao, K. T. Pillai, I. Lieberwirth and A. K. Tyagi, Eur. J. Inorg. Chem., 2013, 14, 2640–2651 CrossRef.
- M. Zheng, P. Wang, X. Zhi, K. Yang, Y. Jiao, J. Duan, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 14936–14944 CrossRef CAS PubMed.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, E9261–E9270 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden, S.-F. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. G. de Arquer, R. K. Miao, C. P. O'Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z.-Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C.-S. Tan, D.-H. Nam, R. Quintero-Bermudez, T.-T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- H. Y. An, L. F. Wu, L. D. B. Mandemaker, S. Yang, J. de Ruiter, J. H. J. Wijten, J. C. L. Janssens, T. Hartman and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 16576–16584 CrossRef CAS PubMed.
- Y. W. Jiang, X. Y. Wang, D. L. Duan, C. H. He, J. Ma, W. Q. Zhang, H. J. Liu, R. Long, Z. B. Li, T. T. Kong, X. J. Loh, L. Song, E. Y. Ye and Y. J. Xiong, Adv. Sci., 2022, 9, 2105292 CrossRef CAS PubMed.
- F. Q. Yu, X. Liu, L. L. Liao, G. M. Xia and H. M. Wang, Small, 2023, 2301558 CrossRef CAS PubMed.
- C. Liu, M. M. Wang, J. Y. Ye, L. B. Liu, L. G. Li, Y. H. Li and X. Q. Huang, Nano Lett., 2023, 23, 1474–1480 CrossRef CAS PubMed.
- M. Liu, Q. Y. Wang, T. Luo, M. Herran, X. Y. Cao, W. R. Liao, L. Zhu, H. M. Li, A. Stefancu, Y.-R. Lu, T.-S. Chan, E. Pensa, C. Ma, S. G. Zhang, R. Y. Xiao and E. Cortés, J. Am. Chem. Soc., 2024, 146, 468–475 CrossRef CAS PubMed.
- S. Q. Zhu, T. H. Li, W.-B. Cai and M. H. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- S. H. Min, X. Xu, J. X. He, M. Sun, W. L. Lin and L. T. Kang, Small, 2024, 2400592, DOI:10.1002/smll.20240059.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- Y. Kim, S. Park, S.-J. Shin, W. Choi, B. K. Min, H. Kim, W. Kim and Y. J. Hwang, Energy Environ. Sci., 2020, 13, 4301–4311 RSC.
- H. Luo, B. Li, J. G. Ma and P. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202116736 CrossRef CAS PubMed.
- J. Q. Feng, L. M. Wu, S. J. Liu, L. Xu, X. N. Song, L. B. Zhang, Q. G. Zhu, X. C. Kang, X. F. Sun and B. X. Han, J. Am. Chem. Soc., 2023, 145, 9857–9866 CrossRef CAS PubMed.
- W. C. Ma, S. J. Xie, T. T. Liu, Q. Y. Fan, J. Y. Ye, F. F. Sun, Z. Jiang, Q. H. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- Z. M. Wei, J. Ding, X. X. Duan, G.-L. Chen, F.-Y. Wu, L. Zhang, X. J. Yang, Q. Zhang, Q. Y. He, Z. Y. Chen, J. Huang, S.-F. Hung, X. Yang and Y. M. Zhai, ACS Catal., 2023, 13, 4711–4718 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02170c |
| This journal is © The Royal Society of Chemistry 2024 |