
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKey structural features to favour imines over hydrates in water: pyridoxal phosphate as a muse†
Ferran
Esteve
 *,
Tanguy
Rieu
and
Jean-Marie
Lehn
*
*,
Tanguy
Rieu
and
Jean-Marie
Lehn
*
Laboratoire de Chimie Supramoléculaire, Institut de Science et d’Ingénierie Supramoléculaires (ISIS), Université de Strasbourg, 8 allée Gaspard Monge, Strasbourg, 67000, France. E-mail: estevefranch@unistra.fr; lehn@unistra.fr
First published on 11th June 2024
Abstract
Imination reactions in water represent a challenge not only because of the high propensity of imines to be hydrolysed but also as a result of the competing hydrate formation through H2O addition to the aldehyde. In the present work we report a successful approach that allows for favouring imitation reactions while silencing hydrate formation. Such remarkable reactivity and selectivity can be attained by fine-tuning the electronic and steric structural features of the ortho-substituents of the carbonyl groups. It resulted from studying the structure–reactivity relationships in a series of condensation reactions between different amines and aldehydes, comparing the results to the ones obtained in the presence of the biologically-relevant pyridoxal phosphate (PLP). The key role of negatively-charged and sterically-crowding units (i.e., sulfonate groups) in disfavouring hydrate formation was corroborated by DFT and steric-hindrance calculations. Furthermore, the best-performing aldehyde leads to higher imine yields, selectivity and stability than those of PLP itself, allowing for the inhibition of a PLP-dependent enzyme (transaminase) through dynamic aldimine exchange. These results will increase the applicability of imine-based dynamic covalent chemistry (DCvC) under physiological conditions and will pave the way for the design of new carbonyl derivatives that might be used in the dynamic modification of biomolecules.
Introduction
Imine-based dynamic covalent reactions have offered remarkable advantages in a wide range of fields.1–3 The reversible nature of aldimine covalent bonds allows for thermodynamically-driven dynamic rearrangements and error-checking processes,4 with the species (or mixture) that presents the lowest free energy predominating in equilibrium under specific conditions.5–8 Such dynamicity proved useful for the development of systems displaying a behaviour of higher complexity.9–11 Nevertheless, the high propensity of imines to be hydrolysed in aqueous media has hampered their application under physiological conditions.12 As a consequence, many research efforts have been devoted in the last years toward the implementation of chemical methods that increase the stability of imines in water, mainly using covalent/supramolecular multivalency and hydrophobic effects.13–17 In nature, imitation reactions are achieved in the sophisticatedly-arranged active sites of enzymes, using pyridoxal phosphate (PLP) as the aldehyde.18 The mode-of-action of this cofactor has been extensively studied as it plays an essential role in numerous enzymatic processes (e.g., racemizations, transaminations, aldol cleavages, and decarboxylations).19,20 Briefly, the pyridine ring acts as an electron sink to enhance the electrophilic character of the carbonyl group; the –OH group in ortho-position to the carbonyl unit is involved in amine activation and imine stabilization through H-bonding/electrostatic attractions; and the phosphate sidechain mainly acts as the anchoring scaffold to the protein's active site through multiple supramolecular interactions.21–23 Thus, the high efficiency of PLP-assisted transformations suggests that aldimine formation and transimination reactions can be attained under physiological conditions when the system is properly designed. Despite that a handful of studies showed how supramolecular and electronic interactions can be used to increase imine stability,24–28 the structural features to favour aldimine over the competing hydrate formation in water have been quite overseen.Herein we report a successful approach to increase imine yields while preventing the hydration of the aldehyde by taking advantage of the supramolecular, electronic, and steric preferences of each reaction component (aldehyde, amine, hydroxide/water), intermediate (hemiaminal), and product (hydrate, imine, iminium). Besides, the best-performing aldehyde acts as an inhibitor of the glutamic-pyruvic transaminase likely through dynamic aldimine exchange with the PLP cofactor.
Results and discussion
One notes that increasing the electrophilic character of the aldehyde (e.g., electron-withdrawing substituents) is the most commonly used method to increase imine yields. However, this approach also favours the H2O addition to the activated carbonyl that generates the hydrate, decreasing the aldimine selectivity (Fig. 1A). We envisaged that the introduction of negatively-charged bulky groups near the carbonyl unit may disfavour the hydrate formation due to electrostatic repulsions and crowding effects. Accordingly, sulfonate groups were selected, also taking into account their electron-withdrawing character that could further favour imine formation.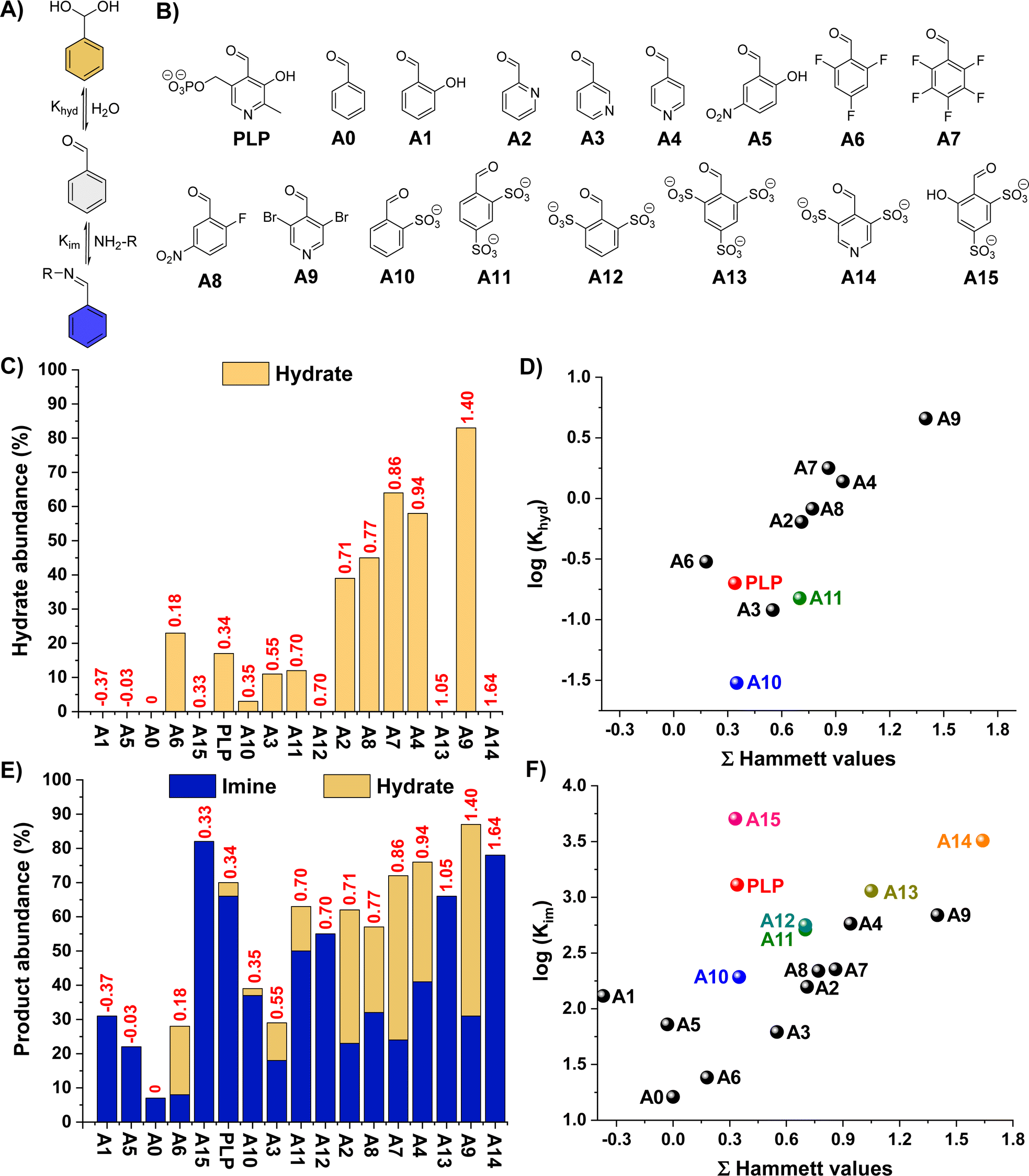 | ||
| Fig. 1 (A) Hydration and imine formation equilibrium. (B) Chemical structures of the aldehydes studied in this work (C) hydrate abundances (%) and (D) Hammett plot of the log(Khyd) for the different aldehydes. Khyd for A1, A5, A0, A12, A13, A14 and A15 could not be calculated because no hydrate was detected. (E) Hydrate-imine product abundances (%) and (F) Hammett plot of the log(Kim) for the reaction between the aldehydes and B1. The results have been sorted with increased ΣHammett scores (red values in C and E). Results for PLP and aldehydes of major interest have been coloured. Hydration reaction conditions: 5 mM aldehyde, D2O, pD 7.0 (50 mM PBS), 295 K, 1 h. Imination reaction conditions: 5 mM aldehyde and 5 mM B1, D2O, pD 7.0 (50 mM PBS), 295 K, 3 h. Product abundances determined by 1H NMR spectroscopy (500 MHz, 295 K). Error in NMR measurements: 5%. | ||
The propensity of a series of benzaldehyde derivatives (AX, Fig. 1B) and PLP to form hydrates was first assayed in D2O (5 mM aldehyde, pD 7.0, 50 mM PBS). Aldehydes A12–A15 were then synthesized in one-step following reported procedures (see ESI† for details).29 The Hammett plot of the apparent hydration equilibrium constants (Khyd) showed the expected increase in hydrate formation for the aldehydes containing more electron-withdrawing substituents (higher Hammett values) with a slope of 1.7 (e.g., A0 < A6 < A2 < A8 < A4 < A9, Fig. 1C and Table S1;† see also Fig. 1D),30,31 in good agreement with previous reports.32 Interestingly, however, the species containing two sulfonate groups in ortho-position led to significantly lower Khyd than aldehydes with similar Hammet values (see for instance A11vs.A2 in Fig. 1D), suggesting a destabilizing effect of these negatively-charged bulky groups towards hydrate formation. The effect of pD on the aldehyde hydration was also assessed. Dissolving the aldehydes at pD 0.6 resulted in the expected increase in hydrate formation for PLP, A2, A3, A4, and A9 due to the protonation of their pyridine nitrogen site (Fig. S1†). In contrast, pyridinium A14 was barely hydrated under these conditions despite its great Hammett score (3.27), stressing that the sulfonate groups were disfavouring aldehyde hydration. The hydrate abundance of the other aldehydes did not significantly change at such acidic pD. An increase in the pD of the solution to 10.6 led to minor differences when compared to the results obtained at pD 7.0 (Fig. S2†). At pD 13.6, hydrate abundances ≥95% were obtained for A2, A3, A4, A6, A7, A8, and A9 (Fig. S3†), but no hydration was observed for PLP, A1, A5, and A10–A15, corroborating the key role of bulky and/or negatively-charged ortho-substituents in preventing hydrate formation.
We then screened the imine formation reactions by studying the apparent equilibrium constants (Kim) for the cholamine chloride (B1) addition to the different aldehydes, since its terminal –NH2 group is barely protonated at such pD.33 In general, the Kim increased with higher Hammet scores. Notwithstanding, the non-linearity of the results gave insight into the key structural features to favour imines over hydrates (Fig. 1E and F, see also Fig. S4 and S5†):
(i) Aldehydes with small and neutral electron-withdrawing groups (i.e., A2–A4, A6–A9) do not effectively form imines due to significant competing hydrate formation (Fig. 1F; see also grey fitting in Fig. S5†).
(ii) Aldehydes with bulky and negatively-charged ortho-substituents (i.e., A10–A14) barely form hydrates and give high Kim, leading to remarkable imine-to-hydrate selectivities (Fig. S4;† see also brown fitting in Fig. S5†). In addition, these negative groups might also be involved in the stabilization of the potential iminium cations through attractive electrostatic interactions and H-bonding.22
(iii) Salicylaldehyde derivatives (viz.A1, A5 and PLP) promote relatively high Kim despite their low Hammett values of −0.37, −0.03 and 0.34, respectively (Fig. 1F; see also yellow fitting in Fig. S5†).34 This behaviour is ascribed to the presence of intramolecular H-bonding and electrostatic forces that stabilize the aldimine/aldiminium.20,22,23,35
(iv) The combination of (ii) and (iii), as in the case of A15, leads to the highest Kim with excellent aldimine selectivities (Fig. 1F and S4†).
One notes that the abundance of hydrate and imine when using aldehydes with small and neutral electron-withdrawing groups is rather similar, indicating that the difference in energies of formation for such products is small (e.g., A9, A4, A7, A8). On the other hand, this energy difference is much greater in favor of the imine/iminium for aldehydes containing ortho-substituents that can stabilize such aldimine/aldiminium bonds by means of supramolecular interactions (see for instance DFT section below).
The Kim dependence on the pD was also assayed to evaluate the applicability of the sulfonate-containing aldehydes at different pH ranges. Remarkably, the Kim for A15 was almost 10-times higher than that of PLP all over the biologically-relevant pD range (i.e., 6.5–7.8), with imine abundances >90% (Fig. S6†). At lower pD values (<5.5), the addition of B1 to the different aldehydes was hampered by the complete protonation of the terminal –NH2 of the amine (Fig. S7 and S8†). As expected, higher pD decreased the protonation of B1 and resulted in greater Kim for A0, in agreement with previous reports.23 In contrast, aldehydes containing sulfonate (viz.A10, A12, A13, A13 and A15) and/or phenolate (A1, A5, PLP and A15) units in ortho-position showed a decrease in imine formation at pD > 8.5 (Fig. S7 and S8†). We hypothesized that these findings could be related to the iminium/imine equilibrium and its effect on the dynamic covalent reaction. The high δ values (>8.6 ppm) observed for the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– signals of these aldehydes suggested the presence of the corresponding iminium cations, in line with reported results.23 We thus attempted to determine the pKa of the iminium cations (pKa,iminium) by 1H-NMR spectroscopy through the δ changes in the –CHN– signal (Fig. S9†). However, the only species that experienced a significant shift in the –CHN– signal were A5 and PLP/A15, giving pKa,iminium values of ca. 8 and 10.5, respectively. For these aldehydes, the Kim drastically dropped once the pD of the solution overtook their pKa,iminium, suggesting a much higher thermodynamic stability of the iminium cations over that of the imines. In contrast, the –CHN– signal for the B1-condensation products with the aldehydes A10, A11, A12, A13 and A14 barely shifted even at quite high pD, but their Kim still decreased to almost 0. Hence, we rationalized that the pD value where the Kim dropped corresponded to the pKa,iminium, and that the stability of the unprotonated imines must be lower than those of PLP and A15, as no NMR shift indicative of imine formation was observed at pD > pKa,iminium.
N– signals of these aldehydes suggested the presence of the corresponding iminium cations, in line with reported results.23 We thus attempted to determine the pKa of the iminium cations (pKa,iminium) by 1H-NMR spectroscopy through the δ changes in the –CHN– signal (Fig. S9†). However, the only species that experienced a significant shift in the –CHN– signal were A5 and PLP/A15, giving pKa,iminium values of ca. 8 and 10.5, respectively. For these aldehydes, the Kim drastically dropped once the pD of the solution overtook their pKa,iminium, suggesting a much higher thermodynamic stability of the iminium cations over that of the imines. In contrast, the –CHN– signal for the B1-condensation products with the aldehydes A10, A11, A12, A13 and A14 barely shifted even at quite high pD, but their Kim still decreased to almost 0. Hence, we rationalized that the pD value where the Kim dropped corresponded to the pKa,iminium, and that the stability of the unprotonated imines must be lower than those of PLP and A15, as no NMR shift indicative of imine formation was observed at pD > pKa,iminium.
DFT calculations were performed for the hydrate, hemiaminal, imine and iminium derived from A12 (sulfonate groups in both ortho-positions) to shed light on the iminium-to-hydrate selectivity observed experimentally (Fig. 2A). The energetic profiles showed endergonic processes for the formation of the species presenting a Csp3 hybridization: hydrate and hemiaminal. This indicated that the sterically-crowded microenvironment of the carbonyl unit preferred a Csp2 planar conformation rather than the bulkier Csp3 tetrahedral configurations of the hydrate and hemiaminal.36 The preference for C–O vs. C–N release/cleavage from the tetrahedral intermediates may also involve stereoelectronic effects.37,38 Whereas the imine A12B1 presented a subtly exergonic free energy of formation (−1.5 kcal mol−1), a much stronger stabilization was observed on generation of the iminium derivative (ΔG = −13.6 kcal mol−1), in accordance with the experimental results. Such exergonic energies were assigned to the recovery of the sterically-suitable Csp2 hybridization in the A12–B1 condensation products, with additional electrostatic, H-bonding and n → π* interactions stabilizing the iminium cation.27,39
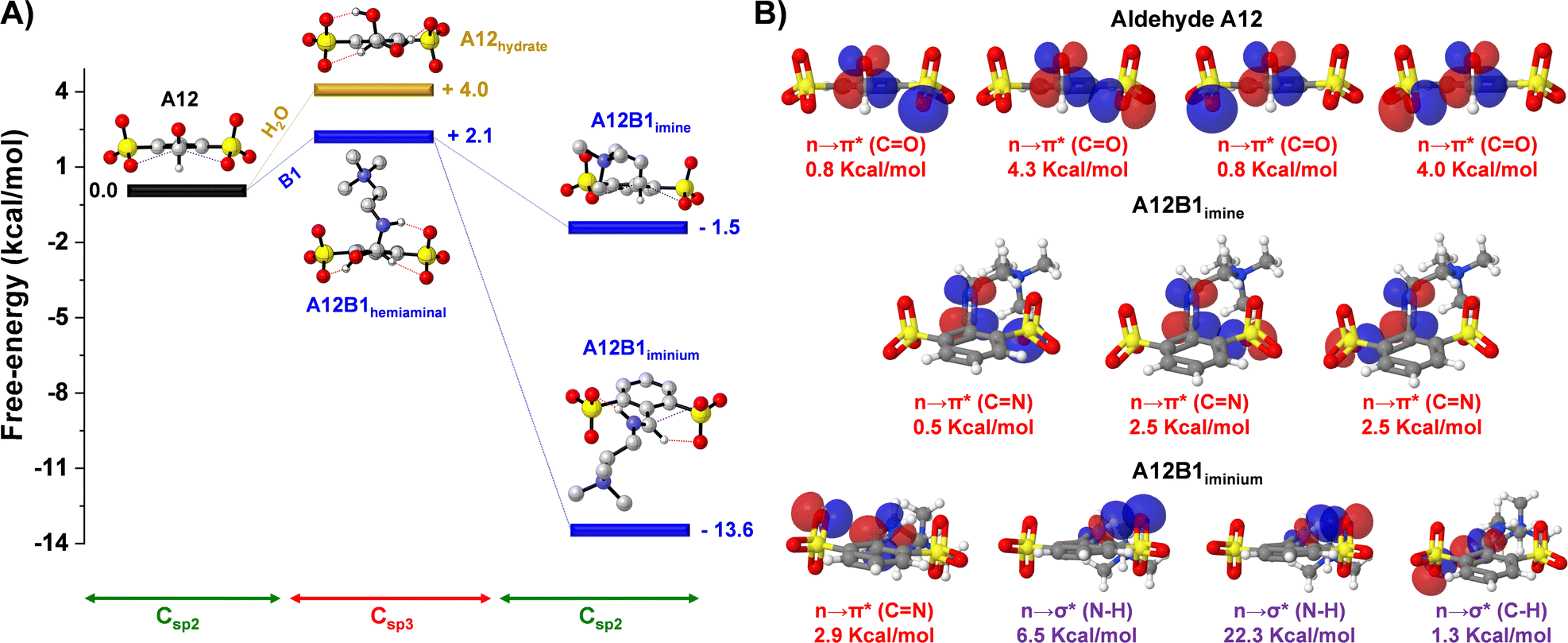 | ||
| Fig. 2 (A) DFT calculated energy profile (b3lyp/6-311g(d,p), PCM = water) for the generation of the intermediate -hemiaminal- and product -hydrate, imine and iminium-of the reaction between A12-H2O (yellow)/A12–B1 (blue). Discontinuous lines in red and purple show the H-bonds and n → π* interactions, respectively. (B) Optimized structures of A12, imine A12B1 and iminium A12B1, with NBOs and energies (kcal mol−1) listed. | ||
The role of the n → π* and H-bond interactions was further studied through natural bond orbital (NBO) analyses to estimate the interaction energies according to second-order perturbation theory (ΔE(2)).26 Two oxygen lone pairs of each of the sulfonate groups (4 lone pairs in total) were overlapping with the antibonding orbital of the carbonyl group in A12, with NBO energies of 0.8, 4.3, 0.8 and 4.0 kcal mol−1 (n → π* (CO) in Fig. 2B; see also Table S2†). The sum (9.9 kcal mol−1) was significantly higher than the one observed for the sulfonate-imine n → π* (CN) interactions (sum = 5.5 kcal mol−1). These values are in agreement with the aldehydes being better electron acceptors than their imines. A different scenario was found for the A12B1 iminium cation. This species was mainly stabilized by a strong hydrogen bonding between the sulfonate group and the acidic proton of the iminium unit (n → σ* (N–H) = 28.8 kcal mol−1), together with a weaker H-bond between the iminium CH and the lone pair of the other ortho-sulfonate substituent (n → σ* (C–H) = (1.3 kcal mol−1)), which also contributed through a sulfonate-iminium n → π* (CN) interaction (2.9 kcal mol−1). Therefore, the low Kim observed for the sulfonate-containing species at high pD values (pD > pKa,iminium) was likely the result of the stronger n → π* interactions for the aldehyde than for the imine derivatives that shifted the equilibrium back to the reagents once the iminium could not be generated.
We envisaged that such bulky and negatively-charged substituents near the electrophilic site might also affect the kinetic preferences of the products.40,41 The steric hindrance of the carbonyl unit was estimated using the SambVca web server, which calculates buried volumes and steric maps (Fig. 3A; see ESI† for details).42,43 Results suggested a remarkably sterically-hindered microenvironment for species presenting sulfonate groups in both ortho-positions, namely A12, A13 and A14, which should experience slower nucleophilic additions. In fact, the CHO buried volumes for such aldehydes represented ca. 27% of the total space, providing a ≈ 9-fold increase in comparison with the value calculated for A0 (3%, Fig. 3A). In terms of the imine/iminium formation, 1H NMR monitoring over time revealed slower rates for the reaction between B1 and A12/A13/A14, needing ca. 3 h to reach the equilibrium state (Fig. 3B). In contrast, aldehydes with CHO buried volumes <20% (i.e., PLP, A10, A11, A15) attainted the equilibrium in less than 10 min, in good accordance with the expected fast imitation reactions in water.12
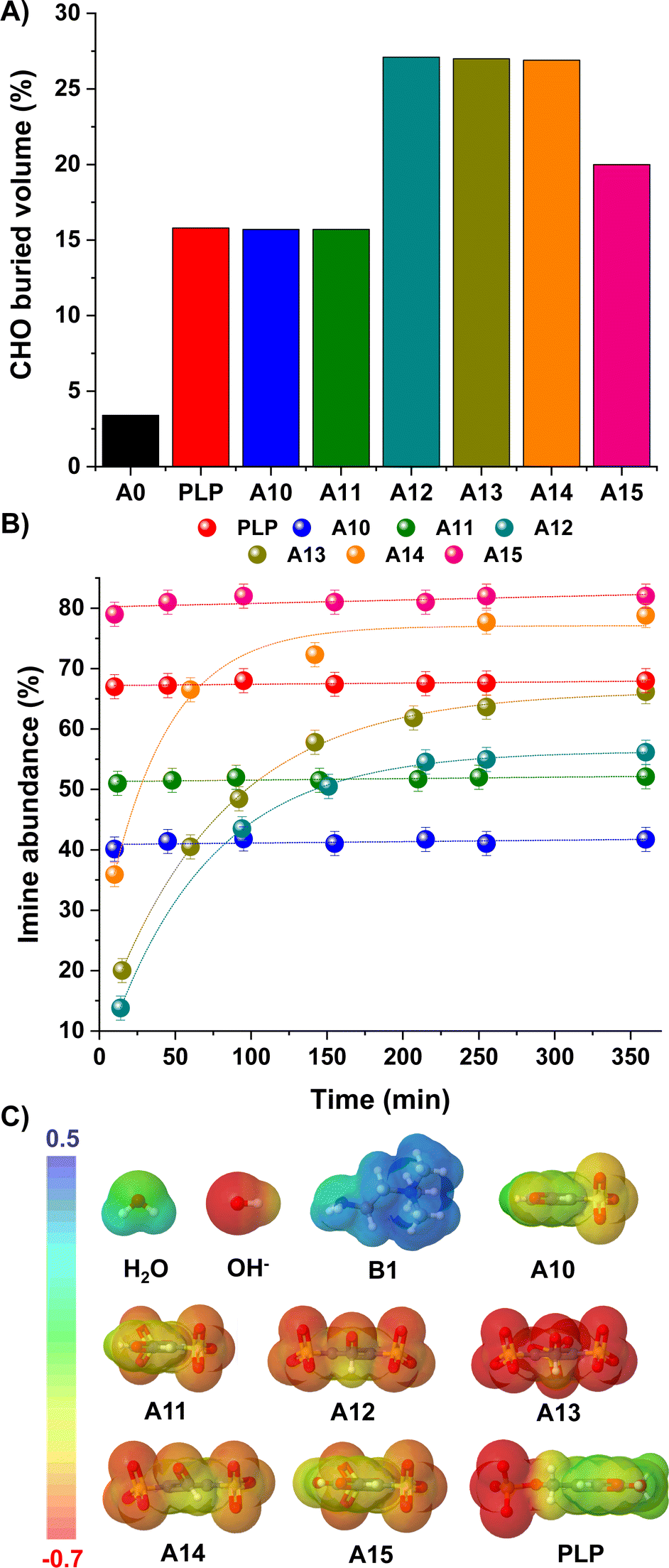 | ||
| Fig. 3 (A) Buried volumes (%) calculated for the carbonyl group of aldehydes A0, PLP, A10–A15. Values calculated using SambVca web server (see ESI†).42,43 (B) Kinetic profiles for the imine formation reaction between B1 and aldehydes PLP and A10–A15. Imine abundances (%) determined by 1H NMR spectroscopy (500 MHz, D2O, pD 7.0, 50 mM PBS, 295 K). Concentration: 5 mM for each component. (C) EDS (b3lyp/6-311g(d,p), PCM = water) for aldehydes A10–15/PLP and nucleophiles H2O/OH−/B1. The isosurface density values range from −0.7 to 0.5 a.u. in all cases. | ||
Concerning the aldehyde hydration, the kinetic profiles were too fast to be measured due to the huge excess of H2O present in the media. Yet, one should note that the presence of strong electrostatic repulsions between the electron-rich O atom of the OH−/H2O and the negatively-charged sulfonate groups may slow down the generation of the hydrate, but without affecting the thermodynamic outcome. Electron density surfaces (EDS) of the aldehydes (A10–15, PLP) and nucleophiles (H2O, OH−, B1) were calculated to evaluate this assumption (Fig. 3C). In the case of the nucleophiles, the electron density of OH− -and to a lesser degree H2O– was substantially more negative than that of B1, in line with the number of lone pairs present in the nucleophilic site (3 for OH−, 2 for H2O and 1 for B1). On the other hand, the negative electron density surrounding the aldehydes increased in the order: A10 ≈ PLP < A11 ≪ A15 < A12 ≈ A14 < A13.
The imine formation dependence on the protonation state of the amines was then evaluated for the best-performing aldehydes PLP, A14 and A15. Results for A14 revealed a drastic decrease in Kim upon reaction with amines containing <10% unprotonated –NH2 groups at pD 7.0 (see orange bars for B3, B4, Val and B5, Fig. 4).33 For example, A14 did not react with Val and B5, and imine yields of ca. 10% were attained with B3 and B4 (pKa values of 9.1 and 9.6, respectively; see Fig. S10 and Table S3†). In contrast, aldehyde A15 gave high imine abundances even when reacted with amines presenting a pKa value >10 (e.g., B5). In a similar manner, PLP was able to react with such amines despite the high degree of protonation of the –NH2 groups at physiological pH, suggesting that the ortho-OH group must be activating the amines for their nucleophilic attack.44 To our delight, A15 was able to render higher Kim than PLP in all cases, stressing its remarkable reactivity (Fig. 4). Moreover, the stability of PLP-derived imines was lower than that of A15, as evidenced by the appearance of side-product signals in the 1H NMR spectrum of PLPB1 after 120 h (Fig. S11 and S12†). The imitation reactions of A14, A15 and PLP with Lys-derivatives were also assayed. As predicted, aldehyde A14 was only able to react with the Nα of LysOMe due to its relatively low –NH2 protonation at pD 7.0 (50%, Fig. S13†),13,45 not forming any imines with the Nε of Lys and LysOMe nor with the Nα of Lys (unprotonated –NH2 < 2%, Table S3; Fig. S14†). On the other hand, both PLP and A15 reacted with the Nα and Nε of LysOMe and Lys, with a slightly preferential aldimine formation at the Nα in both cases. The reaction with Nε was further corroborated using AcNαLysOMe as the nucleophile, observing that only PLP and A15 gave aldimines.
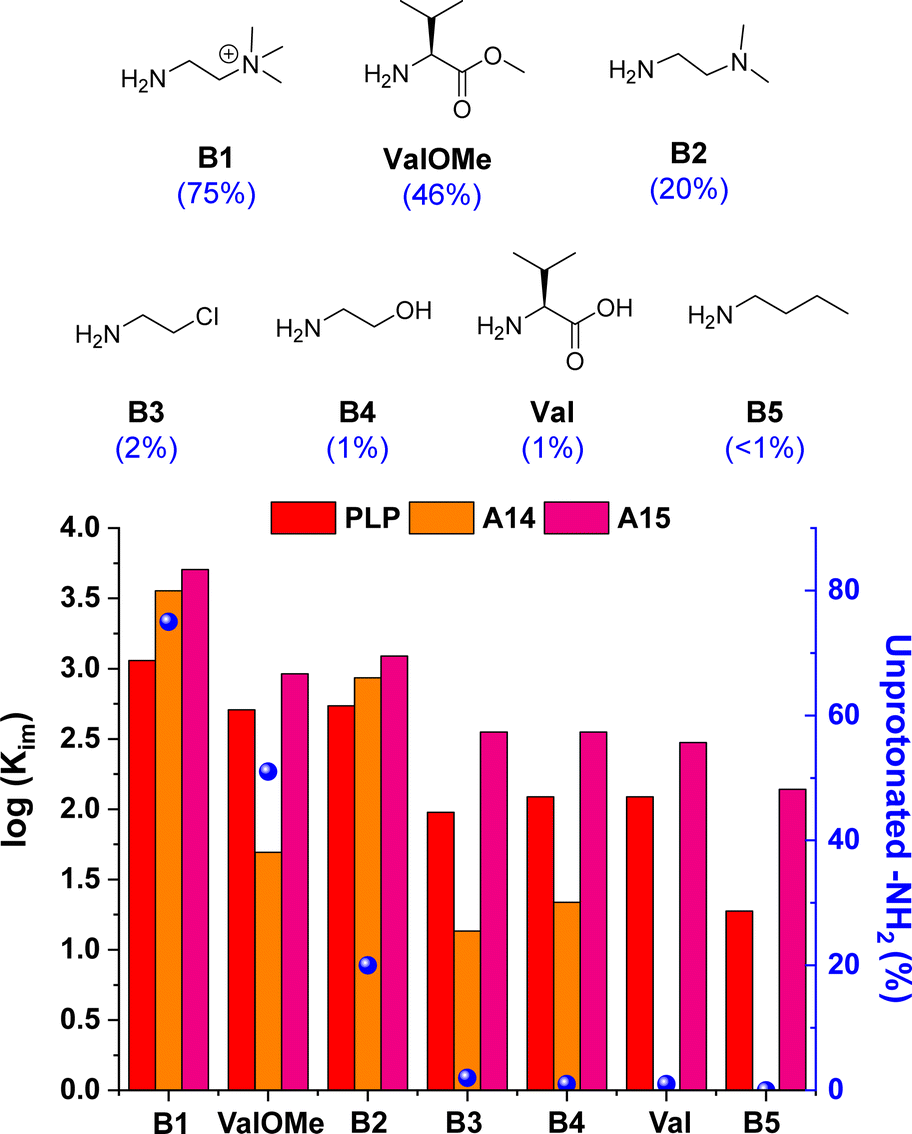 | ||
Fig. 4 Chemical structures (above) and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kim (below) for the amine screening using PLP/A14/A15. Component abundances determined by 1H NMR spectroscopy (500 MHz, D2O, pD 7.0, 50 mM PBS, 295 K). Concentration: 5 mM for each component. The numbers in brackets correspond to the unprotonated –NH2 group at pD 7.0. See Table S3† for pKa and protonation degree of amines. See Fig. S7† for imine abundances (%). Kim (below) for the amine screening using PLP/A14/A15. Component abundances determined by 1H NMR spectroscopy (500 MHz, D2O, pD 7.0, 50 mM PBS, 295 K). Concentration: 5 mM for each component. The numbers in brackets correspond to the unprotonated –NH2 group at pD 7.0. See Table S3† for pKa and protonation degree of amines. See Fig. S7† for imine abundances (%). | ||
The dynamic character of A15-derived aldimines was also evaluated with a simple re-arrangement experiment, observing that when B1 (5 mM final concentration) was introduced into a solution containing A15B5 (5 mM mixture of B5 and A15), B5 was released and A15B1 was formed as the major product (Fig. S15†). This aldimine interconversion occurred instantaneously in the time range of the experiment, stressing the outstanding reversibility of A15-derived aldimines under physiological conditions.
All these results paved the way for studying the regulation of a PLP dependent enzyme (i.e., Glutamic-Pyruvic Transaminase from porcine heart, GPT) with aldehyde A15.46–50GPT is catalytically-active in amino acid-D2O exchange reactions in the presence of pyruvate under slightly basic conditions.51 Thus, we studied the L-alanine-D2O exchange reaction in an NMR tube containing L-alanine (90 mM), pyruvate (1.5 mM), potassium phosphate buffer (100 mM, pD 7.8) and 7 units of GPT (ca. 1 μM). The reaction course was easily followed by 1H NMR observing the deuterium exchange of both α and β hydrogens of L-alanine (black points in Fig. 5A; see also Fig. S16†). The experiment in the absence of GPT showed no reaction (grey points in Fig. 5A). As expected, the addition of a large excess of PLP and A14 did not change the catalytic activity of GPT (red and orange points in Fig. 5A, respectively), since PLP is already present as the cofactor and A14 does not form imines with Nε-Lys (see Fig. S13†). In contrast, 200 eq. of A15 triggered a remarkable inhibition of the enzyme (ca. 90%). Although lower amounts of A15 led to a less efficient inhibition, the IC50 for this aldehyde was about 15 μM (ca. 15 eq). To support that the inhibition observed was indicative of A15-PLP aldimine exchange, we studied the amount of A15 required to replace PLP from the preformed PLPAcNαLysOMe aldimine under related experimental conditions (pD 7.8). The presence of 0.7 eq. of A15 resulted in a significant PLP replacement (42%), but 4 eq. of A15 were needed to reach an aldimine exchange >90% (Fig. S17†). The higher number of equivalents needed to replace PLP by A15 in the active site of GPT was assigned to a supramolecular mismatch between the para-sulfonate group of A15 and the carboxylate group of the aspartic acid residue (see Fig. S18† for the example of an analogous PLP-complex of the Human alanine aminotransferase 2, PDB: 3IHJ).
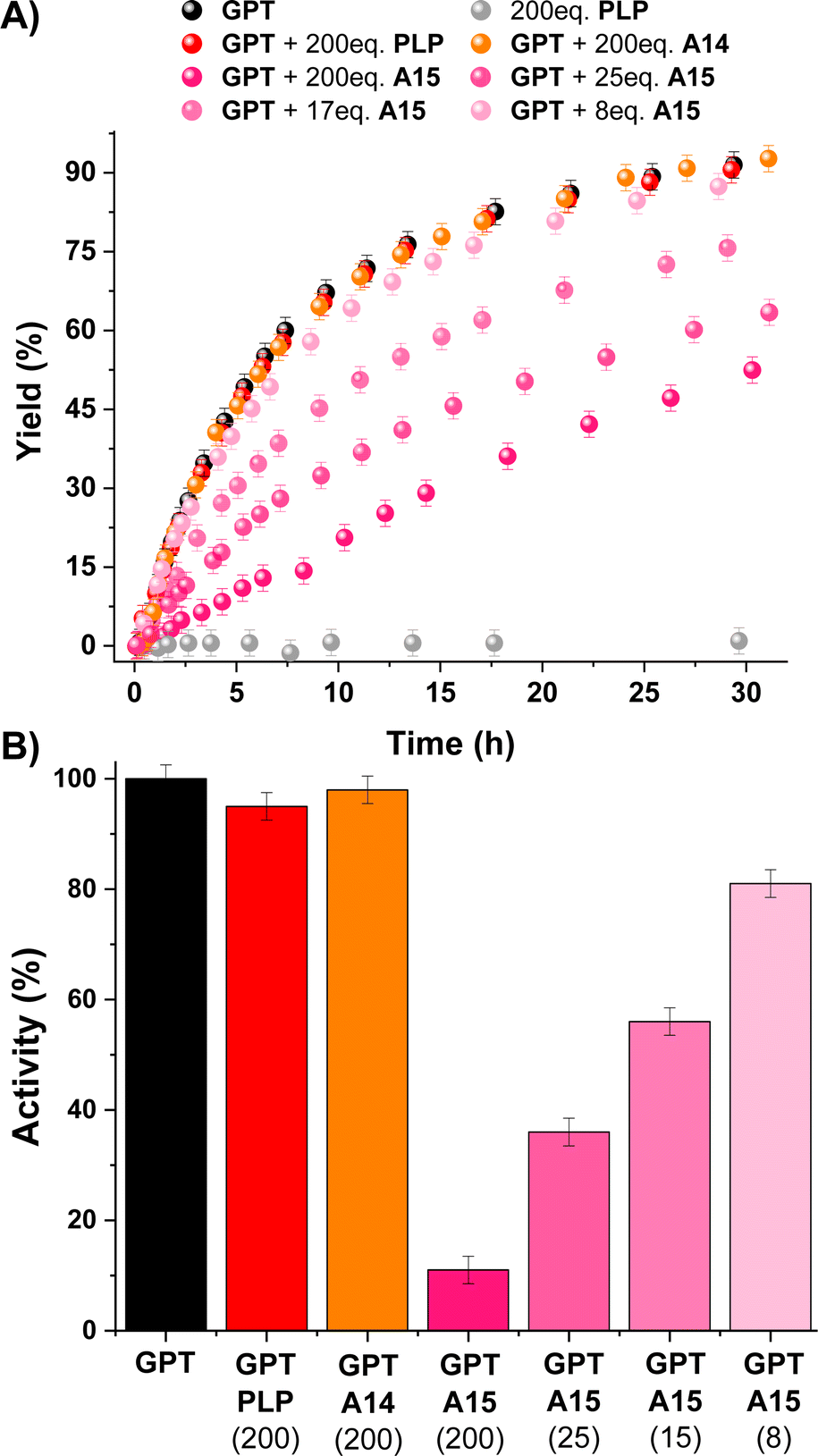 | ||
| Fig. 5 (A) Kinetic profiles for the L-alanine-D2O exchange reactions. Colour code: black (GPT + pyruvate), grey (PLP + pyruvate), red (GPT + PLP + pyruvate), orange (GPT + A14 + pyruvate), pink (GPT + A15 + pyruvate). (B) GPT activity observed for the different inhibition experiments. Black column corresponds to the neat GPT activity. Number in brackets describe the equivalents of the aldehydes introduced (also the concentration in μM). Reaction conditions: D2O, pD 7.8, 100 mM PBS, 295 K. Concentrations: L-alanine (90 mM), pyruvate (1.5 mM), GPT (1 μM). | ||
Conclusions
In conclusion, we have evaluated the hydration and imitation reactions of different aldehydes in aqueous media, comparing the results with the ones attained in the presence of the biologically-relevant PLP. Aldehydes A12–A15 resulted in remarkable Kim with complete silencing of hydrate formation (Khyd) when reacted with B1, despite the low concentrations employed. The mode-of-action to attain excellent imine vs. hydrate selectivities relies on the interplay between: (i) high Hammett values to increase carbonyl electrophilic character; (ii) sterically crowding units to disfavour the Csp2 hybridization of hydrate and hemiaminals; and (iii) negatively-charged sites to stabilize the iminium cations through electrostatic, H-bonding and n → π* interactions. The imine yields for the reaction between the best-performing aldehydes (i.e., PLP, A14 and A15) and amines presenting a high –NH2 protonation were also evaluated. The results obtained in the presence of A15 surpassed the ones of PLP, postulating A15 as the most active aldehyde in imitation reactions under physiological conditions reported to date. In addition, A14 can be used for the selective Nα functionalization of peptides since it does not react with the Nε of Lys-derivatives.Preliminary studies on the A15 conjugation to a model protein suggested the suitability of this aldehyde to replace PLP from the active site through dynamic aldimine exchange, allowing for the inhibition of the enzymatic activity. All these results, taken together, will facilitate the application of imine chemistry in water, with a particular interest in the bioorthogonal modification of proteins with dynamic covalent bonds.
Data availability
The authors confirm that the data supporting the findings of this study are available within the ESI.† Additional experimental data are available on request from the corresponding author (F. E.).Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Conceptualization: F. E. and J. M. L. Data curation: F. E. and T. R. Formal analysis: F. E. Funding acquisition: J. M. L. Investigation: T. R. (supporting) and F. E. (lead). Methodology: F. E. Project administration: J. M. L. Resources: J. M. L. Supervision: F. E. and J. M. L. Validation: F. E. Visualization: T. R. (supporting) and F. E. (lead). Writing – original draft: F. E. Writing – review and editing: T. R. (supporting), F. E., and J. M. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC (Advanced Research grant SUPRADAPT 290585), the University of Strasbourg Institute for Advanced Study (USIAS), and Fondation Jean-Marie Lehn. F. E. acknowledges Fundación Ramón Areces for a postdoctoral fellowship. T. R. thanks the CSC Graduate School funded by the French National Research Agency (CSC-IGS ANR-17-EURE-0016) for a PhD fellowship. The authors thank Dr J. L. Schmitt, Dr G. Ragazzon, Dr A. Osypenko, Dr Z. Yang, Dr S. Jung, B. Kozibroda, M. J. Aguilera, and C. Antheaume for helpful discussions.Notes and references
- M. R. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- J. Yu, M. Gaedke and F. Schaufelberger, Eur. J. Org Chem., 2023, 26, e2022011 Search PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- K. Acharyya, S. Mukherjee and P. S. Mukherjee, J. Am. Chem. Soc., 2013, 135, 554–557 CrossRef CAS PubMed.
- Y.-Q. Zou, D. Zhang, T. K. Ronson, A. Tarzia, Z. Lu, K. E. Jelfs and J. R. Nitschke, J. Am. Chem. Soc., 2021, 143, 9009–9015 CrossRef CAS PubMed.
- Z. Yang, F. Esteve, C. Antheaume and J.-M. Lehn, Chem. Sci., 2023, 14, 6631–6642 RSC.
- M. Ovalle, M. Kathan, R. Toyoda, C. N. Stindt, S. Crespo and B. L. Feringa, Angew. Chem., Int. Ed., 2023, 62, e202214495 CrossRef CAS PubMed.
- A. Seoane, R. J. Brea, A. Fuertes, K. A. Podolsky and N. K. Devaraj, J. Am. Chem. Soc., 2018, 140, 8388–8391 CrossRef CAS PubMed.
- S. P. Afrose, C. Mahato, P. Sharma, L. Roy and D. Das, J. Am. Chem. Soc., 2022, 144, 673–678 CrossRef CAS PubMed.
- F. Esteve, B. Altava, E. García-Verdugo, S. V. Luis and J.-M. Lehn, Chem, 2022, 8, 2023–2042 CAS.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- F. Esteve, F. Rahmatova and J.-M. Lehn, Chem. Sci., 2023, 14, 10249–10257 RSC.
- Y. Lei, Z. Li, G. Wu, L. Zhang, L. Tong, T. Tong, Q. Chen, L. Wang, C. Ge, Y. Wei, Y. Pan, A. C. H. Sue, L. Wang, F. Huang and H. Li, Nat. Commun., 2022, 13, 3557 CrossRef CAS PubMed.
- Y. Lei, Q. Chen, P. Liu, L. Wang, H. Wang, B. Li, X. Lu, Z. Chen, Y. Pan, F. Huang and H. Li, Angew. Chem., Int. Ed., 2021, 60, 4705–4711 CrossRef CAS PubMed.
- Y. Chen, Y. Lei, L. Tong and H. Li, Chem.–Eur. J., 2022, 28, e202102910 CrossRef CAS PubMed.
- K. Meguellati, A. Fallah-Araghi, J. C. Baret, A. El Harrak, T. Mangeat, C. M. Marques, A. D. Griffiths and S. Ladame, Chem. Commun., 2013, 49, 11332–11334 RSC.
- A. C. Eliot and J. F. Kirsch, Annu. Rev. Biochem., 2004, 73, 383–415 CrossRef CAS PubMed.
- J. Liang, Q. Han, Y. Tan, H. Ding and J. Li, Front. Mol. Biosci., 2019, 6, 4 CrossRef CAS PubMed.
- E. F. Oliveira, N. M. F. S. A. Cerqueira, P. A. Fernandes and M. J. Ramos, J. Am. Chem. Soc., 2011, 133, 15496–15505 CrossRef CAS PubMed.
- W. R. Griswold and M. D. Toney, J. Am. Chem. Soc., 2011, 133, 14823–14830 CrossRef CAS PubMed.
- J. P. Richard, T. L. Amyes, J. Crugeiras and A. Rios, Biochim. Biophys. Acta, 2011, 1814, 1419–1425 CrossRef CAS PubMed.
- J. Crugeiras, A. Rios, E. Riveiros and J. P. Richard, J. Am. Chem. Soc., 2009, 131, 15815–15824 CrossRef CAS PubMed.
- S. Jia, H. Ye and L. You, Org. Chem. Front., 2022, 9, 3966–3975 RSC.
- K. B. Muchowska, D. J. Pascoe, S. Borsley, I. V. Smolyar, I. Mati, C. Adam, G. S. Nichol, K. B. Ling and S. L. Cockroft, Angew. Chem., Int. Ed., 2020, 59, 14602–14608 CrossRef CAS PubMed.
- H. Chen, H. Ye, Y. Hai, L. Zhang and L. You, Chem. Sci., 2020, 11, 2707–2715 RSC.
- H. Zheng, H. Ye, X. Yu and L. You, J. Am. Chem. Soc., 2019, 141, 8825–8833 CrossRef CAS PubMed.
- P. M. S. D. Cal, J. B. Vicente, E. Pires, A. V. Coelho, L. F. Veiros, C. Cordeiro and P. M. P. Gois, J. Am. Chem. Soc., 2012, 134, 10299–10305 CrossRef CAS PubMed.
- T. Fiala, J. Wang, M. Dunn, P. Šebej, S. J. Choi, E. C. Nwadibia, E. Fialova, D. M. Martinez, C. E. Cheetham, K. J. Fogle, M. J. Palladino, Z. Freyberg, D. Sulzer and D. Sames, J. Am. Chem. Soc., 2020, 142, 9285–9301 CrossRef CAS PubMed.
- J. H. Blanch, J. Chem. Soc. B, 1966, 937–939 RSC.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- R. A. McClelland and M. Coe, J. Am. Chem. Soc., 1983, 105, 2718–2725 CrossRef CAS.
- Note: 75% of unprotonated –NH2 group for B1 at pH 7.4. Value calculated using Marvin JS software. See Table S3† for pKa and protonation degrees of the different amines considered in this work..
- Note: Hammett values of ionizable groups might change depending on the pH of the solutions. See Table S1† for the different values used in this work..
- S. M. So, L. Mui, H. Kim and J. Chin, Acc. Chem. Res., 2012, 45, 1345–1355 CrossRef CAS PubMed.
- J. M. Sayer and W. P. Jencks, J. Am. Chem. Soc., 1977, 99, 464–474 CrossRef CAS.
- J.-M. Lehn, G. Wipff and H. B. Bürgi, Helv. Chim. Acta, 1974, 57, 493–496 CrossRef CAS.
- J.-M. Lehn and G. Wipff, Helv. Chim. Acta, 1978, 61, 1274–1286 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- Note: The potential proton transfer events (either intramolecular or with the H2O solvent molecules) occurring during each reaction step hampered the successful characterization of the transition states involved..
- J. Hine and F. A. Via, J. Am. Chem. Soc., 1971, 94, 190–194 CrossRef.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS.
- J. P. Richard, T. L. Amyes, J. Crugeiras and A. Rios, Curr. Opin. Chem. Biol., 2009, 13, 475–483 CrossRef CAS PubMed.
- H. Jiang, W. Chen, J. Wang and R. Zhang, Chin. Chem. Lett., 2022, 33, 80–88 CrossRef CAS.
- T. Sahu, M. Chilamari and V. Rai, Chem. Commun., 2022, 58, 1768–1771 RSC.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- M. M. Zegota, T. Wang, C. Seidler, D. Y. W. Ng, S. L. Kuan and T. Weil, Bioconjugate Chem., 2018, 29, 2665–2670 CrossRef CAS PubMed.
- L. Purushottam, S. R. Adusumalli, U. Singh, V. B. Unnikrishnan, D. G. Rawale, M. Gujrati, R. K. Mishra and V. Rai, Nat. Commun., 2019, 10, 2539 CrossRef PubMed.
- L. Purushottam, S. R. Adusumalli, M. Chilamari and V. Rai, Chem. Commun., 2017, 53, 959–962 RSC.
- U. M. Babu and R. B. Johnston, Biochem. Biophys. Res. Commun., 1974, 58, 460–466, DOI:10.1016/0006-291x(74)90387-8.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, main text support figures and tables, copies of 1H, 13C NMR, and HRMS spectra, DFT cartesian coordinates. See DOI: https://doi.org/10.1039/d4sc02206h |
| This journal is © The Royal Society of Chemistry 2024 |