
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArresting dissolution of two-dimensional metal–organic frameworks enables long life in electrochemical devices†
Gopi M. R.
Dontireddy
a,
Satya Prakash
Suman
a,
Jose L.
Merino-Gardea
a,
Tianyang
Chen
 b,
Jin-Hu
Dou
*c and
Harish
Banda
*a
b,
Jin-Hu
Dou
*c and
Harish
Banda
*a
aDepartment of Chemistry and Biochemistry, The University of Texas at El Paso, El Paso, Texas 79968, USA. E-mail: hbanda1@utep.edu
bDepartment of Chemical Engineering, Stanford University, Stanford, California 94305, USA
cSchool of Materials Science and Engineering, Peking University, Beijing, 100871, China. E-mail: doujinhu@pku.edu.cn
First published on 31st May 2024
Abstract
Two-dimensional conjugated metal–organic frameworks (2D cMOFs) are emerging as promising materials for electrochemical energy storage (EES). Despite considerable interest, an understanding of their electrochemical stability and the factors contributing to their degradation during cycling is largely lacking. Here we investigate three Cu-based MOFs and report that the dissolution of 2D cMOFs into electrolytes is a prevalent and significant degradation pathway. Several factors, such as the inherent solubility of ligands in electrolyte solvents and the duration of charge–discharge cycling exert a strong influence on the dissolution process. When these factors combine within a MOF, severely limited cycling stability is observed, with dissolution accounting for up to 80% of capacity degradation. Conversely, excellent cycling stability is observed when testing a Cu-MOF with a sparingly soluble ligand within an optimized potential window. Overall, these findings represent essential insights into the electrochemical stability of 2D cMOFs, offering crucial guidelines for their targeted development in EES applications.
Introduction
Two-dimensional conjugated metal–organic frameworks (2D cMOFs) are an emerging class of electrically conducting, porous crystalline materials.1–3 Typically, 2D cMOFs host multitopic π-conjugated aromatic ligands in square planar MX4 (X = O, NH, or S) linkages that repeat and create porous 2D sheets. Various 2D cMOFs have been synthesized using ligands with core structures ranging from benzene4–12 to larger structures13–17 like triphenylene,18–22 coronene,23 phthalocyanine,24,25 and hexa-cata-hexabenzocoronene.26 This structural versatility, coupled with favorable physical properties and the potential for redox activity from both ligands and metal nodes, has prompted the exploration of 2D cMOFs as electrode materials for electrochemical energy storage (EES). Several 2D cMOFs have demonstrated different EES mechanisms, including electric double-layer storage, battery-like faradaic processes, and rapid pseudocapacitance.27–29 Organic ligands that would otherwise dissolve into the electrolytes are considered to be immobilized within 2D frameworks and participate in the EES processes.4,30 Notably, Cu-based MOFs have attracted significant attention due to their accessible redox activity on both ligands and Cu nodes (Cu2+/Cu+ redox couple), leading to enhanced EES.30–37 However, investigations into the factors that determine the electrochemical stability of 2D cMOFs and lead to their capacity loss during charge–discharge cycling are rare.3 A comprehension of the degradation pathways in 2D cMOFs is crucial for devising strategies to enhance their stability, meeting the thresholds of thousands to hundreds of thousands of charge–discharge cycles needed for EES applications in batteries and supercapacitors, respectively.Previous studies on 2D cMOFs for EES primarily evaluated their electrochemical stability by examining the crystallinity of cycled electrodes using powder X-ray diffraction (PXRD) analyses.27,33 Typically, the retention or loss of crystallinity in PXRD patterns of cycled electrodes is directly correlated to capacity loss. While these studies are useful in providing a rapid diagnosis of the cycled electrodes, they do not reveal underlying causes of capacity loss. Indeed, an amorphous-like PXRD pattern may result from various phenomena. For instance, processes such as the pulverization of microcrystalline electrode powders, dissolution of electrode material into electrolytes, and crystalline-to-amorphous phase transformations during charge–discharge cycling can all reduce crystallinity in the PXRD patterns. However, each of these processes can influence capacities differently. Other studies examining 2D cMOFs in two-electrode symmetric cells have provided consolidated observations of capacity losses at the cell level.28,36 Although these studies are useful for simulating practical devices, they cannot provide a detailed understanding of individual electrode-level processes. Overall, understanding capacity losses and the underlying degradation pathways is essential for the targeted development of 2D cMOFs for EES.
Herein, we examine the electrochemical charge storage behavior and cycling stability of three catecholate-based Cu-MOFs, which demonstrate reversible redox activity on the [CuO4] units. Through comprehensive post-mortem analyses of the electrolytes, we find that the primary cause of capacity loss during cycling is the dissolution of MOFs. Ligands with high solubility in the electrolytes result in rapid degradation of MOF performance, whereas ligands with low solubility promote stable cycling. A control of the experimental conditions and the electrochemical parameters is shown to exert a crucial influence on the dissolution, and the cycling stability of 2D cMOFs.
Results
Three Cu-based MOFs were synthesized with increasingly larger aromatic cores progressing from tetrahydroxy-1,4-benzoquinone (THQ) to 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) and 2,3,7,8,12,13-hexahydroxytetraazanaphthotetraphene (HHTT) using the reported procedures.10,19,38,39 (Schemes 1–3) Powder X-ray diffraction patterns (PXRDs) of the synthesized powders confirmed the crystallinity and phase purity of the three Cu-MOFs (Fig. S1†). In all cases, copper ions (a mixture of Cu2+ and Cu+) bond with two neighboring ligands, forming square planar [CuO4] secondary building units that repeat to generate planar, porous sheets. These sheets are stacked in eclipsed or slip-stacked arrangements, thereby creating 1D pores (Cu3HHTT2 shown in Fig. 1a).38 The electrochemical behavior of the three MOFs was investigated in Swagelok-type cells using a three-electrode cell configuration and electrodes prepared with 80% active materials (details in methods). Because the focus of this report is on revealing general degradation pathways for 2D cMOFs, electrochemical tests on all MOFs were performed in similar potential windows above 2.4 V vs. Li+/Li or −0.7 V vs. Ag/AgCl. These potential ranges mainly accommodate the redox processes that occur on the [CuO4] units and do not involve deep reduction of aromatic cores.12,22,31–34,36 The redox activity of Cu3THQ2 and Cu3HHTP2 were previously studied in these cathodic potentials, however, Cu3HHTT2 has not been explored.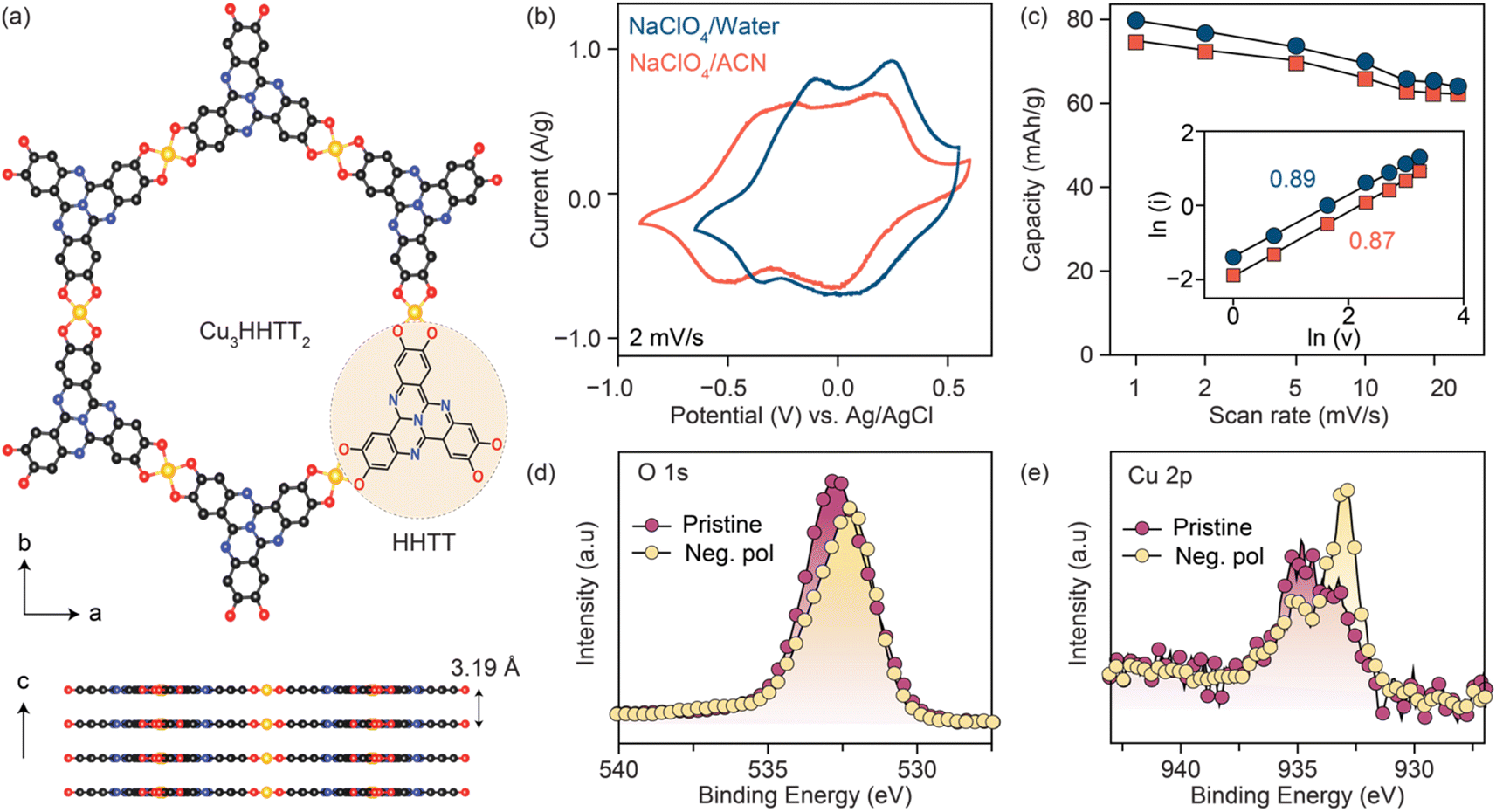 | ||
Fig. 1 (a) Crystal structure of Cu3HHTT2 shown in ab plane and along the c axis. (b) Cyclic voltammetry curves of Cu3HHTT2 were recorded at a scan rate of 2 mV s−1 in 0.5 M NaClO4 solutions of water and acetonitrile (ACN). (c) Specific capacities were calculated from CVs recorded at scan rates ranging from 1–25 mV s−1 in NaClO4/water and NaClO4/ACN electrolytes. The inset shows power-law analysis and b values of 0.89 and 0.87 for Cu3HHTT2 in NaClO4/water and NaClO4/ACN, respectively. High-resolution ex situ X-ray photoelectron spectra of (d) O 1s and (e) Cu 2p of pristine and negatively polarized Cu3HHTT2 (−0.75 V vs. Ag/AgCl). A notable decrease in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Cu2+ components indicates a reduction of both ligand and copper nodes under negative polarization. O and Cu2+ components indicates a reduction of both ligand and copper nodes under negative polarization. | ||
Cyclic voltammograms (CVs) of Cu3HHTT2 recorded at a scan rate of 2 mV s−1 in aqueous and acetonitrile (ACN) electrolytes (0.5 M NaClO4) both displayed two sets of reversible redox peaks and matched well with a theoretical capacity of ∼80 mA h g−1 calculated for a three-electron reduction per Cu3HHTT2 (molecular weight 1010 g mol−1, ESI Table 1). Moreover, CVs recorded over a range of scan rates exhibited excellent power capability, with a capacity of ∼63 mA h g−1 at 25 mV s−1 (Fig. 1c and S2†). Additionally, an evaluation of the electrode kinetics using scan rate versus peak current analysis (eqn (1), where i is the peak current, v is the scan rate, and a and b are adjustable parameters) found b values close to 0.9 in both aqueous and non-aqueous electrolytes. These b values, close to unity, indicate that the charge storage in Cu3HHTT2 can be characterized as a rapid, pseudocapacitive process.40
| i(v) = avb | (1) |
The origins of the two distinct redox events observed for Cu3HHTT2 in NaClO4/ACN were investigated by analyzing X-ray photoelectron spectra (XPS) of the negatively polarized electrodes under ex situ conditions (Fig. 1d, e and S3†). A comparison of the high-resolution O 1s XPS data of the pristine MOF and the negatively polarized sample (−0.75 V vs. Ag/AgCl) reveals a decrease in intensity for the CO component (532.3 eV) and an increase in the C–O component (531.5 eV), confirming redox activity on the ligand moieties in Cu3HHTT2 (Fig. 1d, deconvolution shown in S3†). Similarly, an analysis of the high-resolution Cu 2p XPS data of the pristine MOF and the negatively polarized sample at −0.75 V vs. Ag/AgCl (Fig. 1e, deconvolution shown in S3†) indicated an obvious enhancement in the relative composition of Cu+ species (peaks at 932.7 and 952.2 eV for Cu 2p3/2 and Cu 2p1/2, respectively) over Cu2+ (peaks at 934.4 and 954.6 eV for Cu 2p3/2 and Cu 2p1/2, respectively). These results indicate that the three-electron reduction of Cu3HHTT2 occurs both on the HHTT moieties and the Cu nodes, consistent with previous reports on Cu-MOFs.15,31,33
The electrochemical stability of Cu3HHTT2 was investigated through galvanostatic cycling with potential limitation (GCPL) studies in both aqueous and ACN-based electrolytes. Charge–discharge profiles at a current density of 750 mA g−1 (or ∼3 C rate, where 1 C equals full discharge in one hour) delivered discharge capacities of ∼80 mA h g−1 with an average discharge voltage of −0.1 V vs. Ag/AgCl in both electrolytes (Fig. 2a). Moreover, excellent power performance was observed with discharge capacities of ∼65 mA h g−1 at a high current density of 2 A g−1 or ∼25 C rate (Fig. S4 and S5†). Repeated cycling of Cu3HHTT2 in aqueous electrolytes at current densities of 250, 500, and 750 mA g−1 exhibited remarkable stability over hundreds of cycles, with nearly full retention of discharge capacities (Fig. 2b). However, cycling in NaClO4/ACN resulted in a significant decrease in discharge capacities, with ∼70% of the initial capacity lost within 350, 500, and 950 cycles at current densities of 250, 500, and 750 mA g−1, respectively. Moreover, impedance spectra recorded in between the GCPL cycles at a current density of 750 mA g−1 indicated little to no changes, emphasizing that the degradation in capacities observed during GCPL cycling is not caused by increasingly resistive electrodes (Fig. S6†).
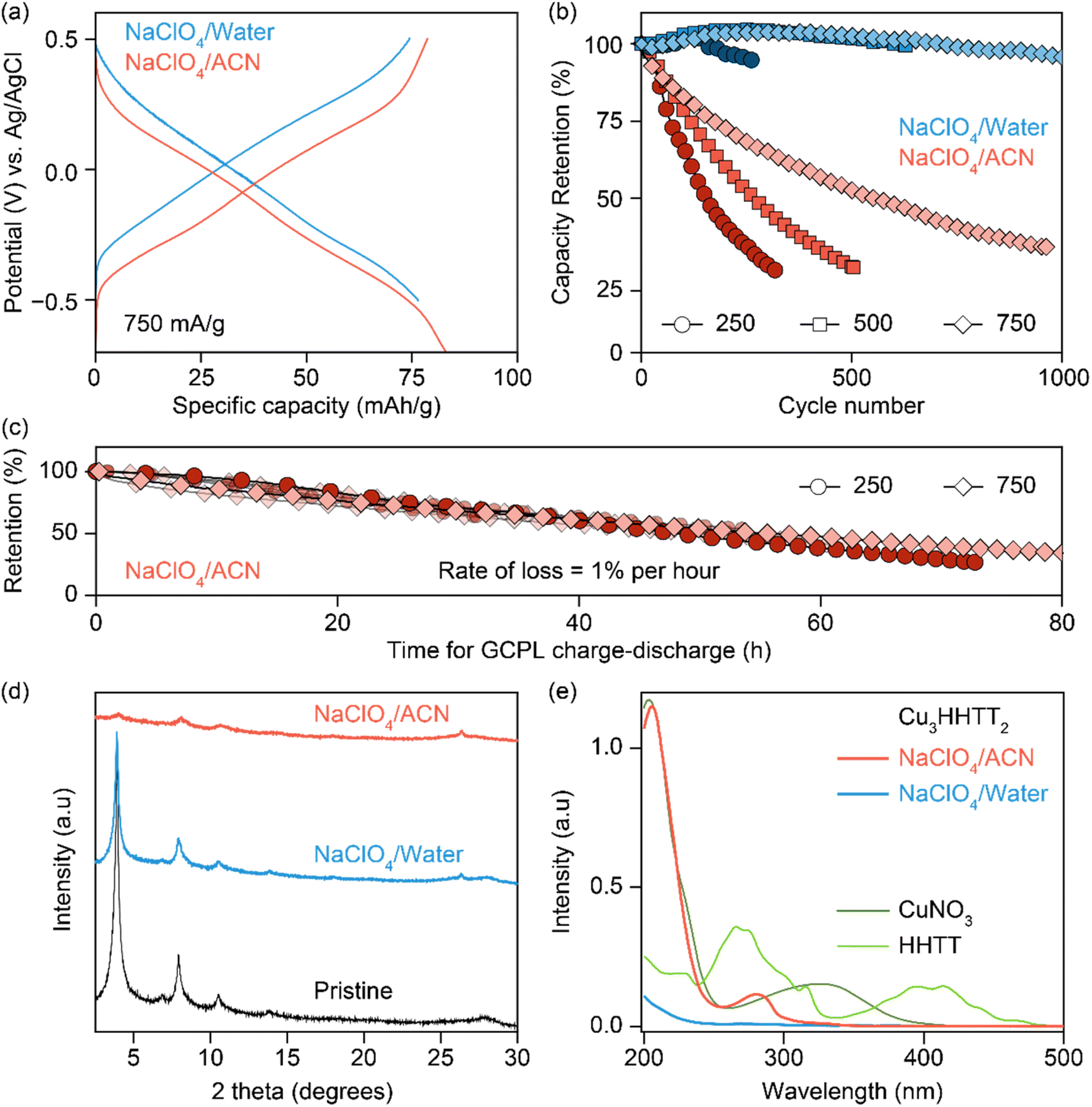 | ||
| Fig. 2 (a) Charge–discharge profiles of Cu3HHTT2 recorded at a current density of 750 mA g−1 in NaClO4/water and NaClO4/ACN solutions. (b) Capacity retention versus the number of cycles for galvanostatic cycling with potential limitation (GCPL) at current densities of 250, 500, and 750 mA g−1 in NaClO4/water and NaClO4/ACN. (c) Capacity retention versus cumulative time of GCPL cycling in NaClO4/ACN is shown for six cells, three each at 250 and 750 mA g−1. All six cells display a similar degradation rate of 1% per hour. (d) Powder X-ray diffraction patterns of pristine and cycled Cu3HHTT2 electrodes. Electrodes cycled in NaClO4/water and NaClO4/ACN display a retention and loss of crystallinity, respectively. An additional peak at ∼26° in the electrodes corresponds to the carbon paper substrate. (e) Ultraviolet-visible absorption spectra of electrolytes retrieved from cycled cells of Cu3HHTT2 in NaClO4/water and NaClO4/ACN, also shown are absorption spectra for solutions of HHTT and CuNO3 in ACN. | ||
It is notable that the conventional representation of cycling data, where capacity retention is plotted against the number of cycles, can potentially lead to misinterpretation, suggesting greater stability at higher cycling rates. Interestingly, an alternative representation of cycling data, plotting capacity retention against the total time of cycling, revealed strikingly similar degradation profiles for Cu3HHTT2 at different current densities. Specifically, results from the charge–discharge cycling of six cells in NaClO4/ACN indicated a consistent capacity loss rate of 1% per hour (Fig. 2c). Overall, these results underscore the distinct cycling stability behaviors of Cu3HHTT2 between aqueous and ACN-based electrolytes and emphasize that the total duration of cycling governs the extent of its degradation in NaClO4/ACN.
Electrode dissolution and capacity loss
Analysis of cycled Cu3HHTT2 electrodes using PXRD indicated that their crystallinity remained intact after 100 hours of cycling in an aqueous electrolyte (Fig. 2d). In contrast, electrodes cycled and degraded in NaClO4/ACN exhibited a notable decrease in crystallinity, resembling typical observations associated with degraded MOF electrodes. However, while these analyses are valuable in assessing the final state of cycled electrodes, they do not provide insights into the underlying causes of capacity loss. Ultraviolet-visible (UV-vis) spectroscopy was employed to examine the electrolyte solutions retrieved from cycled and disassembled cells. The UV-vis spectra of electrolytes obtained from cells cycled in NaClO4/ACN showed prominent absorption peaks at 210 and 280 nm (Fig. 2e). In contrast, electrolytes from cells cycled in aqueous conditions exhibited reduced intensity around 200 nm. Notably, the pristine electrolytes themselves lack absorption features within the 200–500 nm range (Fig. S7a†), indicating that the observed absorption in cycled electrolytes stems from additional species introduced during cycling. Interestingly, a comparison of the UV-vis spectra of CuNO3 and HHTT in ACN with cycled electrolytes reveals that the peaks at 210 and 280 nm corresponded to the presence of copper and HHTT, respectively. Furthermore, control experiments that recorded UV-vis spectra of sonicated solutions of Cu3HHTT2 in ACN did not show a similar presence of copper and HHTT (Fig. S7b†). These tests indicate that the observation of copper and HHTT in the electrolytes from cycled cells is not due to poor adhesion of MOF particles that may break off from the electrode substrate and enter the electrolyte solution. Taken together, these findings strongly suggest that Cu3HHTT2 disintegrates and dissolves from the electrodes into the electrolytes during cycling. Additionally, the differences in cycling stabilities and absorption spectra between NaClO4/water and NaClO4/ACN may then be attributed to the higher solubility of HHTT in ACN. Indeed, further GCPL studies on Cu3HHTT2 in electrolyte mixtures of water and ACN revealed a direct correlation, where a higher content of ACN led to rapid degradation of capacities (Fig. S8 and S9†). Overall, these results confirm that the dissolution of a MOF governs its cycling stability, and the choice of electrolyte solvent, such as ACN versus water, is crucial.The validity of observations regarding the degradation and dissolution of Cu3HHTT2 was probed in two other Cu-based MOFs – Cu3THQ2 and Cu3HHTP2. THQ and HHTP are two commonly used organic ligands with smaller aromatic cores compared to HHTT. While HHTP and HHTT exhibit similar solubilities (∼0.1 mg mL−1) in water, THQ is twenty times more soluble (1.97 mg mL−1) than both (Fig. 3a, details in methods). In ACN, THQ and HHTP (∼0.66 mg mL−1) are three times more soluble than HHTT (0.22 mg mL−1). GCPL studies on Cu3THQ2 and Cu3HHTP2 revealed significant degradation within 12 hours of cycling in both aqueous and ACN-based electrolytes (Fig. 3b and S10†). Specifically, Cu3THQ2 degraded by 50% within the first two hours of cycling. UV-vis analysis of the retrieved electrolytes from cycled cells indicated strong absorption at wavelengths corresponding to the presence of Cu, THQ, and HHTP, respectively (Fig. S11 and S12†). Taken together, the cycling behavior and the UV-vis absorption spectra of Cu3THQ2 and Cu3HHTP2 validate that dissolution is a common pathway for the degradation of 2D cMOFs. Furthermore, a correlation emerges between ligand solubility and the performance of its corresponding MOF in an electrolyte.
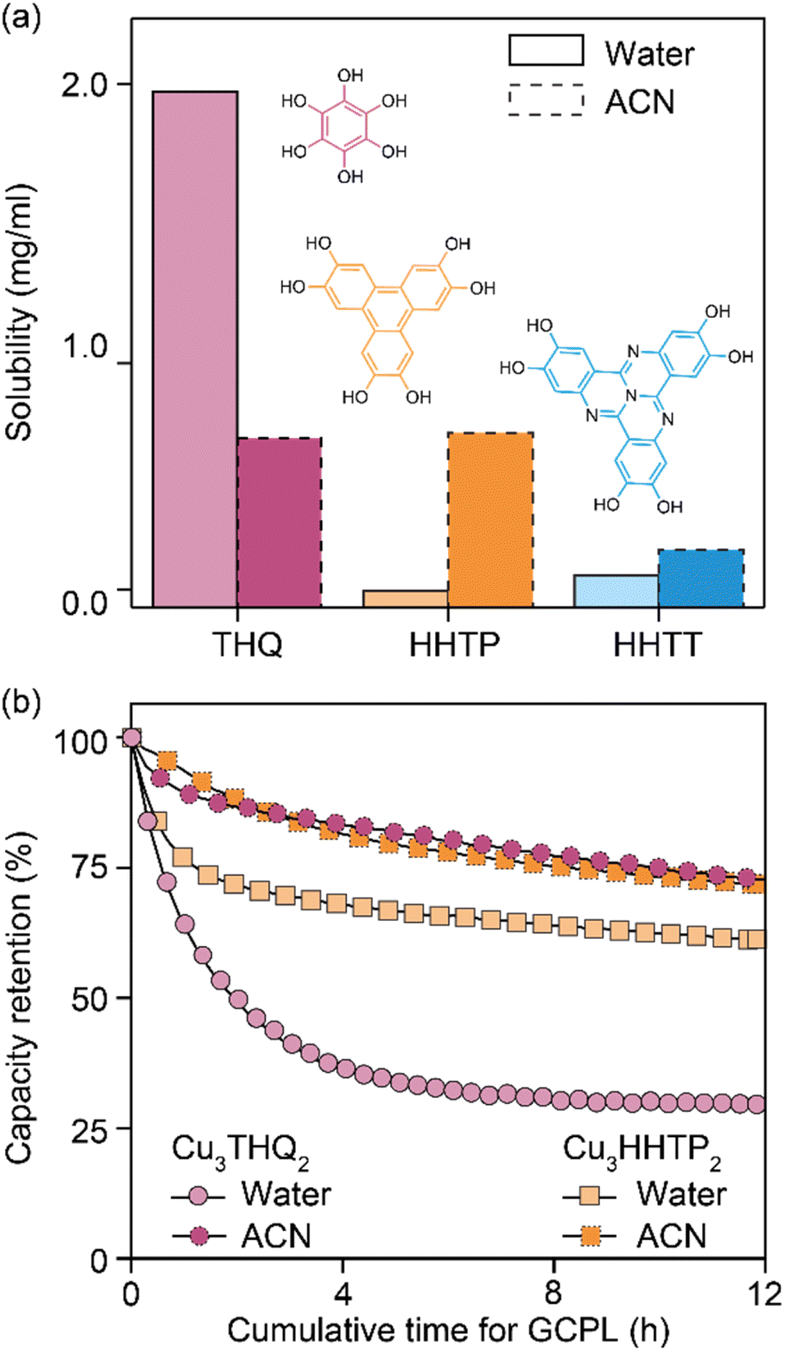 | ||
| Fig. 3 (a) Solubilities of THQ, HHTP, and HHTT in acetonitrile (ACN) and water, measured at ambient conditions. (b) Capacity retention versus cumulative time of GCPL cycling for Cu3THQ2 and Cu3HHTP2 at a current density of 750 mA g−1 in NaClO4/water and NaClO4/ACN. | ||
Quantifying degradation
The contribution from electrode dissolution to the overall capacity loss during the cycling of 2D cMOFs was investigated by quantifying dissolved species in the electrolyte using UV-vis and inductively coupled plasma mass spectrometry (ICP-MS). Cu3HHTP2 was selected for analysis due to its prominence in prior studies.33,36 UV-vis spectra of degraded Cu3HHTP2 cells in ACN electrolytes were compared to standard solutions prepared by mixing CuNO3 and HHTP in ACN (in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, Fig. 4a, details in methods, Fig. S13†). A quantitative assessment of four Cu3HHTP2 cells, each experiencing a loss of around 50% of their initial capacity, revealed that nearly 30% of the electrode masses were dissolved in the electrolytes. In essence, an average of 60% (ranging between 45–75%) of the capacity losses in Cu3HHTP2 cells could be directly linked to electrode dissolution (Fig. 4b). Additionally, ICP-MS analyses of the electrolytes from degraded Cu3HHTP2 cells identified a significant presence of copper, also attributing 50% (ranging between 30–80%) of the capacity losses to this dissolution process. Overall, both UV-vis and ICP-MS analyses highlight electrode dissolution as the predominant, if not primary, mode of degradation in Cu3HHTP2.
:2 ratio, Fig. 4a, details in methods, Fig. S13†). A quantitative assessment of four Cu3HHTP2 cells, each experiencing a loss of around 50% of their initial capacity, revealed that nearly 30% of the electrode masses were dissolved in the electrolytes. In essence, an average of 60% (ranging between 45–75%) of the capacity losses in Cu3HHTP2 cells could be directly linked to electrode dissolution (Fig. 4b). Additionally, ICP-MS analyses of the electrolytes from degraded Cu3HHTP2 cells identified a significant presence of copper, also attributing 50% (ranging between 30–80%) of the capacity losses to this dissolution process. Overall, both UV-vis and ICP-MS analyses highlight electrode dissolution as the predominant, if not primary, mode of degradation in Cu3HHTP2.
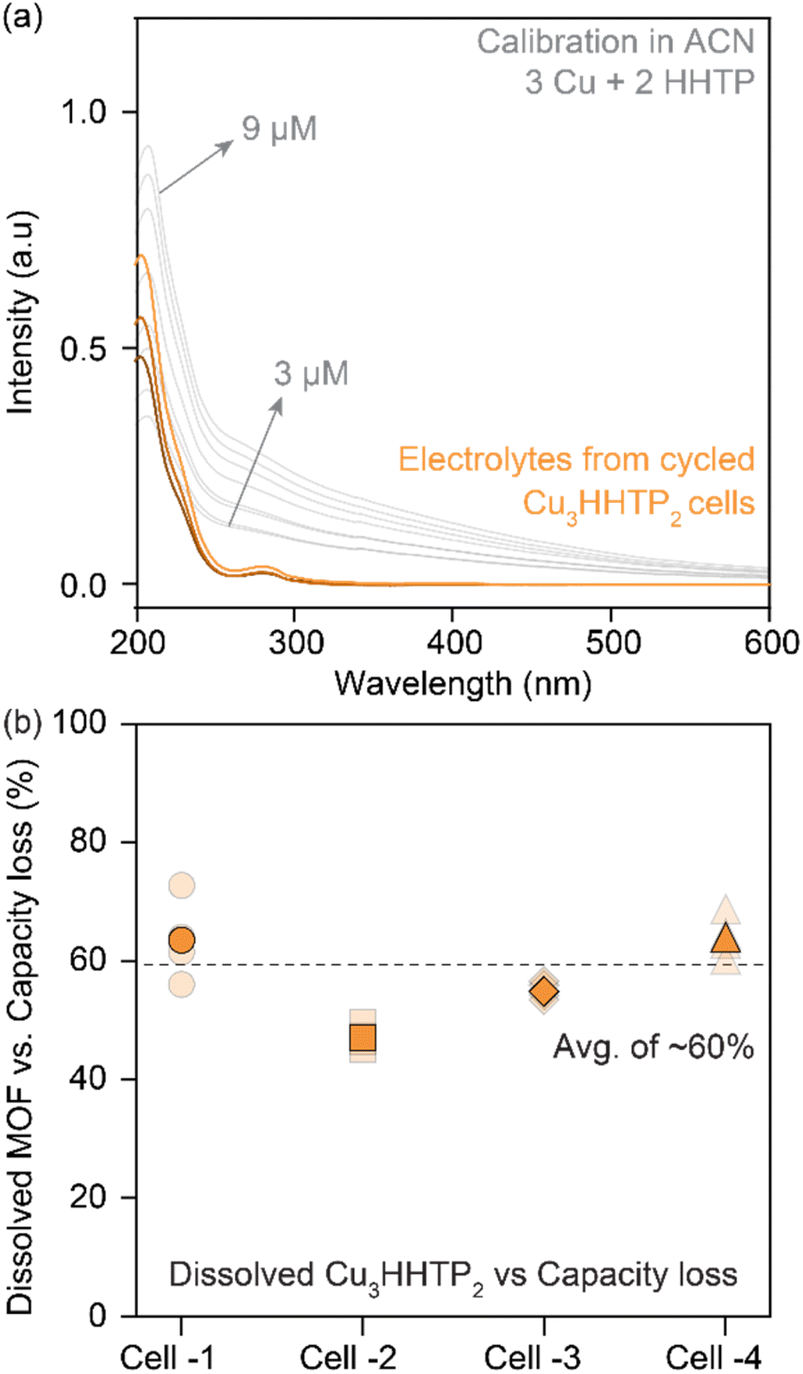 | ||
| Fig. 4 (a) Ultraviolet-visible absorption spectra of electrolytes retrieved from cycled cells of Cu3HHTP2 in NaClO4/ACN. Also shown are the absorption spectra for calibration solutions prepared by mixing CuNO3 and HHTT in a ratio of 3:2 in ACN. (b) Quantitative estimation of the dissolved Cu3HHTP2 in electrolytes retrieved from four cells. An average of 60% of the total degradation can be attributed to electrode dissolution during cycling. | ||
Electrochemical quartz crystal microbalance (EQCM) was used to identify and examine electrode dissolution of 2D cMOFs under operando conditions (Fig. S14†). A comparison was made between the degradation of Cu3THQ2 and Cu3HHTT2 in aqueous electrolytes, representing two contrasting scenarios of cycling stability. Both MOFs were drop-casted as slurry mixtures onto gold-coated quartz crystals for EQCM tests, targeting a mass loading of 20 μg per electrode (details in methods). Initial capacities, calculated from the CVs recorded at 10 mV s−1 (Fig. 5a), were ∼10 and ∼20 mA h g−1 for Cu3THQ2 and Cu3HHTT2 respectively in a shorter stable electrochemical potential window of 500 mV from −0.1 to 0.4 V vs. Ag/AgCl. These capacities fell within the expected range from studies in Swagelok cells in corresponding potential windows, validating the relevance of the obtained results to the MOF performances. Continuous cycling of CVs revealed a rapid 68% capacity loss for Cu3THQ2 and comparatively stable performance for Cu3HHTT2. Frequency profiles obtained during the first 36 CV cycles (or cycling for 1 hour) displayed a significant increase for Cu3THQ2 and a relatively flat profile for Cu3HHTT2 (Fig. 5b). Specifically, Cu3THQ2 showed an average frequency increase (Δf) of ∼115 Hz across three trials, whereas Cu3HHTT2 exhibited an average Δf of ∼5 Hz. According to the principles governing the frequency of oscillating crystals and the Sauerbery equation for EQCM (refer to ESI†), an increasing frequency (positive Δf) corresponds to a decreasing mass (negative Δm) of the crystal. These findings confirm a physical loss of electrode mass, indicating electrode dissolution during MOF cycling. Furthermore, calibration of the EQCM setup enabled the conversion of Δf ∼115 Hz for Cu3THQ2 to a Δm of −5.5 μg, corresponding to a ∼45% loss of active material. A comparison between the capacity loss from electrochemical measurements (68%) and the mass loss from EQCM measurements (45%) provided conclusive evidence supporting electrode dissolution as the primary mechanism for capacity loss in Cu3THQ2. Moreover, the rate of capacity loss in Cu3THQ2, and the corresponding increases in resonating frequency of the electrode, are highest during the first few cycles of CVs. This observation resembles the rapid capacity loss noted for Cu3THQ2 in the GCPL tests (Fig. 3b). A scheme representing the rapid electrode dissolution in Cu3THQ2 and relative stability in Cu3HHTT2 is shown in Fig. 5c.
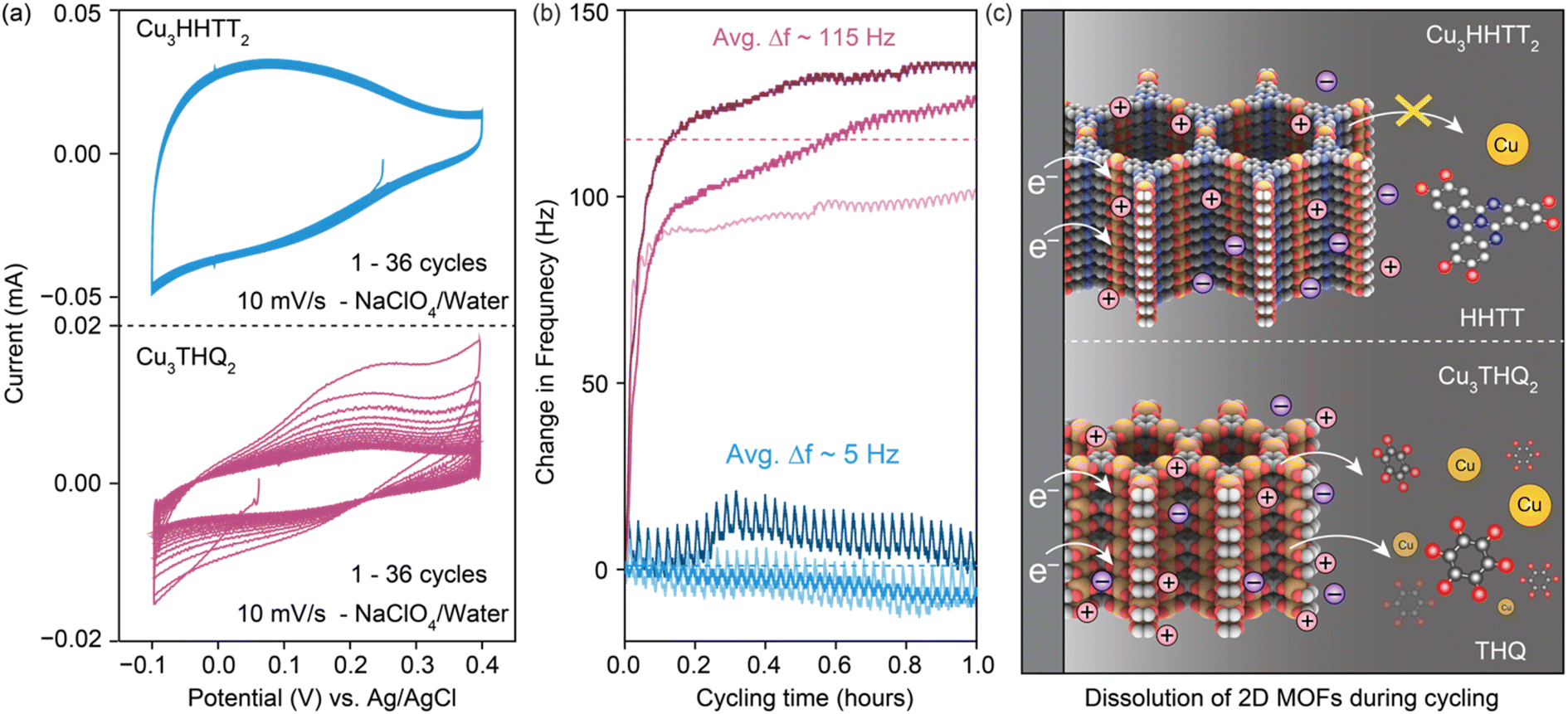 | ||
| Fig. 5 (a) Cyclic voltammetry at a scan rate of 10 mV s−1 and (b) corresponding electrochemical quartz crystal microbalance responses from Cu3HHTT2 and Cu3THQ2 in 0.5 M NaClO4/water over 36 cycles. Cu3THQ2 displayed a decrease in capacity (65%) and an increase in frequency (Δf ∼115 Hz). The increase in frequency corresponds to a mass loss of ∼5.5 μg, or ∼45% of the initial deposition. On the other hand, Cu3HHTT2 displayed relatively stable capacities and Δf ∼5 Hz. (c) Schematic depiction of the stability and dissolution of Cu3HHTT2 and Cu3THQ2 during electrochemical cycling. | ||
Factors affecting degradation
The experimental and electrochemical parameters that govern the dissolution of Cu3HHTT2 were investigated to optimize its cycling stability for EES applications. First, testing Cu3HHTT2 cells in ACN after resting for more than 100 hours showed no degradation in its capacities, indicating sufficient insolubility of the pristine MOF in the electrolyte solution (Fig. S15†). Second, subjecting Cu3HHTT2 cells in ACN to a potentiostatic condition (constant voltage or CSTV) of −0.70 V vs. Ag/AgCl with intermittent GCPL cycling (2 cycles after every two hours under polarization) resulted in a rapid degradation behavior, faster than the GCPL condition alone (Fig. 6a and S16†). These findings indicate a significant influence of externally applied polarization and the cumulative duration of polarization during cycling on Cu3HHTT2 dissolution. This observation also aligns with the earlier data on the cycling behavior of Cu3HHTT2 under GCPL conditions, where tests at various current densities resulted in a consistent capacity loss of 1% per hour of cycling (Fig. 2c).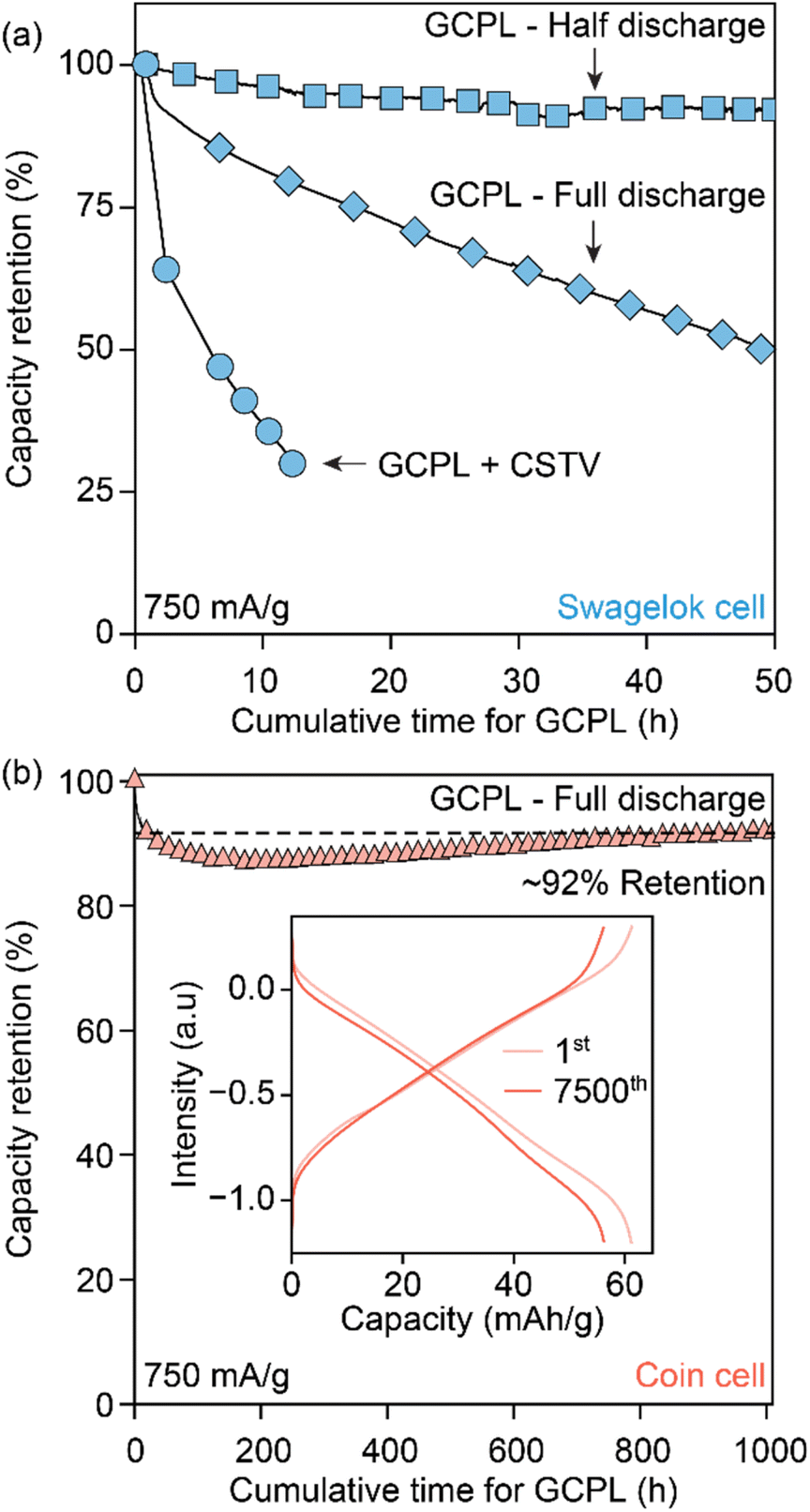 | ||
| Fig. 6 (a) Cycling of Cu3HHTP2 in NaClO4/ACN under different electrochemical conditions in Swagelok-type cells. GCPL cycling to half and full discharge states correspond to a current density of 750 mA g−1 between 0.50 to −0.10 V and 0.50 to −0.75 V vs. Ag/AgCl, respectively. GCPL + CSTV corresponds to subjecting Cu3HHTP2 to a constant potential of −0.70 V vs. Ag/AgCl, with intermittent GCPL cycling at 750 mA g−1 in the full potential window. (b) GCPL cycling of Cu3HHTP2 in a full potential window using NaClO4/ACN electrolyte and a coin cell setup. Swagelok and coin cell setups use different electrolyte volumes of 2 mL and 50 μL, respectively. | ||
The influence of external polarization on the electrochemical stability of Cu3HHTT2 was further investigated through studies in different potential windows. GCPL cycling of Cu3HHTT2 in NaClO4/ACN within the potential window of 0.5 to −0.1 V vs. Ag/AgCl (Fig. 6a, S17 and S18† labelled as half discharge), delivered remarkably stable cycling relative to the cycling performed in the full window between 0.5 to −0.7 V vs. Ag/AgCl (Fig. 2b). Cycling Cu3HHTT2 over 100000 CV cycles or 1500 hours in this half-discharge window has also resulted in extraordinary stability (Fig. S19†). Ex situ XPS analyses were then performed on the half and full discharged states of Cu3HHTT2 to understand its stable cycling in the half-discharge window (Fig. S20†). A comparison with the pristine MOF revealed that the reduction of Cu2+/Cu+ predominantly occurs in the second half of the discharge, suggesting that a high relative content of Cu2+ aids cycling till the half-discharged state. When this significant finding was then tested in Cu3HHTP2, which is known to also exhibit Cu2+/Cu+ reduction, cycling in both half and full discharge windows resulted in similar degradation patterns, (Fig. S21†).
These seemingly contrasting results in two MOFs, obtained in loosely defined half and full discharge potential windows, were then carefully analyzed by applying stepwise electrochemical polarization to both Cu3HHTT2 and Cu3HHTP2. Specifically, increasingly negative potentials (with 100 mV intervals) were applied for 2 h with intermediate GCPL cycling to identify the “on-set” potentials for capacity degradation in both MOFs (details in Fig. S22†). Both MOFs displayed similar capacity retention patterns versus applied potentials, wherein, three distinct regions can be identified. Constant potential polarizations in regions 1 and 3 resulted in minor changes to the capacities, whereas polarizations in region 2 led to a significant drop. Notably, the potentials that make up regions 1–3 are specific to each MOF. Nevertheless, the application of polarization can cause more changes to a MOF, beyond chemical reduction alone. A recent report on Cu3HHTP2 has revealed a reversible expansion and contraction of lattice during ion sorption in its pores.33 The influence of the mechanical strain originating from such structural changes on cycling stability will likely vary between different MOFs.
Avoiding redox contributions from region 2 for enhanced capacity retention leads to lower storage capacities for MOFs. We sought to maintain the redox activity in Cu3HHTT2 while addressing its dissolution through control of the experimental parameters. Specifically, HHTT has a low solubility of 0.22 mg mL−1 or 0.52 nM in ACN, which should, in theory, rapidly saturate the electrolyte with HHTT, preventing continuous dissolution of Cu3HHTT2. Notably, all electrochemical data discussed so far were obtained in Swagelok-type cells that accommodate significant electrolyte volume (∼2 mL) that likely facilitates continuous dissolution of MOF during cycling (a picture of a Swagelok cell is shown in Fig. S23a†). We hypothesized that lowering the amount of electrolyte per active material mass could lead to a saturation of HHTT in the electrolyte, enabling longer cycling life. Indeed, tests performed in Swagelok cells using increasing electrolyte volumes of 1–15 mL mg−1 of active material have delivered greater capacity retention in cells with lower electrolyte volumes (Fig. S24†). Furthermore, tests performed in coin cells (a picture of a coin cell is shown in Fig. S23b†), which enabled the use of significantly less amount of electrolyte (50 μL or 0.05 mL mg−1), delivered remarkable stability over 7500 cycles (or 1000 hours of continuous operation) compared to the tests in the Swagelok cells (Fig. 6b). The distinct performance of Cu3HHTP2 in different cell designs that accommodate different electrolyte volumes, while also differing in other aspects such as the pressure between electrodes and the area of the electrode surface, perhaps explain the different cycling performances noted for Cu3HHTP2 (ref. 33) and Cu3THQ2 (ref. 31 and 34) in this work and the previous reports. Overall, these studies further reaffirm that dissolution represents the primary mode of degradation for 2D cMOFs and the low solubilities of ligands in electrolytes could be leveraged for enhanced cycling stability.
Discussion
The discovery of electrode dissolution as the primary mode of electrochemical degradation for 2D cMOFs raises questions on what causes dissolution and how it proceeds. Although the combination of UV-vis, EQCM, and ICP-MS techniques has provided a qualitative and quantitative estimation of MOF dissolution, the true identity of the dissolved species remains unclear. An understanding of the dissolved species is essential in describing possible mechanisms for MOF dissolution. Nevertheless, the success of strategies aimed at saturating dissolution by using less electrolyte identifies low ligand solubility as a key design principle for future MOF design. Additionally, the series of stepwise electrochemical polarizations of cMOFs have identified a range of potentials (region 2) that exert the most detrimental influence on their cycling stability. Although ex situ characterizations have identified redox reactions to occur on both the ligand and metal nodes in this range of potentials, further investigations involving in situ and operando studies are necessary to capture the chemical and structural changes at the MX4 linkages at these potentials. However, the dynamic and reversible nature of MX4 linkages,41–43 and the possibility of transient metal mediation in ligand-centered reduction processes,11 together complicate the determination of their true influence on the cycling stability of cMOFs. Furthermore, it may also be construed as inappropriate to treat metal and ligand reduction as isolated processes when the material itself is electronically delocalized through π-d overlaps.6,43 Overall, these factors present a need for deeper investigations into the effects of external polarization on MOF stability.Although it is reasonable to expect that the conclusions drawn here may extend to other systems, such attempts must consider the limitations of the choice of materials and the experimental conditions used here. Specifically, the three MOFs investigated here share [CuO4] linkages, and are studied under reductive potentials in water and ACN-based solutions. MOFs with different MX4 linkages and their behavior under oxidative or purely capacitive electrochemical conditions may differ from this work. Additionally, several parameters such as the particle size, morphology (scanning microscopy images of Cu3HHTT2 in Fig. S25†), electrode composition, type of binder, cell setup, mass loading, and electrolyte volume can all affect the cycling stability of a MOF.44,45 Notably, the dissolution of MOFs shares similarities with the well-studied dissolution of organic electrode materials (OEMs), presenting opportunities for shared solutions.46–53 Nevertheless, while the dissolution of OEMs typically provides strong visual cues, only Cu3THQ2 among the three MOFs studied here showed color in the electrolytes (Fig. S26†).
Conclusions
The electrochemical stability of 2D cMOFs and the potential mechanisms for their degradation during repeated charge–discharge cycling were investigated in three Cu-based 2D cMOFs. Cu3HHTT2, with a sparingly soluble ligand in aqueous electrolytes, delivered excellent cycling stability over 100000 cycles or 1500 hours of operation. On the other hand, Cu3THQ2 and Cu3HHTP2 exhibited rapid degradation within the first 12 hours of their cycling. A comprehensive qualitative and quantitative analysis revealed that electrode dissolution is the primary mode of degradation for all three Cu-MOFs. A combination of UV-vis, ICP-MS, and EQCM studies revealed that up to 80% of the capacity loss in a MOF can proceed via electrode dissolution. Several factors that govern dissolution are investigated and solutions for mitigating dissolution are presented. Overall, these findings offer important design principles for targeted development and application of 2D cMOFs in EES.
Data availability
Data is made available upon a reasonable request.Author contributions
J.-H. D. and H. B. conceived the project. G. M. R. D. and H. B. developed the methodology. G. M. R. D., S. P. S., J. L. M.-G. performed all experimental syntheses and investigations. T. C. performed XPS data collection and analysis. G. M. R. D., S. P. S., and H. B. prepared all visualizations. H. B. acquired the funding and supervised the project. G. M. R. D. and H. B. prepared the original draft. All authors have contributed to editing the manuscript and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge access to the core instrumentation facilities provided by the Department of Chemistry and Biochemistry at the University of Texas, El Paso (UTEP). This work used the X-ray Photoelectron Spectrometer at UTEP funded by the National Science Foundation MRI program under award number NSF 2216473. This work is supported by the startup grant provided to H. B. by the University of Texas at El Paso and the Rising Star Award granted to H. B. by the University of Texas system. The authors thank Dr Mahesh Narayan for access to the UV-vis spectrometer, Dr Dino Villagran for insightful discussions, and Dr Alejandro Metta for technical support and training.References
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed
.
- M. Yu, R. Dong and X. Feng, J. Am. Chem. Soc., 2020, 142, 12903–12915 CrossRef CAS PubMed
- C. N. Hong, A. B. Crom, J. I. Feldblyum and M. R. Lukatskaya, Chem, 2023, 9, 798–822 CAS
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS PubMed
- J.-H. Dou, L. Sun, Y. Ge, W. Li, C. H. Hendon, J. Li, S. Gul, J. Yano, E. A. Stach and M. Dincă, J. Am. Chem. Soc., 2017, 139, 13608–13611 CrossRef CAS PubMed
- K. Sakaushi and H. Nishihara, Acc. Chem. Res., 2021, 54, 3003–3015 CrossRef CAS PubMed
- H. Banda, J.-H. Dou, T. Chen, N. J. Libretto, M. Chaudhary, G. M. Bernard, J. T. Miller, V. K. Michaelis and M. Dincă, J. Am. Chem. Soc., 2021, 143, 2285–2292 CrossRef CAS PubMed
- H. Banda, J.-H. Dou, T. Chen, Y. Zhang and M. Dincă, Angew. Chem., 2021, 133, 27325–27331 CrossRef
- Z. Wu, D. Adekoya, X. Huang, M. J. Kiefel, J. Xie, W. Xu, Q. Zhang, D. Zhu and S. Zhang, ACS Nano, 2020, 14, 12016–12026 CrossRef CAS PubMed
- J. Park, A. C. Hinckley, Z. Huang, D. Feng, A. A. Yakovenko, M. Lee, S. Chen, X. Zou and Z. Bao, J. Am. Chem. Soc., 2018, 140, 14533–14537 CrossRef CAS PubMed
- D. Rambabu, A. E. Lakraychi, J. Wang, L. Sieuw, D. Gupta, P. Apostol, G. Chanteux, T. Goossens, K. Robeyns and A. Vlad, J. Am. Chem. Soc., 2021, 143, 11641–11650 CrossRef CAS PubMed
- Z. Wang, G. Wang, H. Qi, M. Wang, M. Wang, S. Park, H. Wang, M. Yu, U. Kaiser, A. Fery, S. Zhou, R. Dong and X. Feng, Chem. Sci., 2020, 11, 7665–7671 RSC
- J. Liu, Y. Zhou, Z. Xie, Y. Li, Y. Liu, J. Sun, Y. Ma, O. Terasaki and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 1081–1086 CrossRef CAS PubMed
- J. Yin, N. Li, M. Liu, Z. Li, X. Wang, M. Cheng, M. Zhong, W. Li, Y. Xu and X.-H. Bu, Adv. Funct. Mater., 2023, 33, 2211950 CrossRef CAS
- M. Yang, X. Zeng, M. Xie, Y. Wang, J.-M. Xiao, R.-H. Chen, Z.-J. Yi, Y.-F. Huang, D.-S. Bin and D. Li, J. Am. Chem. Soc., 2024, 146, 6753–6762 CrossRef CAS PubMed
- P. Apostol, S. M. Gali, A. Su, D. Tie, Y. Zhang, S. Pal, X. Lin, V. R. Bakuru, D. Rambabu, D. Beljonne, M. Dincă and A. Vlad, J. Am. Chem. Soc., 2023, 145, 24669–24677 CAS
- Y. Lu, Z. Hu, P. Petkov, S. Fu, H. Qi, C. Huang, Y. Liu, X. Huang, M. Wang, P. Zhang, U. Kaiser, M. Bonn, H. I. Wang, P. Samorì, E. Coronado, R. Dong and X. Feng, J. Am. Chem. Soc., 2024, 146, 2574–2582 CrossRef CAS PubMed
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS
- D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dincă, J. Am. Chem. Soc., 2014, 136, 8859–8862 CrossRef CAS PubMed
- S. Bi, H. Banda, M. Chen, L. Niu, M. Chen, T. Wu, J. Wang, R. Wang, J. Feng, T. Chen, M. Dincă, A. A. Kornyshev and G. Feng, Nat. Mater., 2020, 19, 552–558 CrossRef CAS PubMed
- L. Guo, J. Sun, W. Zhang, L. Hou, L. Liang, Y. Liu and C. Yuan, ChemSusChem, 2019, 12, 5051–5058 CrossRef CAS PubMed
- R. Dong, Z. Zhang, D. C. Tranca, S. Zhou, M. Wang, P. Adler, Z. Liao, F. Liu, Y. Sun, W. Shi, Z. Zhang, E. Zschech, S. C. B. Mannsfeld, C. Felser and X. Feng, Nat. Commun., 2018, 9, 2637 CrossRef PubMed
- P. Zhang, M. Wang, Y. Liu, S. Yang, F. Wang, Y. Li, G. Chen, Z. Li, G. Wang, M. Zhu, R. Dong, M. Yu, O. G. Schmidt and X. Feng, J. Am. Chem. Soc., 2021, 143, 10168–10176 CrossRef CAS PubMed
- P. Zhang, M. Wang, Y. Liu, Y. Fu, M. Gao, G. Wang, F. Wang, Z. Wang, G. Chen, S. Yang, Y. Liu, R. Dong, M. Yu, X. Lu and X. Feng, J. Am. Chem. Soc., 2023, 145, 6247–6256 CrossRef CAS PubMed
- J. Zhang, G. Zhou, H.-I. Un, F. Zheng, K. Jastrzembski, M. Wang, Q. Guo, D. Mücke, H. Qi, Y. Lu, Z. Wang, Y. Liang, M. Löffler, U. Kaiser, T. Frauenheim, A. Mateo-Alonso, Z. Huang, H. Sirringhaus, X. Feng and R. Dong, J. Am. Chem. Soc., 2023, 145, 23630–23638 CrossRef CAS PubMed
- D. Feng, T. Lei, M. R. Lukatskaya, J. Park, Z. Huang, M. Lee, L. Shaw, S. Chen, A. A. Yakovenko, A. Kulkarni, J. Xiao, K. Fredrickson, J. B. Tok, X. Zou, Y. Cui and Z. Bao, Nat. Energy, 2018, 3, 30–36 CrossRef CAS
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed
- M. R. Lukatskaya, D. Feng, S.-M. Bak, J. W. F. To, X.-Q. Yang, Y. Cui, J. I. Feldblyum and Z. Bao, ACS Nano, 2020, 14, 15919–15925 CrossRef CAS PubMed
- J. Yan, Y. Cui, M. Xie, G.-Z. Yang, D.-S. Bin and D. Li, Angew. Chem., Int. Ed., 2021, 60, 24467–24472 CrossRef CAS PubMed
- Q. Chen, O. Adeniran, Z.-F. Liu, Z. Zhang and K. Awaga, J. Am. Chem. Soc., 2023, 145, 1062–1071 CrossRef CAS PubMed
- K. W. Nam, S. S. Park, R. dos Reis, V. P. Dravid, H. Kim, C. A. Mirkin and J. F. Stoddart, Nat. Commun., 2019, 10, 4948 CrossRef PubMed
- J. M. Wrogemann, M. J. Lüther, P. Bärmann, M. Lounasvuori, A. Javed, M. Tiemann, R. Golnak, J. Xiao, T. Petit, T. Placke and M. Winter, Angew. Chem., Int. Ed., 2023, 62, e202303111 CrossRef CAS PubMed
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X.-Q. Yang, E. Hu, Y. Cao, J. Sun, Y. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef CAS PubMed
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 18769–18776 CrossRef CAS PubMed
- J. W. Gittins, C. J. Balhatchet, Y. Chen, C. Liu, D. G. Madden, S. Britto, M. J. Golomb, A. Walsh, D. Fairen-Jimenez, S. E. Dutton and A. C. Forse, J. Mater. Chem. A, 2021, 9, 16006–16015 RSC
- Y. Chen, Y. Zhang, Q. Huang, X. Lin, A. Zeb, Y. Wu, Z. Xu and X. Xu, ACS Appl. Energy Mater., 2022, 5, 7842–7873 CrossRef CAS
- J.-H. Dou, M. Q. Arguilla, Y. Luo, J. Li, W. Zhang, L. Sun, J. L. Mancuso, L. Yang, T. Chen, L. R. Parent, G. Skorupskii, N. J. Libretto, C. Sun, M. C. Yang, P. V. Dip, E. J. Brignole, J. T. Miller, J. Kong, C. H. Hendon, J. Sun and M. Dincă, Nat. Mater., 2021, 20, 222–228 CrossRef CAS PubMed
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed
- J. Liu, D. Xie, X. Xu, L. Jiang, R. Si, W. Shi and P. Cheng, Nat. Commun., 2021, 12, 3131 CrossRef CAS PubMed
- J. L. Obeso, M. T. Huxley, C. Leyva, J. Gabriel Flores, N. Martín-Guaregua, M. Viniegra, J. Aguilar-Pliego, J. Antonio de los Reyes, I. A. Ibarra and R. A. Peralta, Coord. Chem. Rev., 2023, 496, 215403 CrossRef CAS
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS PubMed
- J. W. Gittins, C. J. Balhatchet, S. M. Fairclough and A. C. Forse, Chem. Sci., 2022, 13, 9210–9219 RSC
- S. P. Suman, G. M. R. Dontireddy, T. Chen, J. Wang, J.-H. Dou and H. Banda, ACS Energy Lett., 2024, 1572–1580 CrossRef CAS
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS
- Y. Lu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142 CrossRef CAS PubMed
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef
- J. Wang, A. E. Lakraychi, X. Liu, L. Sieuw, C. Morari, P. Poizot and A. Vlad, Nat. Mater., 2021, 20, 665–673 CrossRef CAS PubMed
- H. Banda, D. Damien, K. Nagarajan, M. Hariharan and M. M. Shaijumon, J. Mater. Chem. A, 2015, 3, 10453–10458 RSC
- T. Chen, H. Banda, J. Wang, J. J. Oppenheim, A. Franceschi and M. Dincă, ACS Cent. Sci., 2024, 10, 569–578 CrossRef CAS PubMed
- T. Chen, H. Banda, L. Yang, J. Li, Y. Zhang, R. Parenti and M. Dincă, Joule, 2023, 7, 986–1002 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02699c |
| This journal is © The Royal Society of Chemistry 2024 |