
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpontaneous ligand loss by soft landed [Ni(bpy)3]2+ ions on perfluorinated self-assembled monolayer surfaces†
Hugo Y.
Samayoa-Oviedo
 a,
Harald
Knorke
b,
Jonas
Warneke
*bc and
Julia
Laskin
*a
a,
Harald
Knorke
b,
Jonas
Warneke
*bc and
Julia
Laskin
*a
aDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: jlaskin@purdue.edu; Tel: +1-765-494-5434
bWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, 04103 Leipzig, Germany. E-mail: Jonas.warneke@uni-leipzig.de
cLeibniz Institut für Oberflächenmodifizierung (IOM), Permoserstraße 15, 04318 Leipzig, Germany
First published on 19th June 2024
Abstract
Transition metal (TM) complexes are widely used in catalysis, photochemical energy conversion, and sensing. Understanding factors that affect ligand loss from TM complexes at interfaces is important both for generating catalytically-active undercoordinated TM complexes and for controlling the degradation pathways of photosensitizers and photoredox catalysts. Herein, we demonstrate that well-defined TM complexes prepared on surfaces using ion soft landing undergo substantial structural rearrangements resulting in ligand loss and formation of both stable and reactive undercoordinated species. We employ nickel bipyridine (Ni-bpy) cations as a model system and explore their structural reorganization on surfaces using a combination of experimental and computational approaches. The controlled preparation of surface layers by mass-selected deposition of [Ni(bpy)3]2+ cations provides insights into the chemical reactivity of these species on surfaces. Both surface characterization using mass spectrometry and electronic structure calculations using density functional theory (DFT) indicate that [Ni(bpy)3]2+ undergoes a substantial geometry distortion on surfaces in comparison with its gas-phase structure. This distortion reduces the ligand binding energy and facilitates the formation of the undercoordinated [Ni(bpy)2]2+. Additionally, charge reduction by the soft landed [Ni(bpy)3]2+ facilitates ligand loss. We observe that ligand loss is inhibited by co-depositing [Ni(bpy)3]2+ with a stable anion such as closo-dodecaborate dianion, [B12F12]2−. The strong electrostatic interaction between [Ni(bpy)3]2+ and [B12F12]2− diminishes the distortion of the cation due to interactions with the surface. This interaction stabilizes the soft landed cation by reducing the extent of charge reduction and its structural reorganization. Overall, this study shows the intricate interplay of charge state, ion surface interactions, and stabilization by counterions on the structure and reactivity of metal complexes on surfaces. The combined experimental and computational approach used in this study offers detailed insights into factors that affect the integrity and stability of active species relevant to energy production and catalysis.
Introduction
The adsorption of molecules or ions is a powerful strategy for designing efficient functional interfaces.1–5 Recent advances in the fabrication of efficient electrocatalysts, photovoltaic, and photoelectrochemical devices rely on the control of the chemical stability of redox mediators at complex interfaces.6–9 Transition metal (TM) complexes are widely used as photosensitizers in photovoltaic devices10 and as active species for electrocatalytic water and CO2 reduction reactions11–13 and semihydrogenation of alkynes.14 All of these processes rely on interactions of TM complexes with interfaces. Loss of ligands from TM complexes adsorbed onto surfaces that generates undercoordinated species is one of the key degradation pathways of functional interfaces.12,15–17 At the same time, ligand loss from a TM complex is necessary to generate undercoordinated species of interest to electrocatalysis.14,18,19 The design of stable functional interfaces and efficient electrocatalysts require a detailed understanding of the role of the surface on the properties of the adsorbed analytes. Because of the complexity of solid/air and solid/liquid interfaces, understanding the impact of TM complex interactions with interfaces on their degradation and catalytic activity presents a significant challenge.14,20One approach to studying the properties of complex molecules and ions on surfaces employs their controlled deposition from the gas phase using a technique known as ion soft landing. Ion soft landing enables the preparation of well-defined ionic interfaces by the deposition of mass-selected ions onto surfaces.21–24 The emergence of electrospray ionization (ESI), a soft ionization technique that enables transferring of intact ions from solution into a mass spectrometer, has facilitated the deposition of a broad range of ions for the analysis of their structures and reactivities on substrates.25–32 For example, scanning tunneling microscopy of cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)-ruthenium(II) on a Au(III) surface revealed that its gas-phase geometry is distorted on the surface to maximize ligand–surface interactions.33 In another study, soft landed vanadium-benzene sandwich clusters were shown to orient on a CH3-terminated self-assembled monolayer on gold (HSAM) to maximize interactions between the aromatic rings of the cluster and methylene groups of the HSAM.34
Charge reduction by soft landed ions is another important phenomenon that affects their properties on surfaces.35,36 The accumulation of cations on surfaces facilitates charge reduction or neutralization through either proton detachment or electron recombination.37,38 The deposition of mass-selected ions on surfaces enables the study of how the charge state of ions influences their structures32,39 and reactivity on surfaces.40,41 Herein, we demonstrate that both structural reorganization and charge reduction contribute to the spontaneous loss of ligands from [Ni(bpy)3]2+, a model system for Ni(II) complexes commonly used in photoredox catalysis and electrocatalytic applications.42 It has been demonstrated that the electrochemical reduction of [Ni(bpy)3]2+ in solution may result in ligand loss from the complex,8,43 but this process at interfaces has not been previously explored.
In this study, we provide first fundamental insights into the role of ion–surface interactions on the structure and stability of [Ni(bpy)3]2+ on self-assembled monolayer (SAM) surfaces. We have previously demonstrated that an analogous [Ru(bpy)3]2+ complex does not undergo substantial charge reduction44 and remains intact after soft landing on surfaces.45 In a striking contrast with [Ru(bpy)3]2+, this study shows that soft landed [Ni(bpy)3]2+ dissociates generating [Ni(bpy)] and [Ni(bpy)2] species (presumably in different charge states). Our experimental observations indicate that both geometry reorganization after soft landing and charge reduction contribute to the observed dissociation of [Ni(bpy)3]2+. These results are supported by density functional theory (DFT) calculations of model systems that enable qualitative understanding of the effect of the surface on the structure of the deposited species. Based on our experimental observations and DFT calculations, we propose that soft landed [Ni(bpy)3]2+ cations undergo conformational changes to maximize their interaction to surfaces. These changes reduce ligand binding energy and facilitate ligand loss from the fully coordinated species. Remarkably, co-deposition of [Ni(bpy)3]2+ with stable anions substantially reduces the extent of dissociation of this complex on surfaces indicating that this approach may be used to preserve the fully coordinated Ni-bpy ions on surfaces. The results of this study provide a better understanding of the factors that control the structure and reactivity of TM complexes at interfaces.
Experimental section
Chemicals
Nickel(II) sulfate hexahydrate (NiSO4·6H2O) was purchased from Fisher Scientific (Waltham, MA). 2,2′-Bipyridine (bpy) and 1H,1H,2H,2H-perfluorodecanethiol (CF3(CF2)7(CH2)2SH, FSAM) were purchased from MilliporeSigma (St. Louis, MO). 2,2′-Dipyridyl-d8 (bpyD) was purchased from CDN Isotopes (Pointe-Claire, Quebec, Canada). Cesium dodecafluoro-closo-dodecaborate Cs2B12F12 salts were purchased from MilliporeSigma (St. Louis, MO). Solutions of these analytes were prepared by dissolving them in methanol (HPLC grade, Fisher Scientific).ESI-MS measurements
Direct infusion electrospray ionization mass spectrometry (ESI-MS) measurements were typically performed on an Agilent 6560 ion mobility quadrupole time-of-flight (QTOF) mass spectrometer (Santa Clara, CA, USA). A syringe pump (Legato 101, KDScientific, Holliston, MA) with a 1.0 mL Hamilton (Reno, NV) syringe is used to propel the solvent with the analytes at a flow rate of 0.3 μL min−1 through a fused silica capillary (Polymicro, 150 μm O.D., 50 μm I.D.) to the mass spectrometer inlet where they are ionized by ESI. The ESI voltage was −4.0 kV, fragmentor voltage was 400 V, and octupole RF peak-to-peak voltage was 750 V. Although these settings on the QTOF instrument are tuned to prevent collisional activation of ions in the front end of the instrument, we still observed some in-source fragmentation of [Ni(bpy)3]2+. To evaluate the extent of fragmentation occurring in the QTOF we also acquired mass spectra using an Agilent 6230B time-of-flight (OTOF) mass spectrometer (Santa Clara, CA, USA) with a milder ESI source. Analytes were introduced into the mass spectrometer inlet in a similar fashion to the one described for the QTOF. The ESI voltage was −3.5 kV, fragmentor voltage was 100 V, skimmer voltage was 80 V, and octupole RF peak-to-peak was 550 V. Data acquisition and analysis was performed using Agilent Qualitative Navigator software and ChemCalc online tool.46Surface preparation
FSAM on a gold surface was prepared following literature procedure.47 Briefly, a 10 × 10 mm gold coated silicon wafer (500 Å Au layer and 50 Å chromium adhesion layer, Angstrom Engineering, ON, Canada) was cleaned using a UV ozone cleaner for 20 min and immersed in a glass scintillation vial containing a 50% v/v thiol solution in ethanol. The monolayer was assembled for at least 12 h and then ultrasonically washed in methanol for 10 min. The surface was rinsed with ethanol, dried under nitrogen, and mounted onto a sample holder for soft landing experiments.Ion soft landing experiments
Soft landing experiments were performed using a specially-designed instrument described elsewhere.45 Briefly, ions are generated in an ESI interface by spraying a 150 μM solution of an analyte. For ESI, a high voltage is applied to the syringe needle containing the solution, which is delivered to the instrument inlet by a syringe pump at a flow rate of 1 μL min−1. Charged microdroplets generated in this process are transferred into vacuum through two stainless steel heated inlets (1.0 mm I.D.) maintained at 160 °C where desolvation takes place. Two independent ESI interfaces for producing ions of the same or opposite polarity are used in this system. Both ion beams are introduced orthogonally into a high-pressure ion funnel (HPF), followed by a low-pressure funnel (LPF), and transferred into a collision cell (CC), which can be operated either as an ion guide or as an ion trap. After the CC, the ion beam is transferred through a 90° bent ion guide (BF), which helps eliminate neutral molecules coming from the ionization source along the instrument axis. Ions leaving the ion guide are transferred through a conductance limit into the soft landing chamber. In this chamber, maintained at 8 × 10−6 torr, ions are mass-selected using a quadrupole mass filter (9.5 mm rod diameter, 4.137 mm inscribed radius, Extrel CMS, Pittsburgh, PA) and focused by an einzel lens onto a surface. All the voltages applied to different elements of the instrument are provided by a Modular Intelligent Power Sources (MIPS) system from GAA Custom Electronics LLC (Kennewick, WA).The surface is located approximately 1 cm away from the einzel lens. A metal mask with a 3 mm diameter aperture is placed in front of the surface to confine the deposition area. The ion current on the surface is measured using a picoammeter (RBD Instruments, Bend, OR). All samples were prepared by depositing about 2.1 × 1013 ions (35 pmol) onto a 3 mm diameter area, which corresponds to a full monolayer coverage. The kinetic energy of the ions is determined by the potential applied to the CC bias.45 To minimize ion fragmentation during deposition, we used a low landing kinetic energy of 5 eV/z, which was achieved by applying a retarding potential to the surface.
ESI of a Ni(bpy)3SO4 solution generates both [Ni(bpy)3]2+ and its fragment ion produced by neutral ligand loss, [Ni(bpy)2]2+. We used low RF peak-to-peak voltage in the HPF to minimize the extent of fragmentation of [Ni(bpy)3]2+. Table S1 in the ESI† provides a summary of typical pressures and voltage settings for ion transmission of the [Ni(bpy)3]2+. Typical ion currents of mass-selected [Ni(bpy)3]2+ was 1.0 nA. Co-deposition of Ni-bpy cations with [B12F12]2− anions (typical ion current: −0.9 nA) was performed using a voltage switching approach described in a previous study by our group.48 In these experiments, a custom-designed automated script that controls the MIPS system is used to switch between the optimized voltage settings of the corresponding analyte ions. The duration of each sub-deposition event and total deposition time are selected to reach a desired ion coverage for each ion.
Surface analysis using nano-DESI
Nanospray desorption electrospray ionization (nano-DESI)49 experiments were performed using a custom-designed source interfaced with the QTOF mass spectrometer. Surface analysis was performed using acetonitrile as the extraction solvent. The nano-DESI probe is assembled by forming a liquid bridge between the primary and nanospray fused silica capillaries (Polymicro, 150 μm O.D., 50 μm I.D.). A syringe pump (Fusion 101A, Chemyx, Stafford, TX) with a 1.0 mL Hamilton (Reno, NV) syringe is used to propel the solvent at a flow rate of 0.3 μL min−1 through the nano-DESI probe. Analytes extracted into the liquid bridge are delivered to the mass spectrometer inlet through the nanospray capillary and ionized by ESI. The voltage settings are the same as the ones used for the ESI-MS measurements. The surface is mounted onto a sample holder and positioned using a motorized XYZ stage (Zaber Technologies Inc., Vancouver, BC, Canada). The XYZ stage is controlled by a custom-designed Labview program.50 We performed line scans through the deposition area by scanning the sample under the nano-DESI probe at 20 μm s−1 while acquiring mass spectra. To obtain the mass spectrum of the surfaces, the chromatograms of the line scans were integrated over the deposition spot and subtracted from the blank. To probe the reactivity of soft landed ions, we acquired nano-DESI line scans using ACN containing 3 μM bpyD. Data acquisition and analysis was performed using Agilent Qualitative Navigator software and ChemCalc online tool.46 Some measurements were also performed on a Q-Exactive HF-X Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose, CA), which affords milder source settings that prevent ion fragmentation in the ion transfer region of the instrument. The electrospray voltage was 4.0 kV and the RF drive of the ion transfer funnel was set to 10% to diminish in-source fragmentation. Data acquisition and analysis was performed using Xcalibur software (Thermo Fisher Scientific) and Chemcalc online tool.46Theoretical calculations
Quantum chemical calculations performed using Gaussian 16 C.02.51 Geometry optimization of the investigated systems were performed using DFT calculations with the B3LYP52 functional and Ahlrich-type basis set, def2SVPP.53,54 In order to correct for the known underestimation of dispersion interactions by B3LYP, we applied the Grimme correction (GD3BJ).55 Vibrational frequency calculations were performed to confirm that the structures correspond to minima on the potential energy surface. To qualitatively describe the structural changes that different Ni-bpy complexes undergo on a surface, we used hydrogen terminated diamond-like surface.Results and discussion
In solutions where bpy ligand is present in more than three-fold excess with respect to Ni2+, the formation of the fully coordinated [Ni(bpy)3]2+ species is preferred over the formation of its undercoordinated analogs due to the high stability of the complex (log(K1) = 7.1, log(K2) = 6.7, and log(K3) = 6.1).56 The undercoordinated species may be formed either by decreasing the amount of the ligand in solution below the Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bpy 1:3 stoichiometric ratio or by electroreduction of [Ni(bpy)3]2+.8,57 Similarly, gas phase studies indicate that the fully coordinated [Ni(bpy)3]2+ is stable and ∼200 kJ mol−1 are required to remove one bpy ligand from this cation.58 Based on the energy requirement for generating undercoordinated Ni-bpy complexes, it is not surprising that these species are elusive in the condensed phase. In this work, we examined the stability of [Ni(bpy)3]2+ on surfaces using soft landing of mass-selected ions. Using nano-DESI MS analysis, we observed extensive dissociation of these ions on surfaces resulting in formation of undercoordinated species containing one or two ligands.
:bpy 1:3 stoichiometric ratio or by electroreduction of [Ni(bpy)3]2+.8,57 Similarly, gas phase studies indicate that the fully coordinated [Ni(bpy)3]2+ is stable and ∼200 kJ mol−1 are required to remove one bpy ligand from this cation.58 Based on the energy requirement for generating undercoordinated Ni-bpy complexes, it is not surprising that these species are elusive in the condensed phase. In this work, we examined the stability of [Ni(bpy)3]2+ on surfaces using soft landing of mass-selected ions. Using nano-DESI MS analysis, we observed extensive dissociation of these ions on surfaces resulting in formation of undercoordinated species containing one or two ligands.
ESI-MS of Ni-bpy solutions
To establish a baseline for surface characterization using nano-DESI MS, we first examined the distribution of Ni-bpy species in solutions prepared by mixing NiSO4 and bpy in different ratios. For these measurements, a small droplet (∼20 μL) of the mixture was placed on top of an FSAM surface. The surface with the droplet was placed in front of a mass spectrometer; a small fused silica capillary was used to sample from the liquid droplet and transfer the solution into a mass spectrometer. In this approach, the ionization step is the same as in nano-DESI analysis, which enables a direct comparison with the analysis conducted for the soft landed samples.Fig. 1 shows ESI-MS spectra of Ni-bpy solutions obtained using two different mass spectrometers: QTOF and OTOF characterized by harsher and softer source conditions, respectively. Table 1 provides a summary of the assignments and Table S1† contains a full list of the measured and calculated m/z of all the species observed in this and subsequent experiments. Both mass spectra are quite complex and contain an abundant peak of [Ni(bpy)3]2+ (1). It is remarkable that although the solution contains a sufficient amount of bpy ligand to form [Ni(bpy)3]2+ (1), this ion is not the most abundant species in both mass spectra. Instead, the mass spectrum obtained using QTOF (Fig. 1a) is dominated by the peak corresponding to [Ni(bpy)2]2+ (2). Meanwhile, [Ni(bpy)2(CH3CN)2]2+ (17) is the most abundant peak in the mass spectrum obtained using milder source conditions of OTOF (Fig. 1b). This peak corresponds to the undercoordinated complex with two bpy ligands and two solvent (CH3CN) molecules coordinated to the metal center. It is reasonable to assume that solvent molecules are weakly bound to the complex and are readily removed by in-source fragmentation in the QTOF. We propose that peak (2) in the spectrum obtained using QTOF (Fig. 1a) is generated through in-source fragmentation of peak (17) formed under milder source conditions of OTOF (Fig. 1b). Other peaks observed in Fig. 1 (4–6 and 18–24) correspond to adducts of (2) with solvent molecules and carboxylates present in the solvent. The presence of (17) and multiple adducts of [Ni(bpy)2]2+, which are particularly pronounced in Fig. 1b may be attributed to complex equilibria either in the bulk solution or during the ionization process,59,60 in which some coordinating species present in the solvent displace bpy from the metal center. Despite the complexity of this system, these results indicate that surface analysis using nano-DESI MS should be able to detect intact (1) when it is present on a surface. Although the abundance ratio of (1) and (2) must be treated with caution, changes in the relative abundance of (1) and (2) observed using the same instrument are meaningful. Unless otherwise noted, mass spectra presented in this work were acquired using QTOF due to its high mass resolution and ability to perform tandem mass spectrometry (MS2) experiments, which were used for the identification of some of the species present on surfaces and in solutions.
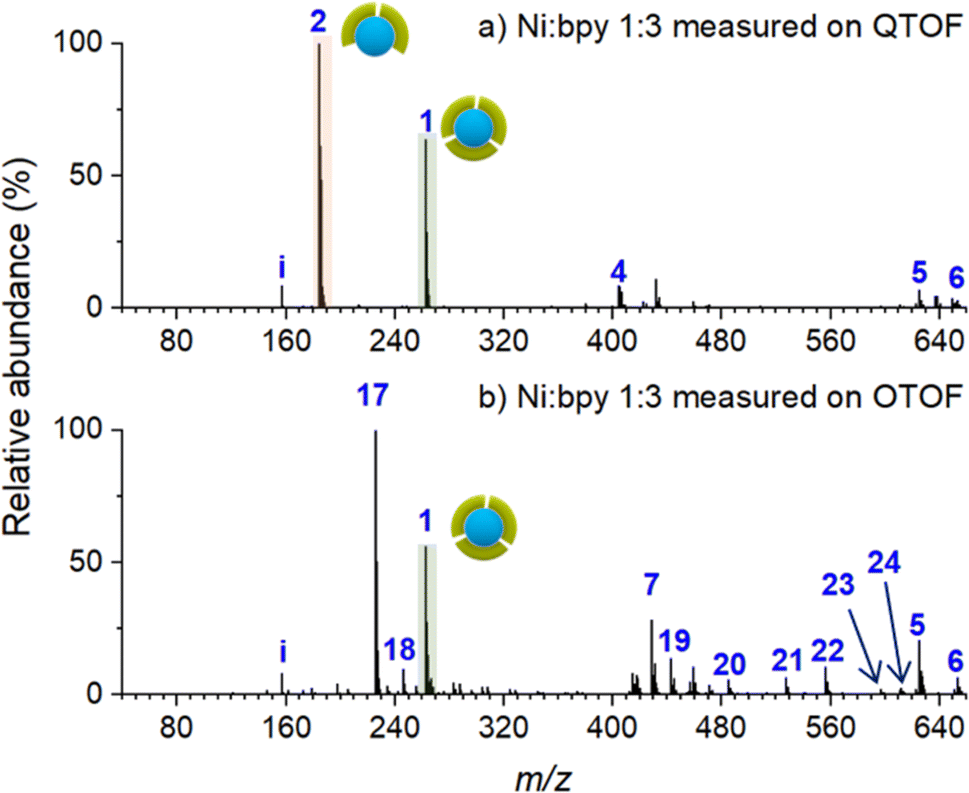 | ||
| Fig. 1 ESI-MS of solutions prepared by mixing NiSO4 and bpy in a 1:3 molar ratio measured on the (a) QTOF (harsher conditions) and (b) OTOF (softer conditions) instruments. The pictograms symbolize the number of ligands coordinated to Ni. Green symbols represent the bpy ligand. | ||
| Label | Assignment |
|---|---|
| 1 | [Ni(bpy)3]2+ |
| 1a | [Ni(bpy)2(bpyD)]2+ |
| 1b | [Ni(bpy)(bpyD)2]2+ |
| 1c | [Ni(bpyD)3]2+ |
| 2 | [Ni(bpy)2]2+ |
| 2a | [Ni(bpy)(bpyD)]2+ |
| 2b | [Ni(bpyD)2]2+ |
| 3 | [Ni(bpy)]2+ |
| 4 | [Ni(bpy)2Cl]+ |
| 5 | [Ni(bpy)2(C16H31O2)]+ |
| 6 | [Ni(bpy)2(C18H35O2)]+ |
| 7 | [Ni]+ |
| 8 | [Ni(bpy)(CH3CN)]2+ |
| 9 | [Ni(bpy)]+ |
| 10 | [Ni(bpy)(OH)]+ |
| 11 | [Ni(bpy)(Cl)]+ |
| 12 | [Ni(bpy)(C3H5O3)]+ |
| i | [bpy]+ |
| ii | C9H19N2+ |
Soft landing of [Ni(bpy)3]2+
An ex situ nano-DESI mass spectrum of [Ni(bpy)3]2+ soft landed at 5 eV/z is shown in Fig. 2a. The mass spectrum shows a distribution of ions related to soft-landed species all of which are listed in Table 1. In addition to peaks corresponding to soft landed species, the mass spectrum contains peaks corresponding to background molecules on the surface or in the extraction solvent which are not labeled for simplicity. The same mass spectrum shown over a wider m/z range is provided in Fig. S1a.† We observe that [Ni(bpy)3]2+ (1) is significantly lower in abundance (lower than 30%) than [Ni(bpy)2]2+ (2) and [Ni(bpy)]2+ (3) ions that are dominant species in the mass spectrum shown in Fig. 2a. This is a striking finding given that [Ni(bpy)3]2+ is one of the major species in the ESI-MS spectra of Ni-bpy solutions shown in Fig. 1. Furthermore, [Ni(bpy)]2+ (3) is a new species that was not observed in the mass spectra of solutions shown in Fig. 1.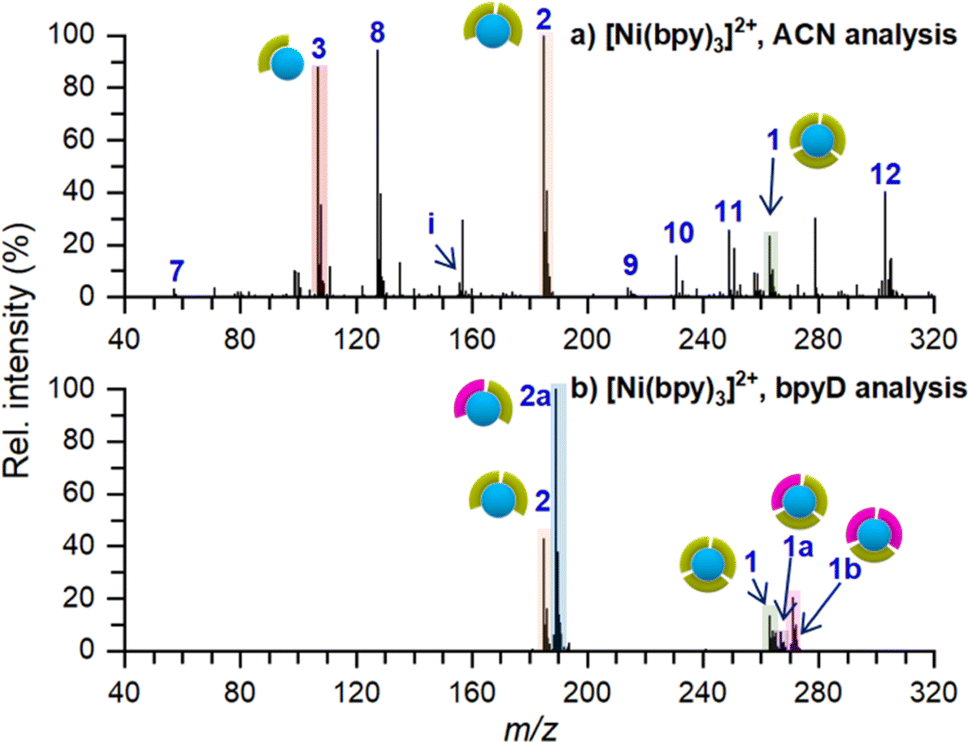 | ||
| Fig. 2 Ex situ nano-DESI MS of the deposition spots of [Ni(bpy)3]2+ analyzed with (a) ACN and (b) 3 μM bpyD in ACN as the nano-DESI solvent. Ion assignments are listed in Table 1. The pictograms symbolize the number of ligands coordinated to Ni. Green and pink symbols represent bpy and bpyD, respectively. Peaks originating from the solvent are not marked in the spectra for simplicity. | ||
The low abundance of [Ni(bpy)3]2+ in the mass spectrum of the deposition spot (Fig. 2a) indicates that the stability of this ion is substantially reduced on the surface and that it undergoes losses of one or two ligands to form [Ni(bpy)2]2+ and [Ni(bpy)]2+, respectively. The mass spectrum in Fig. 2a also contains several peaks corresponding to products of reactions between [Ni(bpy)]2+ and background molecules present in the nano-DESI solvent. For example, ion (8) is an adduct of [Ni(bpy)]2+ with CH3CN and ions (10–12) are reaction products with anions present in the solvent such as OH−, Cl−, and lactate, respectively. The high reactivity of [Ni(bpy)]2+ is consistent with the expected tendency of an undercoordinated metal center to recover its coordination shell. The mass spectrum also contains some free Ni+ (7), [Ni(bpy)]+ (9), and bpy+ (i) ions. It is important to note that most of the features observed in the mass spectrum of soft landed [Ni(bpy)3]2+ (Fig. 2a) are absent in the ESI-MS spectra of Ni-bpy solutions shown in Fig. 1. Specifically, neither [Ni(bpy)]2+ (3) nor [Ni(bpy)]2+-containing species (8, 10–12) are observed in Fig. 1 while in Fig. 2a they constitute a majority of the Ni-containing species. These observations show that the observed dissociation of [Ni(bpy)3]2+ into [Ni(bpy)]2+ cannot be attributed to the ESI process. Collectively, the results presented in Fig. 2a indicate that soft landing of [Ni(bpy)3]2+ generates a distribution of products on the surface, which is attributed to the loss of ligands by the soft landed cation.
It is important to distinguish which of the chemical species in Fig. 2a are present on the surface and which ones are formed during nano-DESI analysis. For example, some [Ni(bpy)3]2+ observed in Fig. 2a could be formed by the reaction of [Ni(bpy)2]2+ and bpy after the extraction into the nano-DESI solvent. To examine which of the species observed in Fig. 2a are present on the surface, we performed a reactive nano-DESI analysis using 3 μM bpyD in ACN as the extraction solvent. The rationale for choosing bpyD is that this molecule is a deuterated analog of the bpy ligand and has a high binding affinity to the metal center. If [Ni(bpy)3]2+ were not present on the surface and instead were formed in the nano-DESI solvent, the high concentration of bpyD in the solvent would eliminate [Ni(bpy)3]2+ and promote the formation of the fully coordinated complex containing both bpy and bpyD ligands such as [Ni(bpy)2(bpyD)]2+. In addition, we anticipate that bpyD added to the solvent will effectively compete with background molecules for binding to any undercoordinated Ni-bpy species, thus reducing the abundance of adducts (8) and (10–12) in the mass spectrum. To guide our observations, we first acquired ESI mass spectra of a solution prepared by combining 15 μM Ni(bpy)3SO4 with 15 μM bpyD immediately after mixing and 3 hours after preparing the solution. Both mass spectra are presented in Fig. S2† and show that ligand exchange is a slow process and occurs to a significant extent only 3 hours after preparing the solution. It follows, that if species with bpyD are observed in the reactive nano-DESI analysis of the surface, they are either formed by ligand pickup or ligand exchange of the undercoordinated metal centers, but not through ligand exchange by the fully coordinated [Ni(bpy)3]2+.
The nano-DESI mass spectrum of soft landed [Ni(bpy)3]2+ obtained using bpyD in the solvent is presented in Fig. 2b. The same mass spectrum shown over a wider m/z range is provided in Fig. S1b.† To ensure that the result of this measurement is comparable with the result shown in Fig. 2a, we acquired both mass spectra by alternating the acquisition between regular and reactive nano-DESI in adjacent line scans across the same deposition spot. The addition of bpyD to the extraction solvent significantly simplifies the observed product distribution by eliminating ions (3), (7), (9), and the products of reaction with background molecules (8, 10–12). This is consistent with the expectation that bpyD effectively competes with molecules and ions in the solvent for binding to the undercoordinated Ni-containing species. We observe that the reactive nano-DESI analysis generates several [Ni(bpy)x(bpyD)y]2+ species. Specifically, we observe an abundant [Ni(bpy)(bpyD)]2+ (2a) in the region of the spectrum containing doubly ligated complexes. Meanwhile, both [Ni(bpy)2(bpyD)]2+ (1a) and [Ni(bpy)(bpyD)2]2+ (1b) are observed in the m/z region containing the fully coordinated Ni-bpy species. The high abundance of bpyD-containing species indicates that ligand exchange and ligand pickup for the undercoordinated species are fast compared to the timescale of the analysis. When comparing Fig. 2a and b, [Ni(bpy)3]2+ is still observed even when bpyD is present in the solvent, which confirms that this ion is already present on the surface and not formed during the nano-DESI analysis. Fig. 2b also shows ion (1a) which is likely formed through ligand pickup from (2). In addition (2a), is now the most abundant species in the mass spectrum in Fig. 2b. We propose that (2a) is formed by the reaction of (3) with bpyD or ligand exchange in (2).
Collectively, the results shown in Fig. 2 indicate that [Ni(bpy)3]2+ precursors soft landed onto an FSAM surface undergo one or two ligand losses to form [Ni(bpy)2]2+ and [Ni(bpy)]n+ (n = 1, 2). This striking observation is in contrast with the high stability of [Ni(bpy)3]2+ in both the solution (log(β3) = 19.9)56 and gas phase (ligand dissociation energy of ∼200 kJ mol−1).58 Previous studies by our groups and others have explored the physicochemical processes on surfaces that affect the structure and/or reactivity of soft landed ions.33,61 Based on these studies, we consider three main processes that could rationalize the observed dissociation of soft landed [Ni(bpy)3]2+: (1) vibrational excitation of the precursor ion during its collision with the surface (crash landing), (2) charge reduction resulting in change of the coordination geometry of the ion and, (3) structural rearrangement induced by ion–surface interactions. In the following sections, we will discuss the contribution of these three processes to the product distribution generated by soft landing of [Ni(bpy)3]2+.
Effect of the landing kinetic energy on the stability of [Ni(bpy)3]2+ on surfaces
Ion soft landing defined as intact deposition of ions is one of several pathways that ions can undergo after colliding with a surface.21 Although ion soft landing is the dominant process at low kinetic energies, ions can also undergo scattering, reactive landing, and surface-induced dissociation at higher kinetic energies.21,62 The observed dissociation of the soft landed [Ni(bpy)3]2+ (Fig. 2a) may be attributed to the vibrational excitation of the ion upon collision with the surface (crash landing).25,63,64 To investigate the effect of the kinetic energy, we compared product distributions obtained after soft landing of [Ni(bpy)3]2+ at 2 eV/z and 15 eV/z. Nano-DESI mass spectra of surfaces prepared by depositing the precursor ion at 2 eV/z and 15 eV/z are shown in Fig. 3a and b, respectively. Similar to the results obtained at 5 eV/z (Fig. 2a), the deposition of [Ni(bpy)3]2+ at 2 eV/z (Fig. 3a) generates abundant [Ni(bpy)2]2+ (2) and [Ni(bpy)]2+ (1) species whereas [Ni(bpy)3]2+ (3) is present in less than 20% abundance on the surface. The only notable difference between the two mass spectra shown in Fig. 3 is that no intact (3) ions are present on the surface prepared using 15 eV/z deposition (Fig. 3b). This difference may be attributed to crash landing of [Ni(bpy)3]2+ at elevated kinetic energies. The similarity of the composition of the surfaces prepared using soft landing at 2 eV/z (Fig. 3a) and 5 eV/z (Fig. 2a) indicates that the observed fragment distribution generated by soft landing of [Ni(bpy)3]2+ at these kinetic energies cannot be attributed to collisional activation. In addition, although crash landing of [Ni(bpy)3]2+ could generate [Ni(bpy)2]2+, the formation of [Ni(bpy)]2+ at the time of collision is highly unlikely owing to the high barrier associated with the removal of two ligands from [Ni(bpy)3]2+.58 It is remarkable that the singly coordinated species is abundant even when low kinetic energies are used for the deposition (Fig. 2a and 3a); an observation that will be addressed later in the text. In summary, both the similarity between product distributions on surfaces prepared using deposition at 2 eV/z and 5 eV/z and the increase in the extent of fragmentation at 15 eV/z indicate that crash landing is not a dominant process contributing to the observed dissociation of [Ni(bpy)3]2+ deposited onto surfaces at lower kinetics energies.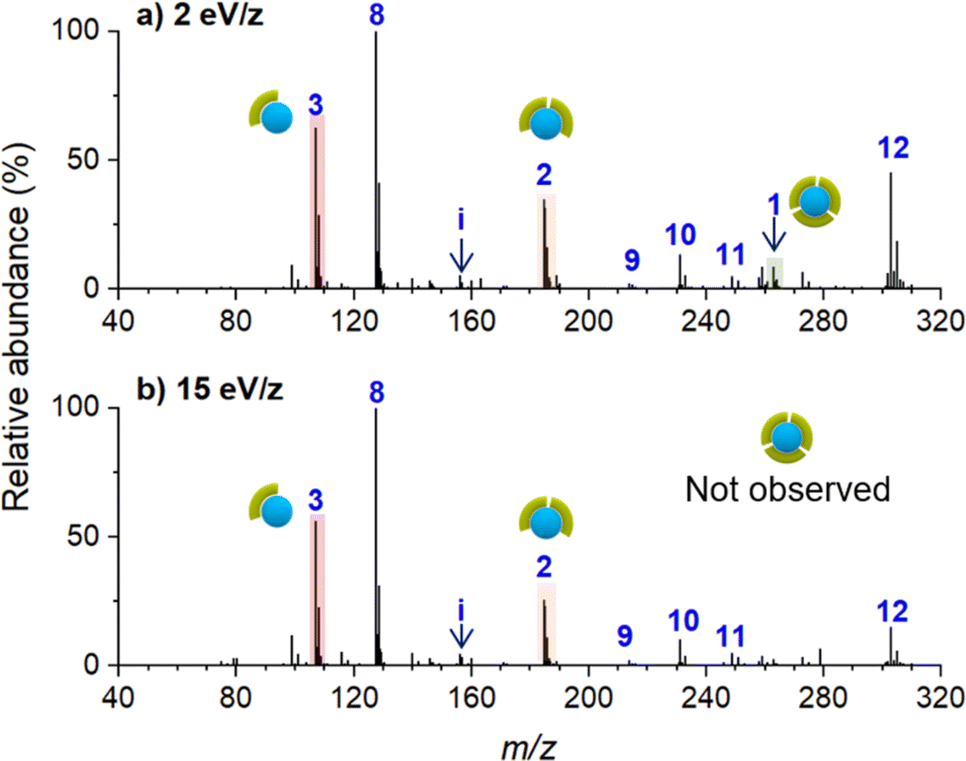 | ||
| Fig. 3 Ex situ mass spectra of [Ni(bpy)3]2+ deposited at (a) 2 eV/z and (b) 15 eV/z. | ||
Charge reduction of [Ni(bpy)3]2+ on the surface
The observed dissociation of [Ni(bpy)3]2+ on surfaces may be attributed to changes in the coordination geometry of the metal center induced by a change in its oxidation state. Solution phase studies have shown that the electrochemical reduction of [Ni(bpy)3]2+ produces the unstable [Ni(bpy)3]0 species which undergoes ligand loss to form [Ni(bpy)2]0 and bpy.8,57 We hypothesize that soft landed [Ni(bpy)3]2+ ions get partially or fully reduced on the surface which results in ligand loss. In ion soft landing experiments, ion accumulation on top of a SAM surface results in the formation of charge-imbalanced layers.22 Charge accumulation generates a potential build-up across the SAM interface, which may cause charge reduction of soft landed ions once the potential exceeds the dielectric barrier provided by the SAM (approximately 2.4 × 1011 doubly charged ions deposited onto a 3 mm diameter area representing 1% of a full monolayer coverage).65 Charge reduction of soft landed cations of peptides,32,66 gold clusters,67–69 and organometallics40,44,70 has been previously explored. These studies have demonstrated that loss of charge by soft landed cations involves either electron capture or proton loss on surfaces. Based on the solution phase electrochemical studies referenced earlier, we hypothesize that charge reduction of [Ni(bpy)3]2+ may result in ligand losses from the complex.8,57 We have previously demonstrated that charge reduction is most efficient in the center of the deposition area generated by a Gaussian ion beam.67–69 This phenomenon was attributed to the more efficient accumulation of charge in the center of the spot, which facilitates electron tunneling and charge reduction of the deposited cations. It is reasonable to assume that products of surface reactions initiated by charge reduction are localized closer to the center of the deposition spot than products generated by other processes. To test this assertion, we examined line profiles showing the variations in the abundance of different species obtained across the deposition spot using nano-DESI MS. We note that a direct observation of charge-reduced species on surfaces using ex situ nano-DESI MS is challenging because the species present on the surface can be reionized during the analysis. We infer processes associated with charge reduction from the observed differences in the localization of different species across the deposition spot revealed by line profiles.Fig. 4 shows line profiles of [Ni(bpy)3]2+ (1), [Ni(bpy)2]2+ (2), [Ni(bpy)]2+ (3), and [Ni(bpy)]+ (9) across the deposition area. We observe that the signal of [Ni(bpy)]+ (9) is abundant in the center of the deposition spot (between 2.8 and 5.2 mm) and depleted at the edges. Because [Ni(bpy)]2+ (3) is colocalized with (9), we conclude that (3) is likely generated through the reionization of [Ni(bpy)]+ or [Ni(bpy)]0 during the analysis. In contrast, we observe that [Ni(bpy)3]2+ (1) is depleted with respect to the expected Gaussian distribution in the center of the deposition spot and is more abundant at the edges. This distribution is consistent with previously published localization of soft landed ligated cationic gold clusters that undergo charge reduction in the regions of higher ion accumulation on a SAM surface.69 As indicated earlier, a larger amount of ions is accumulated in the center of the deposition spot as compared to the edges. Cations deposited onto areas that accumulate ions faster are more likely to undergo charge reduction67,69 caused by electron tunneling from the gold substrate through SAM. The enhancement of the signals of (3) and (9) and the depletion of the signal of (1) in the center of the deposition area indicate that the formation of singly coordinated Ni-bpy species is likely attributed to charge reduction of [Ni(bpy)3]2+. It is reasonable to assume that when enough [Ni(bpy)3]2+ ions get accumulated, they undergo charge reduction to form [Ni(bpy)3]0, which then undergoes ligand losses to produce mainly [Ni(bpy)]0. This species is oxidized either upon exposure to air or during the nano-DESI analysis and observed as ions (9) and (3).
 | ||
| Fig. 4 Line scan profiles of (a) [Ni(bpy)]+, (b) [Ni(bpy)]2+, (c) [Ni(bpy)3]2+, and (d) [Ni(bpy)2]2+ observed in the mass spectrum in Fig. 2a. The diameter of the deposition area containing Ni-bpy species is approximately 6.0 mm and is centered at 0.0 mm. The area where charge reduction was observed is between the blue dashed lines. Despite using a 3.0 mm diameter mask to confine the ion beam, the retarding potential applied to the surface likely defocuses the ion beam. We note that the signal tails outside the region where analytes are expected to be observed. Ni-bpy species are “sticky” and likely get adsorbed on the walls of the secondary nano-DESI capillary. They also have a high ionization efficiency such that the small amount of the extracted analytes temporarily retained in the secondary capillary of the nano-DESI setup generates a measurable signal that causes some “carry-over” from the center of the deposition spot during the line scan. The signals disappear ∼30 s after the nano-DESI probe is lifted off the sample. | ||
The line profile of [Ni(bpy)2]2+ (2) resembles the line profile of (1) at the edges but does not show as much flattening of the signal at the center of the deposition spot as (1). Fig. S3† shows the abundance ratio of (1)/(2) across the line scan which confirms that there is a difference in the line profiles of (1) and (2) beyond the experimental error. Fig. S4† shows the mass spectra of the regions at the center and outskirts of the deposition spot demonstrating that these two areas have distinct chemical composition. Since there is a slightly higher abundance of (2) at the center of the deposition spot, we infer that charge reduction plays a role in the formation of this product. The presence of (2) at the edges of the deposition spot (compared to (9) and (3) that are depleted) suggests that another process is responsible for the efficient formation of (2) across the entire deposition area. We propose that this process involves structural rearrangement of soft landed [Ni(bpy)3]2+on the surface resulting in formation of a stable [Ni(bpy)2]2+ product through the loss of one ligand as discussed later in the text.
Co-deposition experiments with [B12F12]2−
As described earlier, an excess surface charge is generated as ions accumulate on top of a SAM surface. When this charge surpasses the dielectric barrier of the SAM, electron tunneling from the gold surface through the SAM becomes efficient. We hypothesize that soft landed Ni-bpy cations may undergo either partial charge reduction or complete neutralization on the surface, resulting in the formation of complexes with Ni(I) or Ni(0), respectively. The change in the oxidation state of the metal center may affect the geometry of the complex.71,72 For the soft landed [Ni(bpy)3]2+, this change in geometry may result in ligand losses and the formation of [Ni(bpy)]0 species.To explore the effect of charge reduction on the product distribution on the surface, we designed an experiment in which we co-deposited [Ni(bpy)3]2+ with [B12F12]2− anions. This anion was selected based on its structural and electronic stability that make it chemically inert.73,74 We have previously demonstrated that co-deposition of cations with stable perhalogenated-closo-dodecaborate anions ([B12X12]2−, X = F, Cl, Br, I) may be used to inhibit charge reduction by soft landed cations.48 In a co-deposition experiment, a relatively low number of cations is continuously deposited onto the surface to achieve a coverage that is below the threshold for charge reduction.22,65,75 At this point, the polarity of the instrument is reversed to allow the transmission of negative ions to the surface. When the number of negative charges deposited on the surface equals the number of positive charges, the submonolayer is said to be charge balanced and charge reduction of ions is inhibited. The alternating co-deposition of cations and anions (polarity switching) is continued until the desired ion coverage is obtained. In this study, we used polarity switching48 to deposit 2.1 × 1011 ions (∼1.5% of a monolayer) of each doubly charged species in each segment onto an FSAM surface for 100 cycles to obtain a total ion coverage of 2.1 × 1013 ions (∼1 monolayer). The experiment was designed to maintain ion coverage deposited in each segment below the threshold at which charge reduction takes place on FSAM surfaces.22,65,75 Surfaces prepared using this approach were analyzed ex situ using nano-DESI MS as described earlier. Similar to previous experiments, we acquired alternating line scans across the deposition spot using ACN and 3 μM bpyD in ACN as extraction solvents. The results of these measurements are shown in Fig. 5. The same mass spectra over a wider m/z range are shown in Fig. S5.†
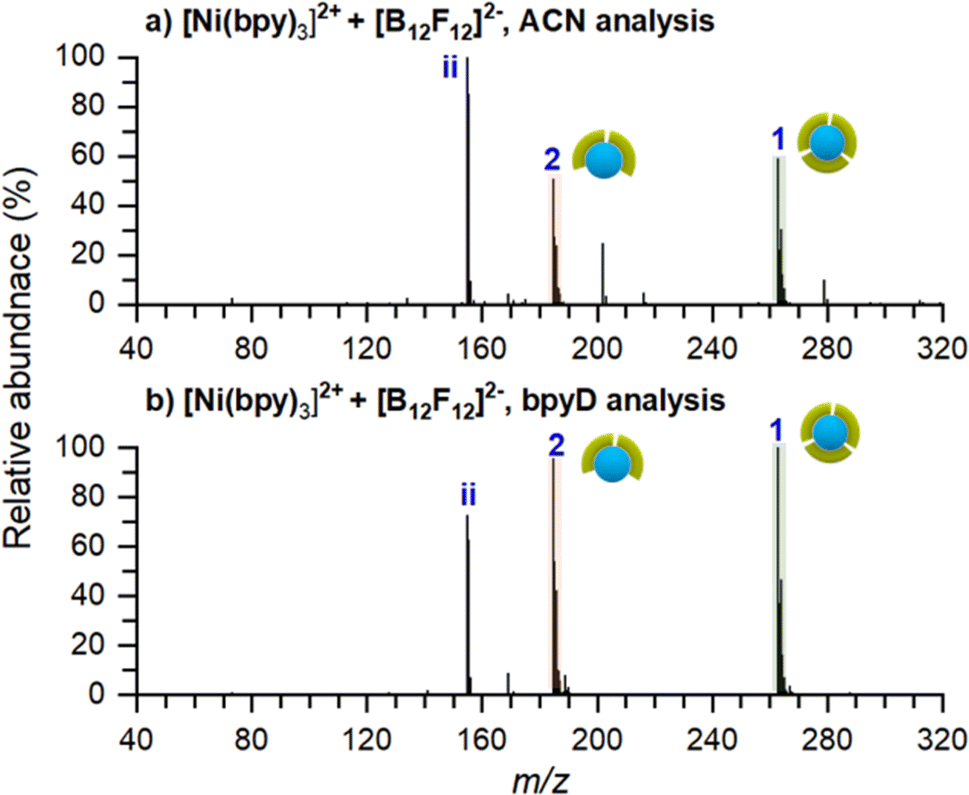 | ||
| Fig. 5 Ex situ MS of the co-deposition experiments of [Ni(bpy)3]2+ with [B12F12]2− measured with (a) ACN and (b) 3 μM bpyD in ACN as the nano-DESI solvent. The pictograms symbolize the number of ligands coordinated to Ni. Green symbols represent bpy. No species containing bpyD are observed in panel (b). Peaks originating from the solvent are not marked in the spectra for simplicity. | ||
In stark contrast with the result obtained for the deposition of [Ni(bpy)3]2+ without the anion (Fig. 2a), we observe an abundant peak of intact [Ni(bpy)3]2+ on the surface prepared by co-deposition of [Ni(bpy)3]2+ and [B12F12]2− (Fig. 5a). This observation indicates that the presence of [B12F12]2− anions on the surface hinders ligand loss from [Ni(bpy)3]2+. Furthermore, peaks corresponding to singly ligated Ni-bpy and its reaction products with molecules and ions in the solvent, which were abundant in Fig. 2a are absent in the mass spectrum shown in Fig. 5a. In fact, the mass spectrum in Fig. 5a closely resembles the one acquired from a solution containing Ni and bpy in a 1:3 molar ratio (Fig. 1a). We note that mass spectra in Fig. 5 contain a new abundant species assigned as C9H19N2+ (ii). The fact that this ion appears in these mass spectra, regardless of the composition of the nano-DESI solvent, indicates that it does not originate from species present in the solvent. Previous studies by our groups have shown that organic molecules present in the soft landing chamber are co-adsorbed with soft landed anions during the soft landing experiment.76–78 The line scan used to obtain the average mass spectrum shown in Fig. 5a (Fig. S6†) indicates that (ii) is localized in the deposition area. Furthermore, the mass spectrum of the surface in negative mode (Fig. S7†) shows that (ii) and [B12F12]2− form ion pair complexes. While we cannot provide further insights into the origin of this ion observed in co-deposition experiments with [B12F12]2−, we hypothesize that it does not play a role in the stabilization of [Ni(bpy)3]2+ on the surface. This is further confirmed by the results of co-deposition with other anions that are presented in the next section in which ion (ii) is not observed on the surface.
The result of surface analysis using reactive nano-DESI is shown in Fig. 5b. Unlike the result obtained without the anion (Fig. 2b), there are no discernible products corresponding to bpyD ligand pick-up in the spectrum. The absence of species containing bpyD in the spectrum of a surface prepared by co-depositing [Ni(bpy)3]2+ and [B12F12]2− confirms that no singly-ligated species that pick up bpyD from the solvent are present on the surface. It follows, that the anion stabilizes [Ni(bpy)3]2+ on the surface. Because [Ni(bpy)3]2+ undergoes slow ligand exchange in solution (Fig. S2†), no ligand exchange or pick up by this ion occurs on the timescale of the analysis. Collectively, our results indicate that the co-deposition of [Ni(bpy)3]2+ with [B12F12]2− anions prevents charge reduction of the cations and inhibits the dissociation of [Ni(bpy)3]2+ into [Ni(bpy)]n+ (n = 0,1) on the surface.
Co-deposition of [Ni(bpy)3]2+ with other anions
To confirm that the stabilizing role of [B12F12]2− on [Ni(bpy)3]2+ is not related to the inherent properties of the anion, we performed two co-deposition experiments with different anions. In these experiments we used either [B12Br12]2− which is structurally similar to [B12F12]2− but has a larger size and lower surface charge density or Keggin-polyoxotungstate ([PW12O40]3−, [WPOM]3−), an anion with high electron density, higher charge state, and larger size in comparison with [B12X12]2−. Co-deposition experiments of [Ni(bpy)3]2+ with these anions were conducted as described earlier. Ex situ mass spectra of these surfaces are shown in Fig. 6. Regardless of the identity of the anion, an abundant [Ni(bpy)3]2+ peak is observed on the surface. This observation confirms that co-deposited anions inhibit charge reduction of the cation and help preserve the intact [Ni(bpy)3]2+ on the surface. Other minor differences between these spectra and the spectrum of the surface prepared using [B12F12]2− (Fig. 5a), such as the presence of [Ni(bpy)]2+ in Fig. 6a and the intensity ratios of [Ni(bpy)2]2+ and [Ni(bpy)3]2+, can be attributed to a slight misalignment between positive and negative ion beams and differences in the analysis conditions.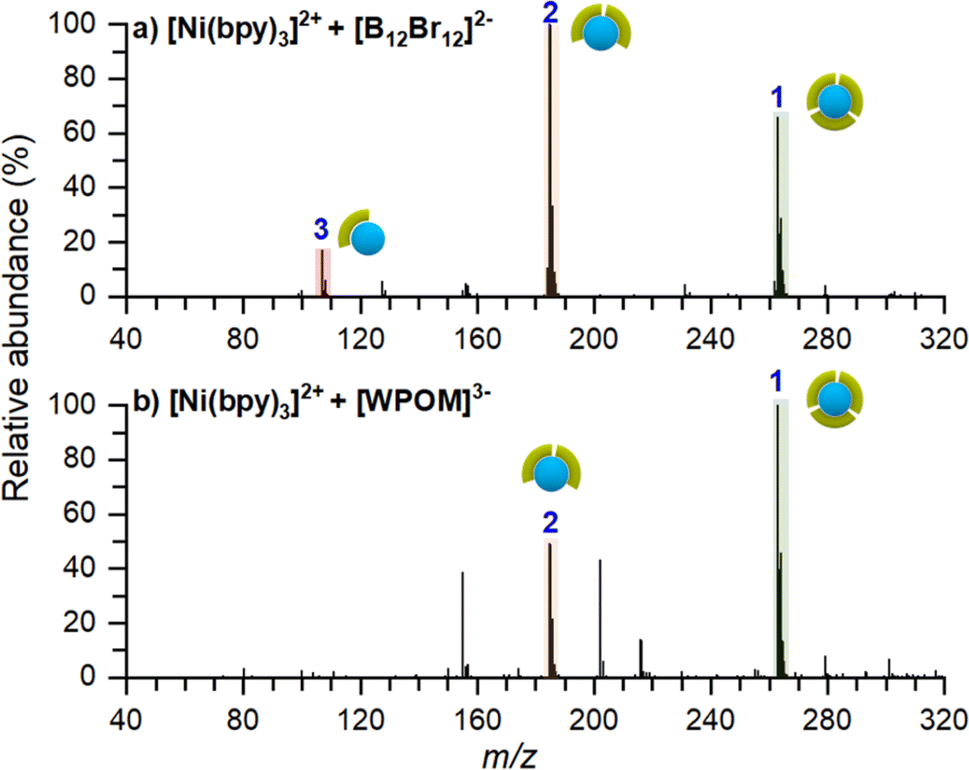 | ||
| Fig. 6 Ex situ MS of surfaces prepared by co-depositing [Ni(bpy)3]2+ with (a) [B12Br12]2−, and (b) [WPOM]3−. Both surfaces show an increased abundance of [Ni(bpy)3]2+ and only minor products of dissociation. | ||
Summary of the observed species on surfaces prepared by soft landing of [Ni(bpy)3]2+
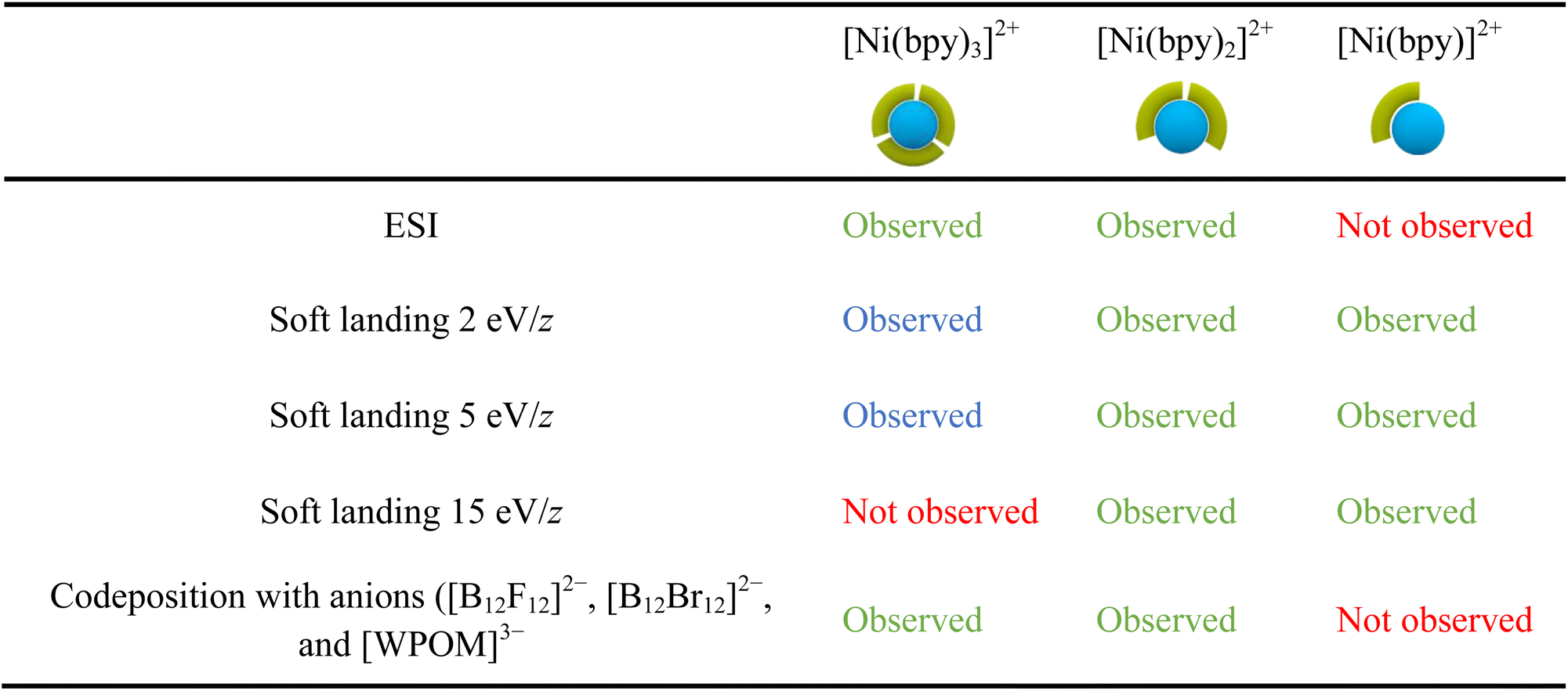
Table 2 provides a summary of the major species observed on surfaces prepared in this work. The results obtained using ESI of Ni-bpy solutions are also included for comparison. In Table 2, chemical species observed with a relative abundance lower than 25% are marked in blue to highlight their low abundance, while species observed in high abundance (>25%) are marked in green. We note that the experimental conditions, such as humidity and the positioning of the nano-DESI probe relative to the MS inlet, can affect the relative abundances observed in the mass spectra. Nevertheless, the 25% threshold offers a fair qualitative description of the data.
|
Table 2 shows that the ESI mass spectrum of a solution containing NiSO4 and bpy primarily displays [Ni(bpy)3]2+ and [Ni(bpy)2]2+ ions, with no presence of [Ni(bpy)]2+. This observation indicates that [Ni(bpy)]2+ is not produced during the electrospray process. Meanwhile, nano-DESI mass spectra of surfaces prepared by soft landing of [Ni(bpy)3]2+ at lower landing kinetic energies (2 and 5 eV/z) contain a low-abundance peak corresponding to intact [Ni(bpy)3]2+, which is not observed on surfaces prepared using a higher kinetic energy of 15 eV/z due to vibrational excitation of [Ni(bpy)3]2+ upon collision with the surface. Instead, mass spectra of surfaces prepared by depositing [Ni(bpy)3]2+ contain abundant signals of [Ni(bpy)2]2+ and [Ni(bpy)]2+ ions. As discussed earlier, [Ni(bpy)]2+ is likely formed through the charge reduction of [Ni(bpy)3]2+. Meanwhile, [Ni(bpy)2]2+ is formed through the loss of a ligand from [Ni(bpy)3]2+ promoted by geometry rearrangement of this ion on the surface. Finally, the codeposition of [Ni(bpy)3]2+ with anions prevents charge reduction of the cation on the surface. The mass spectrum of this surface reveals abundant [Ni(bpy)3]2+ and [Ni(bpy)2]2+ ions, a pattern resembling the results obtained using ESI MS of the solution containing NiSO4 and bpy.
Confirming structural transformations of Ni-bpy on surfaces using theoretical calculations
As previously suggested, the structural rearrangement of soft landed [Ni(bpy)3]2+ is a major process that facilitates the dissociation of the cation on surfaces. Because it is difficult to study this phenomenon experimentally, we conducted a computational study to evaluate this assertion. We used a flat, hydrogen-terminated surface (C54H72) as a model system of an inert interface. Geometry optimization of [Ni(bpy)3]2+ and [Ni(bpy)2]2+ both in the gas phase and in the presence of the surface was carried out to gain further insights into the possible geometry distortion of [Ni(bpy)3]2+ and the stabilization of the undercoordinated [Ni(bpy)2]2+. It should be noted that no quantitative conclusions can be drawn from this model. The interaction of an ion with the model interface used in this study is dominated by van der Waals forces and is therefore expected to underestimate the influence of the real surface on the structure of the complexes. A complete treatment of a surface must include the interfacial properties of the FSAM, image charge generated by soft landed ions in a conductive grounded surface used in the experiment (the computational model uses a total charge state of 2+), and the electronic band structure of the metal surface. A reliable treatment of these parameters would require the development and validation of a large-scale model, which is beyond the scope of this experimentally focused study. Nevertheless, if the comparatively weak interactions included in the simplified model result in the distortion of [Ni(bpy)3]2+ and the stabilization of [Ni(bpy)2]2+ on the surface, it is plausible to expect that a more comprehensive model that takes into account stronger interactions would likewise predict a similar or even greater structural change.The structures of gaseous [Ni(bpy)3]2+ and [Ni(bpy)2]2+ have been previously reported.58 It has been shown that in the absence of solvent, the most stable structure of [Ni(bpy)3]2+ is a triplet state in an octahedral geometry. We have recalculated the energy differences between the singlet and triplet states using a larger basis set (def2-TZVPP), adding the Grimme correction for B3LYP, and including the zero-point energy correction. For [Ni(bpy)3]2+ the triplet state is preferred to the singlet state by 119 kJ mol−1. Meanwhile, the singlet state of [Ni(bpy)2]2+ is 36 kJ mol−1 lower in energy than the triplet state.
The structures of [Ni(bpy)2]2+ and [Ni(bpy)3]2+ on a model surface are shown in Fig. 7a and b, respectively. Remarkably, the interaction with the surface causes a significant distortion of both [Ni(bpy)3]2+ and [Ni(bpy)2]2+ structures. Table S3† provides a summary of all the calculated angles. The octahedral [Ni(bpy)3]2+ “flattens out” on the surface. This major geometry distortion likely destabilizes the complex and promotes ligand loss. In contrast, the tetrahedral [Ni(bpy)2]2+ changes its geometry towards an almost square planar complex. To visualize these changes, Fig. 7a and b show the optimized geometries in contact with the interface overlayed with the optimized geometries of the free gas phase ions (pink). In contrast, when [Ni(bpy)3]2+ is paired with a [B12F12]2− counterion (Fig. 7c), the reduced contact of [Ni(bpy)3]2+ with the surface reduces the influence of the interface on its structure. Thus, the model confirms that, aside from charge reduction (omitted in this model), the observed dissociation of [Ni(bpy)3]2+ and stabilization of [Ni(bpy)2]2+ on surfaces may result from the interaction of the doubly charged complexes with the interface. The stabilization of [Ni(bpy)3]2+ by [B12F12]2− could result not only from hindering electron transfer but also from an enhanced geometry preservation of the [Ni(bpy)3]2+ by the anion.
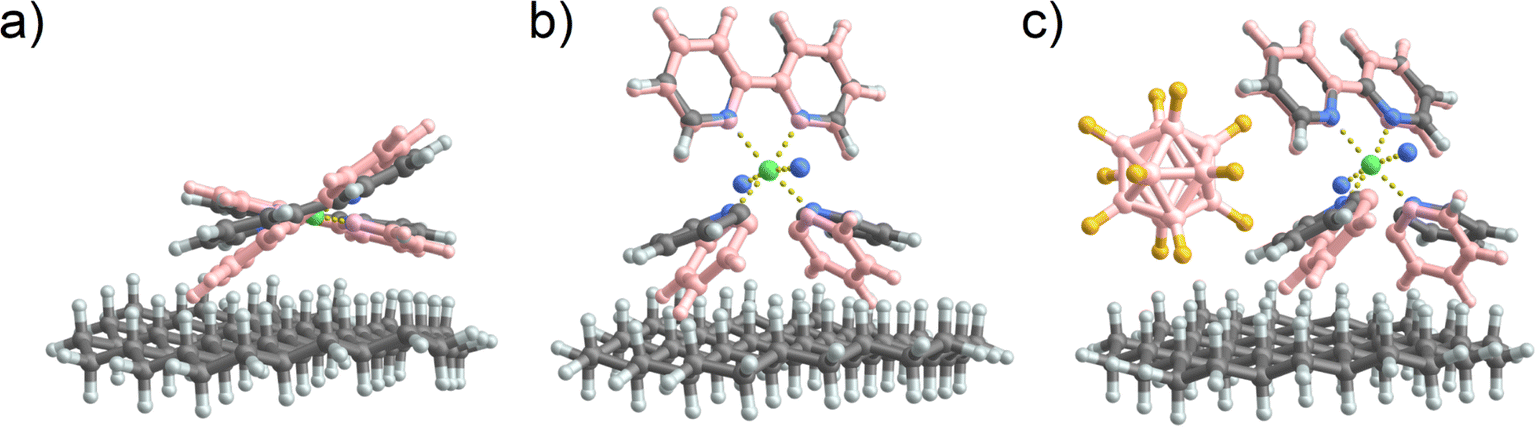 | ||
| Fig. 7 Comparison of the optimized structures of (a) [Ni(bpy)2]2+, (b) [Ni(bpy)3]2+, and (c) [Ni(bpy)3]2+[B12F12]2− in the presence of a small C–H terminated interface. For comparison, the optimized geometries of the isolated gas phase structures are plotted in pink and overlaid with the structures of the ions in contact with the interface. Note that for the bpy ligands pointing to the surface, only one pyridine ring is shown in (b) and (c) for clarity. | ||
Conclusions
In this study, we examine for the first time the effect of charge reduction and structural rearrangement on the stability of well-defined [Ni(bpy)3]2+ ions on surfaces. Our findings show that charge reduction by soft landed [Ni(bpy)3]2+ ions is one of the competing pathways for its dissociation that may generate [Ni(bpy)3]0, which likely undergoes two ligand losses to form [Ni(bpy)]0. We demonstrate that the dissociation of [Ni(bpy)3] into [Ni(bpy)] on surfaces may be inhibited by co-depositing [Ni(bpy)3]2+ with stable anions. Ion soft landing enables co-deposition of ions of opposite polarities to prevent charge reduction of the ions by preventing the potential build up across the interface. Our findings reveal that the interaction of the fully coordinated [Ni(bpy)3]2+ cation with a surface is another competing pathway that induces a significant geometric distortion of the ion, which results in a facile ligand loss to form [Ni(bpy)2]2+. The electrostatic interaction between anions and [Ni(bpy)3]2+ on the surface reduces the distortion of the geometry of the soft landed [Ni(bpy)3]2+ caused by the surface. This study also establishes that co-deposition of cations with stable anions as an effective strategy to suppress cation–surface interactions and prevent charge reduction of the cations. Our findings offer valuable insights for the design of stable functional interfaces using TM complexes.Data availability
Additional details regarding experimental procedures are provided in the ESI.† The link to the database containing the calculated structures is provided in the ESI.†Author contributions
Conceptualization: HYSO, JW, and JL. Funding acquisition: JW and JL. Methodology: HYSO, JW, and JL. Investigation: HYSO and HK. Supervision: JW and JL. Validation: HYSO, JW, and JL. Visualization: HYSO and HK. Writing-original draft: HYSO. Writing-review & editing: HK, JW, and JL.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
JL and HYSO acknowledge support from the Air Force Office of Scientific Research (AFOSR) under grant FA9550-23-1-0137. JW and JL acknowledge the Mercator fellowship for JL by RTG 2721 (DFG Project-ID 443871192) which promoted this collaborative work. JW is grateful to the Volkswagen Foundation for a Freigeist Fellowship. Computations for this work were done with resources of Leipzig University Computing Center.References
- A. Ajith, N. S. Krishnan Gowthaman, S. Abraham John and P. Elango, Direct Adsorption of Graphene Oxide on a Glassy Carbon Electrode: An Investigation of Its Adsorption and Electrochemical Activity, Langmuir, 2023, 39(29), 9990–10000, DOI:10.1021/acs.langmuir.3c00768.
- S. Badieyan, Q. Wang, X. Zou, Y. Li, M. Herron, L. N. Abbott, Z. Chen, G. Neil and E. Marsh, Engineered Surface-Immobilized Enzyme That Retains High Levels of Catalytic Activity in Air, J. Am. Chem. Soc., 2017, 139(8), 2872–2875, DOI:10.1021/jacs.6b12174.
- M. Correira, E. Madeksho and J. Webb, Acetylcholinesterase Adsorption on Modified Gold: Effect of Surface Chemistry on Enzyme Binding and Activity, Langmuir, 2023, 39(29), 9973–9979, DOI:10.1021/acs.langmuir.3c00648.
- T. Bligaard, J. K. Nørskov, S. Dahl, J. Matthiesen, C. H. Christensen and J. Sehested, The Brønsted–Evans–Polanyi Relation and the Volcano Curve in Heterogeneous Catalysis, J. Catal., 2004, 224(1), 206–217, DOI:10.1016/j.jcat.2004.02.034.
- J. Dalmieda, A. Zubiarrain-Laserna, D. Saha, P. Ravi Selvaganapathy and P. Kruse, Impact of Surface Adsorption on Metal–Ligand Binding of Phenanthrolines, J. Phys. Chem. C, 2021, 125(38), 21112–21123, DOI:10.1021/acs.jpcc.1c04509.
- C. E. Housecroft and E. C. Constable, Solar Energy Conversion Using First Row D-Block Metal Coordination Compound Sensitizers and Redox Mediators, Chem. Sci., 2022, 13(5), 1225–1262, 10.1039/D1SC06828H.
- M. Freitag, W. Yang, L. A. Fredin, L. D'Amario, K. M. Karlsson, A. Hagfeldt and G. Boschloo, Supramolecular Hemicage Cobalt Mediators for Dye-Sensitized Solar Cells, ChemPhysChem, 2016, 17(23), 3845–3852, DOI:10.1002/cphc.201600985.
- S. Derien, E. Dunach and J. Perichon, From Stoichiometry to Catalysis: Electroreductive Coupling of Alkynes and Carbon Dioxide with Nickel-Bipyridine Complexes. Magnesium Ions as the Key for Catalysis, J. Am. Chem. Soc., 1991, 113(22), 8447–8454, DOI:10.1021/ja00022a037.
- M. K. Kashif, M. Nippe, N. W. Duffy, C. M. Forsyth, C. J. Chang, J. R. Long, L. Spiccia and U. Bach, Stable Dye-Sensitized Solar Cell Electrolytes Based on Cobalt(II)/(III) Complexes of a Hexadentate Pyridyl Ligand, Angew. Chem., Int. Ed., 2013, 52(21), 5527–5531, DOI:10.1002/anie.201300070.
- C. Wegeberg and S. Wenger, Luminescent First-Row Transition Metal Complexes, JACS Au, 2021, 1(11), 1860–1876, DOI:10.1021/jacsau.1c00353.
- C. Tsay and Y. Yang, Electrocatalytic Hydrogen Evolution under Acidic Aqueous Conditions and Mechanistic Studies of a Highly Stable Molecular Catalyst, J. Am. Chem. Soc., 2016, 138(43), 14174–14177, DOI:10.1021/jacs.6b05851.
- M. Guttentag, A. Rodenberg, C. Bachmann, A. Senn, P. Hamm and R. Alberto, A Highly Stable Polypyridyl-Based Cobalt Catalyst for Homo- and Heterogeneous Photocatalytic Water Reduction, Dalton Trans., 2013, 42(2), 334–337, 10.1039/C2DT31699D.
- B. M. Ceballos and J. Y. Yang, Directing the Reactivity of Metal Hydrides for Selective CO2 Reduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(50), 12686–12691, DOI:10.1073/pnas.1811396115.
- M.-Y. Lee, C. Kahl, N. Kaeffer and W. Leitner, Electrocatalytic Semihydrogenation of Alkynes with [Ni(Bpy)3]2+, JACS Au, 2022, 2(3), 573–578, DOI:10.1021/jacsau.1c00574.
- C. Jeong, C. Coburn, M. Idris, Y. Li, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Understanding Molecular Fragmentation in Blue Phosphorescent Organic Light-Emitting Devices, Org. Electron., 2019, 64, 15–21, DOI:10.1016/J.ORGEL.2018.10.001.
- C. Y. Yang, S. Kang, H. Jeong, H. J. Jang, Y. Lee and J. Y. Lee, Key Host Parameters for Long Lifetimes in Phosphorescent Organic Light-Emitting Diodes: Bond Dissociation Energy in Triplet Excited State, J. Mater. Chem. C, 2020, 8(5), 1697–1703, 10.1039/C9TC05585A.
- A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassani and N. D. McClenaghan, Improving the Photophysical Properties of Copper(I) Bis(Phenanthroline) Complexes, Coord. Chem. Rev., 2008, 252(23), 2572–2584, DOI:10.1016/j.ccr.2008.03.013.
- Y. Kawamata, J. Vantourout, D. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. A. Barman, M. Edwards, A. Garrido-Castro, J. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. Clay, D. Bao, C. Li, J. Starr, C. Garcia-Irizarry, N. S. Sach, H. White, M. Neurock, D. Minteer and S. Baran, Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications, J. Am. Chem. Soc., 2019, 141(15), 6392–6402, DOI:10.1021/jacs.9b01886.
- G. Durin, M.-Y. Lee, M. Pogany, T. Weyhermüller, N. Kaeffer and W. Leitner, Hydride-Free Hydrogenation: Unraveling the Mechanism of Electrocatalytic Alkyne Semihydrogenation by Nickel–Bipyridine Complexes, J. Am. Chem. Soc., 2023, 145(31), 17103–17111, DOI:10.1021/jacs.3c03340.
- J. Beaudelot, S. Oger, S. Peruško, T.-A. Phan, T. Teunens, C. Moucheron and G. Evano, Photoactive Copper Complexes: Properties and Applications, Chem. Rev., 2022, 122(22), 16365–16609, DOI:10.1021/acs.chemrev.2c00033.
- J. Cyriac, T. Pradeep, H. Kang, R. Souda and G. Cooks, Low-Energy Ionic Collisions at Molecular Solids, Chem. Rev., 2012, 112(10), 5356–5411, DOI:10.1021/cr200384k.
- G. E. Johnson, D. Gunaratne and J. Laskin, Soft- and Reactive Landing of Ions onto Surfaces: Concepts and Applications, Mass Spectrom. Rev., 2016, 35(3), 439–479, DOI:10.1002/mas.21451.
- S. Rauschenbach, M. Ternes, L. Harnau and K. Kern, Mass Spectrometry as a Preparative Tool for the Surface Science of Large Molecules, Annu. Rev. Anal. Chem., 2016, 9(1), 473–498, DOI:10.1146/annurev-anchem-071015-041633.
- J. Laskin, E. Johnson and V. Prabhakaran, Soft Landing of Complex Ions for Studies in Catalysis and Energy Storage, J. Phys. Chem. C, 2016, 120(41), 23305–23322, DOI:10.1021/acs.jpcc.6b06497.
- K. Anggara, Y. Zhu, M. Delbianco, S. Rauschenbach, S. Abb, P. Seeberger and K. Kern, Exploring the Molecular Conformation Space by Soft Molecule–Surface Collision, J. Am. Chem. Soc., 2020, 142(51), 21420–21427, DOI:10.1021/jacs.0c09933.
- S. Abb, N. Tarrat, J. Cortés, B. Andriyevsky, L. Harnau, J. C. Schön, S. Rauschenbach and K. Kern, Carbohydrate Self-Assembly at Surfaces: STM Imaging of Sucrose Conformation and Ordering on Cu(100), Angew. Chem., Int. Ed., 2019, 58(25), 8336–8340, DOI:10.1002/anie.201901340.
- S. C. Nanita, Z. Takats, R. G. Cooks, S. Myung and D. E. Clemmer, Chiral Enrichment of Serine via Formation, Dissociation, and Soft-Landing of Octameric Cluster Ions, J. Am. Soc. Mass Spectrom., 2004, 15(9), 1360–1365, DOI:10.1016/j.jasms.2004.06.010.
- W. Ran, A. Walz, K. Stoiber, P. Knecht, H. Xu, A. C. Papageorgiou, A. Huettig, D. Cortizo-Lacalle, J. P. Mora-Fuentes, A. Mateo-Alonso, H. Schlichting, J. Reichert and J. V. Barth, Depositing Molecular Graphene Nanoribbons on Ag(111) by Electrospray Controlled Ion Beam Deposition: Self-Assembly and On-Surface Transformations, Angew. Chem., Int. Ed., 2022, 61(14), e202111816, DOI:10.1002/anie.202111816.
- H. Gholipour-Ranjbar, L. Sertse, D. Forbes and J. Laskin, Effect of Ligands on the Reactivity of the Undercoordinated Fragment Ions of Co6S8(PEt3–XPhx)6+ (x = 0–3) Clusters on Surfaces, J. Phys. Chem. C, 2024, 128(20), 8232–8238, DOI:10.1021/acs.jpcc.4c00941.
- F. Yang, M. Moors, D. Anh Hoang, S. Schmitz, M. Rohdenburg, H. Knorke, A. Charvat, X.-B. Wang, K. Yu Monakhov and J. Warneke, On-Surface Single-Molecule Identification of Mass-Selected Cyclodextrin-Supported Polyoxovanadates for Multistate Resistive-Switching Memory Applications, ACS Appl. Nano Mater., 2022, 5(10), 14216–14220, DOI:10.1021/acsanm.2c03025.
- D. Meier, B. Schoof, J. Wang, X. Li, A. Walz, A. Huettig, H. Schlichting, F. Rosu, V. Gabelica, V. Maurizot, J. Reichert, A. C. Papageorgiou, I. Huc and J. V. Barth, Structural Adaptations of Electrosprayed Aromatic Oligoamide Foldamers on Ag(111), Chem. Commun., 2022, 58(64), 8938–8941, 10.1039/D2CC03286D.
- Q. Hu, P. Wang and J. Laskin, Effect of the Surface on the Secondary Structure of Soft Landed Peptide Ions, Phys. Chem. Chem. Phys., 2010, 12(39), 12802–12810, 10.1039/C0CP00825G.
- N. Hauptmann, C. Hamann, H. Tang and R. Berndt, Soft-Landing Electrospray Deposition of the Ruthenium Dye N3 on Au(111), J. Phys. Chem. C, 2013, 117(19), 9734–9738, DOI:10.1021/jp311420d.
- M. Mitsui, S. Nagaoka, T. Matsumoto and A. Nakajima, Soft-Landing Isolation of Vanadium–Benzene Sandwich Clusters on a Room-Temperature Substrate Using n-Alkanethiolate Self-Assembled Monolayer Matrixes, J. Phys. Chem. B, 2006, 110(7), 2968–2971, DOI:10.1021/jp057194v.
- W.-P. Peng, G. E. Johnson, I. C. Fortmeyer, P. Wang, O. Hadjar, R. G. Cooks and J. Laskin, Redox chemistry in thin layers of organometallic complexes prepared using ion soft landing., Phys. Chem. Chem. Phys., 2011, 13(1), 267–275, 10.1039/c0cp01457e.
- P. Su, H. Hu, J. Warneke, M. E. Belov, G. A. Anderson and J. Laskin, Design and Performance of a Dual-Polarity Instrument for Ion Soft Landing., Anal. Chem., 2019, 91(9), 5904–5912, DOI:10.1021/acs.analchem.9b00309.
- J. Warneke, H. Knorke, A. Charvat, K. G. Warneke, Ion Soft-Landing of Permanently Charged Molecular Ions and Their Charge Retention on Surfaces, In Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2022, pp. 1–15, DOI:10.1002/9781119951438.eibc2750.
- J. Laskin, G. E. Johnson, J. Warneke and V. Prabhakaran, From Isolated Ions to Multilayer Functional Materials Using Ion Soft Landing., Angew. Chem., Int. Ed. Engl., 2018, 57(50), 16270–16284, DOI:10.1002/anie.201712296.
- G. Rinke, S. Rauschenbach, L. Harnau, A. Albarghash, M. Pauly and K. Kern, Active conformation control of unfolded proteins by hyperthermal collision with a metal surface., Nano Lett., 2014, 14(10), 5609–5615, DOI:10.1021/nl502122j.
- J. Laskin, P. Wang and O. Hadjar, Soft-Landing of CoIII(Salen)+ and MnIII(Salen)+ on Self-Assembled Monolayer Surfaces, J. Phys. Chem. C, 2009, 114(12), 5305–5311, DOI:10.1021/jp904384q.
- H. Gholipour-Ranjbar, H. Y. Samayoa-Oviedo and J. Laskin, Controlled Formation of Fused Metal Chalcogenide Nanoclusters Using Soft Landing of Gaseous Fragment Ions., ACS Nano, 2023, 17(17), 17427–17435, DOI:10.1021/acsnano.3c05545.
- A. Cagan, D. Bím, B. Silva, P. Kazmierczak, J. McNicholas and G. Hadt, Elucidating the Mechanism of Excited-State Bond Homolysis in Nickel–Bipyridine Photoredox Catalysts, J. Am. Chem. Soc., 2022, 144(14), 6516–6531, DOI:10.1021/jacs.2c01356.
- Y. G. Budnikova, D. G. Yakhvarov, V. I. Morozov, Y. M. Kargin, A. V. Il’yasov, Y. N. Vyakhireva and O. G. Sinyashin, Electrochemical Reduction of Nickel Complexes with 2,2’-Bipyridine, Russ. J. Gen. Chem., 2002, 72(2), 168–172, DOI:10.1023/A:1015401029839.
- J. Laskin and P. Wang, Charge Retention by Organometallic Dications on Self-Assembled Monolayer Surfaces, Int. J. Mass Spectrom., 2014, 365–366, 187–193, DOI:10.1016/j.ijms.2014.01.012.
- H. Y. Samayoa-Oviedo, K.-A. Behrend, S. Kawa, H. Knorke, P. Su, M. Belov, G. Anderson, J. Warneke and J. Laskin, Design and Performance of a Soft-Landing Instrument for Fragment Ion Deposition, Anal. Chem., 2021, 93(43), 14489–14496, DOI:10.1021/acs.analchem.1c03009.
- L. Patiny and A. Borel, ChemCalc: A Building Block for Tomorrow's Chemical Infrastructure, J. Chem. Inf. Model., 2013, 53(5), 1223–1228, DOI:10.1021/ci300563h.
- D. Bain, E. Barry Troughton, Y. Tai Tao, J. Evall, M. Whitesides and G.G. Nuzzo, Formation of Monolayer Films by the Spontaneous Assembly of Organic Thiols from Solution onto Gold, J. Am. Chem. Soc., 2002, 111(1), 321–335, DOI:10.1021/ja00183a049.
- P. Su, H. Hu, J. Warneke, M. Belov, G. Anderson and J. Laskin, Design and Performance of a Dual-Polarity Instrument for Ion Soft Landing, Anal. Chem., 2019, 91(9), 5904–5912, DOI:10.1021/acs.analchem.9b00309.
- P. J. Roach, J. Laskin and A. Laskin, Nanospray Desorption Electrospray Ionization: An Ambient Method for Liquid-Extraction Surface Sampling in Mass Spectrometry, Analyst, 2010, 135(9), 2233–2236, 10.1039/C0AN00312C.
- I. S. Lanekoff, B. Heath, A. Liyu, M. Thomas, P. Carson and J. Laskin, Automated Platform for High-Resolution Tissue Imaging Using Nanospray Desorption Electrospray Ionization Mass Spectrometry, Anal. Chem., 2012, 84(19), 8351–8356, DOI:10.1021/ac301909a.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. a. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. a. Petersson, H. Nakatsuji, X. Li, M. Caricato, a. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, a. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. a. Keith, R. Kobayashi, J. Normand, K. Raghavachari, a. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., G16_B01, Wallin, 2016 Search PubMed.
- A. D. Becke, Density-functional Thermochemistry. I. The Effect of the Exchange-only Gradient Correction, J. Chem. Phys., 1992, 96(3), 2155–2160, DOI:10.1063/1.462066.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence{,} Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305, 10.1039/B508541A.
- F. Weigend, Accurate Coulomb-Fitting Basis Sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065, 10.1039/B515623H.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- W. A. E. McBryde, A Critical Review of Equilibrium Data for Proton- and Metal Complexes of 1,10-Phenanthroline, 2,2′-Bipyridyl and Related Compounds, in A Critical Review of Equilibrium Data for Proton and Metal Complexes of 1,10-phenanthroline, 2,2’-Bipyridyl and Related Compounds, ed. McBryde, W. A. E., Pergamon, 1978, pp. 1–17, DOI:10.1016/B978-0-08-022344-5.50004-8.
- K. Barman, M. A. Edwards, D. P. Hickey, C. Sandford, Y. Qiu, R. Gao, S. D. Minteer and H. S. White, Electrochemical Reduction of [Ni(Mebpy)3]2+: Elucidation of the Redox Mechanism by Cyclic Voltammetry and Steady-State Voltammetry in Low Ionic Strength Solutions, ChemElectroChem, 2020, 7(6), 1473–1479, DOI:10.1002/celc.202000171.
- H. Nose and M. T. Rodgers, Energy-Resolved Collision-Induced Dissociation Studies of 2,2′-Bipyridine Complexes of the Late First-Row Divalent Transition-Metal Cations: Determination of the Third-Sequential Binding Energies, ChemPlusChem, 2013, 78(9), 1109–1123, DOI:10.1002/cplu.201300156.
- K. P. Wise, L. V. Slipchenko and D. Ben-Amotz, Ion-Size Dependent Adsorption Crossover on the Surface of a Water Droplet, J. Phys. Chem. B, 2023, 127(20), 4658–4665, DOI:10.1021/acs.jpcb.3c01797.
- Z. Wei, Y. Li, R. G. Cooks and X. Yan, Accelerated Reaction Kinetics in Microdroplets: Overview and Recent Developments, Annu. Rev. Phys. Chem., 2020, 71(1), 31–51, DOI:10.1146/annurev-physchem-121319-110654.
- S. Johannsen, M. Gruber, C. Barreteau, M. Seredyuk, J. Antonio Real, T. Markussen and R. Berndt, Spin-Crossover and Fragmentation of Fe(Neoim)2 on Silver and Gold, J. Phys. Chem. Lett., 2023, 14(35), 7814–7823, DOI:10.1021/acs.jpclett.3c01551.
- V. H. Wysocki, K. E. Joyce, C. M. Jones and R. L. Beardsley, Surface-Induced Dissociation of Small Molecules, Peptides, and Non-Covalent Protein Complexes, J. Am. Soc. Mass Spectrom., 2008, 19(2), 190–208, DOI:10.1016/j.jasms.2007.11.005.
- G. Rinke, S. Rauschenbach, L. Harnau, A. Albarghash, M. Pauly and K. Kern, Active Conformation Control of Unfolded Proteins by Hyperthermal Collision with a Metal Surface, Nano Lett., 2014, 14(10), 5609–5615, DOI:10.1021/nl502122j.
- J. Laskin and J. H. Futrell, Energy Transfer in Collisions of Peptide Ions with Surfaces, J. Chem. Phys., 2003, 119(6), 3413–3420, DOI:10.1063/1.1589739.
- J. Pflaum, G. Bracco, F. Schreiber, R. Colorado, O. E. Shmakova, T. R. Lee, G. Scoles and A. Kahn, Structure and Electronic Properties of CH3- and CF3-Terminated Alkanethiol Monolayers on Au(111): A Scanning Tunneling Microscopy, Surface X-Ray and Helium Scattering Study, Surf. Sci., 2002, 498(1–2), 89–104, DOI:10.1016/S0039-6028(01)01495-9.
- O. Hadjar, P. Wang, H. J. Futrell and J. Laskin, Effect of the Surface on Charge Reduction and Desorption Kinetics of Soft Landed Peptide Ions, J. Am. Soc. Mass Spectrom., 2011, 20(6), 901–906, DOI:10.1016/j.jasms.2008.12.025.
- E. Johnson, T. Priest and J. Laskin, Charge Retention by Gold Clusters on Surfaces Prepared Using Soft Landing of Mass Selected Ions, ACS Nano, 2011, 6(1), 573–582, DOI:10.1021/nn2039565.
- G. E. Johnson and J. Laskin, Soft Landing of Mass-Selected Gold Clusters: Influence of Ion and Ligand on Charge Retention and Reactivity, Int. J. Mass Spectrom., 2015, 377, 205–213, DOI:10.1016/j.ijms.2014.05.013.
- E. G. Johnson, T. Priest and J. Laskin, Coverage-Dependent Charge Reduction of Cationic Gold Clusters on Surfaces Prepared Using Soft Landing of Mass-Selected Ions, J. Phys. Chem. C, 2012, 116(47), 24977–24986, DOI:10.1021/jp308795r.
- W.-P. Peng, G. E. Johnson, I. C. Fortmeyer, P. Wang, O. Hadjar, R. G. Cooks and J. Laskin, Redox Chemistry in Thin Layers of Organometallic Complexes Prepared Using Ion Soft Landing, Phys. Chem. Chem. Phys., 2011, 13(1), 267–275, 10.1039/C0CP01457E.
- J. Sub Kim, H. J. Reibenspies and Y. M. Darensbourg, Characteristics of Nickel(0), Nickel(I), and Nickel(II) in Phosphino Thioether Complexes: Molecular Structure and S-Dealkylation of (Ph2P(o-C6H4)SCH3)2Ni0, J. Am. Chem. Soc., 1996, 118(17), 4115–4123, DOI:10.1021/ja953686h.
- G. Kuppuraj, M. Dudev and C. Lim, Factors Governing Metal–Ligand Distances and Coordination Geometries of Metal Complexes, J. Phys. Chem. B, 2009, 113(9), 2952–2960, DOI:10.1021/jp807972e.
- J. Warneke, G.-L. Hou, E. Aprà, C. Jenne, Z. Yang, Z. Qin, K. Kowalski, X.-B. Wang and S. Xantheas, Electronic Structure and Stability of [B12X12]2– (X = F–At): A Combined Photoelectron Spectroscopic and Theoretical Study, J. Am. Chem. Soc., 2017, 139(41), 14749–14756, DOI:10.1021/jacs.7b08598.
- J. Warneke, S. Z. Konieczka, G.-L. Hou, E. Aprà, C. Kerpen, F. Keppner, T. C. Schäfer, M. Deckert, Z. Yang, E. J. Bylaska, G. E. Johnson, J. Laskin, S. S. Xantheas, X.-B. Wang and M. Finze, Properties of Perhalogenated {closo-B10} and {closo-B11} Multiply Charged Anions and a Critical Comparison with {closo-B12} in the Gas and the Condensed Phase, Phys. Chem. Chem. Phys., 2019, 21(11), 5903–5915, 10.1039/C8CP05313H.
- L. Romaner, G. Heimel, C. Ambrosch-Draxl and E. Zojer, The Dielectric Constant of Self-Assembled Monolayers, Adv. Funct. Mater., 2008, 18(24), 3999–4006, DOI:10.1002/adfm.200800876.
- J. Warneke, M. E. McBriarty, S. L. Riechers, S. China, M. H. Engelhard, E. Aprà, R. P. Young, N. M. Washton, C. Jenne, G. E. Johnson and J. Laskin, Self-Organizing Layers from Complex Molecular Anions, Nat. Commun., 2018, 9(1), 1889, DOI:10.1038/s41467-018-04228-2.
- J. Warneke, M. Mayer, M. Rohdenburg, X. Ma, J. K. Y. Liu, M. Grellmann, S. Debnath, V. A. Azov, E. Apra, R. P. Young, C. Jenne, G. E. Johnson, H. I. Kenttämaa, K. R. Asmis and J. Laskin, Direct Functionalization of C–H Bonds by Electrophilic Anions, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(38), 23374–23379, DOI:10.1073/PNAS.2004432117.
- F. Yang, K. A. Behrend, H. Knorke, M. Rohdenburg, A. Charvat, C. Jenne, B. Abel and J. Warneke, Anion–Anion Chemistry with Mass-Selected Molecular Fragments on Surfaces, Angew. Chem., Int. Ed., 2021, 60(47), 24910–24914, DOI:10.1002/anie.202109249.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumental settings for the deposition of Ni-bpy species. Measured and expected m/z values of species observed on surfaces shown in this work. Ex situ nano-DESI MS over a wider m/z range of the deposition spots of [Ni(bpy)3]2+ analyzed with (a) ACN and (b) 3 μM bpyD in ACN as the nano-DESI solvent. Mass spectrum of a solution containing 15 μM Ni(bpy)3SO4 and 15 μM bpyD measured right after preparing the solution (top) and after 3 hours of preparing the solution (bottom). Abundance ratio of [Ni(bpy)2]2+/[Ni(bpy)3]2+ across the deposition spot for the deposition of [Ni(bpy)3]2+. Mass spectra of the surface where [Ni(bpy)3]2+ was deposited on two different regions of the deposition area. Ex situ mass spectrum of the surface prepared by co-deposition of [Ni(bpy)3]2+ and [B12F12]2− analyzed with (a) ACN and (b) 3 μM bpyD in ACN as the nano-DESI solvent. Line scans of the mass spectra in Fig. 6a. Calculated angles between two bpy of [Ni(bpy)3]2+, [Ni(bpy)2]2+ and [B12F12]2−[Ni(bpy)3]2+ in the gas phase and in the presence of the model surface C54H72. Visualization of the angle between the planes spanned by two bpy molecules for [Ni(bpy)2]2+ singlet and [Ni(bpy)3]2+ triplet. Link to database containing the calculated structures. See DOI: https://doi.org/10.1039/d4sc02527j |
| This journal is © The Royal Society of Chemistry 2024 |