
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotophysics and photochemistry of thermally activated delayed fluorescence emitters based on the multiple resonance effect: transient optical and electron paramagnetic resonance studies†
Xi
Chen‡
 a,
Lei
Sun‡
bc,
Andrey A.
Sukhanov‡
d,
Sandra
Doria‡
ef,
Laura
Bussotti
e,
Jianzhang
Zhao
*a,
Haijun
Xu
*bc,
Bernhard
Dick
*g,
Violeta K.
Voronkova
*d and
Mariangela
Di Donato
*ef
a,
Lei
Sun‡
bc,
Andrey A.
Sukhanov‡
d,
Sandra
Doria‡
ef,
Laura
Bussotti
e,
Jianzhang
Zhao
*a,
Haijun
Xu
*bc,
Bernhard
Dick
*g,
Violeta K.
Voronkova
*d and
Mariangela
Di Donato
*ef
aState Key Laboratory of Fine Chemicals, Frontier Science Center for Smart Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn
bJiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Key Laboratory of Forestry Genetics & Biotechnology of Ministry of Education, Jiangsu Provincial Key Lab for the Chemistry and Utilization of Agro-Forest Biomass, College of Chemical Engineering, Nanjing Forestry University, Nanjing, 210037, P. R. China. E-mail: xuhaijun@njfu.edu.cn
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, 453002, China
dZavoisky Physical-Technical Institute, FRC Kazan Scientific Center of RAS, Kazan 420029, Russia. E-mail: vio@kfti.knc.ru
eLENS (European Laboratory for Non-Linear Spectroscopy), Via N. Carrara 1, 50019 Sesto Fiorentino (FI), Italy. E-mail: didonato@lens.unifi.it
fICCOM-CNR, Via Madonna del Piano 10-12, 50019, Sesto Fiorentino (FI), Italy
gLehrstuhl für Physikalische Chemie, Institut für Physikalische und Theoretische Chemie, Universität Regensburg, Regensburg 93053, Germany. E-mail: Bernhard.Dick@chemie.uni-regensburg.de
First published on 3rd June 2024
Abstract
The photochemistry of two representative thermally activated delayed fluorescence (TADF) emitters based on the multiple resonance effect (MRE) (DABNA-1 and DtBuCzB) was studied. No significant TADF was observed in fluid solution, although the compounds have a long-lived triplet state (ca. 30 μs). We found that these planar boron molecules bind with Lewis bases, e.g., 4-dimethylaminopyridine (DMAP) or an N-heterocyclic carbene (NHC). A new blue-shifted absorption band centered at 368 nm was observed for DtBuCzB upon formation of the adduct; however, the fluorescence of the adduct is the same as that of the free DtBuCzB. We propose that photo-dissociation occurs for the DtBuCzB-DMAP adduct, which is confirmed by femtosecond transient absorption spectra, implying that fluorescence originates from DtBuCzB produced by photo-dissociation; the subsequent in situ re-binding was observed with nanosecdon transient absorption spectroscopy. No photo-dissociation was observed for the NHC adduct. Time-resolved electron paramagnetic resonance (TREPR) spectra show that the triplet states of DABNA-1 and DtBuCzB have similar zero field splitting (ZFS) parameters (D = 1450 MHz). Theoretical studies show that the slow ISC is due to small SOC and weak Herzberg–Teller coupling, although the S1/T1 energy gap is small (0.14 eV), which rationalizes the lack of TADF.
Introduction
Charge separation (CS) occurring in organic electron donor–acceptor (D–A) dyads upon photoexcitation is of particular interest, because of its significance in fundamental photochemistry studies,1,2 as well as in photovoltaics,3–5 photocatalysis,2,6–8etc. CS and charge recombination (CR) are also involved in some novel organic optical materials; one example is electron D–A dyads showing thermally activated delayed fluorescence (TADF), which are promising emitters for organic light emitting diodes (OLEDs).9–22In typical D–A type TADF emitters, discrete donor and acceptor units are connected via a linker, and the π-conjugation planes of the electron donor and acceptor units usually adopt an orthogonal geometry, making the electron exchange energy (J) of the CS states small. As a result, the 1CS/3CS state energy gap (2J) is reduced because of the poor molecular orbital overlap, which may facilitate the reverse intersystem crossing (rISC).11,16,23–25 In this way, the triplet excitons can be harvested for electroluminescence, through repopulation of the emissive 1CS state. The ISC mechanism operating in orthogonal dyads is often referred to as spin–orbit charge transfer (SOCT) ISC, since in these cases the electron spin angular momentum change occurring during ISC is off-set by the molecular orbital angular momentum change of the electron transfer.26–29 Recently, we highlighted the critical role of dark states, such as the 3LE (LE: localized excited state) and the 3CS states, in the TADF process of D–A dyads,29–32 by using femtosecond and nanosecond transient absorption (fs/ns-TA) spectroscopy. Most of our measurements experimentally supported the three-state theoretical model, invoked for D–A TADF emitters, according to which the delayed fluorescence is believed to occur through a spin-vibronic coupling mechanism.29–34
Conventional D–A TADF emitters for OLED applications suffer from some limitations. For instance, the spatial separation between the HOMO and LUMO, usually respectively confined on the donor and acceptor, leads to poor radiative decay from the 1CS state. Furthermore, the CS character of the emissive state leads to a large fluorescence bandwidth, which is detrimental for applications for information display purposes.35 One alternative to the conventional D–A TADF emitters is to obtain systems with an emissive S1 state presenting reduced CS character. Unfortunately, it is challenging to maintain a small electron exchange energy while minimizing the molecular orbital spatial separation, because the J magnitude increases with increasing orbital overlapping. Recently, some novel molecular structure has been developed to address this challenge, and TADF emitters based on the multiple resonance effect (MRE) have been proposed.35–48
In these compounds, electron-deficient and electron-rich atoms, such as boron and nitrogen, are positioned alternatively, in a way that the HOMO and LUMO of the molecule are confined on different atoms. Since nitrogen exhibits the opposite resonance effect of boron, para-substitution relative to it can enhance the resonance effect and can significantly spatially separate the HOMO and LUMO without the need to introduce discrete electron donor and acceptor units in the molecular structures. These compounds have a small J and present a ‘short-range’ charge separation, in contrast to the conventional D–A TADF emitters, which instead present ‘long range’ change separation. TADF emitters based on the MRE have large S1 → S0 radiative transition oscillator strength and narrow emission bands, because of their rigid π-conjugation frameworks and their non-conventional HOMO–LUMO separation. Their application in OLED devices has been extensively studied and promising results have been obtained;44,46,49–51 nevertheless their photophysical processes are not completely understood. Many reports can be found in the literature concerning the confinement of the excited state wave functions of conventional D–A TADF emitters, especially in their triplet states. Indeed, for these systems, both the 3LE and 3CS states have been well characterized by transient optical spectroscopic methods,25,27,29–31,52–55 as well as with pulsed laser excited time-resolved electron paramagnetic resonance (TREPR) spectroscopy.56–59 For the conventional CS state, the zero field splitting (ZFS) D parameter of the 3CS state is much smaller than that of the 3LE state of the chromophore,1,27,60–62 because the ZFS D parameter depends on the electron spin–spin dipolar interaction magnitude, and it is related to the distance between the two electrons. However, little information is available for the spin–spin dipolar interaction magnitude of the triplet excited state of MRE emitters.35 Moreover, the central B atom of these molecules should behave like a Lewis acid, but little is known about the experimental manifestation of this property, and the relation between the Lewis acidity and the TADF properties.
Herein we selected two representative MRE-TADF emitters, i.e., DABNA-1 and DtBuCzB (Scheme 1). Both compounds have a triangulene core incorporating ortho-substituted boron and nitrogen atoms to promote the MRE. DABNA-1 is one of the first reported MRE TADF emitters37 and DtBuCzB was also reported for application in OLED devices.63 For both compounds, delayed fluorescence was observed when dispersed in films with 3,3′-bis(N-carbazolyl)1,1′-biphenyl (mCBP) as the host material, showing a lifetime of 68.8 μs.63 However, their excited state dynamics was not studied in detail. In this paper we analyzed their photophysical properties using fs/ns-TA spectroscopic methods, as well as TREPR spectroscopy. The Lewis acidity of the boron center was characterized through the formation of Lewis acid-adducts with pyridine derivatives and with a typical N-heterocyclic carbene.
 | ||
| Scheme 1 Molecular structures of the MRE emitters DtBuCzB, DABNA-1, CzB-H, and DABNA-H used in calculations. The molecular structures of the Lewis bases DMAP and NHC used in the study are also presented. | ||
Results and discussion
Experimental
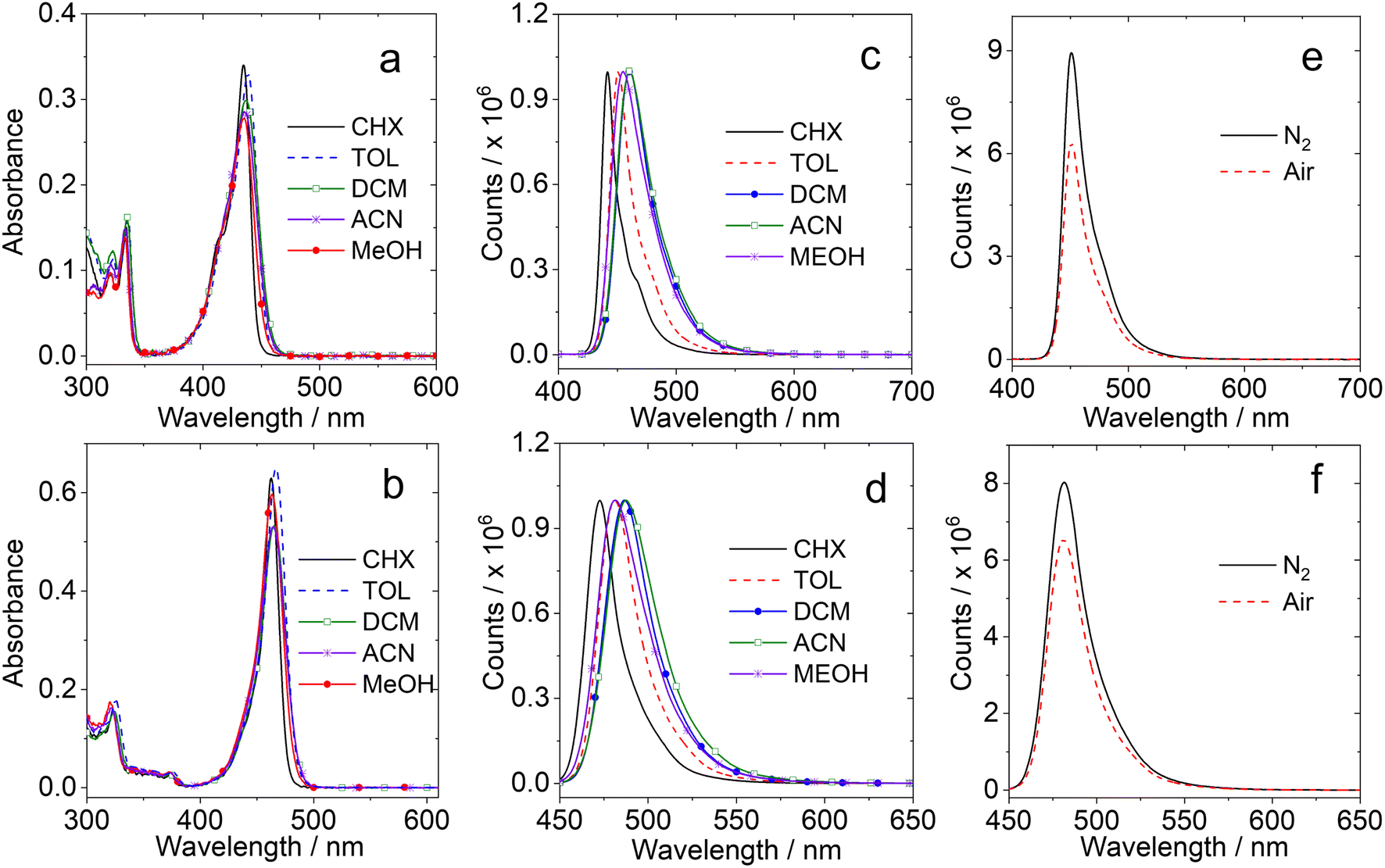 | ||
| Fig. 1 UV-vis absorption spectra of (a) DABNA-1 and (b) DtBuCzB in cyclohexane (CHX), toluene (TOL), dichloromethane (DCM), acetonitrile (ACN), and methanol (MeOH), c = 1.0 × 10−5 M. Normalized fluorescence spectra of (c) DABNA-1 (λex = 335 nm) and (d) DtBuCzB, (λex = 440 nm) in different solvents, and (e) DABNA-1 and (f) DtBuCzB in toluene in different atmospheres. Optically matched solutions were used, A ≈ 0.10. | ||
| λ abs (nm) | ε | λ em (nm) | τ PF (ns) | τ DF (ns) | τ T (μs) | Φ F (%) | Φ Δ (%) | |
|---|---|---|---|---|---|---|---|---|
| a In TOL. Maximal UV-vis absorption wavelength, c = 1.0 × 10−5 M, 20 °C. b Molar absorption coefficient at absorption maxima, ε: 104 M−1 cm−1. c Emission wavelength. d Fluorescence lifetime, λex = 340 nm. e Delayed fluorescence lifetime, λex = 340 nm. f Triplet state lifetime. g Fluorescence quantum yields determined (λex = 333 nm) for DABNA-1 and DtBuCzB in CHX/TOL/DCM/ACN/MeOH, respectively. h Singlet oxygen quantum yields, Ru(bpy)3[PF6]2 was used as the standard compound (ΦΔ = 57% in DCM), λex = 340 nm. i Not observed. | ||||||||
| DABNA-1 | 439 | 3.3 | 451 | 5.7 | —i | 34.3 | 67.1/74.9/83.0/68.7/68.0 | 10.8 |
| DABNA-1-DMAP | 368 | 1.5 | 452 | 5.8 | —i | —i | 89.7 | 0.0 |
| DABNA-1-NHC | 439 | 3.0 | 451 | 5.7 | —i | —i | 12.0 | 0.0 |
| DtBuCzB | 466 | 6.5 | 481 | 4.9 | —i | 38.2 | 83.7/98.5/97.2/92.0/85.2 | 1.0 |
| DtBuCzB-DMAP | 368 | 1.5 | 481 | 4.5 | —i | 13.9 | 85.1 | 0.0 |
| DtBuCzB-NHC | 357 | 3.4 | 383 | 5.4 | 136 | 2.3 | 2.0 | 0.0 |
The full width at half maximum (FWHM) of the absorption band is ca. 983 cm−1 for DABNA-1 and 1382 cm−1 for DtBuCzB. Both compounds do not present charge transfer (CT) absorption bands, although the electron rich and deficient atoms are alternatively positioned.68 The UV-vis absorption spectra of DABNA-1 and DtBuCzB remain the same in different solvents, indicating that the ground state dipole moment is small.69
The fluorescence spectra of the compounds are shown in Fig. 1c and d. The fluorescence emission bands are quite narrow and only show minor changes in solvents with different polarities, as observed for other MRE TADF emitters,65,70 indicating that the emissive S1 state has no conventional intramolecular charge transfer (ICT) character.14,15,71,72
The fluorescence intensity under N2 and air atmospheres are similar (Fig. 1e and f), different from typical D–A TADF emitters, which usually show stronger fluorescence under a N2 atmosphere because triplet states, that are strongly quenched by dioxygen (O2), are involved in the TADF process. The results observed for DABNA-1 and DtBuCzB indicate either that their excited state lifetime is short (less than a few dozens of ns) or that the TADF is not significant. Recently it was proposed that some MRE compounds, such as DABNA-1, do not show TADF behavior in solution, but only in particular solid matrices,40 although they have a small singlet/triplet energy gap ΔES1/T1 = 0.14–0.18 eV.37
Organic boron materials are usually highly reactive and have to be stabilized by steric protection. Planar organic boron compounds with constrained molecular geometry are more stable because of structural constraints. Indeed, the rigidly fixed cyclic skeleton around the boron atom would retard decomposition through the reactions with Lewis basic species, because of the destabilization of a tetra-coordinated intermediate and/or prevention of C–B bond cleavage from the intermediate by the chelating effect.73
It should be noted that the boron centers of both DABNA-1 and DtBuCzB have Lewis acid character and should be able to bind with Lewis bases. The binding can induce notable perturbation of the MRE character, without modification of the molecular structure. The binding with a Lewis base such as DMAP, inducing the formation of a B–N dative bond, will eliminate the MRE, because of the occupation of the vacant p-orbital of the boron atoms. Recently it was shown that DtBuCzB and 5,9-dioxa-13b-boranaphtho[3,2,1-de]anthracene (DOBNA) can interact with Lewis bases such as pyridine, DMAP, and the fluoride ion (F−). DtBuCzB was used as a F− probe, because of its very high binding constant with F− of 1.16 × 105 M−1.74
We analyzed the UV-vis absorption spectral changes of the MRE emitters upon binding with DMAP and NHC (Fig. 2). Upon incremental addition of DMAP into the solution of DtBuCzB in toluene, the strong absorption band of the native DtBuCzB centered at 467 nm as well as the minor band centered at 325 nm decreased in intensity, and a new band centered at 368 nm developed (Fig. 2a). This result is literally the same as the UV-vis evolution upon binding with F− anions.74 Similar results were observed upon addition of NHC (Fig. 2d). The color change in the solution upon formation of the adduct is visible to the naked eye (Fig. 2f).
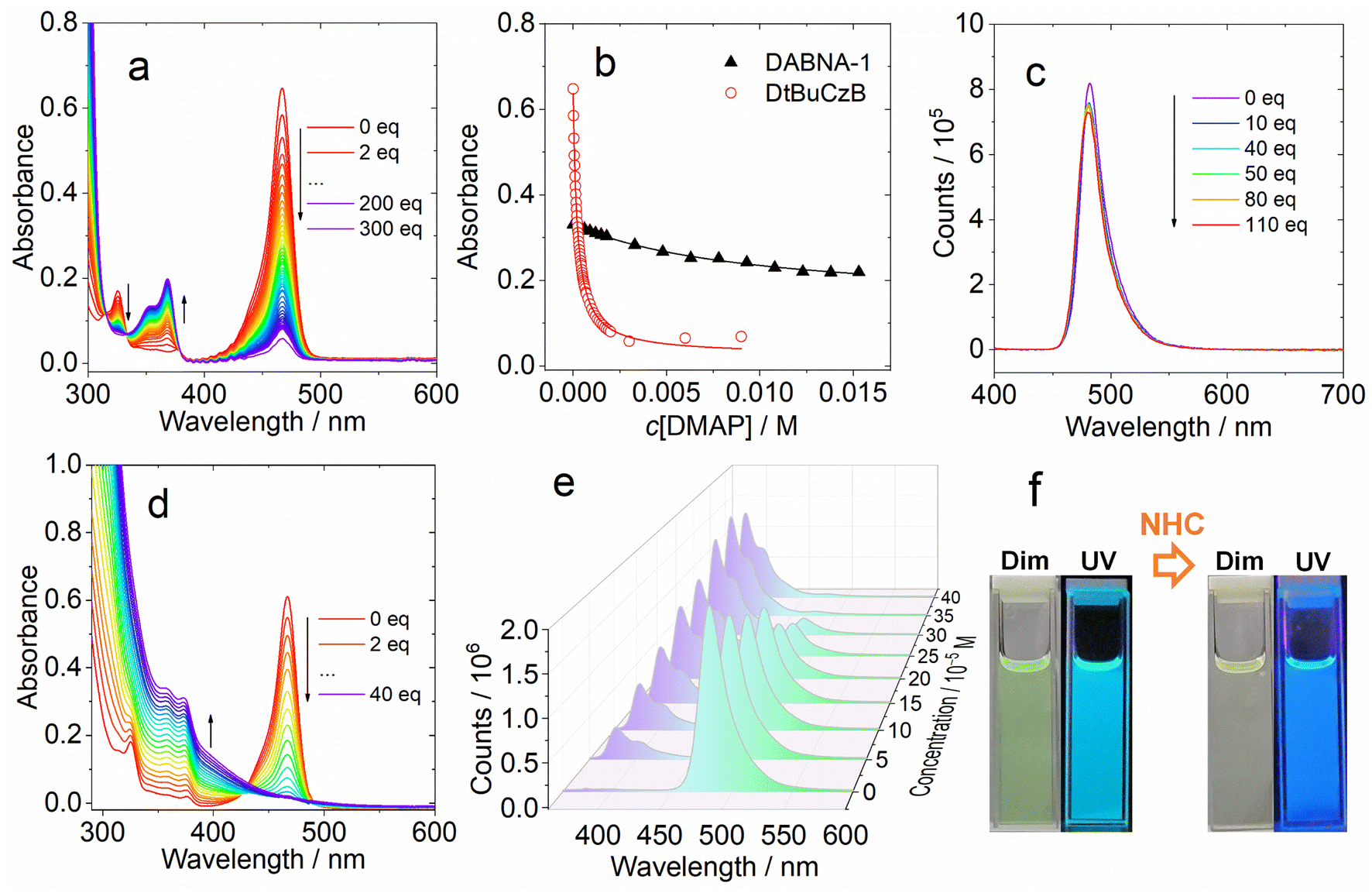 | ||
| Fig. 2 (a) Evolution of the UV-vis absorption spectra of DtBuCzB with an incremental amount of DMAP added. (b) The titration isotherms of DABNA-1 (monitored at 439 nm) and DtBuCzB (monitored at 467 nm). The solid lines are the fitting for the binding constants of DABNA-1 or DtBuCzB toward DMAP. (c) Fluorescence spectra of DtBuCzB with an incremental amount of DMAP added. (d) UV-vis absorption spectra of DtBuCzB with an incremental amount of NHC added. (e) Fluorescence spectra of DtBuCzB with different concentrations of NHC in toluene, λex = 333 nm (isosbestic point of the UV-vis absorption spectra). (f) The digital photographs of the solution of DtBuCzB upon addition of NHC (40 eq.) under dim light and UV light (365 nm) illumination, respectively. c = 1.0 × 10−5 M, 20 °C. | ||
Previously, similar changes were observed for the binding of planar organoboron compounds with Lewis bases such as DMAP.75–77 The Gibbs free energy change (ΔG°) of the binding process with DMAP is −21.1 kJ mol−1 (Table 2). Similar results were observed for DABNA-1, although its binding constant is smaller, with K = (1.06 ± 0.001) × 102 M−1. Previously it was found that DtBuCzB can bind with F anions with a higher binding constant (1.16 × 105 M−1).74 Some analogues containing an O atom instead of N atoms, showing lower electron density in the aromatic rings compared to DABNA-1, have a stronger binding constant with DMAP (4.29 × 104 M−1) compared to DABNA-1.78,79DABNA-1 cannot bind with NHC, showing no significant changes in the UV-vis absorption and fluorescence spectra even upon the addition of 500 equivalents of NHC. For the adduct of DtBuCzB with NHC, a blue-shifted emission band centered at 383 nm was observed, along with the emission of the native DtBuCzB (Fig. 2e). The emission band at 383 nm is attributed to the adduct, while that centered at 481 nm is assigned to the free DtBuCzB. Note the binding of DtBuCzB with NHC is slightly tighter than that with DMAP (Table 2). The evolution of the fluorescence spectra is similar to that found for analogues based on boron-containing dioxygen-bridged molecules.78
| DABNA-1 | DtBuCzB | |||
|---|---|---|---|---|
| K (M−1) | ΔG (kJ mol−1) | K (M−1) | ΔG (kJ mol−1) | |
a ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnK, R ≈ 8.314 J mol−1 K−1, T = 298 K.
b Values are so small as to be negligible. lnK, R ≈ 8.314 J mol−1 K−1, T = 298 K.
b Values are so small as to be negligible.
|
||||
| DMAP | (1.06 ± 0.001) × 102 | −11.55 | (4.51 ± 0.007) × 103 | −20.85 |
| NHC | —b | —b | (5.06 ± 0.03) × 103 | −21.13 |
| DABCO | —b | —b | (4.01 ± 0.006) × 102 | −14.85 |
| 2-Methylpyridine | —b | —b | —b | —b |
Interestingly, we did not observe any variation of the fluorescence of DtBuCzB upon incremental addition of DMAP (Fig. 2c), when exciting the solutions at the isosbestic point (333 nm) of the UV-vis absorption spectrum of the mixture. This observation is unexpected, considering the substantial changes observed in the UV-vis absorption spectrum of the mixture (Fig. 2a), which indicate an efficient formation of the Lewis acid–base adduct. A corresponding decrease in fluorescence intensity would be expected, unless the DtBuCzB-DMAP complex would have exactly the same fluorescence as the free DtBuCzB, which is unlikely. We thus propose that photodissociation may occur upon photoexcitation of the adduct, and that the observed fluorescence is emitted from the photo-released free DtBuCzB.80 Previously, a similar photodissociation was observed for a planar organoboron compound coordinated with a Lewis base (these compounds have no MRE properties).76 Since the ISC of DtBuCzB is not efficient, we assume that photodissociation occurs at the singlet excited state (see later section for the ultrafast transient absorption studies).75 This is different from the scenario of the binding of DtBuCzB with F− anions, for which the fluorescence is strongly quenched,74 possibly because of tight binding, avoiding the photo-dissociation of the adduct. The binding of the MRE emitters with other Lewis bases such as 1,4-diazabicyclo[2.2.2]octane (DABCO) and 2-methylpyridine wasere also studied, and smaller binding constants were observed.
Previously it was found that photodissociation of the adduct formed between a planar trinaphthylborane, having a partially fused structure, and a Lewis base pyridine, generated a dual emission, due to both the adduct and photodissociated borane.75 In our case, although the binding constant is similar, only the fluorescence of the photogenerated DtBuCzB was detected. The prerequisite for emission of the adduct would be a tight binding. For instance, in the case of the adduct of a planar trinaphthylborane with DMAP,75 presenting a very high binding constant of (6.6 ± 1.3) × 106 M−1, no photodissociation was observed.75 The extension of the π-conjugation framework of the MRE TADF emitters DABNA-1 and DtBuCzB may decrease the Lewis acidity of the boron center, thus decreasing the binding constants.81
These examples demonstrate that the occurrence of photodissociation is strongly dependent on the magnitude of the binding constants. Nevertheless, photodissociation was also not observed in the case of an organoboron-phosphine B–P adduct, although its binding constant, K = 61 M−1,76 is smaller. It was also proposed that the photo-generated borane could re-bind with pyridine, but this possibility was not confirmed experimentally, for instance by using ns-TA spectroscopy.75
The binding of borane with Lewis bases has been used for controlling the fluorescence,82–84 and for dynamic modulation of (anti)aromaticity and diradical character of organic diradicaloids.81 For our system, the adduct is formed at room temperature, while for the previously reported borane and phosphine ligand, the adduct can only be formed at low temperature (<270 K).76 In order to quantify the Lewis basicity of the boranes, we carried out Gutmann–Beckett measurements, using Et3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to monitor the shift of the 31P NMR signal.76 For DABNA-1, the chemical shifts changed slightly with Et3PO (e.g. from 52.78 ppm of the planar triangonal to 54.92 ppm of the sp3 hybridized boron center, Fig. S6†), while they changed to a larger extent for DtBuCzB (e.g. from 52.78 ppm of the planar triangonal to 55.67 ppm of the sp3 hybridized boron center, Fig. S7†). For previous compounds the chemical shift changes of 31P after complexation with planar boron compounds were found to be ΔP = 0.03 and 3.56 ppm.76
O to monitor the shift of the 31P NMR signal.76 For DABNA-1, the chemical shifts changed slightly with Et3PO (e.g. from 52.78 ppm of the planar triangonal to 54.92 ppm of the sp3 hybridized boron center, Fig. S6†), while they changed to a larger extent for DtBuCzB (e.g. from 52.78 ppm of the planar triangonal to 55.67 ppm of the sp3 hybridized boron center, Fig. S7†). For previous compounds the chemical shift changes of 31P after complexation with planar boron compounds were found to be ΔP = 0.03 and 3.56 ppm.76
Based on the ns-TA spectral study (see later section), we propose that for the adduct formed between DtBuCzB and NHC, the fluorescence centered at ca. 480 nm is due to the residual free DtBuCzB, and not due to photodissociation of the Lewis acid–base adduct. This agrees with the tight binding between DtBuCzB and the NHC Lewis base. This result is similar to the previously reported binding between planarized borane and DMAP.75 The adduct of an oxygen-bridged planarized triphenylborane with DMAP, showing binding constant of K = (6.6 ± 1.9) × 107 M−1, exhibits very weak fluorescence at room temperature.80
We further measured the fluorescence lifetimes of the compounds in deaerated solutions (Fig. 3 and S16†). The luminescence decay trace of DtBuCzB in deaerated toluene has biexponential character (5.9 ns/99.1%, 244 ns/0.9%, Fig. 3a). Similar results were observed with the solution under an air atmosphere (4.9 ns/99.7%, 506 ns/0.3%). Biexponential decay traces were also observed for DABNA-1 (Fig. 3d). Considering that the long living component of the fluorescence decay is not quenched by air, we conclude that TADF is not significant for DtBuCzB and DABNA-1 in toluene.
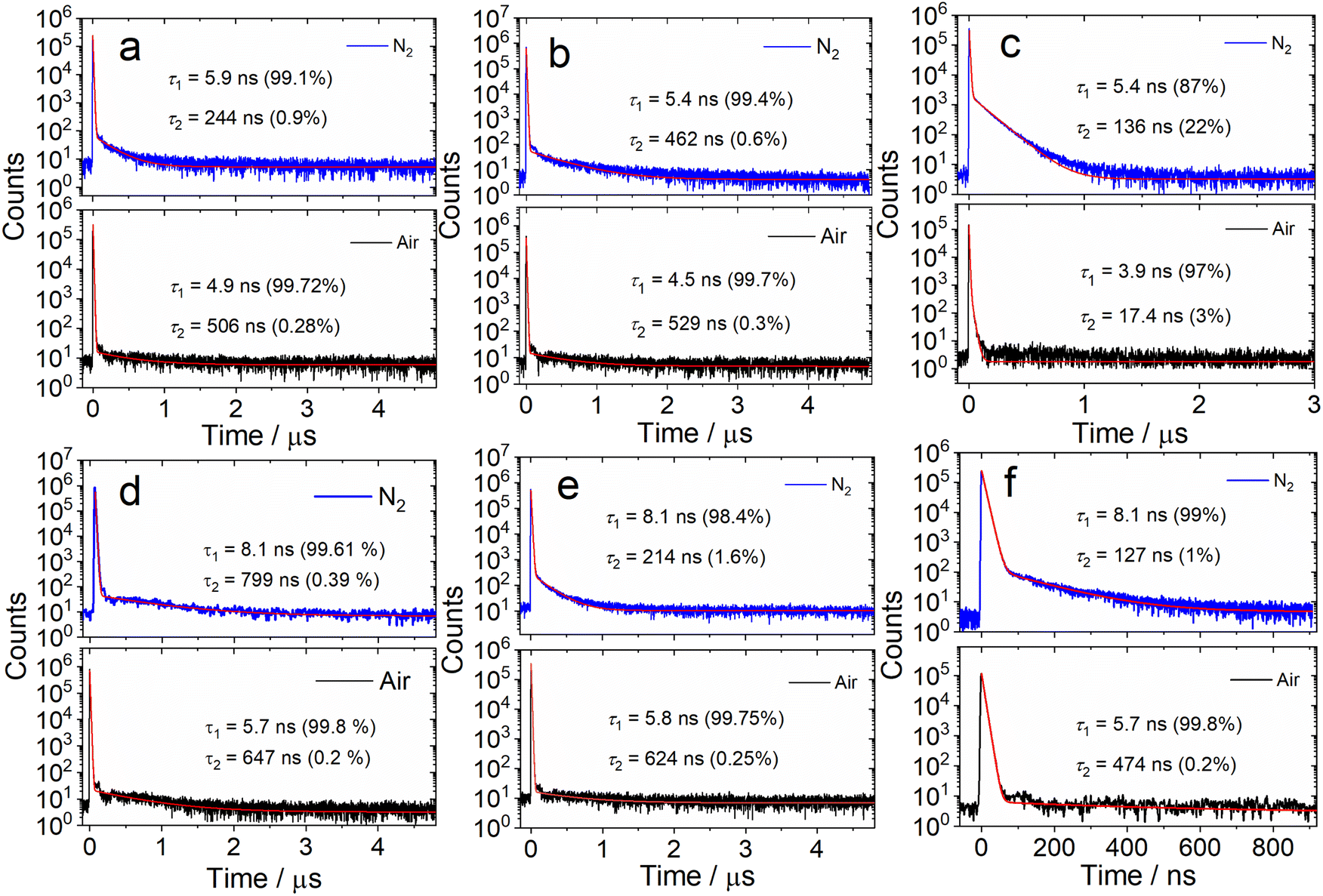 | ||
| Fig. 3 Fluorescence decay traces of (a) DtBuCzB (480 nm); (b) DtBuCzB with DMAP (481 nm); (c) DtBuCzB with NHC (388 nm); (d) DABNA-1 (450 nm); (e) DABNA-1 with DMAP (450 nm); (f) DABNA-1 with NHC (450 nm) in different atmospheres in toluene. Excited with a picosecond pulsed laser, λex = 340 nm, c = 1.0 × 10−5 M, 20 °C. | ||
We attribute the long-lived signal to instrument noise. The fluorescence lifetime of DABNA-1 was reported to be 9.5 ns (in DCM, at a concentration of 2 × 10−5 M),37 while TADF was only observed for DABNA-1 in the doped films in the hole transfer material of 3,3-di(9H-carbazol-9-yl)biphenyl (mCBP. 1W%), with the prompt and delayed fluorescence lifetimes of 8.8 ns and 93.7 μs, respectively.37 Recently Chou et al. proposed that TADF may result from the intermolecular interaction of these MRE-TADF emitters with the host materials in the OLED devices.40
We also studied the fluorescence lifetime of the adducts (Fig. 3b and e). For DtBuCzB, the decay trace of the adduct with DMAP shows biexponential character in toluene under a N2 atmosphere (5.4 ns/99.4%, 462 ns/0.6%, Fig. 3b). Similar results were observed for the aerated solution (4.5 ns/99.7%, 529 ns/0.3%), as well as for the adduct of DABNA-1 with DMAP (Fig. 3e), indicating that there is also no significant TADF for the adducts of DtBuCzB and DABNA-1 with DMAP. Interestingly, the DtBuCzB/NHC adduct shows significant TADF, and a biexponential fluorescence decay trace was observed (Fig. 3c), with lifetimes of 5.4 ns (87%)/136 ns (22%). In the case of the adduct of DABNA-1 with NHC, TADF is not observed, and the fluorescence has a biexponential decay both under a N2 atmosphere (8.1 ns/99%, 127 ns/1%) and in air (5.7 ns/99.8%, 474 ns/0.2%) (Fig. 3f).
We further investigated the luminescence properties of the compounds at low temperatures, measuring the photoluminescence spectra of DtBuCzB and DABNA-1 in a frozen solution of 2-methyltetrahydrofuran (Fig. 4) and 2-methyltetrahydrofuran/iodoethane (2:1, v/v). At 77 K the maximum of the phosphorescence bands are centered at 481 nm and 508 nm, respectively (Fig. S20†). The triplet excited state energy of DtBuCzB (2.56 eV) and DABNA-1 (2.72 eV) is obtained by multi-peak fit analysis of the phosphorescence spectra, and results similar to the literature values are obtained (2.53 eV and 2.59 eV).37,63 Moreover, we found that the luminescence spectra of DtBuCzB and DABNA-1 in the presence of DMAP are blue shifted by 100 nm and 50 nm at 77 K (Fig. 4), implying that at low temperature we only observed the emission of the adduct since photodissociation is substantially suppressed.75
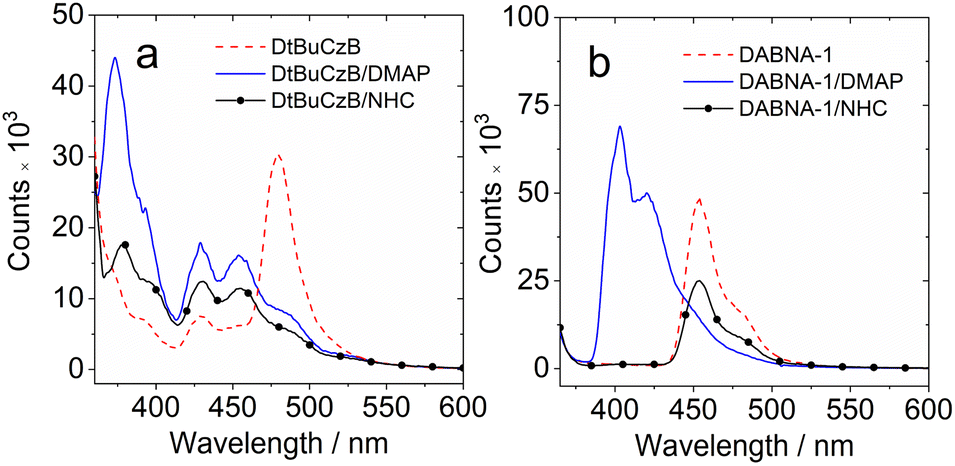 | ||
| Fig. 4 Luminescence spectra of the compounds and the adducts in a frozen solution of (a) DtBuCzB alone, and in the presence of DMAP (50 eq.) or NHC (50 eq.) and (b) DABNA-1 alone and in the presence of DMAP (50 eq.) and NHC (50 eq.) in 2-methyltetrahydrofuran at 77 K. Excited with a picosecond pulsed laser (λex = 340 nm), c = 1.0 × 10−4 M. | ||
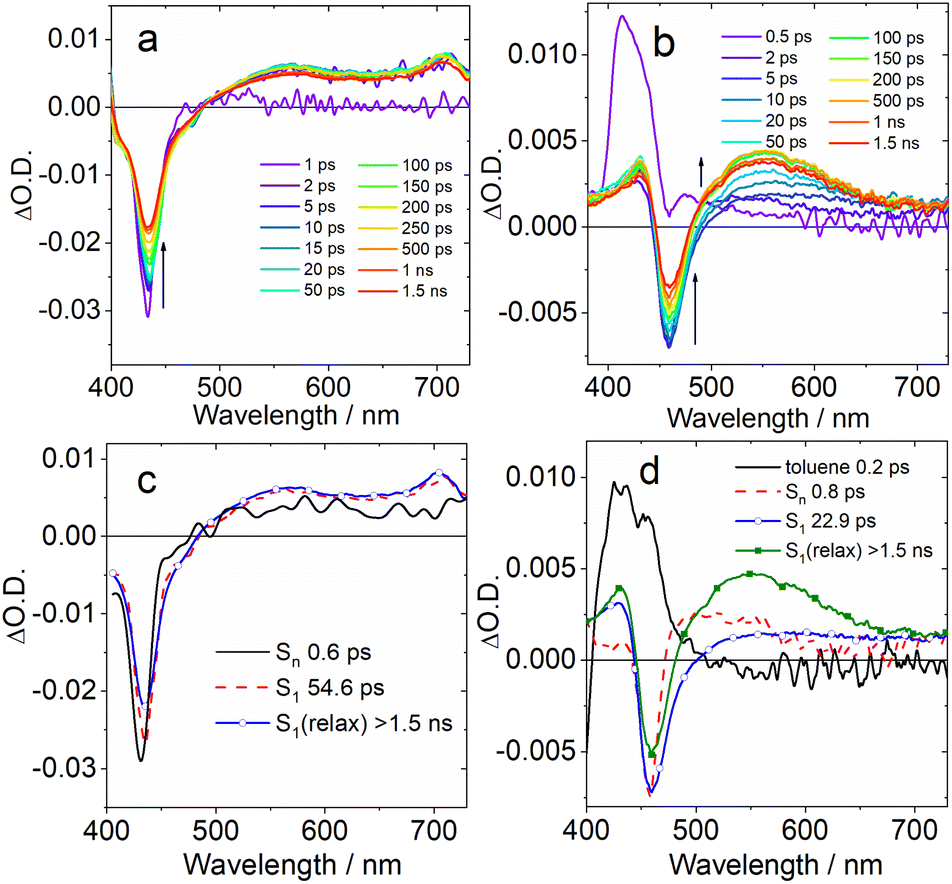 | ||
| Fig. 5 Fs-TA spectra recorded in toluene for (a) DABNA-1 excited at 400 nm and (b) DtBuCzB excited at 340 nm. Panels (c) and (d) present the EADS obtained from global analysis of the transient absorption data. | ||
Selected transient absorption spectra and the evolution associated difference spectra (EADS) obtained from global analysis are reported in Fig. 5. While the transient absorption spectra of conventional TADF emitters have been extensively studied,52,85 the analysis of the excited state dynamics of MRE compounds is rare. The fs-TA spectra of an analogue of DABNA-1 were recently reported, and the results closely match those presented in Fig. 5a.64 In the literature, DABNA-1 was excited at 330 nm, and a rapid internal conversion (IC) from the initially populated Sn to the S1 state was observed.
Upon excitation at 400 nm, the S1 state is directly accessed. A negative band peaked at about 430 nm is observed immediately after excitation (Fig. 5c), ascribed to the convolution of ground state bleaching (GSB) and stimulated emission (SE), which can't be spectrally resolved because of the small Stokes shift. The transient spectrum evolves on a very fast timescale of 0.6 ps: the negative band broadens on its red side and a positive excited state absorption (ESA) band develops in the 500–700 nm region. We attribute this evolution to a fast solvent induced relaxation of the S1 state. In the following 54 ps, a second evolution is noticed, mainly consisting in a further slight red shift of the SE band and an overall intensity decrease of the negative signal.
Previous investigations on a similar compound, both in the visible and IR regions,67 attributed an evolution observed on a similar timescale to structural relaxation, consisting in a planarization of the molecule in the excited state. The assignment is based on a theoretical analysis. In agreement with the previous report, we interpret the evolution between the second and third EADS in a similar way. The final spectral component does not decay on the investigated timescale (1.5 ns).
The transient spectra of DtBuCzB are similar to those of DABNA-1 (Fig. 5b). In this case the sample was excited at 340 nm, thus reaching a higher energy excited state. The intense positive signal observed at a very fast time scale and decaying in about 200 fs is attributed to a solvent response following excitation with UV light (dashed line in Fig. 5d). The following spectral component (black continuous line in Fig. 5d) shows a negative signal peaked at about 458 nm, assigned to GSB, and a positive ESA in the 470–600 nm range. Stimulated emission is not observed on a very fast timescale, since the sample still has to relax towards the S1 emissive state. This process occurs in about 0.8 ps, as shown by the broadening of the negative signal on its red side, caused by the SE build up, the red shift and broadening of the ESA band and the appearance of a second ESA peak at about 430 nm. In the following evolution, occurring in about 22 ps, the red-shifted ESA signal increases in intensity, and the negative GSB/SE signal slightly recovers. Analogous with what was discussed in the case of DABNA-1, we tentatively assign this evolution to a structural relaxation, possibly associated with the planarization of the molecule. Also in this case, the final spectral component lives longer than the timescale accessed with this measurement.
We furthermore studied the photodissociation dynamics of the adducts formed by DABNA-1 and DtBuCzB with DMAP (Fig. 6). The transient spectra of the adduct DABNA-1/DMAP are very similar to those of the isolated DABNA-1, and also upon the addition of a great excess of DMAP in solution (Fig. S25†), except for some changes in the time constants describing the photodynamics (Fig. S23†). For the DtBuCzB-DMAP adduct, whose transient spectra and EADS are reported in Fig. 6, a more complex behaviour is observed.
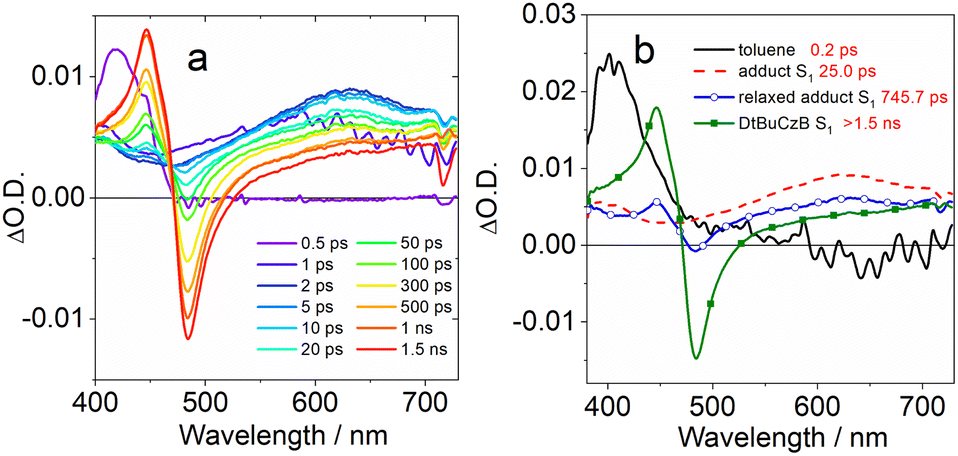 | ||
| Fig. 6 (a) Fs-TA spectra of the DtBuCzB-DMAP (50 eq.) adduct recorded in toluene upon excitation at 370 nm. (b) The EADS obtained from global analysis of the transient absorption data. | ||
Immediately after excitation, we observed an intense positive absorption signal, peaking at about 400 nm (Fig. 6a), which decays on a very fast timescale of about 200 fs, attributed to a fast response of the toluene solvent. The following spectral component, decaying in 25.0 ps, shows up as a very broad and weak positive signal, extending over the full probed spectral range, that we assign to the absorption of the DtBuCzB-DMAP adduct at the S1 state. On this same timescale vibrational and structural relaxation is likely to occur as well. In the following evolution, a positive peak located at 446 nm and a negative peak at 483 nm start to appear superimposed on the broad positive signal of the adduct. The position of these signals closely matches the absorption and emission of DtBuCzB respectively, indicating the occurrence of photodissociation. The intensity of both bands further increases after 745 ps, indicating that the photodissociation has biexponential kinetics.
A possible explanation for biexponential photodissociation dynamics is the existence of different conformations of the adducts in solution, with some of them releasing DMAP more easily. The transient signals of the photogenerated DtBuCzB do not decay on the ns timescale, indicating that the re-binding of the photogenerated DtBuCzB and DMAP is slower. This is reasonable because this intermolecular process is diffusion-controlled, and can take up to microseconds in solution. We further studied the adduct formed by DtBuCzB with the carbene NHC. In this case photodissociation is not observed, because of the stronger binding between the two moieties. Indeed, the transient spectra only show a positive signal decaying on a fast timescale due to solvent response, and a very weak and broad band, similar to the second spectral component observed for the adduct DtBuCzB/DMAP (dotted red line in Fig. 6b), which does not decay on the ns timescale (Fig. S26†).
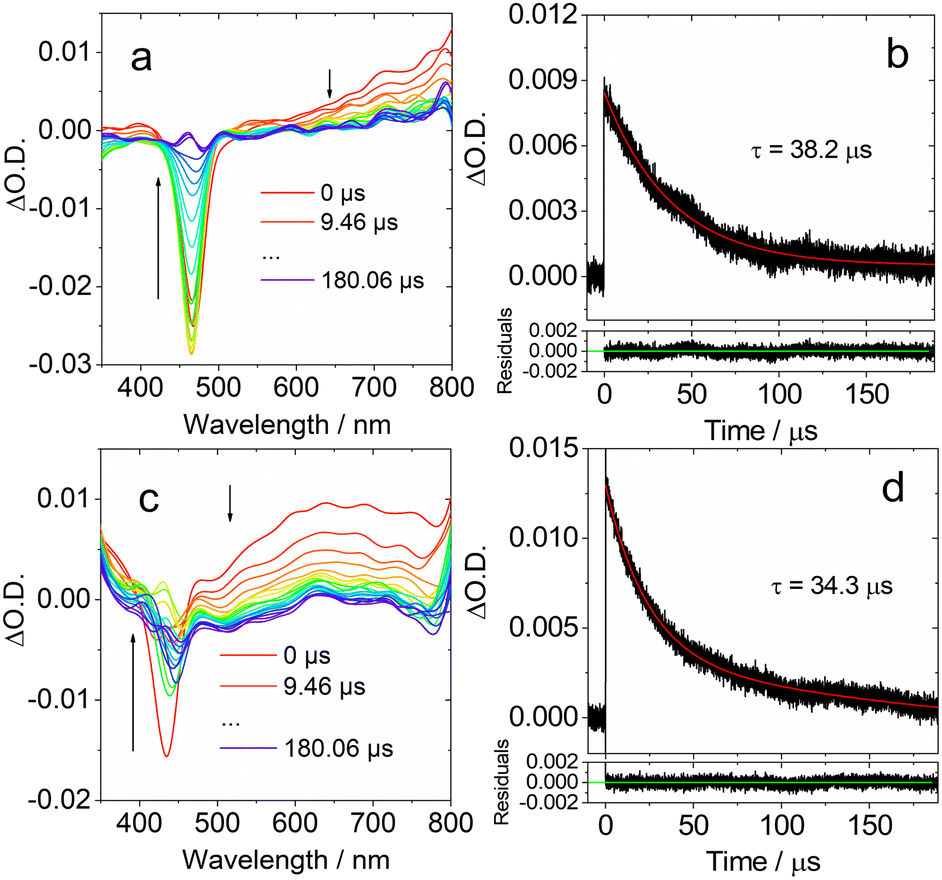 | ||
| Fig. 7 Ns-TA spectra in DCM of (a) DtBuCzB and (c) DABNA-1. Decay traces of (b) DtBuCzB at 750 nm; (d) DABNA-1 at 650 nm. Excited at 440 nm and 420 nm, respectively. c = 2.0 × 10−5 M, 20 °C. | ||
We then studied the dynamic behavior of the adducts of the MRE compounds with Lewis bases. In the case of DtBuCzB in the presence of an excess of DMAP a GSB band centered at 366 nm was observed, in agreement with its steady state absorption spectrum (Fig. 8a). Interestingly, a sharp positive absorption band centered at 465 nm was also observed. The position of this band corresponds very well to the steady state UV-vis absorption band of the free DtBuCzB (Fig. 1).
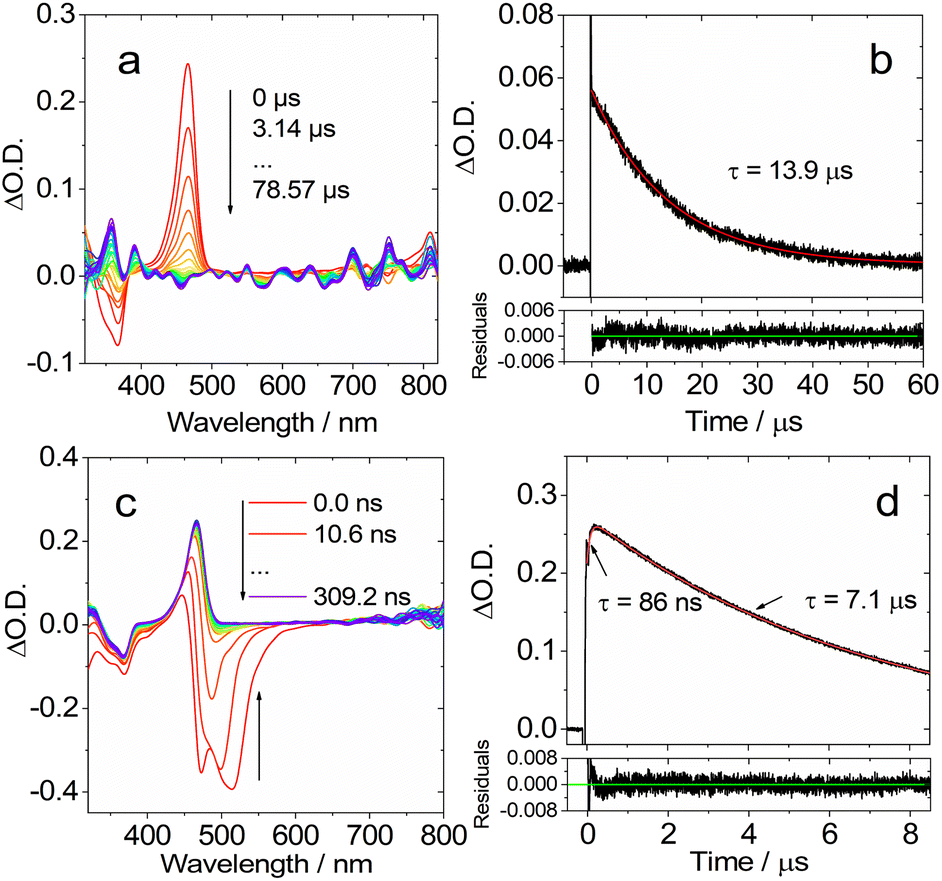 | ||
| Fig. 8 Ns-TA spectra in toluene of DtBuCzB with 4-N,N-dimethylaminopyridine (DMAP. 50 eq.) added. (a) Recorded in the long delay time range of 0–78.6 μs and (c) in the short delay time range of 0–309 ns. Decay traces at 470 nm of DtBuCzB in the presence of DMAP (50 eq.) in toluene: (b) long delay time range and (d) short delay time range. c [DtBuCzB] = 2.0 × 10−5 M, λex = 355 nm, 20 °C. | ||
Based on the fluorescence study of DtBuCzB in the presence of DMAP, and the results of the ultrafast transient absorption measurements presented in the previous section, both indicating the occurrence of photo-dissociation of the bound DMAP (Fig. 2c), the sharp band centered at 465 nm is assigned to the photogenerated free DtBuCzB (Fig. 8a).
In the presence of excess DMAP in the mixture solution, the photo-generated free DtBuCzB becomes a ‘transient’ species, because it re-binds with DMAP quite easily, as indicated by the decay trace at 465 nm in Fig. 8a. The rebinding process has a time constant of 13.9 μs under our specific experimental conditions (Fig. 8b). This time scale agrees with the molecular diffusion kinetics in fluid solution at room temperature. It is worth mentioning that the time required for recombination depends on the experimental conditions, such as the concentration of DMAP, the temperature, the solvent viscosity, etc. (Table S1†).
At the short time scale, the ns-TA spectra of DtBuCzB in the presence of an excess of DMAP show an ESA band centered at 465 nm and a negative band centered at 486 nm, due to stimulated emission (Fig. 8c), similar to the last spectral component in the fs TA spectra (Fig. 6a). The kinetic trace at 470 nm shows a rise component of 86 ns, which indicates the time needed for complete adduct dissociation (Fig. 8d). Previously, the photodissociation of the Lewis acid–base adduct of a planarized trinaphthylborane with a partially fused structure and pyridine was studied with fluorescence spectra, and the re-binding of the photo-generated borane with pyridine was observed, but it was not monitored with ns-TA spectroscopy.75 Recently the binding of DtBuCzB or the dioxygen-bridged analogues with F− anions, DMAP or pyridine, was studied with steady state UV-vis absorption and fluorescence spectra, but not with transient absorption spectra.74,78 We also studied the adduct of DtBuCzB with F− anions with ns-TA spectra (data not shown here), and no photodissociation was observed.
To confirm the occurrence of photodissociation and rebinding, we studied the ns-TA spectra of the DtBuCzB-DMAP mixture in viscous solvents, such as in toluene/silicone oil mixed solution. We obtained similar ns-TA spectra (Fig. S23†), observing a 109 ns rise phase for the kinetic trace at 470 nm, followed by a decay occurring in ca. 17.9 μs (Fig. S28†). These results agree with the expected retarded photo-dissociation as well as the subsequent slower re-binding in a viscous solvent. We also studied the ns-TA spectra of the DtBuCzB-DMAP mixture in the polar solvent acetonitrile, and the results are very similar to those in toluene. In this case, the rise phase of the kinetic trace at 470 nm takes 58 ns, while the decay occurs in 4.5 μs (Fig. S33†). It was furthermore shown that in a polymer film, the relaxation of the S1 state is slower by 3–4 fold as compared to that in fluid solution.64
Moreover, the electron spin selectively of the ISC is manifested by the electron spin polarization (ESP) pattern of the triplet state TREPR spectrum, i.e. either the enhanced absorption or the emission features in the canonical orientation of the molecules in the magnetic field.
Previously, TREPR spectroscopy has been used to characterize the conventional electron D–A dyad TADF emitters.27,29,56,57,59 The triplet states of the two possible representative MRE TADF emitters DABNA-1 and DtBuCzB, were studied with TREPR spectroscopy in a frozen solution at 80 K (Fig. 9). For DABNA-1 and DtBuCzB, a polarized transient TREPR spectrum with E/A features (E: emissive; A: enhanced absorptive) was observed (Fig. 9a and b). Only one triplet was observed regardless of the concentration of these compounds. The spectra of the two compounds slightly differ due to different polarizations, but the parameter D is equal to 1450 MHz for both compounds, and E = −264 MHz and −200 MHz, respectively (Table 3).
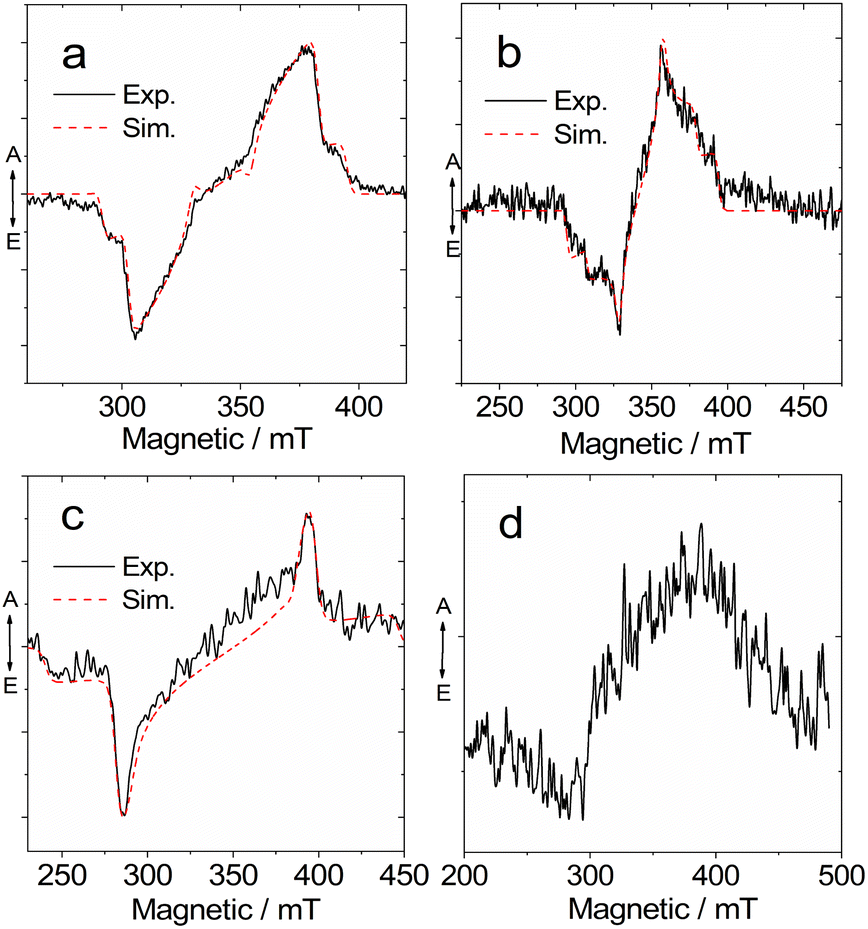 | ||
| Fig. 9 Experimental and simulation TREPR spectra of (a) DABNA-1, (b) DtBuCzB in toluene/2-MeTHF (1:1), (c) DABNA-1 + 50 eq. DMAP, and (d) DtBuCzB + 50 eq. DMAP. At 80 K. c [DABNA-1] = 5.0 × 10−4 M, c [DtBuCzB] = 5.0 × 10−4 M, the laser energy is 1 mJ per pulse at 355 nm. The delay after laser flash (DAF) is 300 ns. | ||
| Sample | D/MHz | E/MHz | Px | Py | Pz | g-Factor |
|---|---|---|---|---|---|---|
|
a Obtained from simulation of the triplet-state TREPR spectra of the indicated molecules in toluene/2-MeTHF (1:1, v/v) at 80 K.
|
||||||
| DABNA-1 | 1450 | −264 | 0.83 | 0.17 | 0 | 2.007 |
| DtBuCzB | 1450 | −200 | 0.44 | 056 | 0 | |
| DABNA-1+DMAP | 2900 | −116 | 0.8 | 0.2 | 0 | |
We also recorded the TREPR spectra of the compounds upon binding with DMAP (Fig. 9c and d). Interestingly, the TREPR spectrum of the DABNA/DMAP adduct corresponds to one type of triplet state (Fig. 9c), and simulations indicate that the ZFS D parameter becomes as large as 2900 MHz, while the E parameter is −116 MHz (Table 3). These changes are in agreement with the optical spectral studies, showing that the size of the π-conjugation framework is reduced upon binding with DMAP, because the vacant p-orbital of the center B atom is occupied upon formation of a B–N dative bond.87,88 At 80 K the photodissociation of the DABNA-1/DMAP adduct is suppressed as also shown by the low temperature luminescence (Fig. 4b). A broader TREPR spectrum of weak intensity was observed for DtBuCzB upon binding with DMAP, probably due to partial photo-dissociation (Fig. 9d).
The boron center of DABNA-1 and DtBuCzB is almost planar (Fig. 10a and b), and the torsion is 11.6° and 9.9°, respectively. These geometries are close to those of the S0 state obtained with restricted DFT (Fig. S32†). The electron spin density surfaces of the T1 state of DABNA-1 and DtBuCzB are presented in Fig. 10c and d. The electron spin density spreads over the entire π-conjugation system.
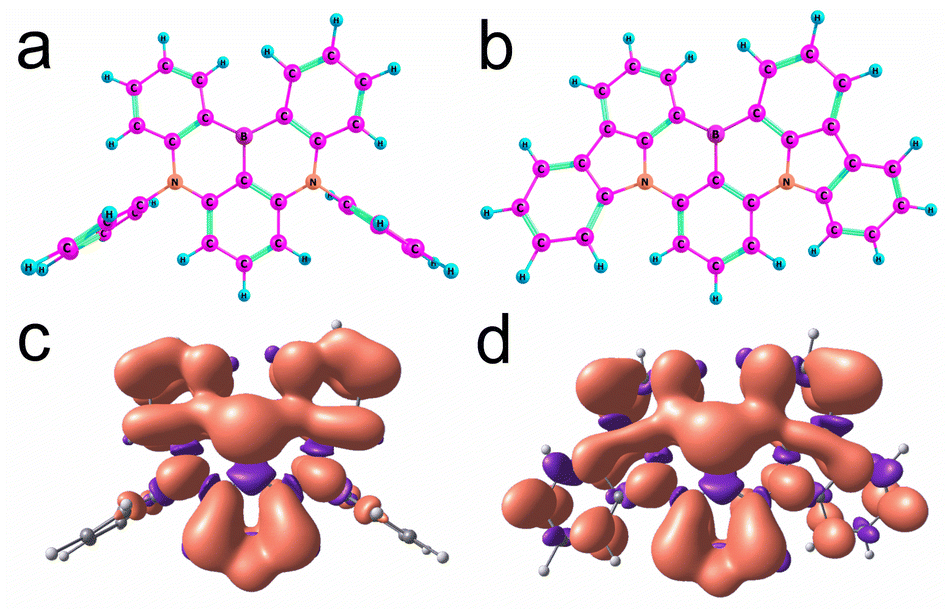 | ||
| Fig. 10 The optimized T1 state geometry of (a) DABNA-1 and (b) CzB-H, and the electron spin-density surfaces of the T1 state of DABNA-1 (c) and CzB-H (d). Calculations with the UB3LYP/def2-SVP method. | ||
To better characterize the triplet state of the B–N compounds, further calculations involving spin–orbit coupling and wavefunction based methods (ROHF = restricted open shell Hartree Fock, CIS = configuration interaction singles, CASSCF = complete active space self consistent field, and NEVPT2 = N-Electron Valence State Perturbation Theory90) were performed with the ORCA programs91,92 employing the def2-SVP basis and in some cases also the def2-TZVP basis. To reduce the computational cost, the t-butyl groups of DtBuCzB were replaced by hydrogen atoms, and this compound was called CzB-H. To check how much the phenyl groups are involved in the electronic excitations in DABNA-1, we also considered DABNA-H where these phenyl groups are replaced by hydrogen atoms, as shown in Scheme 1.
The Zero Field Splitting (ZFS) parameters D and E for the T1 state were computed from UB3LYP/def2-SVP geometry optimizations as well as ROHF/def2-TZVP calculations at these optimized geometries. In contrast to ROHF, UB3LYP suffers from spin contamination (spin angular momentum S2 = ca. 2.015). However, geometries cannot be optimized with ROHF since no gradients are implemented in ORCA. The results are summarized in Table 4.
The optimized geometries of DABNA-H and DABNA-1 have C2 symmetry. For CzB-H we find two stable conformers with C2 and CS symmetry, respectively, with almost the same energies. The calculated values from UB3LYP agree very well with the experimental data, whereas ROHF overestimates the value of D by ca. 20%. The variation of the ZFS D parameter from DABNA-1 to CzB-H agrees with the increased spin delocalization in DtBuCzB, for which the π-conjugation system is larger than that of DABNA-1.
The electronically excited states of DABNA-1 have been previously studied. Methods which consider single electron excitations only, like TD-DFT or CIS, predict energy gaps between S1 and T1 of ca. 0.5 eV which is much larger than the experimental values (ca. 0.15 eV) obtained from the fluorescence and phosphorescence spectra. Hall et al. showed the SCS-CC2 method, which considers correlation through doubly excited configurations, could precisely predict the S1/T1 state energy gap.66 The reason is that the Coulomb correlation effects are substantially larger in the lowest singlet excited state compared to the corresponding triplet. These calculations also confirmed the short-range CT nature of the S1 and T1 states of the MRE emitters.66 When dynamic correlation is considered in the theoretical method, e.g. in SCS-CC2 or CASSCF+NEVPT2, the S2 and T2 states appear much higher in energy. Hence these states should not be involved in ISC and rISC required for TADF. This is different from the calculation results reported previously.93
We used CASSCF(10|10) in combination with NEVPT2 to obtain the energy gap between S1 and T1 states. For DABNA-H and DABNA-1 the CASSCF was averaged over three singlet and two triplet states. A third triplet had to be included for CzB-H since at the CASSCF level the lowest three triplets are almost degenerate. In accordance with the observations of Hall et al., the second order perturbation correction by NEVPT2 lowers S1 much more than T1, bringing both states close together. With the def2-SVP basis the gap is 0.192 eV for DABNA-H, 0.105 eV for DABNA-1, and 0.068 eV for CzB-H. Similar values are obtained with the larger def2-TZVP basis, namely 0.18 eV, 0.075 eV, and 0.170 eV, respectively. In addition, S2 and T2 are shifted to higher energies, making S1 and T1 the only relevant states for TADF.
Since the experimental small S1/T1 state energy gaps are confirmed by these calculations, the absence of TADF in solution must be due to a small ISC rate. ISC rate constants can be calculated with the ESD94,95 (excited states dynamics) module implemented in ORCA. However, consideration of the vibronic interactions (Herzberg–Teller terms) is only possible with single reference methods like TD-DFT or CIS. Inspection of the CASSCF wavefunctions shows that these are dominated by the HOMO–LUMO configuration, and that NEVPT2 results in an energy correction that is smaller than 1% of the total energy. It is hence reasonable to assume that the CIS wavefunction is a good approximation of the CASSCF wavefunction. We optimized the geometries of S1 and T1 with CIS/def2-SVP for DABNA-H, calculated the hessians of both states, and the derivatives of the spin–orbit coupling along all normal coordinates. These data were input into a home-written program that implements the formulae of Baiardi96 for the ISC rate constants. Fig. 11 shows the ISC and rISC rate constants between T1 and S1 as a function of the energy gap. At an experimental energy gap of ca. 1200 cm−1 the calculated rates are 7.6 × 106 s−1 and 3.1 × 104 s−1 for ISC and rISC, respectively. At the calculated energy gap (0.18 eV, CAS(10|10)+NEVPT2/def2-TZVP) the rates of kISC and krISC are 9.4 × 106 s−1 and 2.3 × 104 s−1. Shizu and Kaji found very small rates for kISC and krISC (T1 → S1); therefore the T2 state was considered as an intermediate.93 However, there is no experimental evidence that this T2 state is really near enough to T1 to allow thermal back population. In fact, our rate constant for krISC along T1 → S1 is in good agreement with the experimental result (1.0 × 104 s−1). The calculated rate for kISC is ca. 10% of the decay rate of spontaneous fluorescence, which explains the observed quantum yield of 88% very well. We conclude that the absence of TADF for DABNA-1 in solution40 is due to the rather small spin–orbit coupling between S1 and T1. This is in agreement with our experimental results. The difference between fluorescence and phosphorescence suggests an energy gap of 0.18 eV for DABNA-1 and 0.14 eV for DtBuCzB.
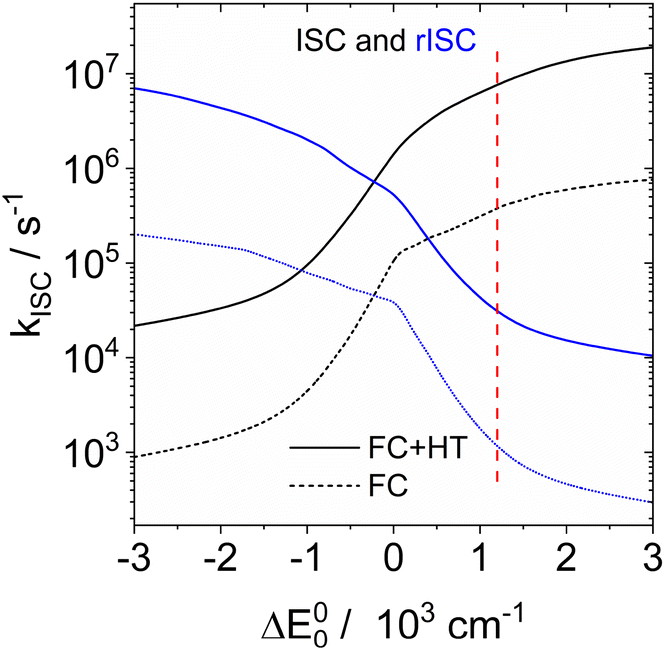 | ||
| Fig. 11 ISC (black curve) and rISC (blue curve) rate constants of DABNA-H calculated with CIS/def2-SVP wavefunctions for the optimized geometries of S1 and T1, as a function of the energy gap. The dashed line is the Franck–Condon contribution, and the solid line includes Herzberg–Teller coupling. The vertical line marks an experimental energy gap of 1200 cm−1. | ||
The MRE idea claims that the positioning of boron and nitrogen atoms as substituents to benzene rings with a relative ortho-arrangement will localize the HOMO and LUMO on different atoms, thus reducing the exchange integral for this pair of atoms. We tested this idea by calculating the exchange integral from the difference of the expectation values of the energies for the HOMO–LUMO singlet and triplet configuration based on the orbitals obtained from the electronic ground state calculated with RHF/def2-SVP. We obtain an energy gap (i.e. twice the exchange integral) between these two configurations of 0.766 eV for DABNA-H, 0.765 eV for DABNA-1, and 0.543 eV for CzB-H. These values are too large for a TADF compound.
Mixing of many singly excited configurations e.g., by the CIS or TD-DFT methods yields similar energy gaps. Apparently, the TADF properties of these compounds cannot be traced to a small exchange integral. Hall et al.66 showed that the energy gaps of these compounds become small when correlations involving doubly and higher excited configurations are included with the SCS-CC2 method. We confirm this conclusion by CASSCF-NEVPT2 calculations. It will hence be very interesting to understand why the MRE design principle proposed by Hatakeyama37 results in the correct prediction of good candidates for TADF although the original idea of small exchange integrals is not confirmed. Based on the above results, the photophysical process of MRE TADF molecules are summarized in Scheme 2.
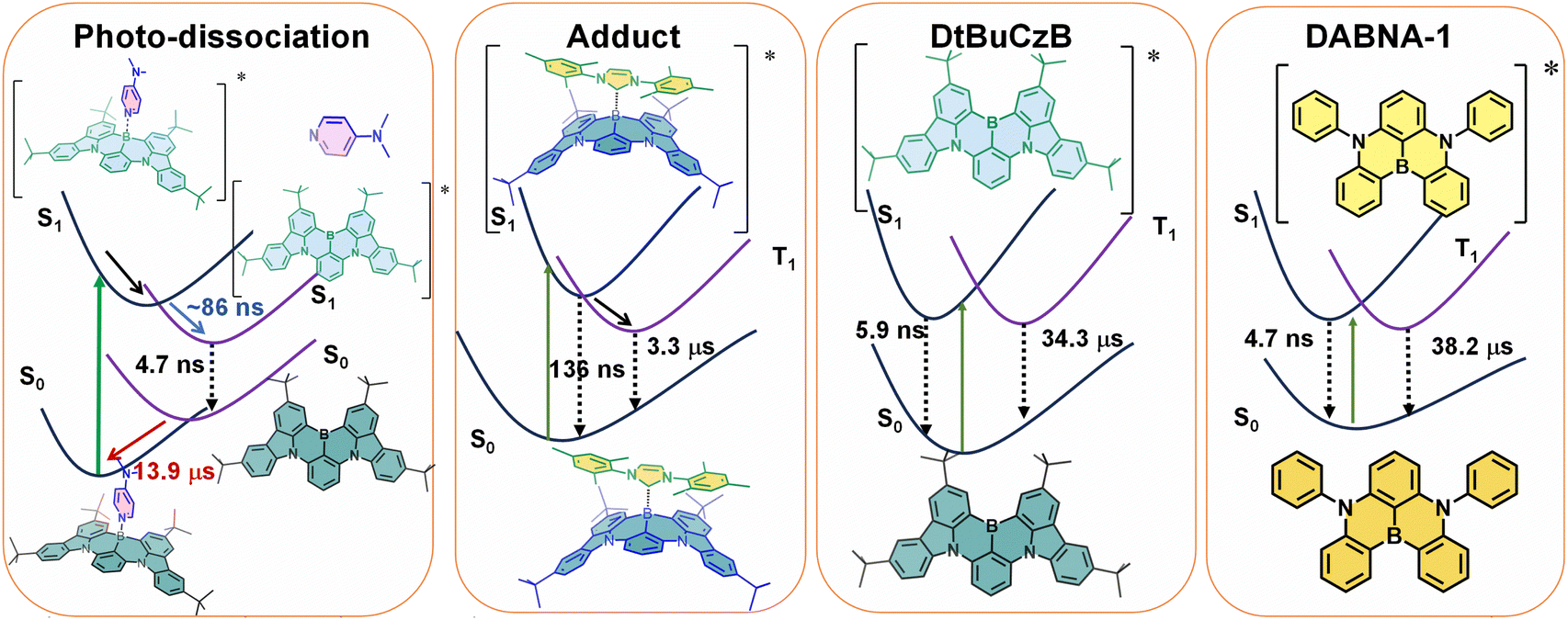 | ||
| Scheme 2 Simplified Jablonski diagram illustrating the photophysical processes involved in DtBuCzB-DMAP, DtBuCzB-NHC, DtBuCzB and DABNA-1 upon photoexcitation. | ||
Conclusions
We studied the photophysics of two representative thermally activated delayed fluorescence (TADF) emitters (DABNA-1 and DtBuCzB) based on the multiple resonance effect (MRE), by using various steady state and transient optical and electron paramagnetic resonance (EPR) spectroscopic methods. For these MRE compounds, the electron exchange energy (J) is proposed to be small, leading to a small ΔES1/T1 energy gap (2J), thus facilitating the reverse intersystem crossing (rISC) and TADF. Previous theoretical computations suggested that the lowest excited state of these compounds has short range charge separation character, with the HOMO and LUMO localized on different atoms in the molecular structure.Spectral studies do not show charge transfer absorption bands, and the fluorescence is independent of the solvent polarity. These features are drastically different from the conventional charge transfer (CT) states in the D–A TADF emitters which contain separate electron donor and acceptor units. A long-lived triplet state (τT = 38.2 μs and 34.3 μs, ΦΔ = 1% and 10%) was observed for these compounds in fluid solution at room temperature; however, no significant TADF was reported. Our computations show that the slow ISC is due to small SOC and small Herzberg–Teller coupling, which rationalizes the lack of TADF although the S1/T1 energy gap is small (0.14 eV). We found that the boron atom in these compounds behaves like a planar Lewis acidic centre and can bind with a Lewis base, such as 4-dimethylaminopyridine (DMAP) and a representative N-heterocyclic carbene (NHC). DtBuCzB shows a higher binding constant with DMAP [(4.51 ± 0.007) × 103 M−1] and NHC [(5.06 ± 0.03) × 103 M−1], in comparison to the binding of DABNA-1 with DMAP [(1.06 ± 0.001) × 102 M−1]. Upon photoexcitation, photo-dissociation of the DMAP adduct occurs and fluorescence from the photo-released native DABNA-1 or DtBuCzB is observed. Photodissociation was confirmed by femtosecond and nanosecond transient absorption (fs/ns-TA) spectroscopy. The dissociation process is slow, taking about 86 ns for completeness. Subsequent re-binding of the MRE molecules with DMAP was observed, which takes ca. 14 μs under our specific experimental conditions, indicating that it is a diffusion-controlled process. No photodissociation was observed for the adduct of DtBuCzB with NHC, which emits at 383 nm (the pristine DtBuCzB emits at 481 nm). Interestingly, for the DtBuCzB/NHC and DABNA-1/NHC adducts, TADF was observed at room temperature (the delayed fluorescence lifetimes are τDF = 136 ns and 127 ns). The triplet excited state of the TADF emitters based on the MRE was also studied by time-resolved electron paramagnetic resonance (TREPR) spectroscopy. The triplet states of the free MRE molecules are characterized by the same parameter D but differ in the degree of orthorhombic distortion. Upon binding of DABNA-1 with DMAP, an increased ZFS D parameter is observed which agrees with the reduced π-conjugation upon binding. These in-depth photophysical studies are helpful for understanding the mechanism of the TADF emitters based on the MRE, as well as for study of fundamental photochemistry of triplet excited states.
Data availability
All relevant data are presented in the main text and/or ESI.†Author contributions
X. Chen, L. Sun, A. A. Sukhanov and S. Doria contributed equally to this work; J. Zhao conceived the research, directed the analysis of the results; X. Chen, L. Sun and H. Xu synthesized the materials; X. Chen performed the UV-vis absorption, fluorescence and nanosecond transient absorption studies and performed parts of the data analysis; A. A. Sukhanov and V. K. Voronkova did the TREPR spectral studies and analyzed the data; S. Doria, L. Bussotti and M. Di Donato did the femtosecond transient absorption studies and analyzed the data; B. Dick performed the calculations with wavefunction based methods and of the ISC rate constants, using ORCA program and analyzed the data; J. Zhao, H. Xu, B. Dick, V. K. Voronkova and M. D. Donato wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z. thanks the National Key Research and Development Program of China (the Ministry of Science and Technology, No. 2023YFE0197600), the NSFC (U2001222), the Research and Innovation Team Project of the Dalian University of Technology (DUT2022TB10), the Fundamental Research Funds for the Central Universities (DUT22LAB610) and the State Key Laboratory of Fine Chemicals for financial support. H. X. thanks the Open Research Fund of the School of Chemistry and Chemical Engineering of Henan Normal University for financial support. A. A. S. and V. K. V. acknowledge financial support from the government assignment for the FRC Kazan Scientific Centre of RAS. M. D. D. thanks the European Union's Horizon 2020 Research and Innovation Program under grant agreement n. 871124 Laser lab-Europe for the support. Lei Sun is grateful for the funding of Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX23_1167).Notes and references
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- S. Fukuzumi, Pure Appl. Chem., 2007, 79, 981–991 CrossRef CAS.
- A. Devižis, J. De Jonghe-Risse, R. Hany, F. Nüesch, S. Jenatsch, V. Gulbinas and J.-E. Moser, J. Am. Chem. Soc., 2015, 137, 8192–8198 CrossRef PubMed.
- A. N. Bartynski, M. Gruber, S. Das, S. Rangan, S. Mollinger, C. Trinh, S. E. Bradforth, K. Vandewal, A. Salleo, R. A. Bartynski, W. Bruetting and M. E. Thompson, J. Am. Chem. Soc., 2015, 137, 5397–5405 CrossRef CAS PubMed.
- Y. Xu, J. Zheng, J. O. Lindner, X. Wen, N. Jiang, Z. Hu, L. Liu, F. Huang, F. Würthner and Z. Xie, Angew. Chem., Int. Ed., 2020, 59, 10363–10367 CrossRef CAS PubMed.
- S. Fukuzumi, H. Kotani, K. Ohkubo, S. Ogo, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2004, 126, 1600–1601 CrossRef CAS PubMed.
- O. Kei and F. Shunichi, Bull. Chem. Soc. Jpn., 2009, 82, 303–315 CrossRef.
- S.-L. Meng, C. Ye, X.-B. Li, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2022, 144, 16219–16231 CrossRef CAS PubMed.
- J. Zhou, P. Chen, X. Wang, Y. Wang, Y. Wang, F. Li, M. Yang, Y. Huang, J. Yu and Z. Lu, Chem. Commun., 2014, 50, 7586–7589 RSC.
- L.-S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- T. Huang, W. Jiang and L. Duan, J. Mater. Chem. C, 2018, 6, 5577–5596 RSC.
- M.-Y. Leung, M.-C. Tang, W.-L. Cheung, S.-L. Lai, M. Ng, M.-Y. Chan and V. Wing-Wah Yam, J. Am. Chem. Soc., 2020, 142, 2448–2459 CrossRef CAS PubMed.
- Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030–1069 RSC.
- Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- B. D. Dherange, M. Yuan, C. B. Kelly, C. A. Reiher, C. Grosanu, K. J. Berger, O. Gutierrez and M. D. Levin, J. Am. Chem. Soc., 2023, 145, 17–24 CrossRef CAS PubMed.
- W.-K. Kwok, L.-K. Li, S.-L. Lai, M.-Y. Leung, W. K. Tang, S.-C. Cheng, M.-C. Tang, W.-L. Cheung, C.-C. Ko, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2023, 145, 9584–9595 CrossRef CAS PubMed.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- D. M. E. Freeman, A. J. Musser, J. M. Frost, H. L. Stern, A. K. Forster, K. J. Fallon, A. G. Rapidis, F. Cacialli, I. McCulloch, T. M. Clarke, R. H. Friend and H. Bronstein, J. Am. Chem. Soc., 2017, 139, 11073–11080 CrossRef CAS PubMed.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- G. Tang, A. A. Sukhanov, J. Zhao, W. Yang, Z. Wang, Q. Liu, V. K. Voronkova, M. Di Donato, D. Escudero and D. Jacquemin, J. Phys. Chem. C, 2019, 123, 30171–30186 CrossRef CAS.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- X. Zhang, X. Liu, M. Taddei, L. Bussotti, I. Kurganskii, M. Li, X. Jiang, L. Xing, S. Ji, Y. Huo, J. Zhao, M. Di Donato, Y. Wan, Z. Zhao and M. V. Fedin, Chem.–Eur. J., 2022, 28, e202200510 CrossRef CAS PubMed.
- X. Zhao and J. Zhao, Chem. Commun., 2022, 58, 7666–7669 RSC.
- X. Zhao, A. A. Sukhanov, X. Jiang, J. Zhao and V. K. Voronkova, J. Phys. Chem. Lett., 2022, 13, 2533–2539 CrossRef CAS PubMed.
- X. Zhang, X. Zhao, K. Ye and J. Zhao, Chem.–Eur. J., 2023, 29, e202203737 CrossRef PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- L. Chen, W.-C. Chen, Z. Yang, J.-H. Tan, S. Ji, H.-L. Zhang, Y. Huo and C.-S. Lee, J. Mater. Chem. C, 2021, 9, 17233–17264 RSC.
- H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581–13585 CrossRef CAS PubMed.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311–9314 CrossRef CAS PubMed.
- I. Kim, K. H. Cho, S. O. Jeon, W.-J. Son, D. Kim, Y. M. Rhee, I. Jang, H. Choi and D. S. Kim, JACS Au, 2021, 1, 987–997 CrossRef CAS PubMed.
- X. Wu, B.-K. Su, D.-G. Chen, D. Liu, C.-C. Wu, Z.-X. Huang, T.-C. Lin, C.-H. Wu, M. Zhu, E. Y. Li, W.-Y. Hung, W. Zhu and P.-T. Chou, Nat. Photonics, 2021, 15, 780–786 CrossRef CAS.
- J.-K. Li, X.-Y. Chen, Y.-L. Guo, X.-C. Wang, A. C. H. Sue, X.-Y. Cao and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 17958–17963 CrossRef CAS PubMed.
- J. Eng and T. J. Penfold, Commun. Chem., 2021, 4, 91 CrossRef PubMed.
- Q. Ou, Y. Shao and Z. Shuai, J. Am. Chem. Soc., 2021, 143, 17786–17792 CrossRef CAS PubMed.
- I. S. Park, H. Min and T. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202205684 CrossRef CAS PubMed.
- X. Cao, K. Pan, J. Miao, X. Lv, Z. Huang, F. Ni, X. Yin, Y. Wei and C. Yang, J. Am. Chem. Soc., 2022, 144, 22976–22984 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, Y. Yamasaki, R. Matsumoto, M. Yoshioka, D. Fukushima, S. Nakatsuka and T. Hatakeyama, J. Am. Chem. Soc., 2022, 144, 106–112 CrossRef CAS PubMed.
- Y. Sano, T. Shintani, M. Hayakawa, S. Oda, M. Kondo, T. Matsushita and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 11504–11511 CrossRef CAS PubMed.
- S. Uemura, S. Oda, M. Hayakawa, R. Kawasumi, N. Ikeda, Y.-T. Lee, C.-Y. Chan, Y. Tsuchiya, C. Adachi and T. Hatakeyama, J. Am. Chem. Soc., 2023, 145, 1505–1511 CrossRef CAS PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468–19472 CrossRef CAS PubMed.
- Y. Liu, X. Xiao, Z. Huang, D. Yang, D. Ma, J. Liu, B. Lei, Z. Bin and J. You, Angew. Chem., Int. Ed., 2022, 61, e202210210 CrossRef CAS PubMed.
- J. M. dos Santos, D. Sun, J. M. Moreno-Naranjo, D. Hall, F. Zinna, S. T. J. Ryan, W. Shi, T. Matulaitis, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, S. L. Warriner, Y. Olivier, M. J. Fuchter and E. Zysman-Colman, J. Mater. Chem. C, 2022, 10, 4861–4870 RSC.
- T. Hosokai, H. Matsuzaki, A. Furube, K. Tokumaru, T. Tsutsui, H. Nakanotani, M. Yahiro and C. Adachi, SID Symposium Digest of Technical Papers, 2016, vol. 47, pp. 786–789 Search PubMed.
- J. Choi, D.-S. Ahn, K. Y. Oang, D. W. Cho and H. Ihee, J. Phys. Chem. C, 2017, 121, 24317–24323 CrossRef CAS.
- W. Zhang, H. Song, J. Kong, Z. Kuang, M. Li, Q. Guo, C.-f. Chen and A. Xia, J. Phys. Chem. C, 2019, 123, 19322–19332 CrossRef CAS.
- Y. Guo, Y. Gao, J. Zhang, C. Wang, Y. Wang, X. Feng and G. Zhao, J. Phys. Chem. C, 2023, 127, 4784–4791 CrossRef CAS.
- T. Ogiwara, Y. Wakikawa and T. Ikoma, J. Phys. Chem. A, 2015, 119, 3415–3418 CrossRef CAS PubMed.
- E. W. Evans, Y. Olivier, Y. Puttisong, W. K. Myers, T. J. H. Hele, S. M. Menke, T. H. Thomas, D. Credgington, D. Beljonne, R. H. Friend and N. C. Greenham, J. Phys. Chem. Lett., 2018, 9, 4053–4058 CrossRef CAS PubMed.
- N. Sharma, M. Y. Wong, D. Hall, E. Spuling, F. Tenopala-Carmona, A. Privitera, G. Copley, D. B. Cordes, A. M. Z. Slawin, C. Murawski, M. C. Gather, D. Beljonne, Y. Olivier, I. D. W. Samuel and E. Zysman-Colman, J. Mater. Chem. C, 2020, 8, 3773–3783 RSC.
- B. H. Drummond, N. Aizawa, Y. Zhang, W. K. Myers, Y. Xiong, M. W. Cooper, S. Barlow, Q. Gu, L. R. Weiss, A. J. Gillett, D. Credgington, Y.-J. Pu, S. R. Marder and E. W. Evans, Nat. Commun., 2021, 12, 4532 CrossRef CAS PubMed.
- Y. Kobori, M. Fuki and H. Murai, J. Phys. Chem. B, 2010, 114, 14621–14630 CrossRef CAS PubMed.
- N. Zarrabi, B. J. Bayard, S. Seetharaman, N. Holzer, P. Karr, S. Ciuti, A. Barbon, M. Di Valentin, A. van der Est, F. D'Souza and P. K. Poddutoori, Phys. Chem. Chem. Phys., 2021, 23, 960–970 RSC.
- X. Xiao, I. Kurganskii, P. Maity, J. Zhao, X. Jiang, O. F. Mohammed and M. Fedin, Chem. Sci., 2022, 13, 13426–13441 RSC.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 CrossRef CAS.
- Y. Gao, Y. Wang, Z. Guo, Y. Wan, C. Li, B. Yang, W. Yang and X. Ma, J. Phys. Chem. B, 2022, 126, 2729–2739 CrossRef CAS PubMed.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- D. Hall, J. C. Sancho-García, A. Pershin, G. Ricci, D. Beljonne, E. Zysman-Colman and Y. Olivier, J. Chem. Theory Comput., 2022, 18, 4903–4918 CrossRef CAS PubMed.
- M. Saigo, Y. Shimoda, T. Ehara, T. Ryu, K. Miyata and K. Onda, Bull. Chem. Soc. Jpn., 2022, 95, 381–388 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley-VCH Verlag GmbH, 2001 Search PubMed.
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 1901627 CrossRef CAS.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J.-L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636–642 CrossRef CAS.
- S. Goto, Y. Nitta, N. O. Decarli, L. E. de Sousa, P. Stachelek, N. Tohnai, S. Minakata, P. de Silva, P. Data and Y. Takeda, J. Mater. Chem. C, 2021, 9, 13942–13953 RSC.
- Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529–4532 CrossRef CAS PubMed.
- Y. Qi, X. Cao, Y. Zou and C. Yang, J. Mater. Chem. C, 2021, 9, 1567–1571 RSC.
- K. Matsuo, S. Saito and S. Yamaguchi, J. Am. Chem. Soc., 2014, 136, 12580–12583 CrossRef CAS.
- N. Ando, T. Yamada, H. Narita, N. N. Oehlmann, M. Wagner and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 9944–9951 CrossRef CAS PubMed.
- F.-W. Gao, R.-L. Zhong, H.-L. Xu and Z.-M. Su, J. Phys. Chem. C, 2017, 121, 25472–25478 CrossRef CAS.
- G. Li, X. Liu, M. Wu, R. Zeng, Q. Li, A. Yuan and C. Shi, Dyes Pigm., 2023, 208, 110805 CrossRef.
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC.
- Y. Kitamoto, F. Kobayashi, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Dalton Trans., 2019, 48, 2118–2127 RSC.
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS PubMed.
- S.-B. Zhao, T. McCormick and S. Wang, Inorg. Chem., 2007, 46, 10965–10967 CrossRef CAS PubMed.
- Y. Sun, Z. M. Hudson, Y. Rao and S. Wang, Inorg. Chem., 2011, 50, 3373–3378 CrossRef CAS PubMed.
- C. J. Berger, G. He, C. Merten, R. McDonald, M. J. Ferguson and E. Rivard, Inorg. Chem., 2014, 53, 1475–1486 CrossRef CAS PubMed.
- M. Saigo, K. Miyata, S. i. Tanaka, H. Nakanotani, C. Adachi and K. Onda, J. Phys. Chem. Lett., 2019, 10, 2475–2480 CrossRef CAS PubMed.
- T. Biskup, Front. Chem., 2019, 7, 10 CrossRef CAS PubMed.
- H. Levanon and J. R. Norris, Chem. Rev., 1978, 78, 185–198 CrossRef CAS.
- S. Richert, C. E. Tait and C. R. Timmel, J. Magn. Reson., 2017, 280, 103–116 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- K. Shizu and H. Kaji, Chem. Commun., 2022, 5, 53 CrossRef CAS PubMed.
- B. de Souza, F. Neese and R. Izsák, J. Chem. Phys., 2018, 148, 034104 CrossRef PubMed.
- B. de Souza, G. Farias, F. Neese and R. Izsák, J. Chem. Theory Comput., 2019, 15, 1896–1904 CrossRef CAS PubMed.
- A. Baiardi, J. Bloino and V. Barone, J. Chem. Theory Comput., 2013, 9, 4097–4115 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02513j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |