
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn interactive dual energy storage mechanism boosts high-performance aqueous zinc-ion batteries†
Shengen
Gong
a,
Meihua
Zhu
a,
Yan
Zhou
a,
Runan
Li
b,
Jianhua
Zhang
b,
Xiaoteng
Jia
*b,
Danming
Chao
 *a and
Caiyun
Wang
*c
*a and
Caiyun
Wang
*c
aCollege of Chemistry, Jilin University, Changchun, 130012, China. E-mail: chaodanming@jlu.edu.cn
bState Key Laboratory of Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun 130012, China. E-mail: xtjia@jlu.edu.cn
cIntelligent Polymer Research Institute, Faculty of Engineering and Information Sciences, University of Wollongong, North Wollongong, NSW 2500, Australia. E-mail: caiyun@uow.edu.au
First published on 5th November 2024
Abstract
Organic materials are promising cathodes for aqueous zinc-ion batteries (AZIBs) due to their cost-effectiveness, environmental friendliness, and tunable structures. However, the energy density of AZIBs remains limited by the inherently low capacity and output voltage of organic cathode materials. To address this challenge, we develop a Mn ion-doped polyaniline (PAM) by harnessing the joint merits of the highly reversible doping process of the conjugated backbone and the unique dissolution–deposition behavior of Mn2+ in ZnSO4 electrolyte. The incorporation of Mn2+ into the PANI backbone facilitates the stabilization of PAM at high potentials by lowering the lowest unoccupied molecular orbital (LUMO) energy level, resulting in enhanced output voltage and cycling stability. This new interactive dual energy storage mechanism, illustrated by density functional theory calculations and ex situ characterization, contributes to the improved capacity by employing a dissolution–deposition storage mechanism. The battery showcases a maximum specific capacity of 496.7 mA h g−1 at an ultra-high working voltage of 2.4 V. And the capacity is 213.2 mA h g−1 when the current density reaches 20 A g−1. This molecular design of the pre-doped PANI cathode and the insight into the groundbreaking dual energy storage mechanism offer a new alternative host for high-performance Zn-organic batteries.
Introduction
Larger-scale energy storage systems are becoming increasingly crucial due to energy shortages and environmental pollution.1–3 Among the most promising candidates, aqueous zinc-ion batteries (AZIBs) stand out due to their intrinsic advantages associated with the Zn anode, such as high theoretical capacity (820 mA h g−1), low redox potential (−0.76 V vs. SHE), and excellent stability in aqueous electrolytes.4,5 The cathode, being the key component of full cells, plays a pivotal role in determining battery performance, including energy density and cycling stability.6–8 Organic redox active materials, composed of sustainable elements, have garnered increased interest owing to their abundant resources.9 More importantly, they possess a flexible molecular structure that can be chemically modified, endowing electrodes with tunable electrochemical performance (i.e., redox potential, doping level, and number of electrons involved).10Polyaniline (PANI), a conventional conducting polymer, has been extensively employed in rechargeable batteries because of its intrinsic conductivity and pseudocapacitive-dominated charge storage mechanism.11,12 The fact that conducting polymers can be charged/discharged is attributed to the doping/dedoping process accompanied by the mobility of anions or cations. However, the PANI cathode in rechargeable batteries is still impeded by the low attainable doping level (i.e., 0.5 holes per monomer ring).13 The electrochemical charge storage of PANI normally depends on the transition from the reduced state (leucoemeraldine salt, LES) to the intermediate state (emeraldine salt, ES) along with the proton doping process, reaching only half of the theoretical capacity (294 mA h g−1).14 Further oxidation to the most oxidized PANI (pernigraniline salt, PNS) requires a potential exceeding the maximum voltage limit of the aqueous electrolyte, leading to irreversible destruction of PANI at high voltage. To make full use of the three redox states of PANI, Sun et al. demonstrated an effective strategy to realize the LES-ES-PNS transition in AZIBs.15 By modulating the ionic activity in the aqueous electrolyte, the redox potential of the ES to PNS reaction is brought within the electrolyte voltage window, endowing PANI with an additional charge transfer process. Although this strategy achieves a high specific capacity (221 mA h g−1), the voltage window of PANI remains narrow (0.5–1.6 V), which prevents it from simultaneously providing both a high capacity and a wide voltage window.
Considering the unique reversible doping/dedoping process of PANI, selecting an appropriate doping strategy, such as anion-dominant doping and cation-dominant doping, is seen as a promising solution to satisfy the above-mentioned requirements.16 By doping an electron-absorbing group (such as –CN, –F, –Cl, and –Br) in the molecular chain, the LUMO energy level of PANI was reduced, thus enhancing the operating voltage.17 However, this anion-dominated doping cannot effectively improve the low intrinsic conductivity due to the entangled molecular chains of PANI. The coordination of cations on imine-type nitrogen atoms can modulate the structural ordering of molecular chains and alleviate the retardation of the protonation/deprotonation process, thereby improving the charge storage capacity.18 Therefore, it is critical to select an appropriate dopant, in addition to good coordination ability, which should have high ion mobility and good ion–solvent interactions with PANI.13 Of the various cations available, the Mn ion has emerged as a suitable dopant due to the comparable hydrated ionic radii of Zn2+ (4.30 Å) and Mn2+ (4.38 Å), facilitating the charge insertion/extraction of Zn ions.19 In particular, the Mn element can effectively regulate the operating voltage due to its abundant multivalent redox potential.20 Meanwhile, MnSO4 is also used as an electrolyte additive that is favorable for capacity and stability enhancement due to the dissolution–deposition process of Mn2+ in the ZnSO4 electrolyte.21 The doping of Mn2+ into PANI is anticipated to contribute to the combined cation storage mechanisms (Zn2+ insertion/extraction and Mn2+ dissolution/deposition), in a manner similar to the presence of manganese dioxide.22 Nevertheless, the incorporation of Mn2+ is normally achieved by mixing active MnO2 nanoparticles with PANI, yielding heterogeneous interfaces.23–25 To avoid phase separation and structural heterogeneity, it remains a significant challenge to develop a new molecular engineering strategy for high-voltage and high-capacity polymer electrodes and elucidate the energy storage mechanism.26,27
Herein, we present a pre-doping method of Mn ions into PANI (PAM) for high-performance AZIBs. The doped Mn ions not only increase the inherent conductivity of PANI but also provide additional active sites. Doping Mn ions contributes to the stabilization of PANI at high voltages, thereby boosting the output voltage. In addition, we reveal an interactive dual energy storage mechanism that relies on the reversible doping of Mn2+ along with H+ and Zn2+ within the PANI host. The proton doping process facilitates the charge storage process and establishes the prerequisites for Zn4SO4(OH)6·H2O (ZSH) formation required for the dissolution–deposition of Mn ions in ZnSO4 electrolytes (similar to a Zn–Mn battery). This work offers a new perspective on understanding the relationship between the cathode material and the electrolyte in the electrochemical process.
Results and discussion
Synthesis and characterization of PAM
We utilized a doping–dedoping–redoping approach to synthesize and design PANI conducting polymers, as illustrated in Fig. 1a. Firstly, the aniline monomer undergoes oxidative polymerization on the surface of carbon cloth (CC) at a low temperature, leading to the formation of PANI. During this stage, the subamino nitrogen atoms within the PANI molecular chain undergo partial protonation through HCl doping, yielding H+-doped PANI (PAH). Although the unprotonated imino in the PAH molecular chain can directly coordinate with MnSO4 hydrothermally to yield PAM, the present study necessitates analysis of the sole effect of Mn2+ doping in PANI that requires the removal of H+. The interaction between the Mn and N centres is a protonation process. Due to the conjugated chemical bonding (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and long-range conjugated structure of PANI, Mn ions can be stored in the quinone nitrogen site (–N) through protonation doping. Due to the principle of electroneutrality, anions can also be doped as counterions at the positively charged nitrogen atoms (–N+). It is worth noting that at this point the doped anion does not form a chemical bond with the nitrogen atom, but rather undergoes charge adsorption. 11,12
N) and long-range conjugated structure of PANI, Mn ions can be stored in the quinone nitrogen site (–N) through protonation doping. Due to the principle of electroneutrality, anions can also be doped as counterions at the positively charged nitrogen atoms (–N+). It is worth noting that at this point the doped anion does not form a chemical bond with the nitrogen atom, but rather undergoes charge adsorption. 11,12
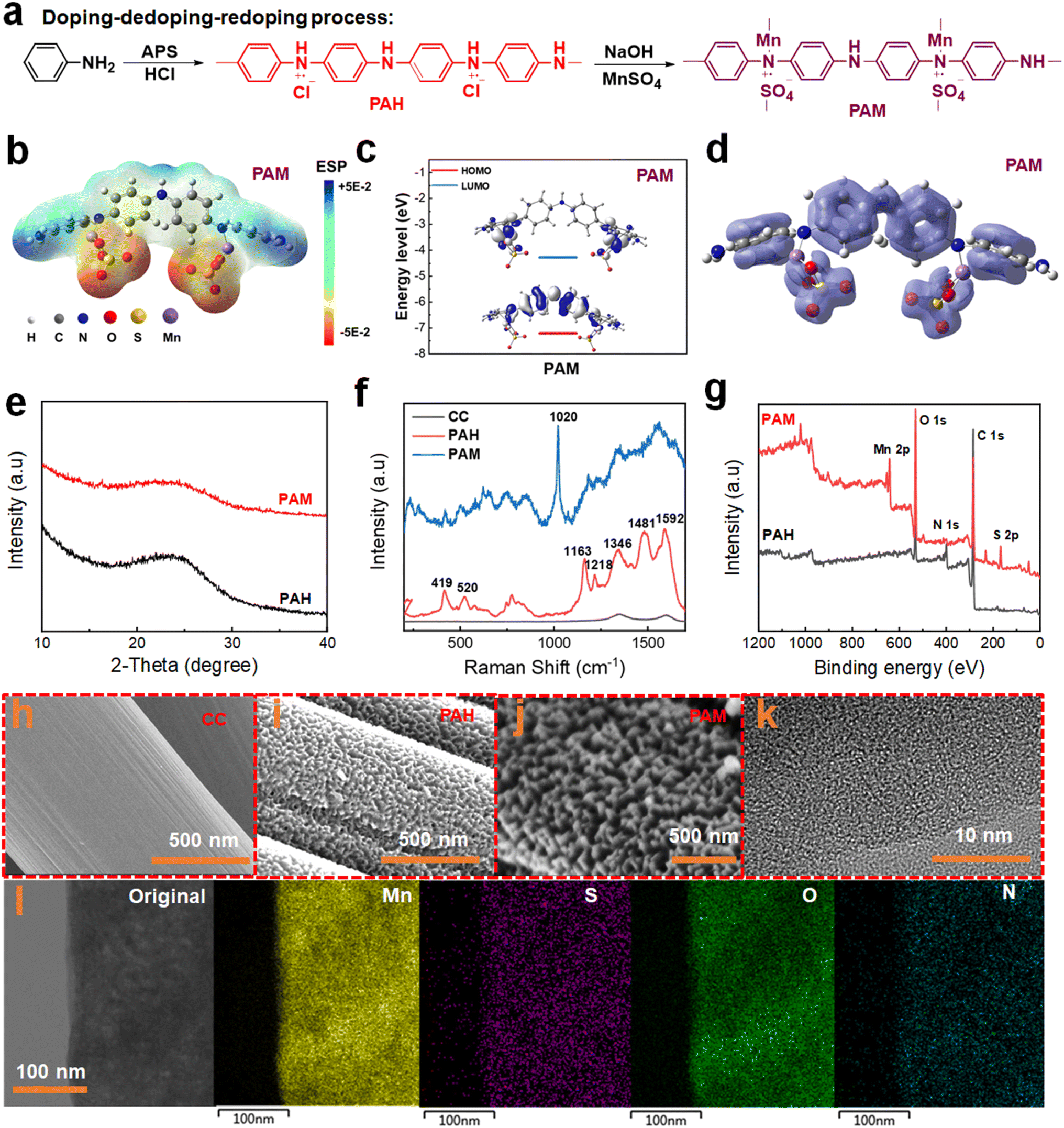 | ||
| Fig. 1 Synthesis, theoretical modeling and characterization of PANI-based electrodes. (a) Synthesis of H+-doped PANI (PAH) and Mn2+-doped PANI (PAM); (b) electrostatic potential (ESP) distribution, (c) calculated energy levels and (d) π-electron localization function image of the optimized PAM monomer; (e) XRD patterns, (f) Raman and (g) XPS spectra of PAH and PAM; SEM images of (h) carbon cloth, (i) PAH and (j) PAM; (k) TEM and (l) mapping images of PAM. | ||
Meanwhile, density functional theory (DFT) calculations reveal that the PAM molecules exhibit a uniform and symmetrical electron cloud distribution (Fig. 1b). This symmetric arrangement prevents the movement of the electron cloud, leading to a decrease in the electronegativity of the molecule and subsequently resulting in a higher discharge voltage. The Mn ion in PAM demonstrates effective coordination with the adjacent imino nitrogen atoms in the PANI molecular chain to form a tetrahedral coordination structure.28 Such a coordination structure will enhance the strength of the aniline unit, inhibiting the excessive volume expansion of the polymer chain. Meanwhile, the energy gap (ΔE) between the LUMO and highest occupied molecular orbital (HOMO) of the PAM material is small, indicating the excellent electronic conductivity of the material (Fig. 1c). The π-electron localization function image of the optimized PAM structure further highlights its pronounced π-conjugation properties (Fig. 1d).28,29
To validate the effective doping of MnSO4, X-ray diffraction (XRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS) measurements are performed to investigate the crystalline phase structure and elemental composition analysis of the PAM electrode material. As shown in Fig. 1e, both PAH and PAM exhibited broad and weak diffraction peaks at around 25° in the XRD spectra, indicative of amorphous carbon.28Fig. 1f presents the Raman spectra of CC, PAH, and PAM. The CC curve presented two characteristic Raman peaks for carbon materials. The peak around 1585 cm−1 represents bulk-phase crystalline graphite and is called the G-band. This peak is the fundamental vibrational mode of graphite crystals. The peak at 1360 cm−1 originates from the vibrations at the edges of the carbon crystalline state of graphite and is called the D-band. The PAH spectrum displays four characteristic Raman peaks at 1549 cm−1, 1504 cm−1, 1339 cm−1, and 1153 cm−1 corresponding to the CC stretching vibration, C–N stretching vibration, C–N+ stretching vibration, and C–H bending deformation vibration, respectively.30 The signature peaks observed at 419 cm−1 and 520 cm−1 in the fingerprint region can be attributed to amine deformation and ring deformation vibrations, respectively. Moreover, the characteristic peak located at 1020 cm−1 is ascribed to the SO vibration resulting from the doping of SO42−.31 The PAM shows a characteristic peak similar to that of PAH in the 1100–1600 cm−1 range. The characteristic Raman peaks of PAH and PAM are listed in Table S1.† When compared to PAH, the characteristic peak of PAM undergoes a noticeable Raman blueshift and peak broadening, suggesting the doping of Mn2+ with the nitrogen atoms in the PANI molecular chain.
In addition, the survey XPS spectra (Fig. 1g) confirmed that Mn and S elements were successfully doped into the PANI molecular chain. The corresponding elemental and valence analysis of each peak in the XPS spectrum of PAM is shown in Fig S1a.† Regarding Mn 2p in PAM (Fig. S1b†), the peaks located at 641.1 eV and 643.8 eV belong to divalent and tetravalent Mn ions, respectively.26,32 The area ratio of the divalent and tetravalent states was 4.3, indicating that the majority of the doped Mn ions exist in the divalent state. The presence of a high binding energy peak at 406.1 eV in the N 1s spectrum (Fig. S2†) is attributed to the N–O bond, indicative of the doping process of O− and amino nitrogen.28 Additionally, two prominent peaks at 167.6 eV and 168.8 eV in the spectra of S 2p (Fig. S3†) correspond to the S 2p3/2 and S 2p1/2 of metal sulfate, respectively.33 These distinctive peaks serve as additional confirmation of the successful incorporation of SO42− into the PANI backbone as a balanced charge counterion.
To verify the reversibility of the PAM materials, the prepared samples are immersed into a 0.1 M HCl solution for 24 h and finally dried at room temperature. XRD analysis of the experimental PAM sample shows clear diffraction peaks belonging to MnSO4, aligning with the standard reference card PDF#39–0602 (Fig. S4†). This observation underscores that the doping process within the PAM material is indeed reversible.
Notably, the peak at 1173 cm−1, attributed to the in-plane bending vibrational mode of CNH+ in the quinone ring upon the addition of Mn ions, shifted to 1104 cm−1 (Fig. S5†). This blueshift in this band is typically attributed to the electronic off-domain in the PANI chain. In addition, the vibrational peaks at 796 cm−1 are attributed to the stretching vibration (CC bending) of the p-disubstituted benzene ring, with a blue shift observed at 753 cm−1. The results indicate that the degree of polariton delocalization in the PAM material exceeded that of PAH, confirming the role of manganese sulfate as a secondary dopant in PANI.33
Furthermore, scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) are used to analyze the evolution of the material microstructure during the doping process and to observe the changes in the morphology. In contrast to the smooth surface of carbon cloth on carbon fibers (Fig. 1h), PAH demonstrates nanocluster structures on its surface (Fig. 1i). Following the dedoping–redoping process, PAM causes the nanocluster structure of PAH to become coarser, as evident in Fig. 1j. HRTEM images of PAM also revealed the absence of crystalline regions, consistent with the broad diffraction peaks observed in the XRD analysis (Fig. 1k). In addition, TEM-EDS images depict a uniform distribution of Mn, S, O, and N elements in the PAM, as shown in Fig. 1l. Next, various experimental conditions are investigated to achieve the optimal electrochemical properties (Fig. S6 and S7†).
Electrochemical kinetics and performance of PAM electrodes
To demonstrate the distinct electrochemical advantages of the cathode material formed using this strategy, Zn ion cells are assembled using PAH, PAM, and PAH-M electrodes for comparison. Notably, PAH-M refers to a Zn//PAH-M cell where PAH serves as the cathode, and a directly added MnSO4 solution functions as the electrolyte. This is used to compare with Zn//PAM pairs formed by indirect doping of MnSO4 on the anode to investigate the effect of the two different approaches on the electrochemical performance. Relative to PAH, the PAM sample displays a significantly larger area enclosed by the CV curve, an additional pair of redox peaks, and reduced polarization (Fig. 2a). This result implies that the introduction of Mn ions contributes to an increase in the capacity of the cell. The redox peaks of both PAH-M and PAM appear similar, hinting at a potentially identical energy storage mechanism. When considering discharge capacity at a current density of 0.5 A g−1, PAM achieves a notable maximum of 388.8 mA h g−1, substantially surpassing the capacities of PAH (112.6 mA h g−1) and PAH-M (290.8 mA h g−1), as depicted in Fig. 2b. Concurrently, the CE of PAM at a current density of 0.5 A g−1, standing at 99.5%, markedly exceeded those of PAH (77.7%) and PAH-M (88.7%). In terms of rate performance, the discharge capacity of PAM is found to be 87.2 mA h g−1 when the current density reaches 20 A g−1, while PAH and PAH-M samples achieve only 22.7 mA h g−1 and 48.3 mA h g−1, respectively. Considering cycle life, PAM, PAH, and PAH-M materials all exhibit successful operation over 500 cycles (Fig. S8†). However, at the same current density, PAM outperforms with a maximum discharge capacity of 175.1 mA h g−1, whereas PAH achieves 139.6 mA h g−1 and PAH-M records 70.1 mA h g−1, showcasing significantly higher capacity. In addition, PAM demonstrates a notably superior CE of 98.6% and capacity retention of 75.7% compared to both PAH (96.5% and 73.6%) and PAH-M (94.8% and 65.9%). The suboptimal cycling performance of PAH-M indicates the instability of the directly added MnSO4 electrolyte. At 600 °C, the remaining mass of PAM was 85.9%, much higher than the 63.4% of PAH (Fig. S9†). This result also showed a significant enhancement in the thermal stability of the doped PAM. | ||
| Fig. 2 Electrochemical performance of PAH, PAM and PAH-M electrodes in 2 M ZnSO4 electrolyte. (a) CV curves, (b) rate capability, (c) EIS curves, (d) LSV curves, and (e) double layer capacitance linear fitting diagram; (f) chemical structures and calculated energy levels of the optimized PAH, PAM and PAH-M monomers. | ||
Further insights into the kinetics of electron transport are obtained using EIS analysis. The impedance data are fitted using Z-view software, revealing Rs and Rct values for the PAM electrode as 1.4 Ω and 14.6 Ω, respectively (Fig. 2c). Conversely, the PAH electrode exhibits Rs and Rct values of 2.7 Ω and 27.4 Ω, respectively, while the PAH-M electrode shows 2.4 Ω and 37.4 Ω. A steeper slope of the straight line of the EIS curve for the PAM electrode indicates high ion diffusion and enhanced ionic conductivity. The lower Rs and Rct and improved ion diffusion capability of the PAM electrode can be ascribed to the higher degree of order and crystallinity resulting from the chelation between Mn ions and molecular chains. These findings collectively suggest that the material synthesized through the strategy of electrolyte storage at the anode exhibits superior electrochemical properties. Pre-doping Mn2+ into polyaniline hinders the chain entanglement, thus enhancing the order and crystallinity. This fostered favorable spatial conditions for the subsequent combined cation storage mechanisms: Zn2+ insertion/extraction and Mn2+ dissolution/deposition. 34
Compared to PANI (Fig. S10a†), both MnO2 and PAM showed a significant discharge plateau (∼1.3 V), indicating that MnO2 provided the main capacity contribution. PAM exhibited a much higher capacity. PAM displayed the lowest Rs and Rct of 1.4 Ω and 14.6 Ω, respectively (Fig. S10b†), compared to PANI (2.6 Ω and 42.6 Ω) and MnO2 (5.6 Ω and 52.3 Ω). The larger slope of the straight-line portion of the EIS curve for PAM indicates higher ionic diffusion and ionic conductivity. The higher ionic diffusion of the PAM cell may be attributed to the manganese ion pre-structuring design, which offers a more conducive ion transport pore structure.
To elucidate the advantages stemming from the strategy of storing the electrolyte in the positive electrode, linear scanning voltammetry (LSV) is employed to examine the electrochemical signal of the cell. When compared to PAH and PAH-M, PAM exhibits a more effective inhibition of hydrogen evolution, which explains their better cycling performance (Fig. 2d). To ascertain whether the PAM generates more active sites, the study involves the electrochemically active specific surface area for the analysis (Fig. S11–S13†). Fig. 2e presents the double-layer capacitance (Cdl) values of 0.024, 0.135, and 0.126 F cm−2 for the PAH electrode, PAM electrode, and PAH-M electrode, respectively, which are determined using the following equation:34
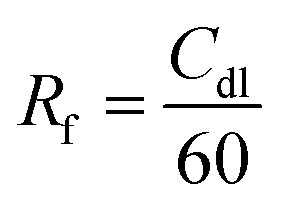 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, and 21000, respectively. This evidence suggests that PAM electrodes possess a greater electrochemically active surface area on their surface, affording more active sites for charge storage during the electrochemical reactions. This phenomenon can be attributed to the emergence of more favorable zinc sites following the doping of PAM electrodes and the increased defects stemming from reduced crystallinity.
500, and 21000, respectively. This evidence suggests that PAM electrodes possess a greater electrochemically active surface area on their surface, affording more active sites for charge storage during the electrochemical reactions. This phenomenon can be attributed to the emergence of more favorable zinc sites following the doping of PAM electrodes and the increased defects stemming from reduced crystallinity.
In addition, we use theoretical calculations to further corroborate that the PAM material possesses an optimal electronic structure. The LUMO value of PAM (−4.27 eV) is lower than that of PAH (−3.25 eV) and PAH-M (−3.57 eV), signifying that PAM exhibits a stronger electron affinity and a higher reduction potential (Fig. 2f). At the same time, the total energy gap (ΔE) between the HOMO and LUMO in PAM materials is the smallest, indicating that their electronic conductivity surpasses that of both PAH and PAH-M. This further supports the notion that increasing the π-electron conjugation of the azirine ring can effectively improve the electronic conductivity of the material.
To gain a deeper understanding of the changes in the energy storage mechanism introduced by this approach, we assemble full cells using the PAH electrode, PAM electrode, and PAH-M electrode as the positive electrode, in conjunction with a negative zinc electrode, respectively, which are then subjected to pseudocapacitance analysis. Fig. 3a–c depict the CV curves of the cells with PAH, PAM, and PAH-M electrodes, respectively, across a voltage window of 0–2.0 V at varying scan rates. Notably, the shape of the CV curves for the PAM electrode and PAH-M electrode remains largely consistent even when the scan rate increases from 0.8 mV s−1 to 10 mV s−1, highlighting their good rate capability. CV tests conducted at different scan rates can provide insights into the complete electrochemical process, facilitating an exploration of the ion transfer and diffusion processes occurring within the electrode. Therefore, we further analyze the ratio of diffusion to capacitance control in the current response at different scan rates using the following equation:35
| i = avb | (2) |
| i(v) = k1v + k2v1/2 | (3) |
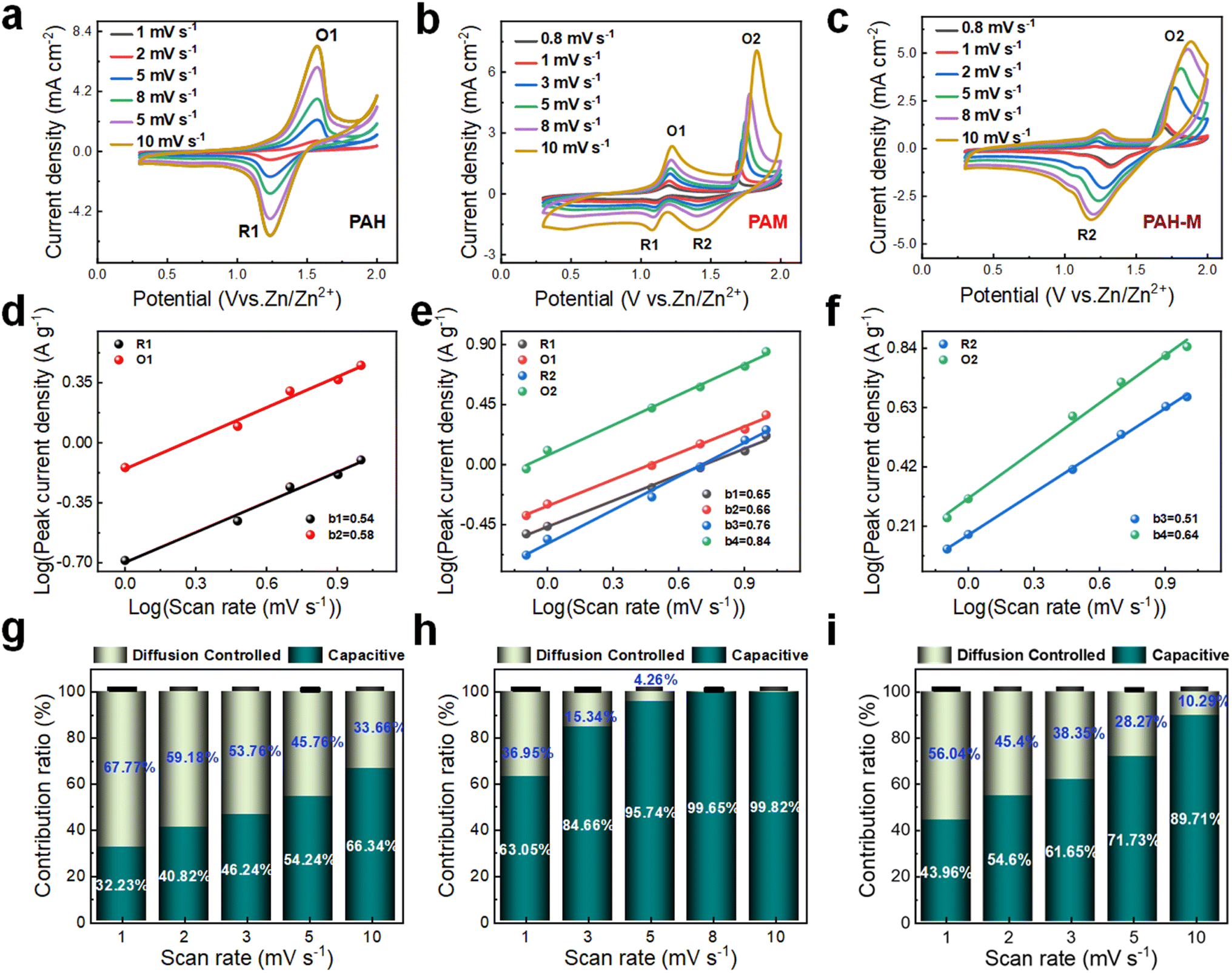 | ||
| Fig. 3 The charge storage kinetics of PAH, PAM and PAH-M electrodes in a half-cell in the voltage window of 0–2.0 V. (a–c) CV curve at different scan rates of 0.8–10 mV s−1; (d–f) Fitting curves and b values; (g–i) Pseudocapacitance contribution at different scan rates. | ||
Electrochemical performance of Zn//PAM cells
Next, to assess the self-discharge behavior of the Zn//PAM cell, the CE is measured after fully charging and discharging the cell to 1.8 V, holding it for 24 h, and then discharging again. As depicted in Fig. 4a, following an initial voltage drop, the Zn//PAM cell maintains a high CE of 94.3% due to the protective effect of the stable structure formed by the redoping process, inhibiting the corrosion and energy dissipation at the interface. To further evaluate the practical applicability resulting from the strategy of electrolyte storage at the cathode, Zn//PAM cells are subjected to constant current charge/discharge long-cycle tests. As illustrated in Fig. 4b, even after 2000 cycles at a current density of 1 A g−1, the cell retains a substantial residual capacity of 90.9%, reducing from 275.1 to 250.2 mA h g−1, while consistently maintaining a CE above 95%. In Fig. 4b, the large fluctuations in the 900–1000th cycle range may be due to current instability.37 The voltage and current of the battery may fluctuate during long cycles due to several factors. As charging and discharging proceed, the electrochemical reactions within the battery produce polarisation, leading to instability in voltage and current. However, MnO2 had only 78.6% capacity remaining under the same conditions (Fig. S14†).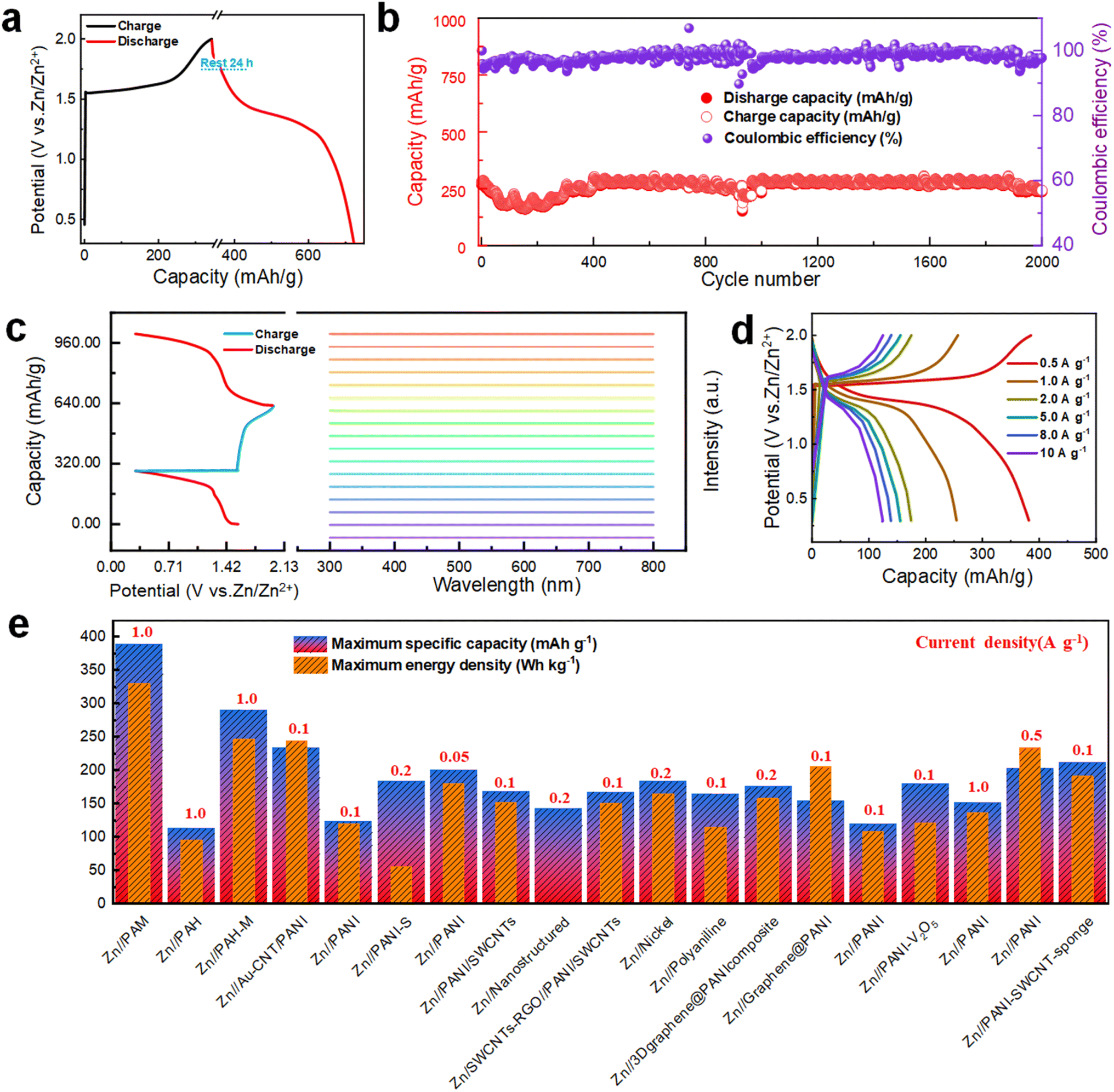 | ||
| Fig. 4 Electrochemical performance of Zn//PAM cells. (a) Self-discharge behavior after resting for 24 h, (b) the capacity retention after 2000 cycles at a current density of 1 A g−1, (c) in situ UV-vis curves of the 2 M ZnSO4 electrolyte during a GCD cycle, (d) GCD curves at different current densities of 1–10 A g−1, and (e) the capacity and energy density comparison among reported PANI-based AZIBs. | ||
To further investigate the stability of the Zn//PAM cell, in situ UV-vis spectroscopy tests were conducted on the electrolyte during the cycling process to monitor the potential dissolution behavior of PAM (Fig. 4c). Notably, no transition of π–π* electrons from the benzene ring in the PAM molecular chain was observed, nor any transitions of n-π electrons between the HOMO of the benzene ring and the LUMO of the quinone ring throughout the cycle. This behavior indicated the insolubility of PAM in all charging/discharging states, ensuring the excellent stability of the cathode during long-term cycling.
At current densities spanning from 0.5 to 20 A g−1, the specific discharge capacities obtained for Zn//PAM cells are 388.8, 254.4, 175.2, 140.2, 123.9, and 62.8 mA h g−1, with CE remaining around 98.6% (Fig. S15†). The charge/discharge curves corresponding to these different current densities are presented in Fig. 4d. Predominantly, two distinct discharge plateaus located around 1.4 V and 1.0 V are observed in the figure. The first discharge plateau makes a more substantial capacity contribution than the second one, which is in accordance with the shape and size of the reduction peak depicted in the CV curve in Fig. 3b. To further exemplify the potential practicality stemming from the strategy of electrolyte storage at the cathode, the specific discharge capacity and energy density of the Zn//PAM cell are compared to those of cells utilizing similar electrode materials (Table S2†). It is evident that the maximum discharge capacity and energy density obtained for the Zn//PAM cell are 388.8 mA h g−1 and 330.5 W h kg−1, respectively, which is considerably higher than those of various cells featuring PANI-based electrode materials as reported in the existing literature (Fig. 4e). To illustrate the feasibility of a full cell with a dual energy storage mechanism, large-capacity Zn//PAM full cells were assembled. As shown in Fig. S16,† after 500 cycles at a current density of 1 A g−1, the cell with a load of approximately 10 mg retained 94.5% residual capacity (1.65–1.56 mA h). Even with a load of up to 20 mg, the cell still retained 83.1% residual capacity (2.72–2.26 mA h). These results show that the mechanism still has great potential at high capacity.
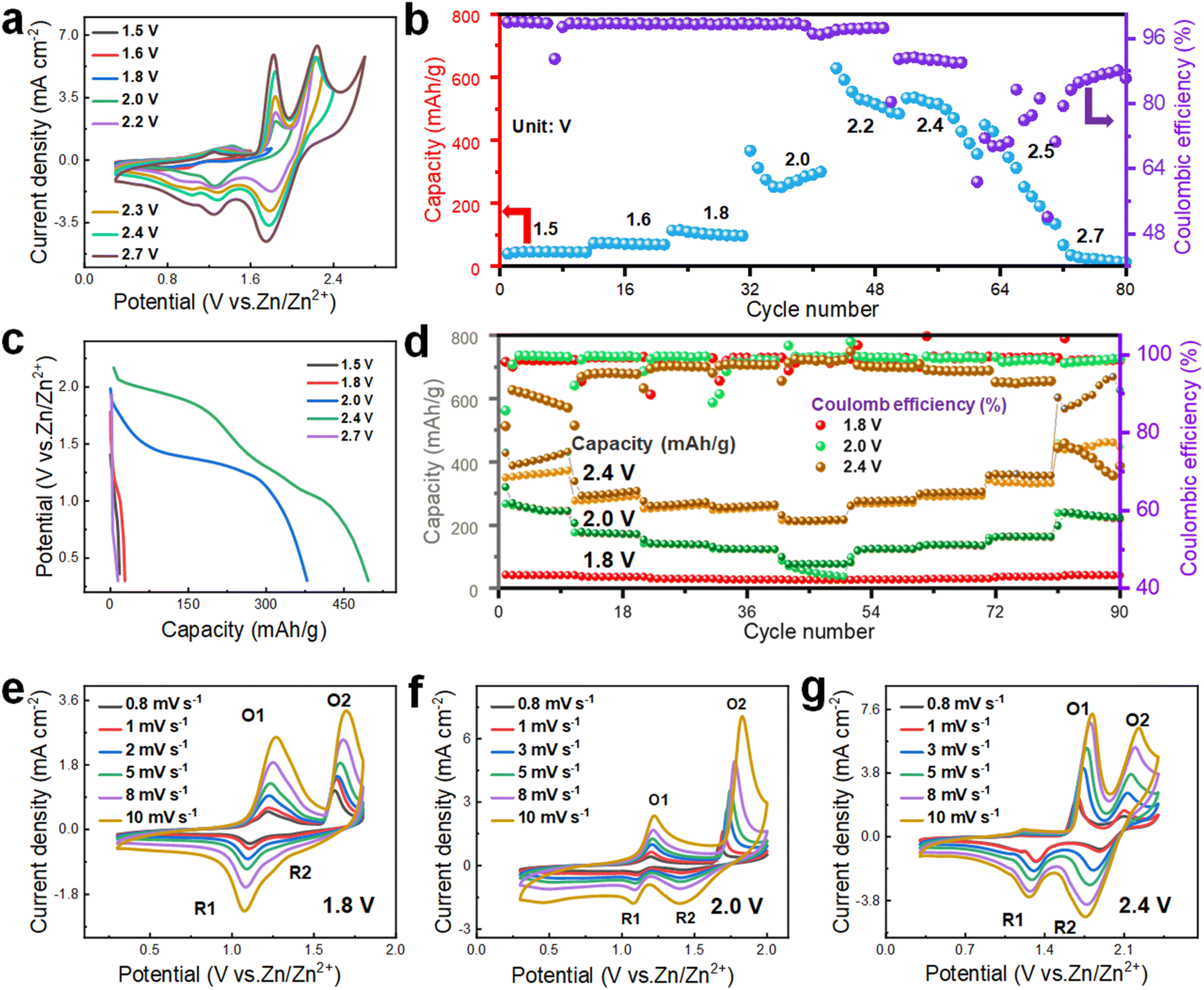
Now, the relationship between voltage and electrochemical performance is explored to ascertain the true potential of the PAM cathode cell. As shown in Fig. 5a, significant polarization spikes only emerge within a voltage range of 0.3–2.7 V, implying that 2.7 V is the suitable voltage for electrochemical testing. To more precisely determine the optimal voltage range, the GCD curves are analyzed at different voltage intervals. As depicted in Fig. 5b, the CE starts to fall below 80% when the voltage exceeds 2.4 V. At the same time, there is no notable increment in capacity. Moreover, when subjected to a voltage of 2.7 V, the battery experiences a short circuit after only a few test cycles, corroborating the observations from the prior CV curve results. From Fig. 5c, it can be seen that the specific capacity at 2.4 V is distinctly the highest. As a consequence, 2.4 V is chosen as the highest voltage for the subsequent electrochemical tests. To validate the feasibility of this remarkably high voltage for practical applications, three maximum operating voltages of 1.8 V, 2.0 V, and 2.4 V are selected for testing rate performance and assessing long-term cycling stability. When evaluating discharge capacities at a current density of 1 A g−1, the capacity reaches 376.8 mA h g−1 at 2.4 V, which surpasses the values at 2.0 V (254.4 mA h g−1) and 1.8 V (66.4 mA h g−1), as illustrated in Fig. 5d. Furthermore, the CE at a current density of 1 A g−1 is notably higher at 2.0 V (99.0%) compared to both 1.8 V (97.7%) and 2.4 V (81.5%). Meanwhile, at a high current density of 20 A g−1, the discharge capacity at 2.4 V reaches 213.2 mA h g−1, which outperforms the capacities at 1.8 V (32.6 mA h g−1) and 2.0 V (87.2 mA h g−1), underscoring the advantageous rate performance. In terms of cycle life, 2.4 V offers a shorter lifespan, maintaining normal operation for only 600 cycles, which is considerably fewer than the 2000 cycles observed at 2.0 V (Fig. S17†). From the given analysis, while 2.4 V yields the highest capacity, the optimal long-term cycling stability and CE are observed at 2.0 V. Although the redox peaks of the CV curves appear similar across the three different upper operating voltages, cells operating at higher voltages within the same range exhibit a larger CV area. This infers that they are capable of providing more discharge capacity, as portrayed in Fig. 5e–f. However, the decomposition voltage of the electrolyte is inadequate to support an excessively high upper operating voltage, leading to diminished long-term cycle life and CE.
 | ||
| Fig. 5 Electrochemical behavior of Zn//PAM cells in different voltage ranges. (a) CV curves in the voltage range from 1.5 V to 2.7 V at a scan rate of 10 mV s−1, (b) capacity and coulombic efficiency at different voltages at a current density of 0.5 A g−1, (c) GCD curves at a current density of 0.5 A g−1; (d) rate capability in the voltage range from 1.8 V to 2.4 V; (e–g) CV curves of cells at different scan rates. | ||
Structural characterization of PAM electrodes during different charge/discharge states
We examine the structural and phase transitions occurring on the surface of the PAM cathode to investigate the charge/discharge electrochemical reactions. Notably, the characteristic XRD diffraction peaks associated with Zn4SO4(OH)6·H2O (ZSH, PDF#33–0690) start to appear during the first lap of the discharge cycle, progressing from PAM-a to PAM-b (Fig. 6a and b). In contrast, the characteristic peak of ZSH in the fully charged state PAM-d disappears during the second charging cycle. Additionally, there is no presence of a pronounced sharp peak, potentially owing to the low crystallinity of ZnxMnO(OH)2 (ZMH). To ascertain whether full charging leads to the formation of ZMH, we devise three comparative cell experiments, as illustrated in Fig. S18.† The outcome reveals that the cell containing the PAH electrode does not produce the yellow-brown precipitate, whereas this precipitate is present in the cell containing PAM and PAH-M electrodes, confirming that the formation of the yellow-brown precipitate requires the participation of Mn ions. To determine the material composition of the precipitate, XRD is utilized for the analysis (Fig. S19†). The yellow-brown precipitate is identified as a combination of Zn2Mn3O8·H2O (PDF#09–0459), MnOOH (PDF#74–1049), and the residual electrolyte ZnSO4·6H2O (PDF#32–1478) on the current collector. This finding somewhat suggests the co-formation of ZMH and ZSH, facilitated by the participation of Mn ions during the charging process. Moreover, during the second discharge (from PAM-d to PAM-f), the characteristic peak of ZSH re-emerges on the PAM cathode, signaling that the entire electrochemical process is highly reversible.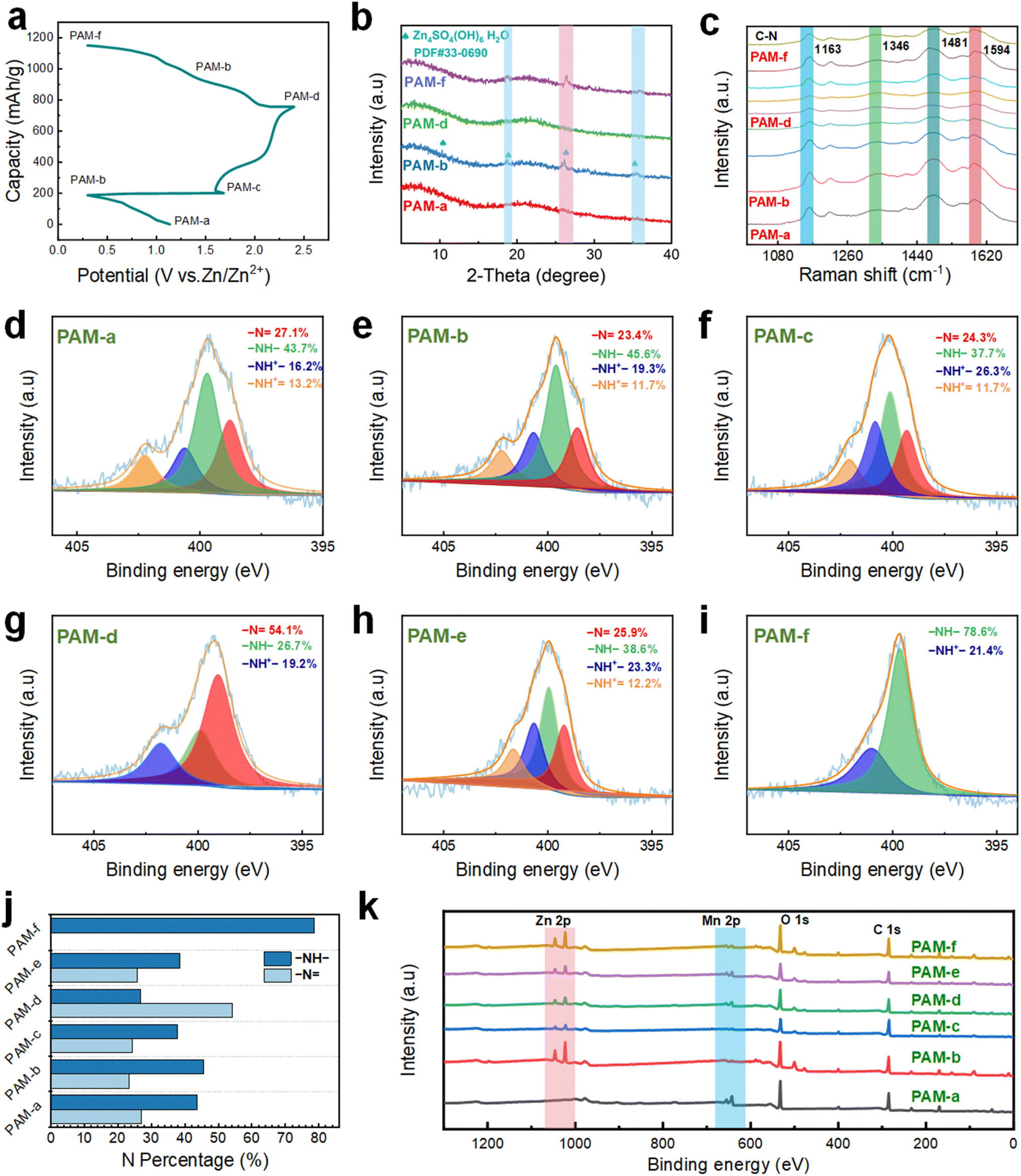 | ||
| Fig. 6 Structural characterization of PAM electrodes during different charge/discharge states. (a) GCD curve during cycling at a current density of 2 A g−1; (b) ex situ XRD and (c) Raman spectra during cycling; (d–e) ex situ N 1s XPS curves during the first cycle of discharge; (f–i) ex situ N 1s XPS curves in the second charge cycle; (j) changes in the bonding components of N during cycling; (k) ex situ XPS spectra of the Zn 2p, Mn 2p, O 1s, and C 1s regions at each stage during cycling. | ||
Considering the limited information provided by the XRD patterns, ex situ Raman spectra are acquired to investigate the evolution process of the PAM cathode. As demonstrated in Fig. S20,† throughout the charging process, three distinct Raman bands gradually appear at 482 cm−1, 570 cm−1, and 647 cm−1, corresponding to the V4 (Mn–O) stretching vibration mode associated with the [MnO6] sheet in layered manganese oxide, the V3 (Mn–O) stretching vibration, and the symmetric stretching vibration of V2 (Mn–O) belonging to the A1g symmetry mode, respectively.38 Noticeably, the Raman band of layered manganese oxide weakens during the initial discharge cycle, indicating a reduction in the content of Mn ions in the PAM cathode. This trend implies that Mn ions are being replaced by Zn and hydrogen ions in the doped PAM cathode during this stage. In addition, the Raman band of layered manganese oxide progressively fades during the second lap of the discharge cycle. Thus, the ex situ Raman spectra reveal the reversible formation and dissolution of layered manganese oxide during the charging and discharging process. As seen in Fig. 6c, the Raman peaks associated with the quinone ring are enhanced during the charging process. Particularly, significant changes are observed in the stretching vibration peak of the CN bond around 1481 cm−1 and the planar vibration of the C–H bond around 1163 cm−1, while other variations are subtler. Moreover, the peak related to the nitrogen cation radical was located near 1346 cm−1 (C–N+♦ mode vibrations), which gradually enhances as the charging continues, suggesting an increase in the quinone ring structure. Besides, the Raman signals related to the benzene ring show enhancement during the second discharge cycle, with apparent changes in the stretching vibration peak of the CC bond near 1594 cm−1 and the stretching vibration peak of the C–N bond around 1260 cm−1, whereas other shifts remain relatively insignificant. This change in the Raman signal implies an increase in the benzene ring structure. Therefore, the ex situ Raman spectra highlight the reversible doping and dedoping mechanism of the PAM cathode throughout the charging and discharging process.
To again corroborate the changes in elemental composition during the electrochemical process, ex situ XPS is employed for the analysis. For this, the basic units of PANI can be divided into three categories: the fully reduced form, known as leucoemeraldine base (LEB); the semi-oxidized form, termed emeraldine base (EB); and the fully oxidized form, referred to as pernigraniline base (PNB). Interestingly, both EB and PNB units can undergo protonation by the anion, resulting in their respective salt forms: emeraldine salt (ES) and pernigraniline salt (PNS). The N 1s spectrum is widely considered one of the most reliable methods to analyze the structural changes in PANI due to doping. Within the N 1s spectrum, four N components are identified as quinone diimine (–N, 399 eV), phenylenediamine (–NH–, 400 eV), protonated amine (–NH+–, 401 eV, monopolariton), and protonated imine (–NH+, 402 eV, dipolariton).39 The last three N components are linked to the oxidation state, whereas the first one is related to the reduction state. During the initial discharge cycle (Fig. 6d and e), there is an observable increase in the content of protonated nitrogen (–NH+– and –NH+) in the fully discharged state (PAM-b), amounting to 31.0%, compared to its pristine state (PAM-a) which is at 29.5%. Conversely, there is a decline in the content of quinone diimine (–N), dropping from 27.2% to 24.3%. Thus, the increment in the content of the N component in the oxidized state indicates the continued doping process of the PAM cathode during the discharge cycle, i.e., the LES-M state. In the second charging cycle (Fig. 6f and g), the content of the quinone diimine component increases by 30.7%, while the content of the N component related to the benzylamine (–NH–) decreases by 18.9% in the fully charged state (PAM-d) compared to the fully discharged state (PAM-b), i.e., the PNS-M state. At the charging points between PAM-c and PAM-d, the content of N components in the oxidised state also showed an increasing trend, which is consistent with the above results (Fig. S21†).
Hence, the enhanced content of the N component in the reduced state indicates that the dedoping of the PAM cathode takes place during the charging process, involving the continued deprotonation of the phenylenediamine structure in the electrolyte, leading to its oxidation to the quinone structure. Furthermore, in the second discharge cycle (Fig. 6h and i), the content of the N component related to phenylenediamine (–NH–) increases to 78.6% in the fully discharged state (PAM-f) compared to the fully charged state (PAM-d), with no presence of the quinone diimine component, indicating the appearance of the LES-M state. During the discharge, the quinone-like structure of the PAM cathode undergoes continuous protonation, reducing to a benzene-like structure, as evidenced by the ultra-high content of the oxidized state of the N component. Both the semi-charged state (PAM-c) and the semi-discharged state (PAM-e) show four nitrogen-containing components, with protonated nitrogen accounting for more than one-third of the total, indicating the ES-M state. In addition, four nitrogen-containing components appear at the discharge point between PAM-e and PAM-f, with more than one-third protonated, which is consistent with the above results (Fig. S22†). As shown in Fig. 6j, the content of components with the benzylamine structure is the highest in both PAM-b and PAM-f of the fully discharged states, suggesting that nitrogen sites undergo reversible reduction during the discharge process. These above results imply that the PAM cathode exhibits a reversible transition from the LES-M state to the ES-M state and from the ES-M state to the PNS-M state, as confirmed by the two pairs of redox peaks. This infers that the PAM cathode undergoes a redox process from the LES-M state to the PNS-M state.
Besides, the elemental variations throughout the reaction process are further analyzed by XPS, shedding light on the overall storage mechanism (Fig. 6k). It is noted that the Zn 2p component appears in the fully discharged state (PAM-b) compared to its pristine state (PAM-a), and there is a marked reduction in the intensity of the Mn 2p component during the first cycle of discharge. The observed variations indicate a dynamic replacement where hydrogen and Zn ions replace the Mn ion in the PAM cathode. As the charging process proceeds (PAM-b to PAM-d), the intensity of both the Zn 2p fraction and the Mn 2p fraction initially decreases and then increases. This is because the oxidation process (deprotonation process) occurs first at the PAM cathode, involving Zn2+, Mn2+, and H+ ions, accompanied by the dedoping of sulfate anions. Subsequently, with the increased concentration of Mn ions in the solution, ZSH transitions into low-crystallinity ZMH. Further, during the discharging phase (from PAM-d to PAM-f), the intensity of both the Zn 2p fraction and the Mn 2p fraction initially drops and later rises. This trend is ascribed to the dissolution of ZMH into its constituents Zn2+ and Mn2+, which is accompanied by the doping of sulfate anions into the PAM cathode.
Furthermore, the Mn 3s XPS spectra are often utilized to assess the chemical valence of manganese oxides during discharging and charging cycles; however, it is important to note that the Mn 3s peak at 84.2 eV significantly overlaps with the Zn 3p peak (Zn 3p3/2 at 88.6–89.2 eV and Zn 3p1/2 at 91.4–92.1 eV).40 Consequently, the Mn 3s spectral peaks are deemed inappropriate for conducting this analysis. Meanwhile, there are three Mn 2p components in the spectra due to Mn2+ (641.6 eV), Mn3+ (642.9 eV), and Mn4+ (645.8 eV) states. In the initial discharge phase (Fig. S23a and b†), a unique satellite peak at 647.0 eV, belonging to Mn2+, appears in the fully discharged state (PAM-b), a peak that is absent for both Mn3+ and Mn4+. A notable rise in the content of the Mn2+ component indicates that Mn3+ and Mn4+ are replaced by Zn and hydrogen ions first, which further implies that the doping of Mn3+ and Mn4+ into the PAM cathode is unstable. As the charging continues (Fig. S23b–d†), the content of the Mn2+ component gradually diminishes, suggesting that Mn2+ is dedoped from the PAM cathode to form a ZMH material composed of Mn3+ and Mn4+. During the subsequent discharge cycle (Fig. S23d–f†), the content of the Mn2+ component progressively rises, reaching 82.7%. This result signifies that the dissolution of ZMH took place, resupplying Mn ions for the doping process of PAM. In the fully discharged state, the Mn ions of both (PAM-b) and (PAM-f) predominantly exist as the Mn2+ fraction (Fig. S23h†), underscoring the reversible nature of the entire charge–discharge process.
Moreover, the O 1s XPS spectra are deconvoluted into three distinct component peaks, representing Mn–O–Mn (531.0 eV), Mn–OH (532.5 eV), and H–O–H (533.3 eV), aiding in the identification of the specific Mn compounds present.41 Notably, the content of the H–O–H component increases from 19.6% to 21.8% due to the formation of ZSH in the first lap of the discharge cycle (Fig. S24a and b†). In the subsequent charging phase (Fig. S24b–d†), the content of the Mn–O–Mn component is around 20.2%, while the H–O–H component showed a decline of 14.6%. This phenomenon is because of the formation of ZMH by combining ZSH with Mn ions. Additionally, in the second discharge cycle (Fig. S24d–f†), both the Mn–OH and H–O–H fractions continue to increase, reaching 55.0% and 29.1%, respectively, implying that 21.8% of the ZMH undergoes conversion to a lower chemical valence state. This finding is consistent with the significant increase in the content of Mn2+ components during the discharge process, as shown in Fig. S23.† The predominant presence of the H–O–H component (attributed to ZSH) in both the fully discharged states (PAM-b) and (PAM-f) further underscores the reversible nature of the entire charge–discharge cycle (Fig. S24h†).
Interactive dual energy storage mechanism
From the above-detailed analysis, it is evident that the system utilizes an ingenious dual complementary energy storage mechanism. To simplify, this mechanism can be divided into two parts. Mechanism I involves energy storage directly through the PANI by doping and dedoping processes involving Zn2+, Mn2+, and H+ ions (Fig. 7a). As illustrated in Fig. 5e, during charging, the fully reduced LES-M initially undergoes oxidation to the semi-oxidized ES-M state, accompanied by the involvement of Zn2+, Mn2+, and H+, along with the dedoping of sulfate anions. This corresponds to the oxidation peak O1 in the CV curve. This is also consistent with the increased N content (Fig. 6j) in the reduced state and the enhanced Raman peak associated with the quinone ring (Fig. 6c). Subsequently, there is a continuous deprotonation process from the semi-oxidized ES-M state to the fully oxidized PNS-M, corresponding to the oxidation peak O2 in the CV curve. At the negative electrode, Zn2+ ions in the electrolyte gain electrons to form Zn, thus allowing the balance of the charge of the electrolyte during the charging process. The charging process is the reverse of this discharging phase.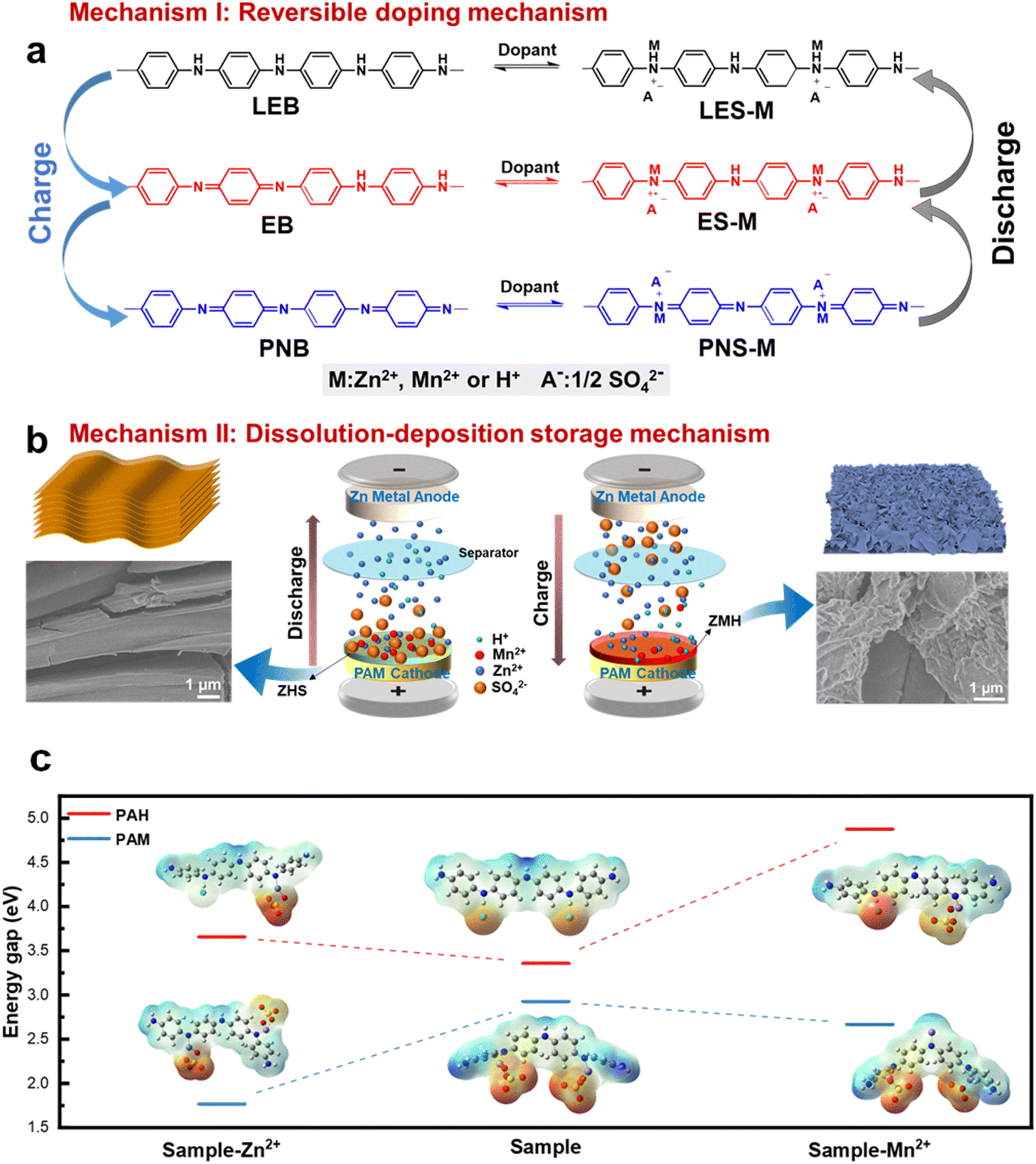 | ||
| Fig. 7 Schematic diagram of the interactive dual energy storage mechanism of PAM. (a) Mechanism I: reversible doping mechanism of Mn2+ along with H+ and Zn+ into the conjugated backbone. (b) Mechanism II: dissolution–deposition storage mechanism of Mn2+ on the cathode surface. (c) The plot of energy gap variation for PAH and PAM sample materials doped with ZnSO4 and MnSO4 electrolytes. | ||
Mechanism II revolves around the dissolution–deposition process where H+ doping from the PAM cathode induces the formation of alkaline zinc sulfate (ZSH), while the de-doping of Mn2+ from the PAM cathode leads to the formation of low-crystallinity ZMH in conjunction with ZSH (Fig. 7b). As the PAM cathode incorporates Mn ions during the electrochemical process, they can be reversibly deposited or dissolved into alkaline compounds along with Zn ions as the pH changes. During the first discharge cycle, hydrogen and Zn ions displace the Mn ion within the PAM cathode. The introduction of hydrogen ions facilitates the creation of a transient localized alkaline environment on the cathode surface, which then transforms the electrolyte ZnSO4 into ZSH. Additionally, during the second charging cycle, the PAM cathode undergoes an oxidation (or deprotonation) process, leading to the dedoping of Zn2+, Mn2+, and H+ alongside sulfate anions. This results in an increased concentration of Mn ions in the solution, causing ZSH to convert into low-crystallinity ZMH, which corresponds to the oxidation peak O2 in the CV curve. This is consistent with the characterization results that the intensity of the Zn 2p and Mn 2p components first decreases (Fig. 6k) and then increases, while the Raman bands of the layered manganese oxides gradually disappear and the Raman peaks associated with the quinone ring are enhanced (Fig. 6c).
Furthermore, during the second discharge cycle, a reduction reaction occurs at both the PAM cathode and the ZMH is generated on its surface. The ZMH thus formed dissolves into Zn2+ and Mn2+ ions, which in turn provide Zn2+, Mn2+, and sulfate anions to the PAM cathode through the doping process. Simultaneously, the dissolution of ZMH and the H+ doping into the PAM cathode create a localized transient alkaline environment, facilitating the transformation of the electrolyte ZnSO4 to basic ZSH again. The entire sequence of the reaction process is as follows:
First discharge cycle:
| PAM + Zn2+ ↔ PAZ + Mn2+ | (4) |
| PAM + 2H+ ↔ PAH + Mn2+ | (5) |
| 4Zn2+ + 6OH− + SO42− + H2O ↔ Zn4SO4(OH)6·H2O | (6) |
Second charging cycle:
| PAH ↔ PA + H+ + e− | (7) |
| PAZ ↔ PA + Zn2+ + 2e− | (8) |
| PAM ↔ PA + Mn2+ + 2e− | (9) |
| Zn4SO4(OH)6·H2O + Mn2+ → ZnxMnO(OH)2 + 4H+ + SO42− | (10) |
Second discharge cycle:
| ZnxMnO(OH)2 + 4H+ → Mn2+ + Zn2+ + H2O | (11) |
| PA + H+ + e− ↔ PAH | (12) |
| 4Zn2+ + 6OH− + SO42− + H2O ↔ Zn4SO4(OH)6·H2O | (13) |
| PA + Zn2+ + 2e− ↔ PAZ | (14) |
| PA + Mn2+ + 2e− ↔ PAM | (15) |
To further confirm the reaction mechanism, we directly studied by SEM images before and after the cycling process. In the first and second GCD cycles, the surface of the PAM electrode clearly shows the formation of ZSH after discharge, which is gradually converted to less crystalline ZMH after charging (Fig. S25†). This is consistent with the results of the above mechanism. The relationship between the mechanisms can be understood as follows: first, the PAM cathode induces the formation of ZSH by doping H+ to regulate the local pH. Through the process of dissolution deposition of Mn2+ in the ZnSO4 electrolyte, ZSH provides a prerequisite for the formation of ZMH, which dissolves into Zn2+ and Mn2+, which in turn provides the PAM cathode with a dopant source of Zn2+ and Mn2+, and the corresponding sulfate anions. The DFT results show that compared to PAH, the PAM samples have lower energy band gaps for the molecular structures obtained after the adsorption of either zinc or manganese ions, suggesting that their molecules have lower impedance, and thus charge storage is more likely to occur (Fig. 7c). This result also effectively demonstrates the structural advantages of PAM in the charge storage process.
Conclusion
In this study, we developed a novel PANI cathode with Mn doping (PAM) via a secondary doping method. The Zn-PAM battery exhibited a superior capacity of 496.7 mA h g−1 at an ultra-high operating voltage of 2.4 V while maintaining stable performance over 600 cycles. It can be attributed to better conductivity, an increased number of active charge storage sites, and a higher pseudocapacitance contribution. The ex situ characterization of the PAM cathode confirmed interactive dual storage mechanisms that mutually enhanced their respective charge storage processes: Mechanism I involved energy storage via the doping/dedoping of Zn2+, Mn2+, and H+; Mechanism II involved a dynamic dissolution/deposition of Mn2+ from the PAM cathode, accompanied by the transition from ZSH to low-crystallinity ZMH. Both Mechanism I and Mechanism II work synergistically, facilitating the enhanced capacity for the PAM cathode. The insights gained from this study could pave the way to re-evaluate and deepen the understanding of the relationship between organic cathodes and electrolytes.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Shengen Gong: conceptualization, software, validation, formal analysis, writing – original draft. Meihua Zhu: conceptualization, methodology, validation, formal analysis, investigation, writing – original draft. Yan Zhou: formal analysis, data curation. Runan Li: formal analysis, visualization. Jianhua Zhang: resources, writing – review & editing. Xiaoteng Jia: resources, writing – review & editing. Danming Chao: conceptualization, resources, writing – review & editing, supervision, funding acquisition. Caiyun Wang: resources, supervision. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22275066, 62371205, and 52103208), Jilin Provincial Science and Technology Department (20240601044RC and 20230402073GH), and Distinguished Young Scholars of Changchun Municipal Science and Technology Project (23YQ16). The authors also thank the Australian National Facility (ANFF) – Materials Node for equipment use.References
- Y. L. Liang and Y. Yao, Designing modern aqueous batteries, Nat. Rev. Mater., 2023, 8, 109–122 CrossRef.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Toward practical aqueous zinc-ion batteries for electrochemical energy storage, Joule, 2022, 6, 1733–1738 CrossRef.
- H. P. Li, C. Guo, T. S. Zhang, P. Xue, R. Z. Zhao, W. H. Zhou, W. Li, A. Elzatahry, D. Y. Zhao and D. L. Chao, Hierarchical Confinement Effect with Zincophilic and Spatial Traps Stabilized Zn-Based Aqueous Battery br, Nano Lett., 2022, 22, 4223–4231 CrossRef PubMed.
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, A high-capacity and long-life aqueous rechargeable zinc battery using a metal oxide intercalation cathode, Nat. Energy, 2016, 1, 16119 CrossRef.
- S. H. Zhu, Y. H. Dai, J. H. Li, C. M. Ye, W. H. Zhou, R. H. Yu, X. B. Liao, J. T. Li, W. Zhang, W. Zong, R. W. Chen, G. J. He, D. L. Chao and Q. Y. An, Cathodic Zn underpotential deposition: an evitable degradation mechanism in aqueous zinc-ion batteries, Sci. Bull., 2022, 67, 1882–1889 CrossRef.
- H. L. Pan, Y. Y. Shao, P. F. Yan, Y. W. Cheng, K. S. Han, Z. M. Nie, C. M. Wang, J. H. Yang, X. L. Li, P. Bhattacharya, K. T. Mueller and J. Liu, Reversible aqueous zinc/manganese oxide energy storage from conversion reactions, Nat. Energy, 2016, 1, 16039 CrossRef.
- Y. X. Tong, Y. Zang, S. D. Su, Y. G. Zhang, J. Z. Fang, Y. Q. Yang, X. M. Li, X. Wu, F. M. Chen, J. H. Hou and M. Luo, Methylene blue intercalated vanadium oxide with synergistic energy storage mechanism for highly efficient aqueous zinc ion batteries, J. Energy Chem., 2023, 77, 269–279 CrossRef.
- Y. Tong, X. Li, S. Su, J. Li, J. Fang, B. Liang, J. Hou and M. Luo, Hydrated lithium ions intercalated V2O5 with dual-ion synergistic insertion mechanism for high-performance aqueous zinc-ion batteries, J. Colloid Interface Sci., 2022, 606, 645–653 CrossRef PubMed.
- Q. Q. Sun, T. Sun, J. Y. Du, K. Li, H. M. Xie, G. Huang and X. B. Zhang, A Sulfur Heterocyclic Quinone Cathode Towards High-Rate and Long-Cycle Aqueous Zn-Organic Batteries, Adv. Mater., 2023, 35, e2301088 CrossRef.
- Z. H. Li, J. Tan, C. Y. Gao, Y. Wang, Y. G. Wang, M. X. Ye and J. F. Shen, Building better aqueous Zn-organic batteries, Energy Environ. Sci., 2023, 16, 2398–2431 RSC.
- C. O. Baker, X. W. Huang, W. Nelson and R. B. Kaner, Polyaniline nanofibers: broadening applications for conducting polymers, Chem. Soc. Rev., 2017, 46, 1510–1525 RSC.
- Q. Zhang, Y. L. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Modulating electrolyte structure for ultralow temperature aqueous zinc batteries, Nat. Commun., 2020, 11, 4463 CrossRef.
- H. Y. Shi, Y. J. Ye, K. Liu, Y. Song and X. Q. Sun, A Long-Cycle-Life Self-Doped Polyaniline Cathode for Rechargeable Aqueous Zinc Batteries, Angew. Chem., Int. Ed., 2018, 57, 16359–16363 CrossRef.
- Y. Liu, Y. Chen, X. Zhang, C. Lin, H. Zhang, X. Miao, J. Lin, S. Chen and Y. Zhang, A high-voltage aqueous rechargeable zinc-polyaniline hybrid battery achieved by decoupling alkali–acid electrolyte, Chem. Eng. J., 2022, 444, 136478 CrossRef.
- W. Wu, Z. Lin, H. Y. Shi, L. Lin, X. Yang, Y. Song, X. X. Liu and X. Sun, Realizing the leucoemeraldine-emeraldine-pernigraniline redox reactions in polyaniline cathode materials for aqueous zinc-polymer batteries, Chem. Eng. J., 2022, 427, 131988 CrossRef.
- H. Lv, Z. Wei, C. Han, X. Yang, Z. Tang, Y. Zhang, C. Zhi and H. Li, Cross-linked polyaniline for production of long lifespan aqueous iron‖organic batteries with electrochromic properties, Nat. Commun., 2023, 14, 3117 CrossRef PubMed.
- Z. Chen, H. L. Cui, Y. Hou, X. Q. Wang, X. Jin, A. Chen, Q. Yang, D. H. Wang, Z. D. Huang and C. Y. Zhi, Anion-chemistry-enabled positive valence conversion to achieve a record-high-voltage organic cathode for zinc batteries, Chem, 2022, 8, 2204–2216 Search PubMed.
- Z. Guo, J. Wang, P. Yu, M. Li, L. Huang, Z. Hu, Y. Wang and Z. Song, Toward Full Utilization and Stable Cycling of Polyaniline Cathode for Nonaqueous Rechargeable Batteries, Adv. Energy Mater., 2023, 13, 2301520 CrossRef CAS.
- M. Huang, X. P. Wang, X. Liu and L. Q. Mai, Fast Ionic Storage in Aqueous Rechargeable Batteries: From Fundamentals to Applications, Adv. Mater., 2022, 34, e2105611 CrossRef.
- F. Zhang, G. Wang, J. Wu, X. Chi and Y. Liu, An Organic Coordination Manganese Complex as Cathode for High-Voltage Aqueous Zinc-metal Battery, Angew. Chem., Int. Ed., 2023, 62, 202309430 CrossRef.
- J. X. Xue, Y. J. Wang, C. Sun, P. Xu, X. X. Fan, J. M. Fan, M. S. Zheng and Q. F. Dong, Suppressing voltage fading and improving cycling stability of Li-rich Mn-based materials by introducing MgSO4, J. Mater. Chem. A, 2020, 8, 22763–22772 RSC.
- H. Chen, C. Dai, F. Xiao, Q. Yang, S. Cai, M. Xu, H. J. Fan and S. J. Bao, Reunderstanding the Reaction Mechanism of Aqueous Zn–Mn Batteries with Sulfate Electrolytes: Role of the Zinc Sulfate Hydroxide, Adv. Mater., 2022, 34, 2109092 CrossRef CAS.
- J. H. Huang, Z. Wang, M. Y. Hou, X. L. Dong, Y. Liu, Y. G. Wang and Y. Y. Xia, Polyaniline-intercalated manganese dioxide nanolayers as a high-performance cathode material for an aqueous zinc-ion battery, Nat. Commun., 2018, 9, 2906 CrossRef.
- C. Zhong, B. Liu, J. Ding, X. Liu, Y. Zhong, Y. Li, C. Sun, X. Han, Y. Deng, N. Zhao and W. Hu, Decoupling electrolytes towards stable and high-energy rechargeable aqueous zinc–manganese dioxide batteries, Nat. Energy, 2020, 5, 440–449 CrossRef CAS.
- W. Gong, B. Fugetsu, W. Mao, A. K. Vipin, I. Sakata, L. Su, X. J. Zhang and M. Endo, Electrochemistry of rechargeable aqueous zinc/zinc-sulphate/manganese-oxide batteries and methods for preparation of high-performance cathodes, J. Mater. Chem. A, 2022, 10, 15415–15426 RSC.
- D. Chao, W. Zhou, C. Ye, Q. Zhang, Y. Chen, L. Gu, K. Davey and S. Z. Qiao, An Electrolytic Zn–MnO2 Battery for High-Voltage and Scalable Energy Storage, Angew. Chem., Int. Ed., 2019, 58, 7823–7828 CrossRef CAS PubMed.
- H. Chen, S. Cai, Y. Wu, W. Wang, M. Xu and S. J. Bao, Successive electrochemical conversion reaction to understand the performance of aqueous Zn/MnO2 batteries with Mn2+ additive, Mater. Today Energy, 2021, 20, 100646 CrossRef CAS.
- Y. C. Chen and Y. B. Xie, Electrochemical Performance of Manganese Coordinated Polyaniline, Adv. Electron. Mater., 2019, 5, 1900816 CrossRef CAS.
- T. Lu and F. W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. C. Ruan, X. L. Xu, X. L. Gao, J. X. Feng, L. H. Yu, Y. H. Cai, X. B. Gao, W. H. Shi, F. F. Wu, W. X. Liu, X. X. Zang, F. Y. Ma and X. H. Cao, Achieving long-cycle-life Zn-ion batteries through interfacial engineering of MnO2-polyaniline hybrid networks, Sustainable Mater. Technol., 2021, 28, e00254 CrossRef CAS.
- J. Rohowsky, K. Heise, S. Fischer and K. Hettrich, Synthesis and characterization of novel cellulose ether sulfates, Carbohydr. Polym., 2016, 142, 56–62 CrossRef CAS PubMed.
- E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling and S. N. Kerisit, XPS determination of Mn oxidation states in Mn(hydr)oxides, Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- G. J. Coyle, T. Tsang, I. Adler, N. Ben zvi and L. Yin, XPS Studies of ion-bombardment damage of transition metal sulfates, J. Electron Spectrosc. Relat. Phenom., 1981, 24, 221–236 CrossRef CAS.
- S. G. Gong, Y. F. Li, Y. Su, B. Li, G. D. Yang, X. L. Wu, J. P. Zhang, H. Z. Sun and Y. F. Li, Construction of Bimetallic Heterojunction Based on Porous Engineering for High Performance Flexible Asymmetric Supercapacitors, Small, 2023, 19, e2205936 CrossRef.
- A. E. Lakraychi and Y. Yao, Designing organic pseudocapacitors through molecular hybridization, Joule, 2023, 7, 858–860 CrossRef.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, Templated nanocrystal-based porous TiO(2) films for next-generation electrochemical capacitors, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS.
- Z. Zhu, Y. Men, W. Zhang, W. Yang, F. Wang, Y. Zhang, Y. Zhang, X. Zeng, J. Xiao, C. Tang, X. Li and Y. Zhang, Versatile carbon-based materials from biomass for advanced electrochemical energy storage systems, eScience, 2024, 4, 100249 CrossRef.
- C. Julien, M. Massot, R. Baddour Hadjean, S. Franger, S. Bach and J. P. Pereira Ramos, Raman spectra of birnessite manganese dioxides, Solid State Ionics, 2003, 159, 345–356 CrossRef CAS.
- W. Wu, Z. Lin, H.-Y. Shi, L. Lin, X. Yang, Y. Song, X.-X. Liu and X. Sun, Realizing the leucoemeraldine-emeraldine-pernigraniline redox reactions in polyaniline cathode materials for aqueous zinc-polymer batteries, Chem. Eng. J., 2022, 427, 131988 CrossRef CAS.
- D. Feng, T. N. Gao, L. Zhang, B. Guo, S. Song, Z. A. Qiao and S. Dai, Boosting High-Rate Zinc-Storage Performance by the Rational Design of Mn2O3 Nanoporous Architecture Cathode, Nano-Micro Lett., 2019, 12, 14 CrossRef.
- W. Chen, G. Li, A. Pei, Y. Li, L. Liao, H. Wang, J. Wan, Z. Liang, G. Chen, H. Zhang, J. Wang and Y. Cui, A manganese–hydrogen battery with potential for grid-scale energy storage, Nat. Energy, 2018, 3, 428–435 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical advantages, structural characterization and mechanism analysis of PAM. See DOI: https://doi.org/10.1039/d4sc05710d |
| This journal is © The Royal Society of Chemistry 2024 |