DOI:
10.1039/D4SC03230F
(Edge Article)
Chem. Sci., 2024,
15, 12026-12035
Chemo-, regio-, and stereoselective tetrafunctionalization of fluoroalkynes enables divergent synthesis of 5-7-membered azacycles†
Received
17th May 2024
, Accepted 22nd June 2024
First published on 24th June 2024
Abstract
Alkyne annulation has been widely used in organic synthesis for the construction of azacycles with unique structural and physicochemical properties. However, the analogous transformation of fluoroalkynes remains a challenge and has seen limited progress. Herein we report a 1,2,3,4-tetrafunctionalization of polyfluoroalkynes for the divergent construction of 5-7-membered (E)-1,2-difluorovinyl azacycles. The use of the fluorine atom as a detachable “activator” not only obviates the use of any transition metal catalysts and oxidizing reagents, but also ensures the [3–5 + 2]-annulation and defluorinative functionalization of fluoroalkynes with high chemo-, regio-, and stereoselectivities. This method exhibits a broad substrate scope, good functional group tolerance, and excellent scalability, providing a modular platform for accessing fluorinated skeletons of medicinal and biological interest. The late-stage modification of complex molecules, the multi-component 1,2-diamination of fluoroalkyne, and the synthesis of valuable organofluorides from the obtained products further highlight the real-world utility of this fluoroalkyne annulation technology.
Introduction
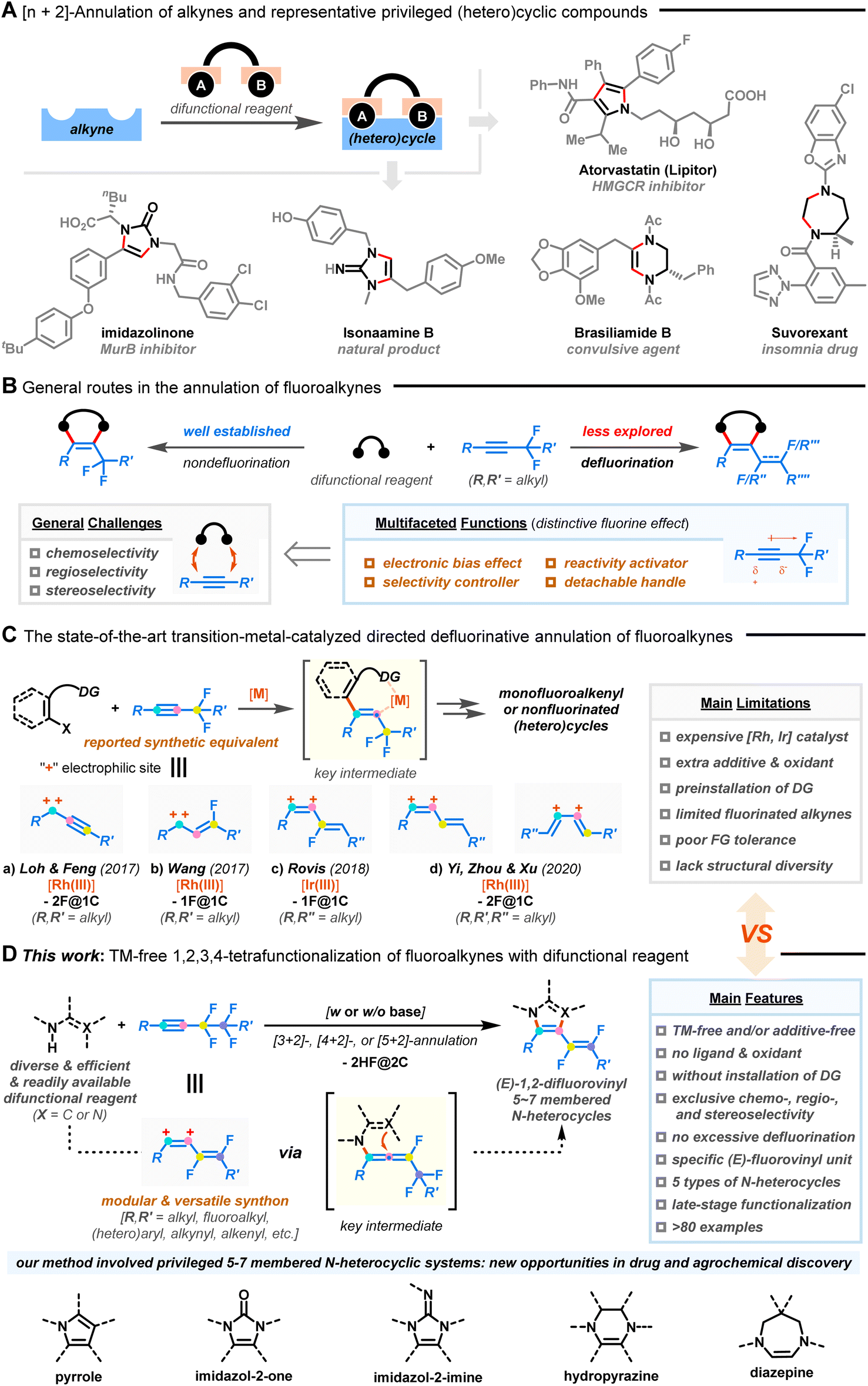
Azacycles have aroused extensive research enthusiasm due to their prevalence among naturally occurring products, FDA-approved drugs, agrochemicals, and materials.1Fig. 1A exemplifies some biologically important N-heterocyclic structures such as pyrrole, imidazole, hydropyrazine, and diazepine derivatives. Nonetheless, there remains a lack of reliable synthetic methodologies for creating fragment libraries of 5-7-membered azacycles with enhanced molecular complexity and improved compatibility with functionalities.2,3
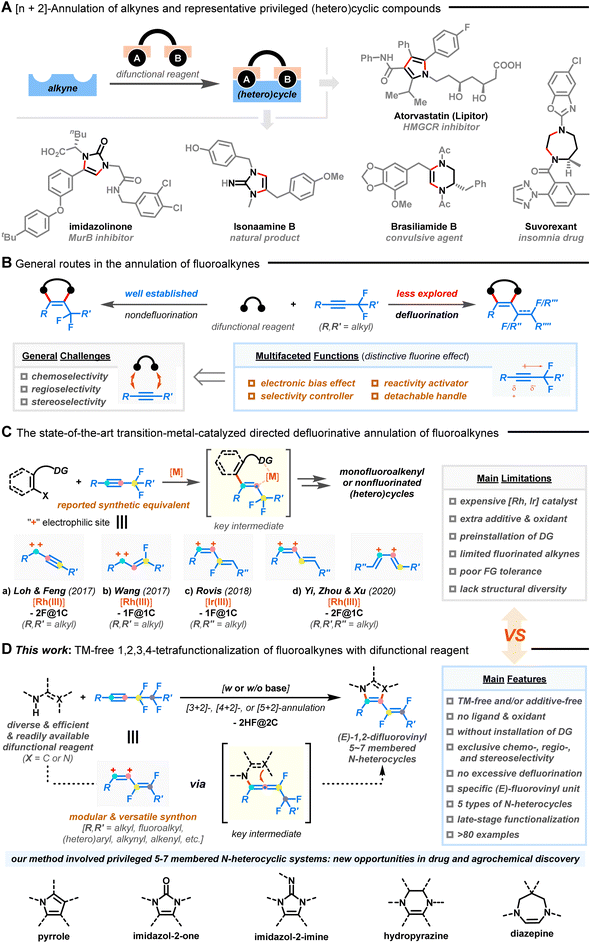 |
| | Fig. 1 Annulation of alkynes for the construction of (hetero)cyclic systems. (A) [n + 2]-annulation of alkynes and representative privileged (hetero)cyclic compounds. (B) General routes for the annulation of fluoroalkynes. (C) The state-of-the-art transition-metal-catalyzed directed defluorinative annulation of fluoroalkynes (“–2F@1C” means two C–F bond cleavages at a single carbon site; “–1F@1C” means one C–F bond cleavage at a single carbon site). (D) Divergent defluorinative [3–5 + 2]-annulation of fluoroalkynes with difunctional reagents (this work). | |
The selective and controllable functionalization of the C–F bond in polyfluorinated compounds has emerged as a complementary method to elegantly access complex, privileged molecules that are challenging to obtain from their nonfluorinated congeners.4–12 In this context, the utilization of fluoroalkynes as coupling partners enriches alkyne chemistry where their multifaceted functions and distinct reactivity could allow nontraditional annulation (Fig. 1B).13,14 Specifically, the presence of neighboring fluorine atoms polarizes the C–C triple bond, imposes steric congestion, and modulates the inherent reaction selectivity/reactivity of the π-system, resulting in chemo- and regioselective transformations.15,16 The easily detachable fluorine atom also eliminates the need for expensive transition metal catalysts and external chemical oxidants, enabling redox-neutral and sustainable processes.17,18 In addition, the availability and versatility of fluorine-functionalized motifs are crucial when taking into account biomedical areas, as they can confer physiochemical and pharmacodynamic advantages to the targeted products.19 However, intermolecular defluorofunctionalization of fluoroalkynes is still scarce.20–22
Recently, defluorinative annulation of gem-difluoromethylene alkynes, pioneered by Loh/Feng,23 Wang,24 Rovis,25 Yi/Zhou/Xu,26,27 and others,28–30 were found to easily allow rapid assembly of diverse heterocyclic skeletons with mono- or nonfluorinated unsaturated side chains (Fig. 1C). Indeed, many of the fluoroalkene products have attracted particular interest due to the combined features of fluorine and alkene.31–33 The key to success lies in transition-metal-assisted regioselective fluoroalkyne insertion, which is followed by β-F elimination and downstream ring closure. Consequently, fluoroalkynes, taking advantage of the fluorine atom as a detachable “activator” and “handle”, show promising abilities as modular C1 (ref. 23 and 24) or C2 (ref. 25−30) synthetic equivalents via tri- or tetrafunctionalization. Although novel and efficient, the advances come with caveats, namely, the use of costly metal catalysts (e.g., Ru and Ir) and extra additives, the need for pre-installed directing groups (DGs), limited substrate variation (R′, R′′, and R′′′ = alkyl groups, Fig. 1C-a–d), poor tolerance of functional groups, and harsh reaction conditions, all of which collectively hinder practical application of the methods.
Furthermore, these processes mainly focused on the manipulation of gem-difluoromethylene alkynes with a single fluorinated carbon atom (F@1C). A convenient procedure for the activation of two C–F bonds on two different fluorinated carbon atoms (F@2C) remains relatively understudied but is highly desirable.34,35 However, the occurrence of steric shielding by fluorine atoms on carbon centers, dipolar resonance structures, competitive multi-activations of C–F/C–C bonds, excessive defluorination, and undesired non-site-selective side reactions restricts the ready transformation of polyfluoroalkynes.
In this report, we describe a chemo-, regio-, and stereoselective [3–5 + 2]-annulation of less-studied polyfluoroalkynes through an alternative, mild, catalyst-free, oxidant-free, DG-free, and divergent sequence of 1,2-carboamination36–39/1,2-diamination40–43 and defluorinative polyfunctionalization (Fig. 1D). Key allenyl intermediates could be generated in the absence of transition-metal catalysts and oxidants,23–30 which allows for the downstream consecutive functionalization of two vicinal C(sp3)–F bonds with ease. A wide range of (E)-1,2-difluorovinyl azacycles with five-, six-, and seven-membered rings, including pyrroles, imidazol-2-ones, imidazol-2-imines, hydropyrazines, and diazepines, were synthesized from readily available difunctional reagents and fluoroalkynes without resorting to substrate prefunctionalization. The strategy also distinguished itself with exceptional substrate scope, good functional group tolerance, and gram-scale ability. More impressively, the integration of a unique fluorovinyl motif with azacycles in one molecule could pave a substantial way for new drug and agrochemical discovery.31–33
Results and discussion
Evaluation of the reaction conditions
The investigation was commenced by testing the model reaction of perfluorobutyl alkyne 1a and DBU (2a) under transition-metal-free conditions (see the ESI for more details, Table S1†). Pleasingly, 22% NMR yield of desired fluorovinyl bicyclic amidine 3, with an E/Z ratio of 7.4/1, could be obtained in DMSO at room temperature for 6 h (entry 1). After systematic optimization of various reaction parameters, such as bases, solvents, reaction temperature, and reaction time, the highest isolated yield (80%) was obtained when performing the reaction in iPrOAc at 70 °C for 15 min (see entry 17, Table S1†). It is worth mentioning that the defluorination process was exclusively and highly chemo-, regio-, and stereoselective (E/Z > 30/1).
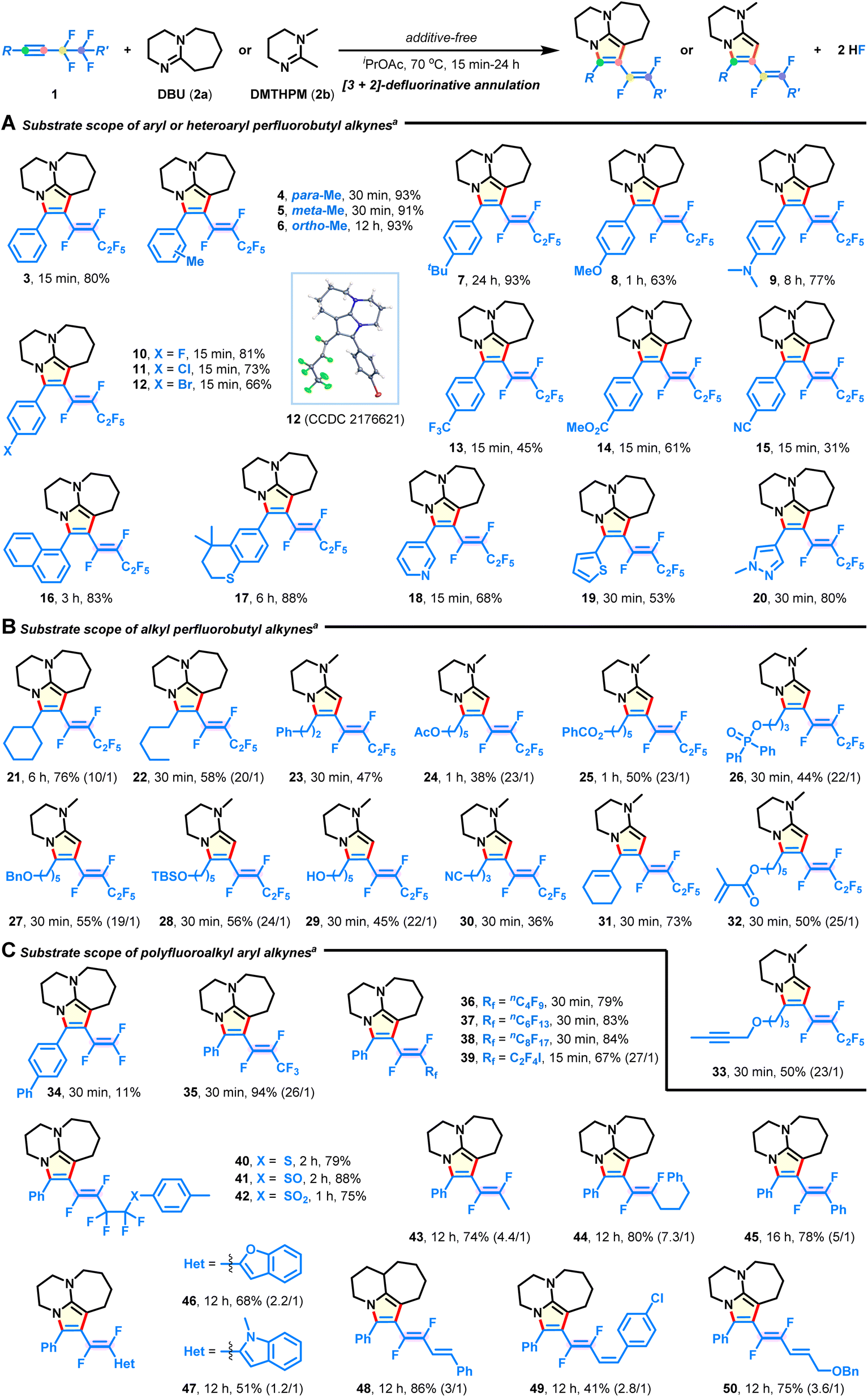
Substrate scope for polyfluoroalkynes
With the optimal conditions in hand, we explored the substrate scope of the intermolecular [3 + 2] annulation with respect to diversely functionalized fluoroalkynes (Fig. 2). First, a series of aryl and heteroaryl perfluorobutyl alkynes with varied substitution patterns could be smoothly converted into the corresponding azacycles in good yields with complete E-selectivity (Fig. 2A, 4–20). Electron-donating (4–9), electron-withdrawing (10–15), functional groups (e.g., ether, amino, halide, trifluoromethyl, ester, and nitrile; 8–15), and heterocycles (e.g., pyridine, thiophene, and pyrazole; 18–20) were well tolerated in the metal-free system, thus providing great flexibility for further post-synthetic modification. Notably, methyl substituents at either the meta- (5) or ortho-position (6) of the phenyl ring did not hinder the reaction efficiency. Transformation of alkyl perfluorobutyl alkynes also successfully occurred (Fig. 2B). The substrates containing strained cyclohexyl (21), acyclic pentyl (22), phenyl (23), acetoxy (24), benzoxy (25), diphenylphosphoryloxy (26), benzyloxy (27), TBS-capped hydroxyl (28), hydroxyl (29), cyano (30), internal alkenyl (31), and terminal alkenyl (32), as well as the alkynyl group (33) were compatible and produced the E-isomeric products, which validated the mildness of the reaction conditions. Moreover, aryl fluoroalkynes possessing length-varied perfluoroalkyl chains (Fig. 2C; C2–C10; 34–38) were found to react with DBU, resulting in the desired products in 11–94% yields. Among them, the low yield of product 34 is due to the instability of the embedded 1,2,2-trifluorovinyl pattern. When synthetically valuable units, such as iodine (39), sulfides (40–42), alkanes (43 and 44), arene (45), heterocycles (46 and 47), and alkenes (48–50), were introduced on the side of the fluoroalkyl group, no significant loss in yield is witnessed, although there is a decrease in E/Z-selectivity in some cases (43–50). In particular, these difluorovinyl azacycles are otherwise unattainable through traditional or Ru/Ir-catalyzed methods,23–30,36 exemplifying the versatility and practicality of the present protocol.
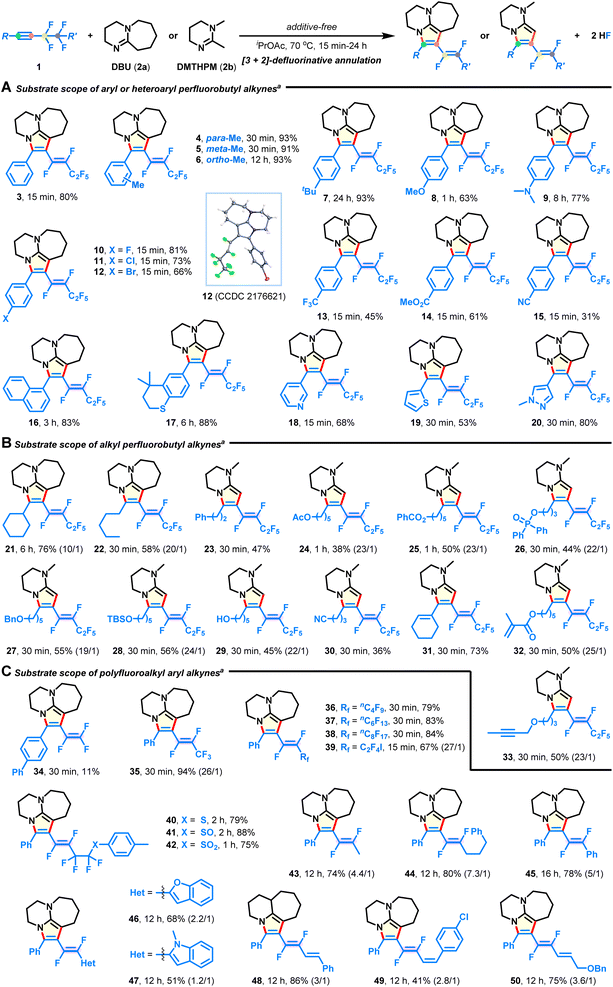 |
| | Fig. 2 Substrate scope of fluoroalkynes. (A) Substrate scope of aryl or heteroaryl perfluorobutyl alkynes. (B) Substrate scope of alkyl perfluorobutyl alkynes. (C) Substrate scope of polyfluoroalkyl aryl alkynes. aReaction conditions: fluoroalkyne 1 (0.3 mmol), DBU (1.05 mmol, 2a) or DMTHPM (1.05 mmol, 2b) in iPrOAc (2 mL) at 70 °C under N2 for 15 min-24 h; E/Z selectivities were >30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 except in some cases where the ratios are given in parentheses. :1 except in some cases where the ratios are given in parentheses. | |
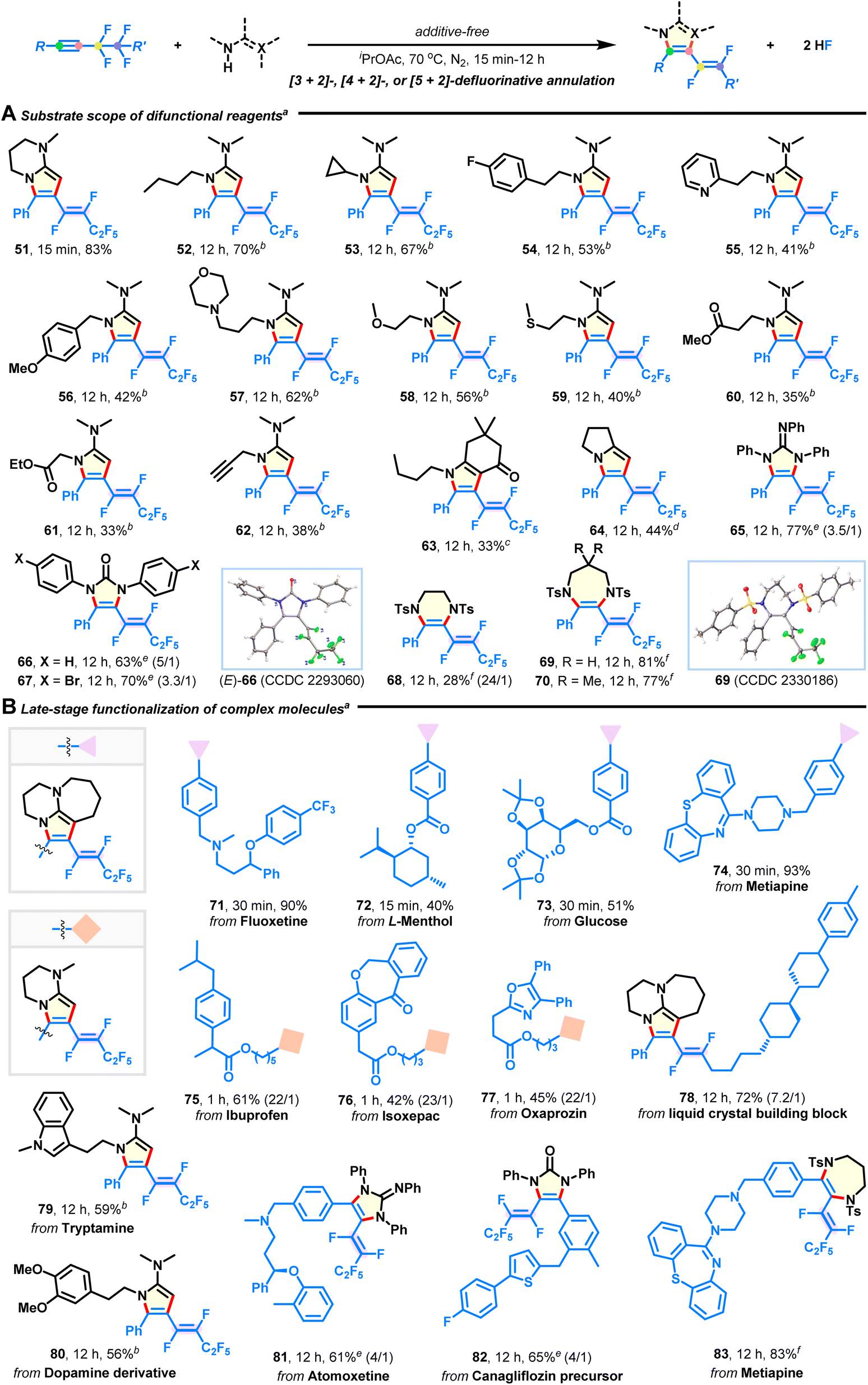
Substrate scope for difunctional reagents
Subsequently, the use of diverse difunctional reagents as competent reaction partners was studied (Fig. 3A). Besides bicyclic derivatives (33–83% yields; 51, 63, 64), pyrrole products with versatile groups were obtained in moderate to good yields with excellent stereoselectivities by using acetamidines derived from commercially available butan-1-amine (52), steric cyclopropanamine (53), (hetero)aryl ethanamines (54 and 55), benzylamine (56), 3-morpholinopropan-1-amine (57), 2-methoxyethan-1-amine (58), 2-(methylthio)ethan-1-amine (59), amino acid esters (60 and 61), and prop-2-yn-1-amine (62). Especially, pyrrole is the core structure of many approved pharmaceuticals. (Hetero)arene, amine, ether, sulfide, ester, and alkyne functionalities did not interfere with intermolecular defluorination. 1,2,3-Triphenylguanidine and 1,3-diarylurea could also be annulated to afford products 65–67 in 63–77% yields, which would otherwise be difficult to synthesize. One apparent advantage of our methodology is that it can be applied to efficient synthesis of 6-7-membered (E)-1,2-difluorovinyl hydropyrazine (68) and diazepines (69–70) through the reaction of 1,2-disulfonamide 2s and 1,3-disulfonamides 2t–u with fluoroalkyne 1a, which significantly enhances product diversity and accessibility. The corresponding structures of products 12, (E)-66, and 69 were characterized by X-ray crystallographic analysis (see ESI†).
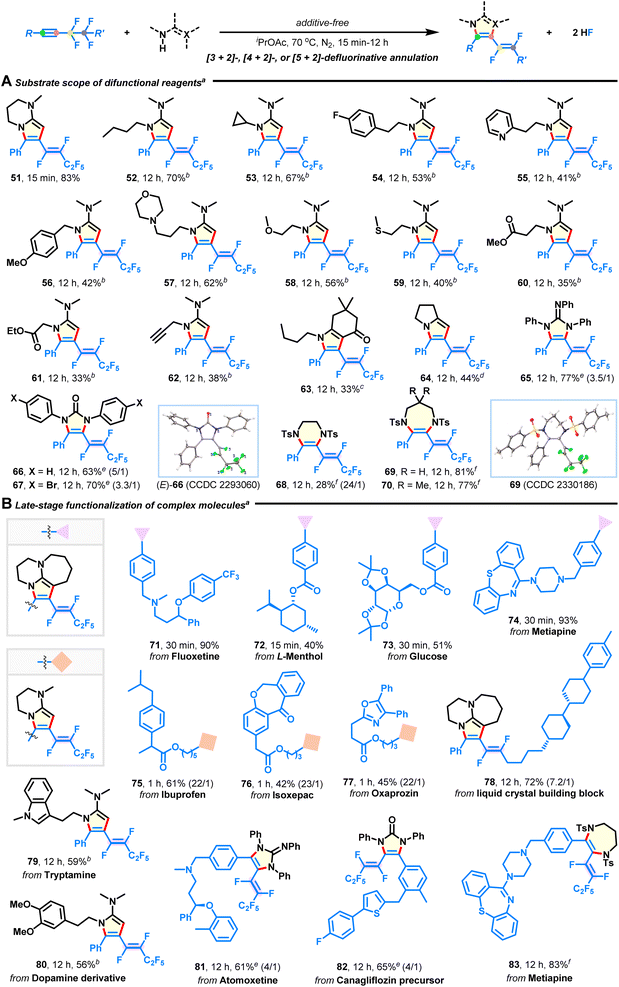 |
| | Fig. 3 Substrate scope of difunctional reagents and late-stage functionalization of complex molecules. (A) Substrate scope of difunctional reagents. (B) Late-stage modification of complex molecules. aReaction conditions: fluoroalkyne (0.3 mmol) and difunctional reagent (1.05 mmol) in iPrOAc (2 mL) at 70 °C under N2 for 15 min to 12 h; E/Z selectivities were >30:1 except in some cases where the ratios are given in parentheses. bUnder neat conditions at 90 °C. cCs2CO3 (1.05 mmol) was used. d1-Methylimidazole (0.75 mmol) was used. eCs2CO3 (0.75 mmol) was used in DMSO at 90 °C. fCs2CO3 (0.9 mmol) was used in DMF at 110 °C. | |
Substrate scope for complex molecules
The broad substrate scope tolerance of this defluorinative technology encouraged us to apply it to late-stage elaboration of more complex compounds (Fig. 3B). To our delight, a large variety of biologically and pharmaceutically relevant molecules, such as fluoxetine (71), L-menthol (72), glucose (73), metiapine (74, 83), ibuprofen (75), isoxepac (76), oxaprozin (77), tryptamine (79), dopamine (80), atomoxetine (81), and canagliflozin (82) were successfully employed to merge with azacyclic frameworks, delivering some intriguing adducts in 40–93% yields with equally excellent stereoselectivities (except products 81 and 82). Furthermore, the liquid crystal (LC) building block (78) was amenable to this reaction, which will provide chemists an attractive alternative for manufacturing new fluorinated LC materials.44
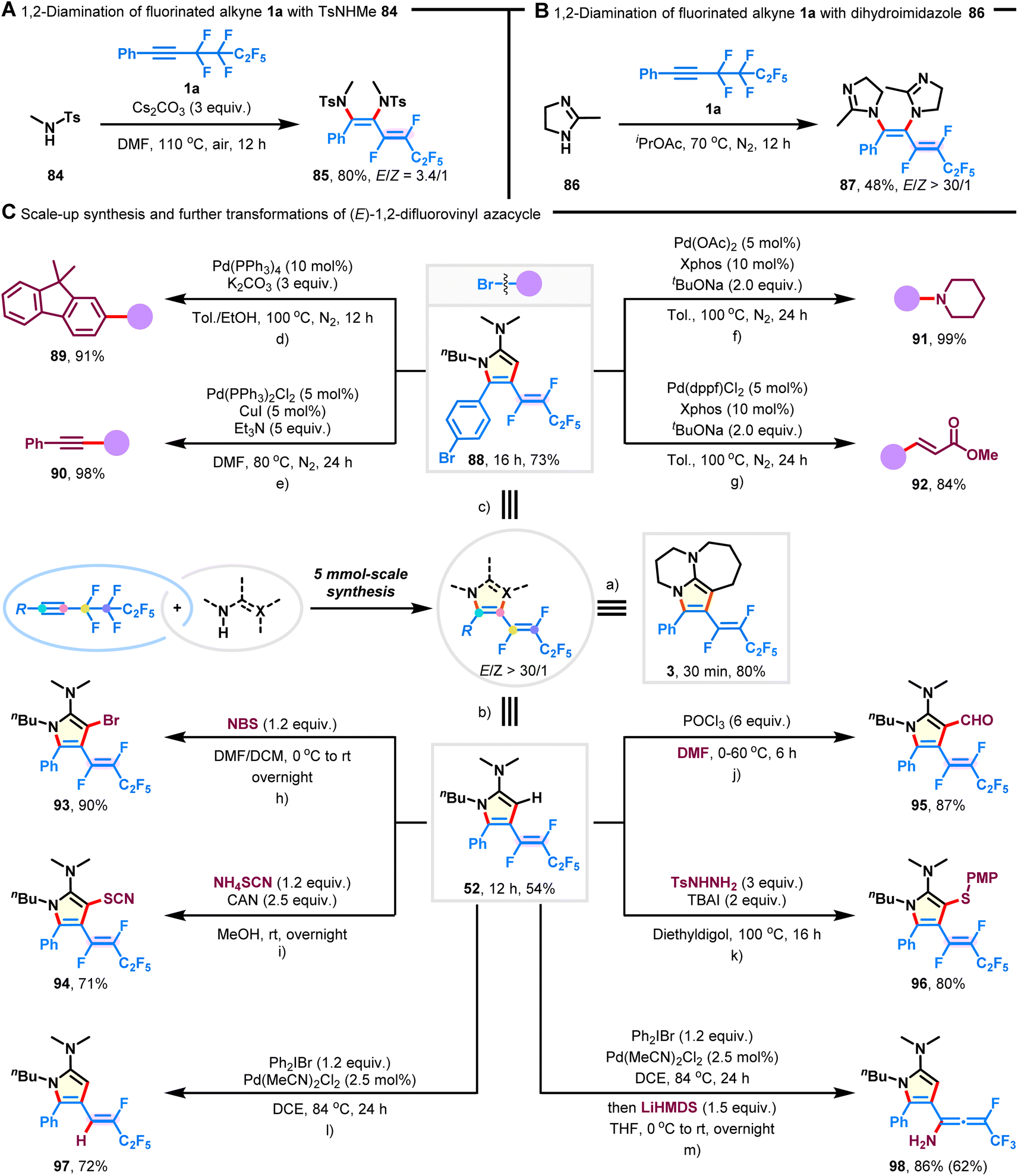
Multi-component 1,2-diamination
To increase the applicability of our technique for accessing 1,2-difluorovinyl scaffolds, we examined the multi-component 1,2-diamination of fluoroalkyne 1a. As shown in Fig. 4A, the desired 1,3-diene-1,2-diamine 85 was synthesized in 80% yield with an E/Z ratio of 3.4/1 when TsNHMe (84) was employed as the amination reagent. Furthermore, cyclic dihydroimidazole (86) showed decreased reactivity, but with high Z-selectivity preserved (Fig. 4B; 48% yield, E/Z > 30/1; 87). It is important to note that competitive hydroaminated products were not observed.45
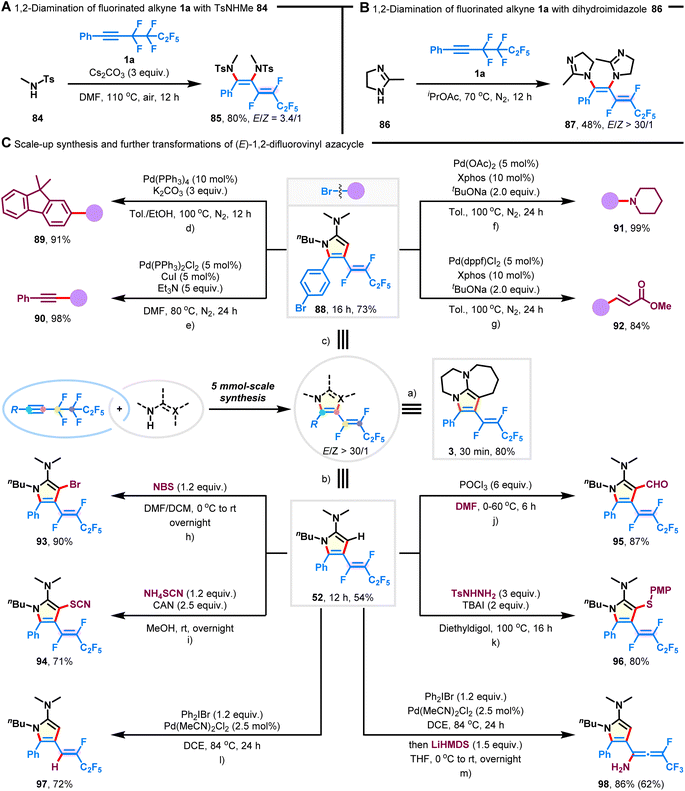 |
| | Fig. 4 Three-component 1,2-diamination, scale-up synthesis, and further transformation of products. (A) Three-component 1,2-diamination of fluoroalkyne 1a with TsNHMe (84). (B) Three-component 1,2-diamination of fluoroalkyne 1a with dihydroimidazole (86). (C) 5 mmol-scale synthesis and further transformations of (E)-1,2-difluorovinyl azacycles. (a) Fluoroalkyne 1a (5 mmol) and DBU (2a) (17.5 mmol) in iPrOAc (30 mL) at 70 °C under N2 for 30 min. (b) Fluoroalkyne 1a (5 mmol) and acetimidamide 2c (17.5 mmol) under neat conditions for 12 h. (c) Fluoroalkyne 1j (5 mmol) and acetimidamide 2c (17.5 mmol) under neat conditions for 16 h. (d) Product 88 (0.3 mmol) and (9,9-dimethyl-9H-fluoren-2-yl)boronic acid (0.6 mmol) were used. (e) Product 88 (0.3 mmol) and phenylacetylene (0.9 mmol) were used. (f) Product 88 (0.3 mmol) and piperidine (0.6 mmol) were used. (g) Product 88 (0.3 mmol) and methyl acrylate (0.9 mmol) were used. (h–l) Product 52 (0.3 mmol) was used. (m) Product 97 (0.3 mmol) was used in the second step; yield in parentheses was over two steps. | |
Scale-up synthesis
The scalability of this procedure was demonstrated by the preparation of azacycles 3, 52, and 88 on a 5 mmol scale, which were isolated without any significant loss in reaction selectivity or yield (Fig. 4C-a–c; 54–80% yield, E/Z > 30/1).
Synthetic applications
Given that the obtained azacycles are decorated with various functionalities, it becomes feasible to introduce more diverse structural features through post-functionalization. The bromide substituent on product 88 could be facilely transformed into valuable motifs, such as fused arene (Fig. 4C-d; 91% yield; 89), alkyne (Fig. 4C-e; 98% yield; 90), amine (Fig. 4C-f; 99% yield; 91), and acrylate (Fig. 4C-g; 84% yield; 92). Particularly, the structure and the configuration of the fluorovinyl group remained intact during these Pd-catalyzed cross-coupling reactions. Besides, product 52 underwent bromination, thiocyanation, formylation, and thioetherification, generating the corresponding pyrrole compounds 93–96 in good yields without the erosion of stereopurity (Fig. 4C-h and k). In addition, one of the C(sp2)–F bonds on the product 52 could be selectively removed in the presence of Pd(MeCN)2Cl2 and Ph2IBr (Fig. 4C-l; 72% yield, 97), and the ready transformation to fluorinated allenyl amine was achieved upon treatment with LiHMDS (Fig. 4C-m; 86% yield, 98).
Mechanistic investigations
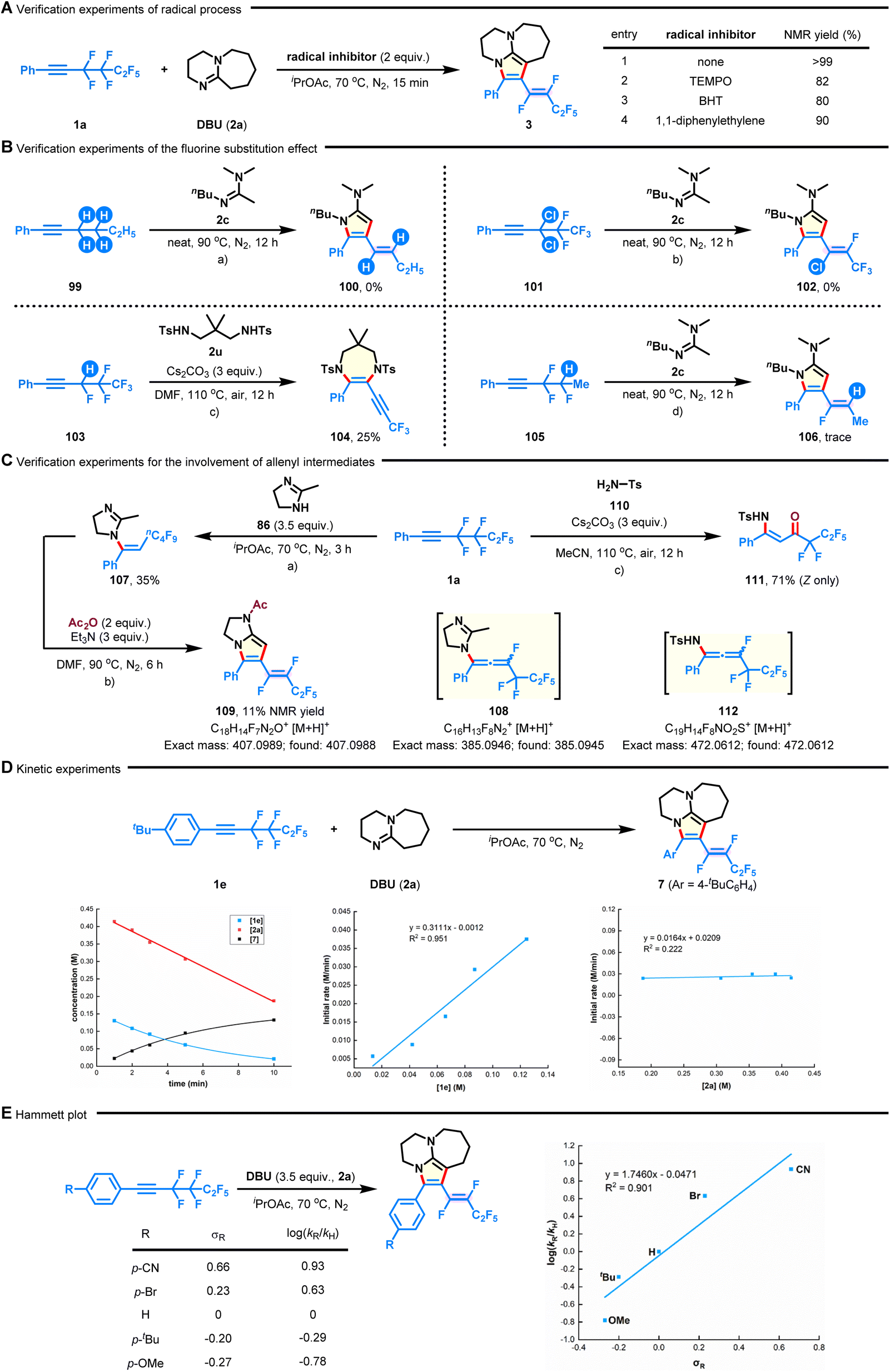
To gain a preliminary understanding of the reaction mechanism, several control experiments were conducted, and the results are shown in Fig. 5. First, the [n + 2]-defluorinative annulation process was not impeded in the presence of TEMPO (tetramethylpiperidine-1-oxyl), BHT (2,6-di-tert-butyl-4-methylphenol), or 1,1-diphenylethylene, revealing that a radical pathway could be excluded at this stage (Fig. 5A). Subsequently, the role of the fluoroalkyl group in this transformation was studied. No desired annulated products were detected in the cases of nonfluorinated hex-1-yn-1-ylbenzene (99) and gem-dichlorine congener 101 under neat conditions (Fig. 5B-a and b). Although the reaction between 1,3-disulfonamide 2u and (3,4,4,5,5,5-hexafluoropent-1-yn-1-yl)benzene (103) successfully delivered 3,3,3-trifluoroprop-1-yn-1-yl azacycle 104 in 25% yield, only a trace amount of product 106 was observed when analogous (3,3,4-trifluoropent-1-yn-1-yl)benzene (105) was used (Fig. 5B-c and d). Together with the experiments described above, these observations demonstrate the indispensability of the perfluoroethane-1,2-diyl group on substrates. It deserves to be mentioned that the formation of product 104 could be attributed to a base-promoted dehydrofluoration of in situ formed α-fluoroalkene.46 Furthermore, by shortening the reaction time of fluoroalkyne 1a with dihydroimidazole (86) to 3 h (Fig. 4B), the hydroaminated product 107 was observed, which could be further converted into bicyclic product 109 in the presence of Ac2O and Et3N (Fig. 5C-a and b). This result means that the initial reaction might proceed through intermolecular hydroamination,45 followed by intramolecular defluorinative annulation, ultimately accomplishing the concessive C–F bond functionalization. Interestingly, when 1a was reacted with TsNH2 (110), it led to the generation of (Z)-N-Ts-enaminone 111 in 71% yield (Fig. 5C-c). The results, combined with the occurrence of possible intermediates 108 and 112 (detected by HRMS analysis, see the ESI† for more details), correlate with the intermediacy of key fluoroallenes in the system.47,48 On the other hand, the plots of kinetic experiments suggested a first-order kinetic dependence on 1e but a zero-order kinetic dependence on 2a (Fig. 5D). The electronic effect of the polyfluoroalkynes (such as 4-OMe, 4-tBu, 4-Br, and 4-CN) on the reaction rate was verified by Hammett studies (Fig. 5E).49 The positive slope (ρ = 1.746) in the Hammett plot might suggest that electron-deficient aryl intermediates would be more favourable during the defluorocyclization process.
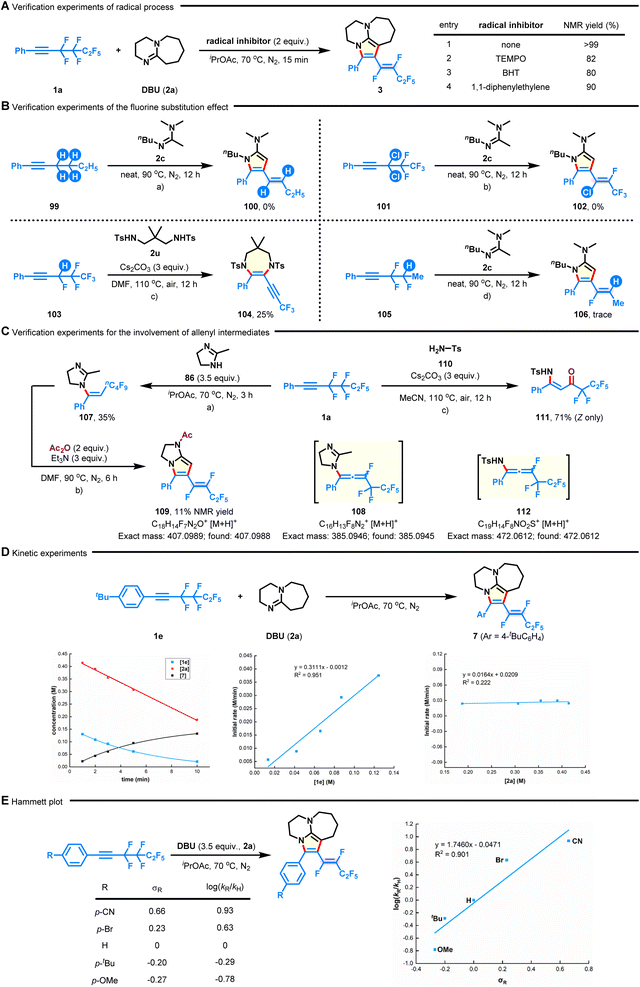 |
| | Fig. 5 Control experiments. (A) Verification experiments of the radical process. (B) Verification experiments of the fluorine substitution effect. (C) Verification experiments for the involvement of allenyl intermediates. (D) Kinetic experiments. (E) Hammett plot of substituted polyfluoroalkynes. | |
Proposed reaction mechanism
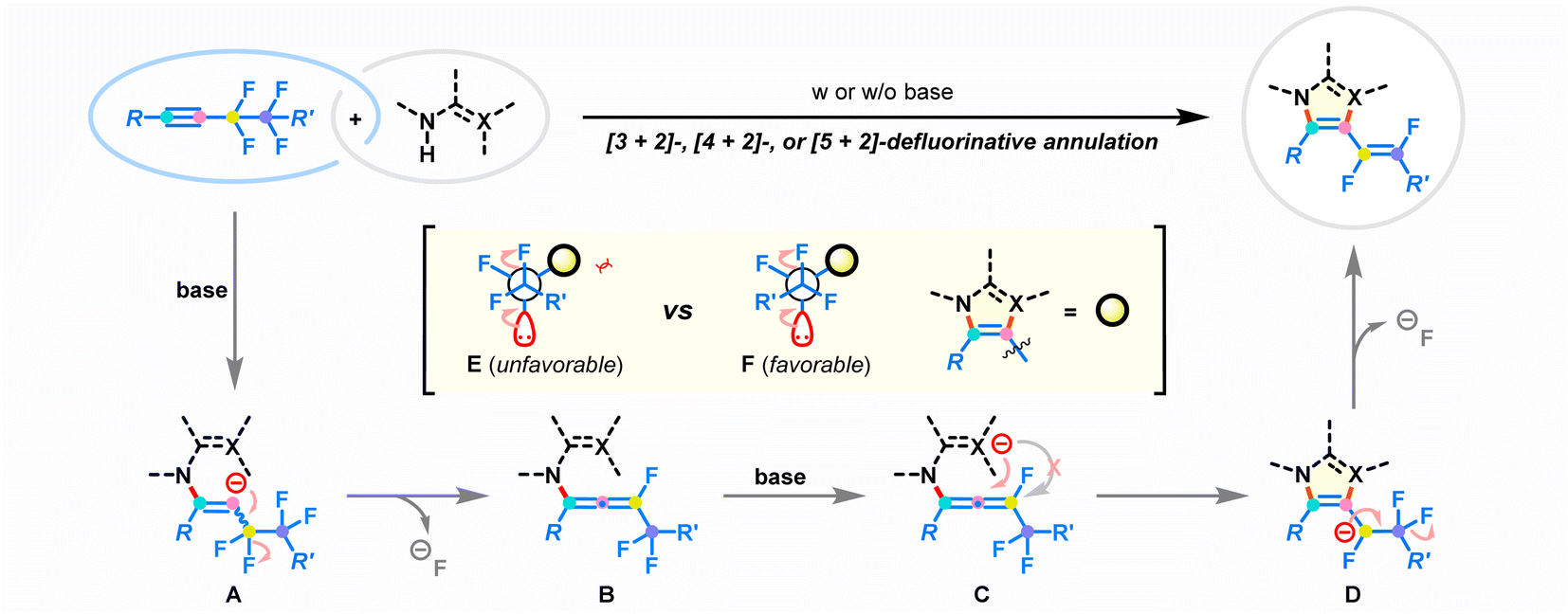
Based on the control experiments and the aforementioned reports,23–30,36,43,45–48 a plausible mechanism for the developed defluorinative annulation is proposed in Fig. 6. Initially, the deprotonation of the amino group in the difunctional reagent enables the nucleophilic attack on fluoroalkynes. The electronic bias of the π-system by the polarization of the adjacent perfluoroethane-1,2-diyl substituent is considered to be the crucial factor that ensures regioselective addition.45 The resulting reactive alkenyl anion A undergoes immediately β-F-elimination to deliver key fluoroallene species B.46,47 At this stage, the strong electron-withdrawing properties of the perfluoroalkyl group and fluorine atom lead to the ensuing intramolecular cyclization to selectively take place at the proximal double bond of allene species B, affording new 5-7-membered azacycles D. Finally, the following defluorinative E1cb elimination at the fluoroalkyl chain results in the stereoselective generation of (E)-1,2-difluorovinyl products. Notably, the steric repulsion between the R′ group and the azacycle core in conformational mode F favors the exclusive production of E-type products. On the other hand, the successive heterolytic cleavage of two vicinal C–F bonds removes electrons from the reaction composite, enabling the annulation to occur in a redox-neutral manner.36–43
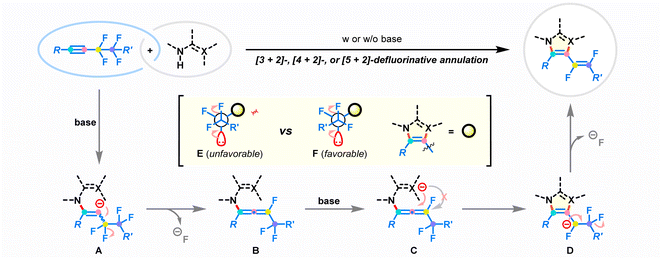 |
| | Fig. 6 Proposed reaction mechanism. | |
Conclusions
In summary, we have developed a modular strategy for the [3–5 + 2]-annulation and 1,2,3,4-tetrafunctionalization of readily accessible polyfluoroalkynes with di-nucleophiles. This method provides practical and scalable access to a wide range of 5-7-membered azacycles with a specific (E)-1,2-difluorovinyl unit. Other notable features of the present strategy include a broad substrate scope, good functional group tolerance, and excellent chemo-/regio-/stereoselectivity. The late-stage modification of complex structures, the multi-component 1,2-diamination of fluoroalkyne, and the synthesis of valuable organofluorides from the obtained products further highlight the real-world utility of this defluorinative annulation technology. The controllable and selective cleavage of two C(sp3)–F bonds at the fluoroalkyl chain contributes to the catalyst-free and oxidant-free heterocycle construction. As such, we believe this intriguing approach would open new avenues and inspire further developments in organic synthetic chemistry and pharmaceutical research.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
X. Q. C. conceived the project. J. W. C., W. J. J., X. Y. H., D. H. G., Z. L. S., and X. Q. C. conducted all experimental work and analyzed the data. The manuscript was written by Z. L. S., K. G., and X. Q. C. with contributions and proofreading from all authors. Z. L .S., K. G., and X. Q. C. directed the project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22001121) and Nanjing Tech University.
Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- C.-V. T. Vo, M. U. Luescher and J. W. Bode, Nat. Chem., 2014, 6, 310–314 CrossRef CAS PubMed.
- Z. Ma, W. Xu, Y.-D. Wu and J. S. Zhou, J. Am. Chem. Soc., 2023, 145, 16464–16473 CrossRef CAS PubMed.
- Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed.
- C. Zhu, M.-M. Sun, K. Chen, H. Liu and C. Feng, Angew. Chem., Int. Ed., 2021, 60, 20237–20242 CrossRef CAS PubMed.
- L. Li, X. Zhang, Y. Ning, X. Zhang, B. Liu, Z. Zhang, P. Sivaguru, G. Zanoni, S. Li, E. A. Anderson and X. Bi, Nat. Commun., 2022, 13, 4280 CrossRef CAS PubMed.
- M. Hu, Y. Liang, L. Ru, S. Ye, L. Zhang, X. Huang, M. Bao, L. Kong and B. Peng, Angew. Chem., Int. Ed., 2023, 62, e202306914 CrossRef CAS PubMed.
- J. Huang, Q. Gao, T. Zhong, S. Chen, W. Lin, J. Han and J. Xie, Nat. Commun., 2023, 14, 8292 CrossRef CAS PubMed.
- Y. Zhang, J. Wang, Y. Guo, S. Liu and X. Shen, Angew. Chem., Int. Ed., 2024, 63, e202315269 CrossRef CAS PubMed.
- H. Qian, Z. P. Cheng, Y. Luo, L. Lv, S. Chen and Z. Li, J. Am. Chem. Soc., 2024, 146, 24–32 CrossRef CAS PubMed.
- M. Chen, Y. Cui, X. Chen, R. Shang and X. Zhang, Nat. Commun., 2024, 15, 419 CrossRef CAS PubMed.
- S. Bonfante, C. Lorber, J. M. Lynam, A. Simonneau and J. M. Slattery, J. Am. Chem. Soc., 2024, 146, 2005–2014 CrossRef CAS PubMed.
- M. C. Pacheco, S. Purser and V. Gouverneur, Chem. Rev., 2008, 108, 1943–1981 CrossRef CAS PubMed.
- S. Arimitsu, Chem. Rec., 2023, 23, e202300021 CrossRef CAS PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224–10239 RSC.
- L. V. Hooker and J. S. Bandar, Angew. Chem., Int. Ed., 2023, 62, e202308880 CrossRef CAS PubMed.
- D. Ge and X.-Q. Chu, Org. Chem. Front., 2022, 9, 2013–2055 RSC.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- S. Arimitsu and G. B. Hammond, J. Org. Chem., 2007, 72, 8559–8561 CrossRef CAS PubMed.
- R. Surmont, G. Verniest and N. De Kimpe, Org. Lett., 2009, 11, 2920–2923 CrossRef CAS PubMed.
- A. Nasr El Dine, D. Gree, T. Roisnel, E. Caytan, A. Hachem and R. Gree, Eur. J. Org Chem., 2016, 2016, 556–561 CrossRef CAS.
- C.-Q. Wang, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2017, 139, 1762–1765 CrossRef CAS PubMed.
- T. Li, C. Zhou, X. Yan and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 4048–4052 CrossRef CAS PubMed.
- F. Romanov-Michailidis, B. D. Ravetz, D. W. Paley and T. Rovis, J. Am. Chem. Soc., 2018, 140, 5370–5374 CrossRef CAS PubMed.
- H. Gao, S. Lin, S. Zhang, W. Chen, X. Liu, G. Yang, R. A. Lerner, H. Xu, Z. Zhou and W. Yi, Angew. Chem., Int. Ed., 2021, 60, 1959–1966 CrossRef CAS PubMed.
- H. Gao, M. Sun, H. Zhang, M. Bian, M. Wu, G. Zhu, Z. Zhou and W. Yi, Org. Lett., 2019, 21, 5229–5233 CrossRef CAS PubMed.
- F. Zhao, J. Qiao, Y. Lu, X. Zhang, L. Dai, X. Gong, H. Mao, S. Lu, X. Wu and S. Liu, Org. Lett., 2021, 23, 5766–5771 CrossRef CAS PubMed.
- Y. Luo, H.-Y. Zhou, Y.-C. Gang and L. Dong, Org. Lett., 2022, 24, 6940–6944 CrossRef CAS PubMed.
- Y. Yang, N. Li, J. Zhao, Y. Jiang, X. Zhang and X. Fan, Adv. Synth. Catal., 2021, 363, 3600–3606 CrossRef CAS.
- S. Couve-Bonnaire, D. Cahard and X. Pannecoucke, Org. Biomol. Chem., 2007, 5, 1151–1157 RSC.
- S. Morand, P. Jubault, J.-P. Bouillon and S. Couve-Bonnaire, Chem.–Eur. J., 2021, 27, 17273–17292 CrossRef CAS PubMed.
- M.-Z. Lu, J. Goh, M. Maraswami, Z. Jia, J.-S. Tian and T.-P. Loh, Chem. Rev., 2022, 122, 17479–17646 CrossRef CAS PubMed.
- R. Doi, M. Yasuda, N. Kajita, K. Koh and S. Ogoshi, J. Am. Chem. Soc., 2023, 145, 11449–11456 CrossRef CAS PubMed.
- C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474–16475 CrossRef CAS PubMed.
- Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572–4576 CrossRef CAS PubMed.
- S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585–9587 CrossRef CAS PubMed.
- H. Xiong, N. Ramkumar, M.-F. Chiou, W. Jian, Y. Li, J.-H. Su, X. Zhang and H. Bao, Nat. Commun., 2019, 10, 122 CrossRef PubMed.
- Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Acc. Chem. Res., 2014, 47, 3665–3678 CrossRef CAS PubMed.
- Y. Fukudome, H. Naito, T. Hata and H. Urabe, J. Am. Chem. Soc., 2008, 130, 1820–1821 CrossRef CAS PubMed.
- B. Yao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2012, 51, 5170–5174 CrossRef CAS PubMed.
- J. Pan, X. Li, F. Lin, J. Liu and N. Jiao, Chem, 2018, 4, 1427–1442 CAS.
- M. Kuroboshi, K. Kanie and T. Hiyama, Adv. Synth. Catal., 2001, 343, 235–250 CrossRef CAS.
- M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240–254 CrossRef CAS PubMed.
- P. Tian, C. Feng and T.-P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef PubMed.
- T. J. O'Connor, B. K. Mai, J. Nafie, P. Liu and F. D. Toste, J. Am. Chem. Soc., 2021, 143, 13759–13768 CrossRef PubMed.
- J. S. Ng and T. Hayashi, Angew. Chem., Int. Ed., 2021, 60, 20771–20775 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General information, experimental details, optimization of reaction conditions, mechanistic studies, characterization data for products, and NMR spectra of products. CCDC 2176621 (product 12), 2293060 [product (E)-66] and 2330186 (product 69). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03230f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zhi-Liang
Shen
a,
Zhi-Liang
Shen