DOI:
10.1039/D4SC02212B
(Edge Article)
Chem. Sci., 2024,
15, 12017-12025
Synergistically activating nucleophile strategy enabled organocatalytic asymmetric P-addition of cyclic imines†
Received
3rd April 2024
, Accepted 4th June 2024
First published on 6th June 2024
Abstract
Herein, we present an attractive organocatalytic asymmetric addition of P-nucleophiles to five-membered cyclic N-sulfonyl imines facilitated by phosphonium salt catalysis, enabling the highly enantioselective synthesis of tri- and tetra-substituted cyclic phosphorus-containing benzosultams. With this protocol, various cyclic α-aminophosphonates were efficiently synthesized with high yields and exceptional enantioselectivities (up to >99% ee) under mild reaction conditions. The utility and practicality of this method were demonstrated through gram-scale reactions and straightforward elaborations. Notably, the success of this approach relies on the deliberate selection of a synergistic organocatalytic system, which helps circumvent foreseeable side effects while handling secondary phosphine oxides (SPOs). Systematic mechanistic studies, incorporating experiments and DFT calculations, have revealed the critical importance of judiciously selecting bifunctional phosphonium salt catalysts for effectively activating P-nucleophiles while stereoselectively controlling the P-attack process.
1. Introduction
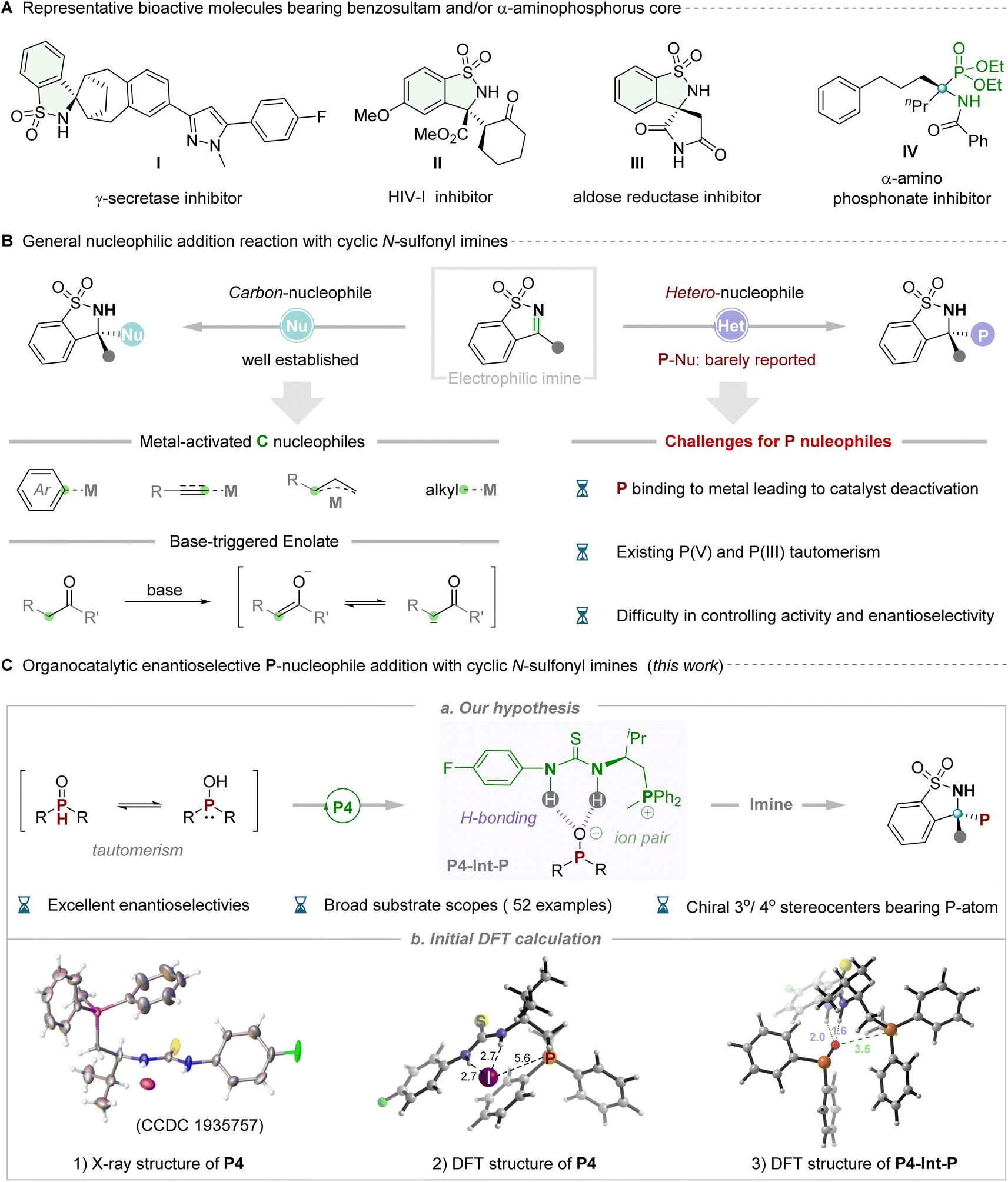
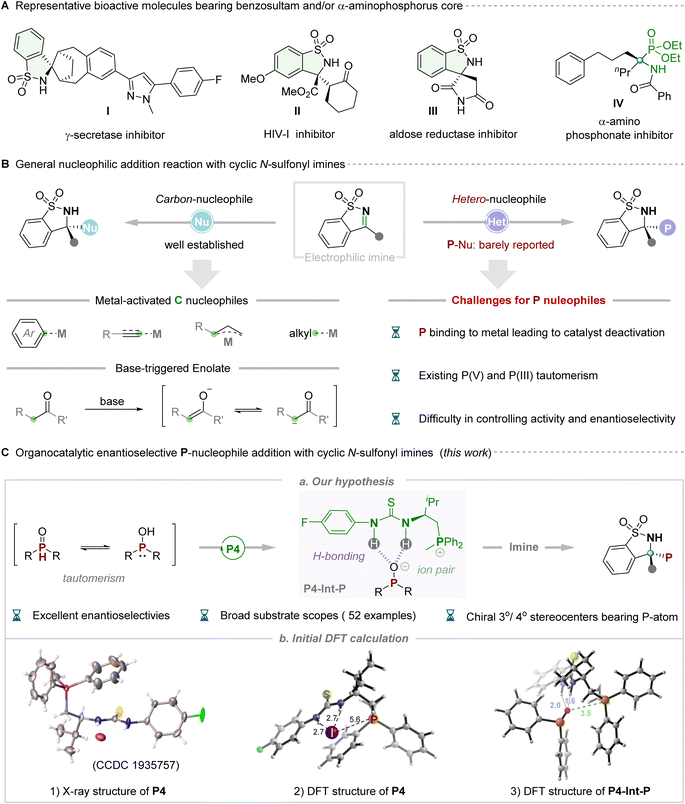
The sulfonamide functional group is recognized as a pivotal pharmacophore, particularly in clinical pharmaceuticals, where optically pure five-membered benzosultams find extensive application as potent inhibitors of γ-secretase, HIV-1, aldose reductase etc.(Fig. 1A, I–III).1 Cyclic N-sulfonyl imine, an exceptionally versatile synthetic precursor, has garnered considerable attention owing to its intriguing structures and associated properties. Chemists primarily concentrate on synthetically manipulating the electrophilic pro-chiral C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of N-sulfonyl imines, which possess multiple heteroatoms (N, O and S), thereby frequently exhibiting or enhancing biological activities.2 Significant advancements have occurred through the utilization of various carbon nucleophiles to react with the imine moiety, facilitating the synthesis of chiral sulfonamides in recent years. In this context, metal-catalyzed, elegantly enantioselective aryl nucleophilic additions utilizing aryl boronic acids or aryl halides as pronucleophilic reagents have been successfully developed by Zhang,3a Xu,3b,c Hayashi3d and Shi.3e Subsequently, strategies employing metal-promoted carbon-nucleophilic addition, utilizing alkyl,4 alkenyl,5 allyl,6 alkynyl7 and other8 nucleophilic reagents, to cyclic N-sulfonyl imines have been extensively employed for the modular assembly of chiral N-substituted quaternary stereocenters. Additionally, sporadic reports in recent years have demonstrated the utilization of ketones and their derivatives to generate the enolate ion through deprotonation under alkaline conditions, facilitating participation in asymmetric nucleophilic addition with cyclic N-sulfonyl imines catalyzed by chiral organic bases.9 Despite such impressive progress, significant limitations are also evident. Moreover, only C-nucleophiles have been utilized, and the majority of these cases necessitated transition metal catalysts. To our knowledge, catalytic asymmetric nucleophilic addition to five-membered cyclic N-sulfonyl imines using heteroatoms, particularly P-atom nucleophilic reagents, has never been reported to date, representing a significant challenge. Challenges include the potential poisoning of metal catalysts by P-nucleophiles, tautomerism between phosphine oxide species (P(V) and P(III)), and difficulties in controlling reactivity and achieving high enantioselectivity. Notably, chiral phosphorus-containing compounds, particularly α-amino phosphates, have been widely employed as candidate pharmacological molecules (Fig. 1A, IV)10 and/or potential chiral ligands/catalysts.11 From this perspective, the incorporation of P-atoms into N-sulfonyl imines to create a new structure bearing multiple hetero-atoms (i.e. O, S, N, and P) is intriguing yet challenging.
N bond of N-sulfonyl imines, which possess multiple heteroatoms (N, O and S), thereby frequently exhibiting or enhancing biological activities.2 Significant advancements have occurred through the utilization of various carbon nucleophiles to react with the imine moiety, facilitating the synthesis of chiral sulfonamides in recent years. In this context, metal-catalyzed, elegantly enantioselective aryl nucleophilic additions utilizing aryl boronic acids or aryl halides as pronucleophilic reagents have been successfully developed by Zhang,3a Xu,3b,c Hayashi3d and Shi.3e Subsequently, strategies employing metal-promoted carbon-nucleophilic addition, utilizing alkyl,4 alkenyl,5 allyl,6 alkynyl7 and other8 nucleophilic reagents, to cyclic N-sulfonyl imines have been extensively employed for the modular assembly of chiral N-substituted quaternary stereocenters. Additionally, sporadic reports in recent years have demonstrated the utilization of ketones and their derivatives to generate the enolate ion through deprotonation under alkaline conditions, facilitating participation in asymmetric nucleophilic addition with cyclic N-sulfonyl imines catalyzed by chiral organic bases.9 Despite such impressive progress, significant limitations are also evident. Moreover, only C-nucleophiles have been utilized, and the majority of these cases necessitated transition metal catalysts. To our knowledge, catalytic asymmetric nucleophilic addition to five-membered cyclic N-sulfonyl imines using heteroatoms, particularly P-atom nucleophilic reagents, has never been reported to date, representing a significant challenge. Challenges include the potential poisoning of metal catalysts by P-nucleophiles, tautomerism between phosphine oxide species (P(V) and P(III)), and difficulties in controlling reactivity and achieving high enantioselectivity. Notably, chiral phosphorus-containing compounds, particularly α-amino phosphates, have been widely employed as candidate pharmacological molecules (Fig. 1A, IV)10 and/or potential chiral ligands/catalysts.11 From this perspective, the incorporation of P-atoms into N-sulfonyl imines to create a new structure bearing multiple hetero-atoms (i.e. O, S, N, and P) is intriguing yet challenging.
 |
| | Fig. 1 Our strategy for the organocatalytic enantioselective P-nucleophilic addition of cyclic N-sulfonyl imines toward phosphorus-containing benzosultams. | |
Returning to the issue of phosphorus nucleophiles, in general, the readily available and bench-stable secondary phosphine oxides (SPOs) were often the preferred choice due to their easy handling and relatively odorless nature.12 In reality, a similar tautomeric equilibrium, like the textbook example of keto–enol tautomerism, can be drawn between the pentavalent oxide P(V) form and the phosphinous acid P(III) form for such phosphorus-containing compounds.13 The flexible tautomeric equilibrium and slippery molecular structure of SPOs make them difficult to selectively control in asymmetric reactions. With the flourishing development of asymmetric catalytic systems in recent decades, the synthetic community has witnessed the progress of the enantioselective P-addition with general imines,14 but the gap of five-membered cyclic N-sulfonyl imines still needs to be filled. Considering the well-known challenge in this area, we recognize that the key to success lies in the rational design and/or selection of an efficient organocatalytic system for its powerful catalytic ability and wide application potential toward synthesis of various important chiral molecules in synthetic chemistry.15 Given our ongoing progress in developing multi- and/or bi-functional phosphonium salt catalysts that have successfully addressed many synthetic challenges and significant targets,16 we anticipate that these highly structurally tunable ion-pair catalysts with multiple hydrogen-bonding interactions with substrates will address the issue of reactivity and stereoselectivity control of P-species. Subsequently, they will participate in nucleophilic addition with cyclic N-sulfonyl imines to afford chiral phosphorus-containing benzosultams (Fig. 1C). Our hypothesis was rationally validated by preliminary analysis of the X-ray structure of thiourea-derived phosphonium salt catalyst P4. In this analysis, the iodine negative ion counterion was observed to be distant from the phosphorus cation center, resulting in weaker binding constants with the thiourea moiety, thus providing an opportunity for exchanging the negative iodine ion with the phosphate anion. Additionally, the key intermediate (P4-Int-P) was formed through a combination of hydrogen bonding and ion pairs within the catalyst, exhibiting stronger hydrogen bonding and electrostatic forces than catalyst P4 according to DFT simulation (Fig. 1C, eqn (3)). We identified a novel activation mode of thiourea-derived phosphonium salt catalysts, wherein the exchanged anion not only acts as the nucleophile in enantiodetermining bond-formation but also accelerates the production of chiral phosphorus-containing benzosultams via hydrogen bonding in phase-transfer catalysis. This approach offers more precise control over stereoselectivity through specific association of the nucleophile with the chiral bifunctional phosphonium salt.17
2. Results and discussion
2.1 Asymmetric P-nucleophilic addition of cyclic N-sulfonyl imines with secondary phosphine oxides
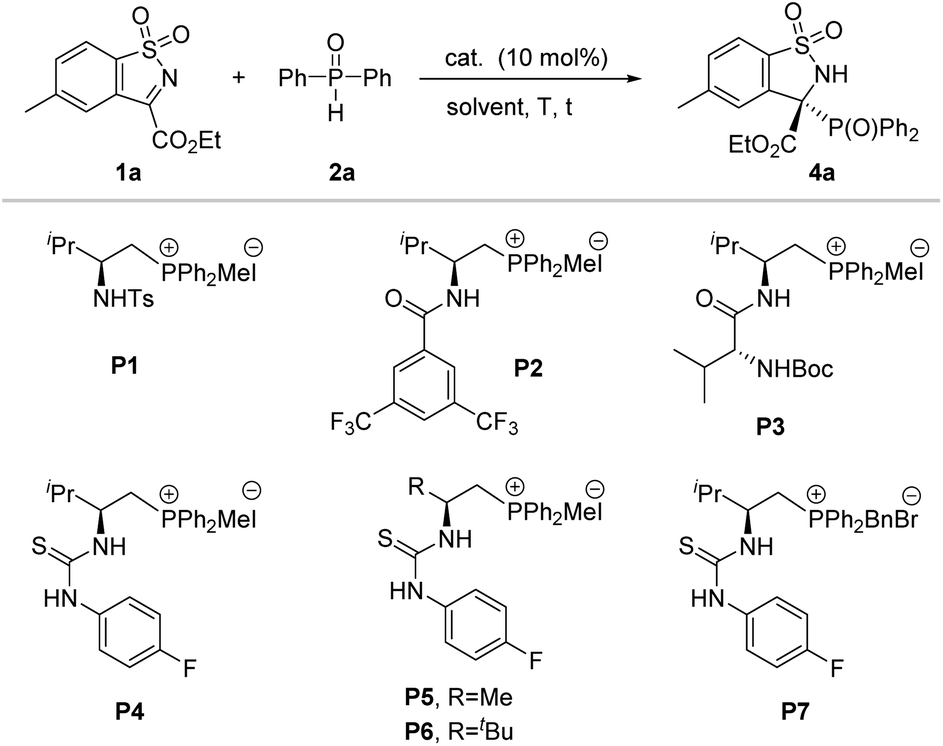
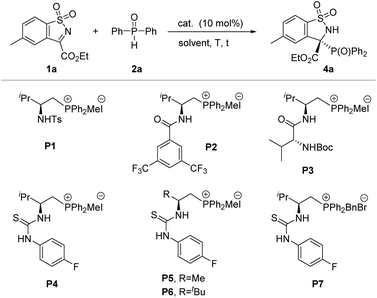
Inspired by the initial studies, we started our investigation to explore the feasibility of this reaction between N-sulfonyl cyclic ketimine 1a and diphenylphosphine oxide 2a in CH2Cl2 at room temperature in the presence of K2CO3. As a result, the desired racemic product 4a was obtained with 98% yield (Table 1, entry 1). Next, when the racemic phosphonium salt P0 was used for this reaction without addition of base, this reaction also proceeded smoothly, affording 4a with 83% yield for 2 hours (Table 1, entry 2). The above-mentioned results implied that the strong background reaction may be involved in this system. Given our previous successful research on the enantioselective control of phosphorus-containing species in asymmetric synthesis within bifunctional phosphonium catalytic systems,16 we chose amide-, dipeptide- and thiourea-derived phosphonium salts as candidate catalysts for their representative hydrogen-bonding and ion-pairing features. Pleasingly, all tested phosphonium salt catalysts were effective in promoting this reaction, furnishing the desired product in high yields and moderate to good enantioselectivities (entries 3–9). In particular, L-valine-derived thiourea-based P4 was discovered to be an excellent catalyst for this transformation, affording the desired product 4a with 93% yield and 90% ee (entry 6). To improve the enantioselectivity, we further screened the solvents (see Table S2† for more details) and proved 1,2-dichloroethane to be the best solvent, affording the desired product in excellent yield (96%) and enantioselectivity (>99% ee) (Table 1, entry 12). Additionally, we further verify the extra base will accelerate the background reaction leading to loss of enantioselectivity (entries 13 and 14). In fact, upon reducing the catalyst loading to 5 mol%, the reaction could maintain good activity, but eroded the enantioselectivity of the product slightly (Table 1, entry 15).
Table 1 Optimization for asymmetric P-nucleophile addition to cyclic N-sulfonyl imines with secondary phosphine oxides
|
|
| Entry |
Cat. |
Base (x equiv.) |
Solvent |
t (h) |
Yield (%) |
ee (%) |
|
Reactions were performed with 1a (0.1 mmol), 2a (0.12 mmol), catalyst (0.01 mmol) in solvent (1.0 mL) at room temperature for 0.5–2.5 h, P0 = Ph2Me2P+I−.
Isolated yield.
The ee value was determined by HPLC analysis on a chiral stationary phase.
1.0 equivalent of K2CO3 was added.
0.5 equivalent of K2CO3 was used.
The catalyst loading was 5 mol%. Ts = 4-Toluenesulfonyl. DCE = 1,2-Dichloroethane.
|
| 1 |
— |
K2CO3 (1.0) |
CH2Cl2 |
1 |
98 |
0 |
| 2 |
P0
|
None |
CH2Cl2 |
2 |
83 |
0 |
| 3 |
P1
|
None |
CH2Cl2 |
2 |
90 |
34 |
| 4 |
P2
|
None |
CH2Cl2 |
2 |
92 |
53 |
| 5 |
P3
|
None |
CH2Cl2 |
2 |
90 |
71 |
| 6 |
P4
|
None |
CH2Cl2 |
2 |
93 |
90 |
| 7 |
P5
|
None |
CH2Cl2 |
2 |
86 |
68 |
| 8 |
P6
|
None |
CH2Cl2 |
2 |
92 |
86 |
| 9 |
P7
|
None |
CH2Cl2 |
2 |
87 |
83 |
| 10 |
P4
|
None |
CHCl3 |
2 |
94 |
73 |
| 11 |
P4
|
None |
Toluene |
2 |
88 |
93 |
| 12 |
P4
|
None |
DCE
|
2
|
95
|
>99
|
| 13 |
P4
|
K2CO3 (1.0) |
DCE |
0.5 |
95 |
<5 |
| 14 |
P4
|
K2CO3 (0.5) |
DCE |
1 |
96 |
75 |
| 15 |
P4
|
None |
DCE |
3 |
92 |
89 |
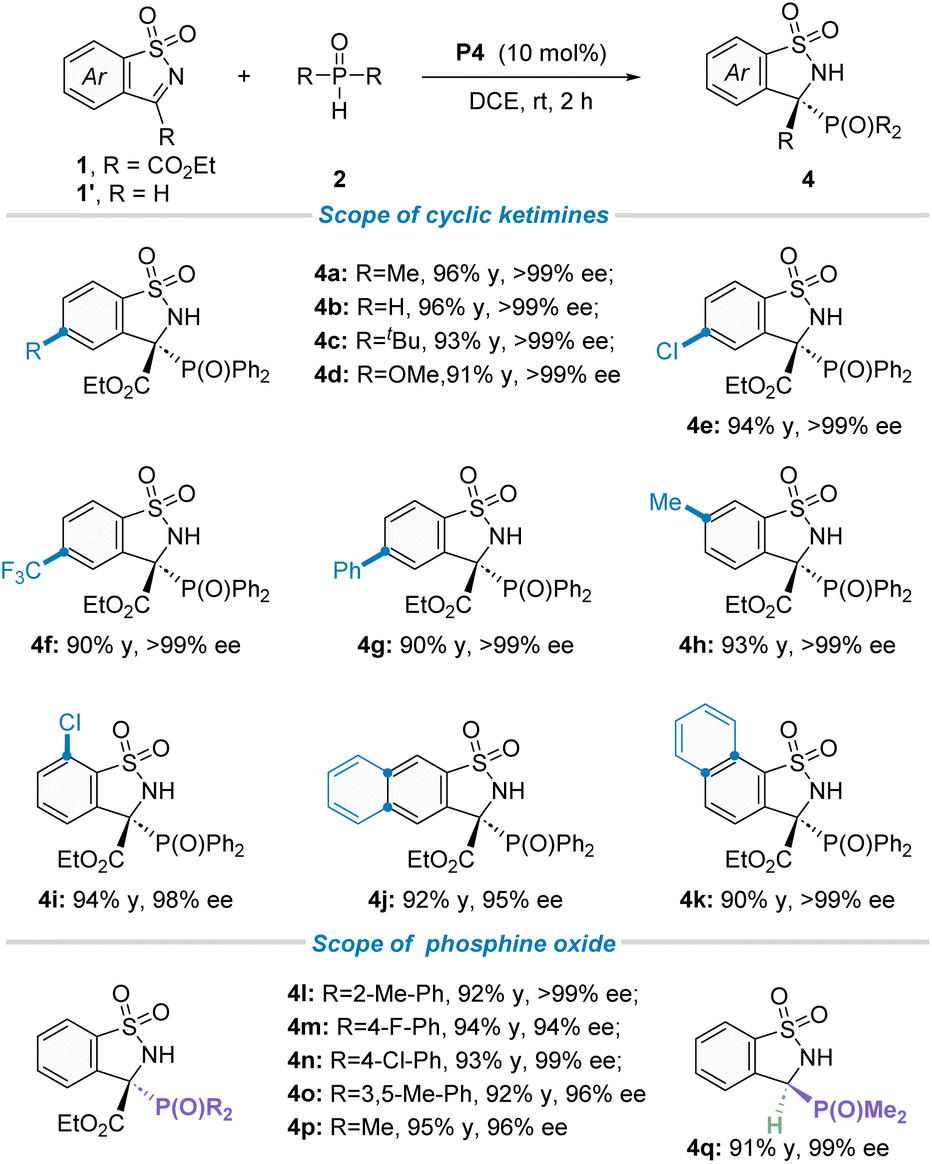
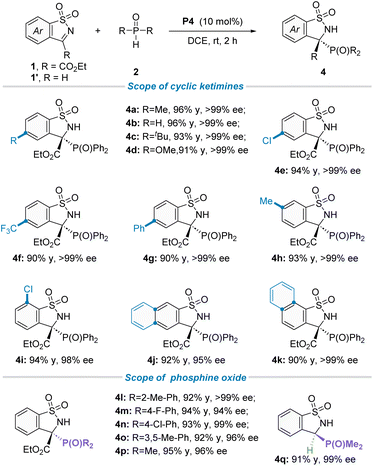
Having established the optimal reaction conditions in hand, we then focused on the generality of this protocol. Firstly, the scope of asymmetric P-nucleophile addition to cyclic N-sulfonyl imines 1 with secondary phosphine oxides was investigated (Table 2). To our delight, an array of cyclic ketimines 1 with electron-donating or electron-withdrawing substituents at the ortho-, meta-, or para-position of the phenyl ring moiety were found to be suitable reaction partners, furnishing the desired products (4a–i) in good yields and excellent enantioselectivities (98 to >99% ee). Additionally, 1-naphthyl and 2-naphthyl substituted cyclic ketimines were also well tolerated under the standard reaction conditions, giving the corresponding products 4j in 92% yield and 95% ee and 4k in 90% yield and >99% ee, respectively. Subsequently, the generality of secondary phosphine oxides for this transformation was evaluated. As shown in Table 2 (down), for diaryl substituted phosphine oxides, the substituent group installed on the phenyl ring did not have an obvious effect on both isolated yields and stereoselectivities, offering the corresponding targets (4l–o) with good yields and outstanding ee values. It is worth noting that dimethyl phosphine oxide has also been proven to be a suitable phosphorus source, not only for ketimine 1a but also for aldimine 1′a, providing the corresponding chiral phosphorus-containing quaternary carbon center product 4p and 3o carbon center product 4q with 98% ee and 99% ee, respectively. To confirm the absolute configuration of these phosphorus-containing benzosultams 4, CD spectra were calculated by the DFT study. The S configuration could be reliably assigned to compound 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19a (see Scheme S3 in the ESI† for more details).
19a (see Scheme S3 in the ESI† for more details).
Table 2 Scope of the asymmetric P-nucleophilic addition of cyclic N-sulfonyl imines with secondary phosphine oxides
|
Reactions were performed with 1/1′ (0.1 mmol), 2 (0.12 mmol), and P4 (0.01 mmol) in DCE (1.0 mL) at room temperature for 2 hours. All yields were isolated yields, and the ee value was determined by HPLC analysis on a chiral stationary phase.
|
|
2.2 Asymmetric P-nucleophilic addition of cyclic N-sulfonyl imines with phosphites
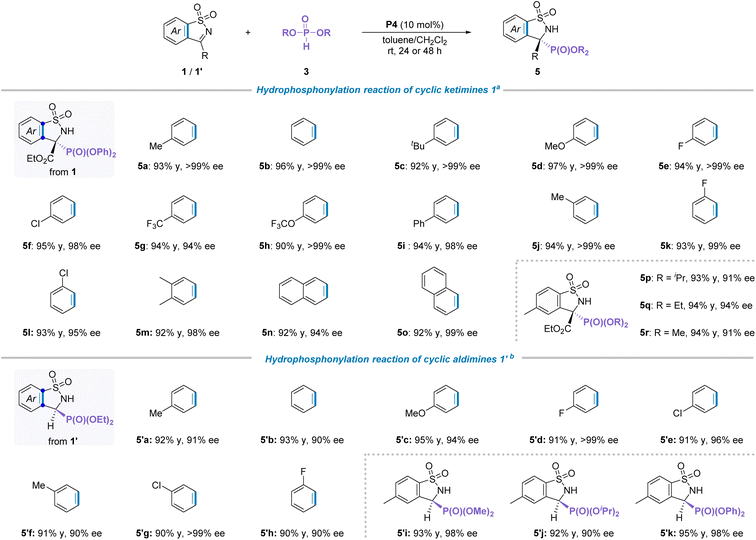
Different types of phosphoryl species have distinct chemical properties and pharmacological activities.18 Particularly, α-amino phosphates and their derivatives usually demonstrated important physiological activity in life.19 Therefore, the generality of phosphites as P-nucleophiles added to cyclic N-sulfonyl imines was explored. Inspired by the above successful results, the hydrophosphonylation reaction between cyclic ketimine 1a and diphenyl phosphite 3a to evaluate catalytic effects in the presence of P4 and 1,2-dichloroethane (DCE) at room temperature was studied, and results show that the desired addition product 5a was obtained in 94% yield and 88% ee. After a series of quick screening of optimization, toluene was found to be the most suitable solvent, affording the corresponding product in 96% yield with >99% ee (see Tables S3 and S4 in the ESI† for more details).
With the identified optimal reaction conditions in hand, we then set out to explore the generality for the scope of this hydrophosphonylation transformation. Firstly, a wide range of cyclic ketimines 1 bearing different substituent groups were investigated (Table 3a). As a result, neither the electronic properties nor the positions of the substituents on the aromatic ring of cyclic imines had an observable influence on the isolated yield and stereoselectivity of the products (5a–5o). Subsequently, the ester groups of phosphites with different sized substituent groups, including methyl, ethyl and isopropyl, were examined. To our delight, all these tested substrates were well compatible with the standard conditions, providing the corresponding products in good yields and enantioselectivities. Afterwards, when the cyclic aldimines 1′ as a model electrophilic reagent combined with diethyl phosphite for this reaction, the optimal solvent was changed to CH2Cl2 with addition of 2.0 equivalents of Cs2CO3 at room temperature in the presence of P4 (see Table S4† for more details). Under these standard reaction conditions, the compatibility for the substituent group was evaluated. As illustrated in Table 3b, a diverse array of cyclic aldimines 1′ regardless of the positions and electronic properties of the substituents on the phenyl ring were perfectly compatible with the standard reaction conditions, providing the corresponding products 5′a–′h in high isolated yields (90–95%) with excellent enantioselectivities (90 to >99%). Moreover, other types of phosphite esters, such as dimethyl, diisopropyl and diphenyl phosphite were also suitable for the reaction and the corresponding adducts 5′i–k were obtained in good yield and ee values. Besides, the absolute configuration of compounds 5 was assigned as S on the basis of analysis of CD spectra calculated by the DFT method.20a The absolute configuration of product 5′ was rationally assigned to be S by comparing the optical rotation of the same compound with previously reported results20b (see the ESI for more details, Scheme S2†).
Table 3 Scope of the asymmetric P-nucleophilic addition of cyclic N-sulfonyl imines with phosphitesa,b
|
Reactions were performed with cyclic ketimines 1 (0.1 mmol), 3 (0.12 mmol), and P4 (0.01 mmol) in toluene (2.0 mL) at room temperature for 24 hours. All yields were isolated yields, and the ee value was determined by HPLC analysis on a chiral stationary phase.
Reactions were performed with cyclic aldimines 1′ (0.1 mmol), 3 (0.12 mmol), P4 (0.01 mmol) and Cs2CO3 (0.2 mmol) in CH2Cl2 (2.0 mL) at room temperature for 48 hours.
|
|
2.3 Asymmetric P-nucleophilic addition of CF3-substituted cyclic N-sulfonyl amines
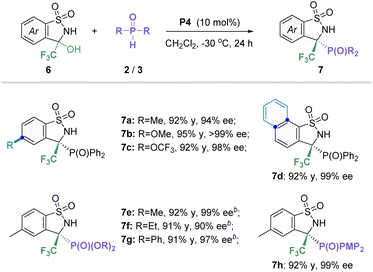
It is a universally accepted fact that the introduction of a trifluoromethyl group into N-heterocyclic molecules can further bring positive effects in the development of agrochemicals or pharmaceuticals.21 Thus, the preparation of the privileged CF3-substituted benzosultam that also contains a useful phosphorus atom is an important endeavor in the synthetic community. Based on the previous literature,22 we envisaged that α-hydroxy, α-trifluoromethyl substituted sulfonamide will in situ generate sulfimide easily, which could serve as a good electrophilic reagent.23 According to the above successful findings, we set out to carry out studies on asymmetric P-nucleophile addition between CF3-substituted cyclic N-sulfonyl amines and P-nucleophiles in the presence of a phosphonium salt catalyst (Table 4). After a quick survey of the reaction conditions, the target product 7a was obtained with 92% yield and 94% ee under the P4 catalyst in CH2Cl2 at −30 °C for 48 h (see Table S5 in the ESI† for more details on conditions optimization). Subsequently, the scope of in situ generated CF3-containing cyclic imine 6 with secondary phosphine oxides was investigated. To our delight, different aryl substituent groups with electron-donating or -withdrawing as well as naphthyl could well tolerate the standard conditions, affording the corresponding products 7b–d in high yields (92–95%) and excellent enantioselectivities (94–99% ee). Furthermore, various phosphites including diaryl and dialkyl phosphites were also found to be good reaction partners, offering the desired products 7e–h in satisfactory yields and enantioselectivities (91–92% yields, 90–99% ee).
Table 4 Asymmetric P-nucleophilic addition of CF3-substituted cyclic N-sulfonyl aminesa
|
Reactions were performed with 6 (0.1 mmol), 2/3 (0.12 mmol), and P4 (0.01 mmol) in CH2Cl2 (3.0 mL) at −30 °C for 24 hours. Isolated yield. The ee value was determined by HPLC analysis on a chiral stationary phase.
At room temperature for 48 hours.
|
|
2.4 Scaled-up synthesis and transformation of the product
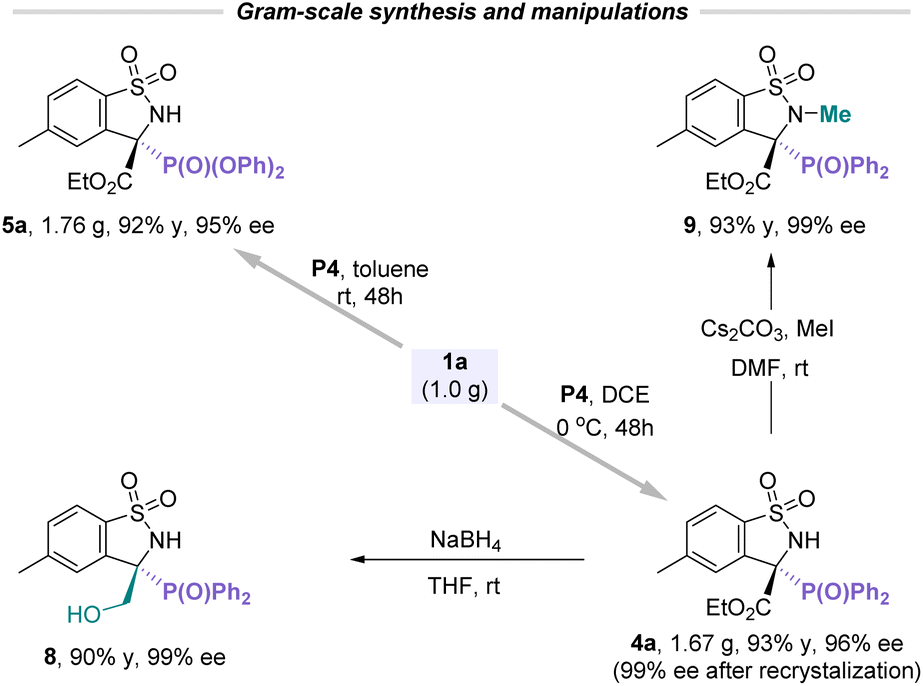
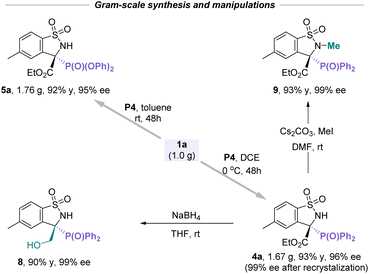
To further validate the scalability and practicality of this catalytic asymmetric protocol, the gram-scale experiments for the P-nucleophile addition with cyclic N-sulfonyl imines proceeded smoothly under the corresponding conditions, providing manifold corresponding products such as 4a and 5a with comparable yields without loss of enantioselectivities (Scheme 1). Moreover, the direct reduction of the ester group of 4a using NaBH4 furnished β-hydroxyl-functionalized compound 8 in excellent yield and enantioselectivity. In addition, the free N–H installed on the chiral benzosultams could be readily N-methylated with iodomethane to afford α-aminophosphonate 9 in high yield and without any loss of the enantiomeric purity, which provides a possibility for further modification on drug molecules.
 |
| | Scheme 1 Scale-up synthesis and manipulation of the product. | |
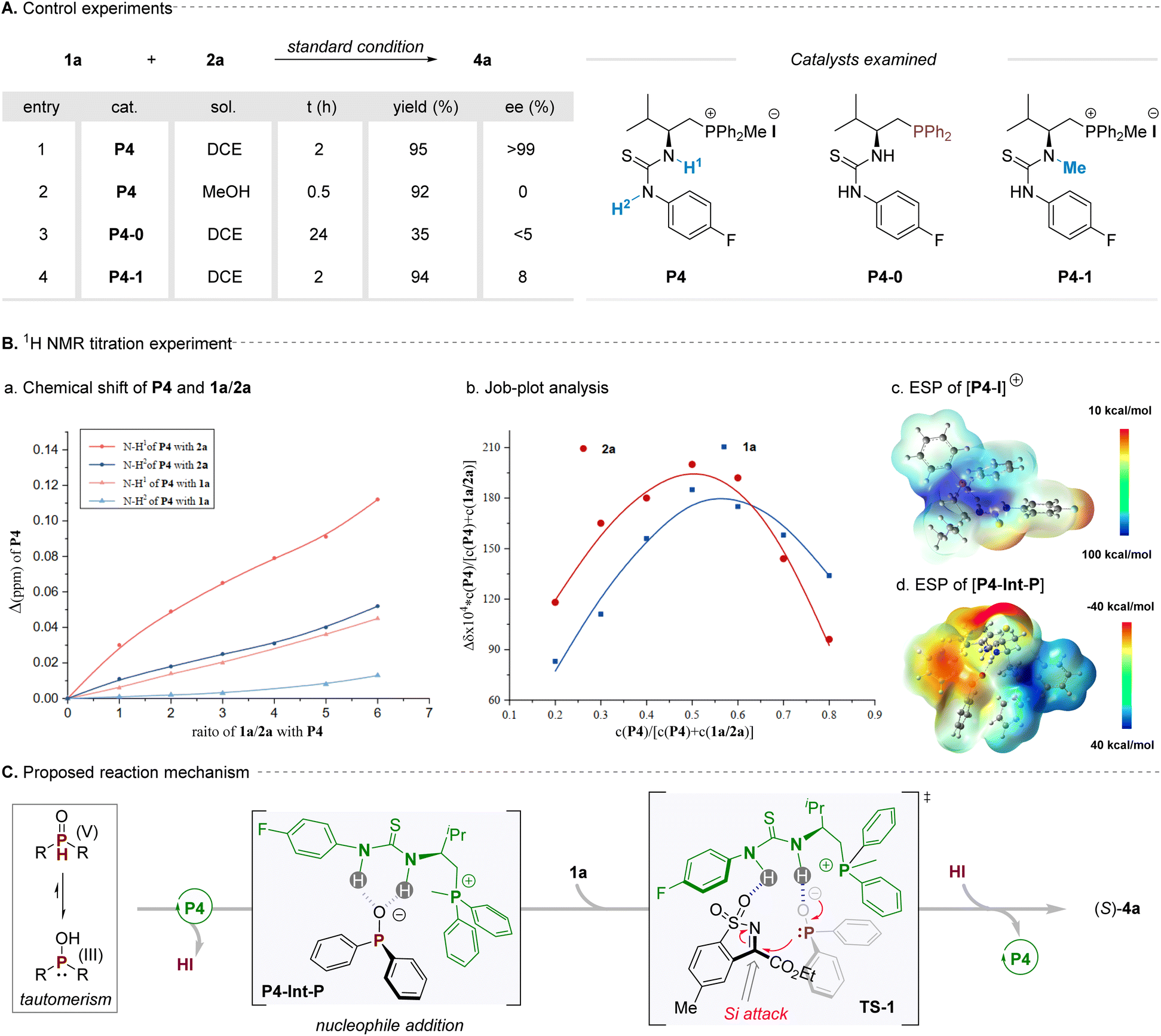
2.5 Mechanistic investigations
To gain insight into the mechanism for this reaction, a series of control experiments were carried out to understand the reaction mechanism, particularly towards the chiral control (Scheme 2A). Firstly, the reaction between cyclic ketimine 1a and diphenylphosphine oxide 2a was performed with bifunctional phosphonium salt P4 in DCE at room temperature for 24 h, affording the corresponding product 4a in 95% yield with >99% ee. When the polar solvent methanol was used, the enantioselectivity of the corresponding product decreased dramatically, which was probably mainly due to hydrogen bonding in systems being weakened or even destroyed (Scheme 2A, entry 2). Besides, when the optimal phosphonium salt catalyst P4 was changed to its corresponding trivalent phosphine (P4-0) under the standard conditions for the reaction, the yield and selectivity of the reaction decreased remarkably, giving a nearly racemic product only with 35% isolated yield (Scheme 2A, entry 3), which also indicated that the ion pairing of the phosphonium salt played an important role in the reaction. Additionally, one of the N–H bonds of the P4 was blocked to form a new methylated catalyst P4-1, which was further used as the catalyst for this model reaction, and the enantioselectivity of the product decreased obviously, with only 8% ee being obtained for the target (Scheme 2A, entry 4). These preliminary results clearly suggested that the H-bonding and ion-pair interactions in this catalytic system are critical factors for asymmetric induction. Subsequently, the 1H NMR titration experiments were performed for gaining further insights to the mechanism, particularly towards the origins of chirality induction for this reaction (Scheme 2B). In this vein, titration of P-nucleophile 2a to the optimal catalyst P4 led to a distinct chemical shift change in the position of both of the thiourea-NH signals of the catalyst. In contrast, upon titration of cyclic N-sulfonyl imine substrate 1a with P4, less pronounced changes were observed. Additionally, Job plot analysis of the addition of 1a or 2a to P4 indicated a 1:1 ratio binding model between the catalyst and substrate (Scheme 2B, b). The above results evidently indicated that the bifunctional phosphonium salt P4 was inclined to combine with the phosphite anion via double H-bonding and ion pair activation. Of note, given the observation that the iodide ion is relatively far from the phosphonium cation via the analysis of X-ray crystal structure for catalyst P4 (Fig. 1C, b1), we envisaged the iodide ion was easily disengaged from the phosphonium salt and could serve as a Brønsted base to remove the proton of P-nucleophiles. Simultaneously, the phosphonium cation particularly possesses a semi-enclosed cavity with an electropositive region as demonstrated by its computed electrostatic potential (ESP) map (Scheme 2B, c), which would be combined with anion species via ion pair interattraction, forming a possible P4-Int-P intermediate, which was also reasonably validated by its electrostatic potential (ESP) map result (Scheme 2B, d). On the basis of these observations, a plausible reaction mechanism of this reaction was proposed in Scheme 2c; the key success for this high reactivity and enantioselectivity for this transformation is the adaptive assembly of the bifunctional phosphonium salt catalyst with P-nucleophiles via H-bonding and ion-pair interactions.
 |
| | Scheme 2 Mechanistic experiments and plausible reaction mechanism. | |
3. Conclusions
In conclusion, we have successfully completed the first highly enantioselective P-nucleophilic addition of five-membered cyclic N-sulfonyl imines by a tunable bifunctional ion-pair catalyst with multiple-hydrogen-bond interaction ability. Under this protocol, three libraries of structurally and functionally diverse enantioenriched cyclic quaternary/tertiary phosphorus-containing benzosultam compounds were efficiently prepared with high isolated yields and excellent enantioselectivities under mild reaction conditions. The key to success is the rational choice of an efficient organocatalytic system that not only avoids predictable side effects upon manipulating SPOs, but also ingeniously reins in the P-nucleophiles with powerful H-bonding and ion-pair interactions. The utility and the practicality of this reaction were demonstrated by gram-scale preparation and facile elaborations. Systematic mechanistic studies including experiments and DFT calculations elucidated the judicious choice of bifunctional phosphonium salt catalysts is critical for the ingenious stereoselective reining in of the P-nucleophiles. This finding is expected to stimulate a more extensive exploration of the catalytic asymmetric construction of phosphorus-containing heterocyclic compounds in synthetic chemistry.
Data availability
All the experimental data are provided in the ESI.† Crystallographic data for P4 has been deposited and available free of charge from the Cambridge Crystallographic Data Centre (CCDC) under NO. 1935757 and can be obtained from https://www.ccdc.cam.ac.uk/.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (22222109, 21921002 and 22101189), National Key R&D Program of China (2018YFA0903500), Beijing National Laboratory for Molecular Sciences (BNLMS202101), Sichuan Science Foundation for Distinguished Young Scholars (2023NSFSC1921), Sichuan Provincial Natural Science Foundation (2022NSFSC1181, 2024NSFSC1122), Fundamental Research Funds from Sichuan University (2020SCUNL108), the Elite Youth Program of the Department of Education of Hunan Province (21B0666) and Fundamental Research Funds for the Central Universities. We also acknowledge the College of Chemistry and the Analytical & Testing Center of Sichuan University, and we thank Dr Jing Li and Dr Dongyan Deng from the College of Chemistry Sichuan University for HRMS and NMR testing, respectively.
Notes and references
-
(a) J. Wrobel, A. Dietrich, S. A. Woolson, J. Millen, M. McCaleb, M. C. Harrison, T. C. Hohman, J. Sredy and D. Sullivan, J. Med. Chem., 1992, 35, 4613–4627 CrossRef CAS PubMed;
(b) G. J. Wells, M. Tao, K. A. Josef and R. Bihovsky, J. Med. Chem., 2001, 44, 3488–3503 CrossRef CAS PubMed;
(c) S. Zhang, L. Li, Y. Hu, Z. Zha, Z. Wang and T.-P. Loh, Org. Lett., 2015, 17, 1050–1053 CrossRef CAS PubMed;
(d) Y.-H. Dong, Q.-W. Ni, S.-T. Ma and Z.-P. Liu, Heterocycles, 2010, 81, 637–648 CrossRef CAS.
-
(a) J. Wrobel, A. S. Dietrich, A. Woolson, J. Millen, M. McCaleb, M. C. Harrison, T. C. Hohman, J. Sredy and D. Sullivan, J. Med. Chem., 1992, 35, 4613–4627 CrossRef CAS PubMed;
(b)
D. C. Baker and B. Jiang, US Pat., 6353112B1, 2002 Search PubMed;
(c)
J. Mao and D. C. Baker, US Pat., 6458962B1, 2003 Search PubMed;
(d)
J. L. Castro, I. J. Pineiro and H. T. Collins, US Pat., 20050014369A1, 2005 Search PubMed;
(e) C. T. Supuran, A. Casini and A. Scozzafava, Med. Res. Rev., 2003, 23, 535–558 CrossRef CAS PubMed.
-
(a) G. Yang and W. Zhang, Angew. Chem., Int. Ed., 2013, 52, 7540–7544 CrossRef CAS PubMed;
(b) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971–974 CrossRef CAS PubMed;
(c) Y. Li, Y.-N. Yu and M.-H. Xu, ACS Catal., 2016, 6, 661–665 CrossRef CAS;
(d) C. Jiang, Y. Lu and T. Hayashi, Angew. Chem., Int. Ed., 2014, 37, 9936–9939 CrossRef PubMed;
(e) J. Xiao, M. Wang, X. Yin, S. Yang, P. Gu, X. Lv, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2023, 62, e202300743 CrossRef CAS PubMed.
-
(a) L.-M. Zhang, W. Luo, J. Fu, Y. Liu and J. Zhang, ACS Catal., 2023, 13, 8830–8837 CrossRef CAS;
(b) G. Li, Y. Zhang, H. Zeng, X. Feng, Z. Su and L. Lin, Chem. Sci., 2022, 13, 4313–4320 RSC.
-
(a) R.-R. Liu, D.-J. Wang, L. Wu, B. Xiang, G.-Q. Zhang, J.-R. Gao and Y.-X. Jia, ACS Catal., 2015, 5, 6524–6528 CrossRef CAS;
(b) Y. Huang, R.-Z. Huang and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 6571–6576 CrossRef CAS PubMed.
-
(a) B. Qiao, Y.-J. Huang, J. Nie and J.-A. Ma, Org. Lett., 2015, 17, 4608–4611 CrossRef CAS PubMed;
(b) L. Wu, Q. Shao, G. Yang and W. Zhang, Chem. Eur. J., 2018, 24, 1241–1245 CrossRef CAS PubMed;
(c) M. Quan, X. Wang, L. Wu, I. D. Gridnev, G. Yang and W. Zhang, Nat. Commun., 2018, 9, 2258–2269 CrossRef PubMed.
-
(a) Z. Ling, S. Singh, F. Xie, L. Wu and W. Zhang, Chem. Commun., 2017, 53, 5364–5367 RSC;
(b) Z. Zeng, F. Yan, M. Dai, Z. Yu, F. Liu, Z. Zhao, R. Bai and Y. Lan, Organometallics, 2022, 41, 270–277 CrossRef CAS;
(c) Y.-L. Li, J.-X. Liu, X.-P. Chen, Y. Zhou, Y.-C. Xiao and F.-E. Chen, Adv. Synth. Catal., 2020, 362, 3202–3207 CrossRef CAS.
- Q. Shao, L. Wu, J. Chen, I. D. Gridnev, G. Yang, F. Xie and W. Zhang, Adv. Synth. Catal., 2018, 23, 4625–4633 CrossRef.
-
(a) H. Zhang, C. Jiang, J.-P. Tan, H.-L. Hu, Y. Chen, X. Ren, H.-S. Zhang and T. Wang, ACS Catal., 2020, 10, 5698–5706 CrossRef CAS;
(b) J. Pan, J.-H. Wu, H. Zhang, X. Ren, J.-P. Tan, L. Zhu, H. Zhang, C. Jiang and T. Wang, Angew. Chem., Int. Ed., 2019, 58, 7425–7430 CrossRef CAS PubMed;
(c) W. Zhuang, S. Saaby and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 4476–4478 CrossRef PubMed;
(d) M. Pareek and R. B. Sunoj, Org. Lett., 2016, 18, 5932–5935 CrossRef CAS PubMed.
-
(a) J. G. Allen, F. R. Atherton, M. J. Hall, C. H. Hassal, S. W. Holmes, R. W. Lambert, L. J. Nisbet and P. S. Ringrose, Nature, 1978, 272, 56–58 CrossRef CAS PubMed;
(b) F. R. Atherton, R. W. Hassall and R. W. Lambert, J. Med. Chem., 1986, 29, 29–40 CrossRef CAS PubMed;
(c) R. Hirschmann, A. B. Smith III, C. M. Taylor, P. A. Benkovic, S. D. Taylor, K. M. Yager, P. A. Sprengler and S. J. Benkovics, Science, 1994, 265, 234–237 CrossRef CAS PubMed;
(d) M. C. Allen, W. Fuhrer, B. Tuck, R. Wade and J. M. Wood, J. Med. Chem., 1989, 32, 1652–1661 CrossRef CAS PubMed.
-
(a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed;
(b) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077–2118 CrossRef CAS PubMed;
(c) J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed.
-
(a) A. Cabré, A. Riera and X. Verdaguer, Acc. Chem. Res., 2020, 53, 676 CrossRef PubMed;
(b) P. Li, Z. A. Sergueeva, M. Dobrikov and B. R. Shaw, Chem. Rev., 2007, 107, 4746–4796 CrossRef CAS PubMed.
-
(a) J. Stawinski and A. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed;
(b) A. Gallen, A. Riera, X. Verdaguer and A. Grabulosa, Catal. Sci. Technol., 2019, 9, 5504–5561 RSC;
(c) J. Stawinski and d. Kraszewski, Acc. Chem. Res., 2002, 35, 952–960 CrossRef CAS PubMed.
-
(a) F. Wang, X. Liu, X. Cui, Y. Xiong, X. Zhou and X. Feng, Chem.–Eur. J., 2009, 15, 589–592 CrossRef CAS PubMed;
(b) X. Cheng, R. Goddard, G. Buth and B. List, Angew. Chem., Int. Ed., 2008, 47, 5079–5081 CrossRef CAS PubMed;
(c) H. Xie, A. Song, X. Zhang, X. Chen, H. Li, C. Sheng and W. Wang, Chem. Commun., 2013, 49, 928–930 RSC;
(d) K. Ogura, I. Isozumi, T. Takehara, T. Suzuki and S. Nakamura, Org. Lett., 2022, 24, 8088–8092 CrossRef CAS PubMed.
-
(a) W. Tan, J.-Y. Zhang, C.-H. Gao and F. Shi, Sci. China Chem., 2023, 66, 966–992 CrossRef CAS;
(b) J. Cheng, S.-H. Xiang and B. Tan, Chin. J. Chem., 2023, 41, 685–694 CrossRef CAS;
(c) F.-T. Sheng, S. Yang, S.-F. Wu, Y.-C. Zhang and F. Shi, Chin. J. Chem., 2022, 40, 2151–2160 CrossRef CAS;
(d) Q.-Q. Hang, S.-F. Wu, S. Yang, X. Wang, Z. Zhong, Y.-C. Zhang and F. Shi, Sci. China Chem., 2022, 65, 1929–1937 CrossRef CAS;
(e) Q.-H. Wu, M. Duan, Y. Chen, P. Yu, Y.-B. Wang, J. Cheng, S.-H. Xiang, K. N. Houk and B. Tan, Nat. Catal., 2024, 7, 185–194 CrossRef CAS.
-
(a) H. Zhang, J. He, Y. Chen, C. Zhuang, C. Jiang, K. Xiao, Z. Su, X. Ren and T. Wang, Angew. Chem., Int. Ed., 2021, 60, 19860–19870 CrossRef CAS PubMed;
(b) J.-P. Tan, K. Li, B. Shen, C. Zhuang, Z. Liu, K. Xiao, P. Yu, B. Yi, X. Ren and T. Wang, Nat. Commun., 2022, 13, 357–369 CrossRef CAS PubMed;
(c) L. Zhu, H. Peng, Y. Guo, J. Che, J.-H. Wu, Z. Su and T. Wang, Angew. Chem., Int. Ed., 2022, 61, e20220246 Search PubMed;
(d) J.-H. Wu, J.-P. Tan, J.-Y. Zheng, J. He, Z. Song, Z. Su and T. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215720 CrossRef CAS PubMed;
(e) S. Fang, Z. Liu and T. Wang, Angew. Chem., Int. Ed., 2023, 47, e202307258 Search PubMed.
-
(a) S. M. Banik, A. Levina, A. M. Hyde and E. N. Jacobsen, Science, 2017, 358, 761–764 CrossRef CAS PubMed;
(b) G. Pupo, A. C. Vicini, D. M. H. Ascough, F. Ibba, K. E. Christensen, A. L. Thompson, J. M. Brown, R. S. Paton and V. Gouverneur, J. Am. Chem. Soc., 2019, 141, 2878–2883 CrossRef CAS PubMed;
(c) D. A. Strassfeld, Z. K. Wickens, E. Picazo and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 9175–9180 CrossRef CAS PubMed.
-
(a) Y. Chen, Z. Yu, Z. Jiang, J.-P. Tan, J.-H. Wu, Y. Lan, X. Ren and T. Wang, ACS Catal., 2021, 11, 14168–14180 Search PubMed;
(b) S. Fang, Z. Liu, H. Zhang, J. Pan, Y. Chen, X. Ren and T. Wang, ACS Catal., 2021, 11, 13902–13912 Search PubMed;
(c) J.-H. Wu, S. Fang, X. Zheng, J. He, Y. Ma, Z. Su and T. Wang, Angew. Chem., Int. Ed., 2023, 62, e2023095 Search PubMed;
(d) S. Fang, J.-P. Tan, J. Pan, H. Zhang, Y. Chen, X. Ren and T. Wang, Angew. Chem., Int. Ed., 2021, 60, 14921–14930 CrossRef CAS.
-
(a) U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed;
(b) M. Dutartre, J. Jugé and S. Bayardona, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
-
(a) H. Yu, H. Yang, E. Shi and W. Tang, Med. Drug Discovery, 2020, 100063 Search PubMed;
(b) A. Mucha, P. Kafarski and Ł. Berlicki, J. Med. Chem., 2011, 54, 5955–5980 CrossRef CAS PubMed.
-
(a) Y.-H. Chen, D.-J. Cheng, J. Zhang, Y. Wang, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 15062–15065 CrossRef CAS PubMed;
(b) Z. Yan, B. Wu, X. Gao, M.-W. Chen and Y.-G. Zhou, Org. Lett., 2016, 18, 692–695 Search PubMed.
- For selected reviews, see:
(a) S. Lectard, Y. Hamashima and M. Sodeoka, Adv. Synth. Catal., 2010, 352, 2708–2732 Search PubMed;
(b) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef CAS PubMed.
-
(a) S. Zhang, L. Cha, L. Li, Y. Hu, Y. Li, Z. Zha and Z. Wang, J. Org. Chem., 2016, 81, 3177–3187 CrossRef CAS PubMed;
(b) S. Zhang, L. Li, Y. Hu, Y. Li, Y. Yang, Z. Zha and Z. Wang, Org. Lett., 2015, 17, 5036–5039 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1935757. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02212b |
| ‡ H. Zhang and J.-P. Tan contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ad,
Xiaoyu
Ren
a,
Fan
Wang
a,
Jia-Yan
Zheng
a,
Jiajia
He
a,
Yu
Feng
ad,
Xiaoyu
Ren
a,
Fan
Wang
a,
Jia-Yan
Zheng
a,
Jiajia
He
a,
Yu
Feng