
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of hydrocarbon belts based on macrocyclic arenes
Guangtan
Fan†
ab,
Zhi
Zhang†
ab,
Guangguo
Wang†
ab,
Li
Shao
 c,
Bin
Hua
*ab and
Feihe
Huang
*ab
c,
Bin
Hua
*ab and
Feihe
Huang
*ab
aStoddart Institute of Molecular Science, Department of Chemistry, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: fhuang@zju.edu.cn
bZhejiang-Israel Joint Laboratory of Self-Assembling Functional Materials, ZJU-Hangzhou Global Scientific and Technological Innovation Center, Zhejiang University, Hangzhou 311215, P. R. China. E-mail: huabin@zju.edu.cn
cDepartment of Materials Science and Engineering, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China
First published on 3rd June 2024
Abstract
Hydrocarbon belts have garnered significant attention due to their intriguing structures, unique properties, and potential applications in supramolecular chemistry and materials science. However, their highly inherently strained structures pose challenges in their synthesis, and the resulting tedious synthesis strategies hinder their large-scale applications. Utilizing unstrained macrocyclic arenes as precursors presents an efficient strategy, allowing for a strain-induction step that mitigates the energy barrier associated with building strain in the formation of these belts. Accessible unstrained macrocyclic precursors play a pivotal role in enabling efficient and large-scale syntheses of highly strained belts, facilitating their broader practical applications. This review provides an overview of the recent advancements in the construction of hydrocarbon belts using accessible macrocyclic arenes as building blocks. The synthetic strategies for these partially and fully conjugated hydrocarbon belts are discussed, along with their unique properties. We hope that this review will inspire the development of novel nanocarbon molecules, opening pathways for emerging areas and applications.
 Guangtan Fan | Guangtan Fan obtained his Master's degree from Wuhan Institute of Technology in 2017. Then he received his PhD in 2021 under the guidance of Prof. Simin Liu from Wuhan University of Science and Technology. In March 2022, he joined Prof. Feihe Huang's group in Zhejiang University as a postdoctoral fellow. His current research interest is focused on the design and synthesis of molecular nanocarbons featuring complex topological structures. |
 Zhi Zhang | Zhi Zhang obtained his Master's degree from Liaoning University in 2018. Then, he joined the Kyushu University and received his PhD in 2021 under the supervision of Prof. Yoshio Hisaeda. Currently, he is a postdoctoral fellow with Prof. Feihe Huang in Zhejiang University. His current research interest is focused on molecular nanocarbons. |
 Guangguo Wang | Guangguo Wang received his PhD from the University of Chinese Academy of Sciences (UCAS) in 2023. He conducted his postdoctoral research at Zhejiang University with Prof. Feihe Huang. His current research interest is focused on cycloparaphenylenes (CPPs). |
 Li Shao | Li Shao joined the laboratory of Prof. Feihe Huang at Zhejiang University and got her PhD in chemistry in 2019. Then she conducted her postdoctoral research at University of Missouri-Columbia with Prof. Jerry L. Atwood. After that, she continued her postdoctoral research at Pacific Northwest National Laboratory (PNNL) with Dr Chun-Long Chen. Currently, she joined the Department of Materials Science and Engineering at Zhejiang Sci-Tech University as a principal investigator. Her research interest is focused on the construction of functional supramolecular polymers based on host–guest chemistry and hierarchical self-assembly. |
 Bin Hua | Bin Hua earned his PhD in Organic Chemistry from Zhejiang University in 2018 under the guidance of Prof. Feihe Huang. He continued as a postdoctoral researcher in the same group until 2020. Subsequently, from 2020 to 2022, he served as a postdoctoral fellow under the supervision of Prof. Niveen M. Khashab at the King Abdullah University of Science and Technology. In March 2022, Bin Hua joined ZJU-Hangzhou Global Scientific and Technological Innovation Center as a research assistant professor. His current research interest is focused on the synthesis of innovative supramolecular host molecules and the exploration of their host–guest chemistry. |
 Feihe Huang | Feihe Huang is Changjiang Scholar Chair Professor of Zhejiang University. His current research is focused on supramolecular polymers and nonporous adaptive crystals (NACs). Awards and honours he has received include Chinese Chemical Society AkzoNobel Chemical Sciences Award, The Cram Lehn Pedersen Prize in Supramolecular Chemistry, Royal Society of Chemistry Polymer Chemistry Lectureship award, and Bruno Werdelmann Lectureship Award. His publications have been cited more than 39740 times. His h-index is 107. He sits/sat on the Advisory Boards of JACS, Chem. Soc. Rev., Chem. Commun., Macromolecules, ACS Macro. Lett., and Polym. Chem. He is an Editorial Board Member of Mater. Chem. Front. |
1. Introduction
Hydrocarbon belts, serving as foundational elements for carbon nanotubes, are unique double-stranded belt-shaped molecules composed of partially and fully saturated benzenoid rings, including fully conjugated beltarenes and their (partially) saturated analogs.1,2 These strained cyclic molecules possess radial p orbitals and come in three types: armchair, zigzag, and chiral, based on the fusion pattern of hexagonal aromatic units.3,4 The category of hydrocarbon belts encompasses aromatic belt compounds that incorporate rings beyond hexagonal ones and/or heteroatoms, offering a wide variety of structural possibilities.5–11 Their distinctive structures, unique properties, and potential applications have captured the keen interest of the research communities. Especially, these belts hold the significant potential to serve as templates or seeds for synthesizing uniform carbon nanotubes with precisely defined structures, marking a significant area of interest within nanocarbon science research.1,12While exploration into synthesizing hydrocarbon belts has spanned half a century, significant progress has only emerged in recent years. The conventional synthetic route to hydrocarbon belts typically involves two crucial steps: (1) preorganization of planar building blocks to form macrocycles; (2) intramolecular coupling of the large rings to form molecular belts.5 The synthesis of hydrocarbon belts poses a major challenge due to their inherent strain, with the conventional fragment preparation method being effective yet tedious.13–15 The utilization of unstrained macrocyclic arenes as precursors presents an efficient and convenient strategy, allowing for a later strain-induction step that mitigates the energy barrier associated with building strain in the formation of these belts. Accessible unstrained macrocyclic precursors such as pillararenes, resorcinarenes, calixarenes, and so on, have demonstrated significant potential in enabling efficient and large-scale syntheses of highly strained belts. These macrocyclic arenes are made up of different aromatic building blocks connected by methylene groups which can be conveniently synthesized through one-pot condensation reaction.16 Then after the intramolecular coupling reaction, the belts were successfully synthesized. Diels–Alder reactions,17–20 aryl–aryl coupling reactions,21 and Scholl reactions22,23 have been effectively utilized in the intramolecular coupling reaction of these hydrocarbon belts. Moreover, with the development of supramolecular and macrocyclic chemistry, a variety of novel macrocyclic arenes have been created, providing a continuous source of raw materials for constructing various new hydrocarbon belts.16,24
In this review, we present challenges and strategies in the synthesis of hydrocarbon belts and then propose the rationality and feasibility of constructing hydrocarbon nanobelts with accessible supramolecular macrocyclic arenes including resorcin[n]arenes, pillar[n]arenes, prism[6]arene, calix[3]carbazole and fluoren[3]arenes. Our focus lies on utilizing various traditional macrocyclic arenes as building blocks and showcasing successful synthesis cases with a hydrocarbon framework from recent years (Fig. 1). We have intentionally excluded discussions on non-traditional macrocyclic arenes for constructing hydrocarbon nanobelts. The goal is to share similar construction strategies and inspire more researchers to delve into this emerging and promising field.
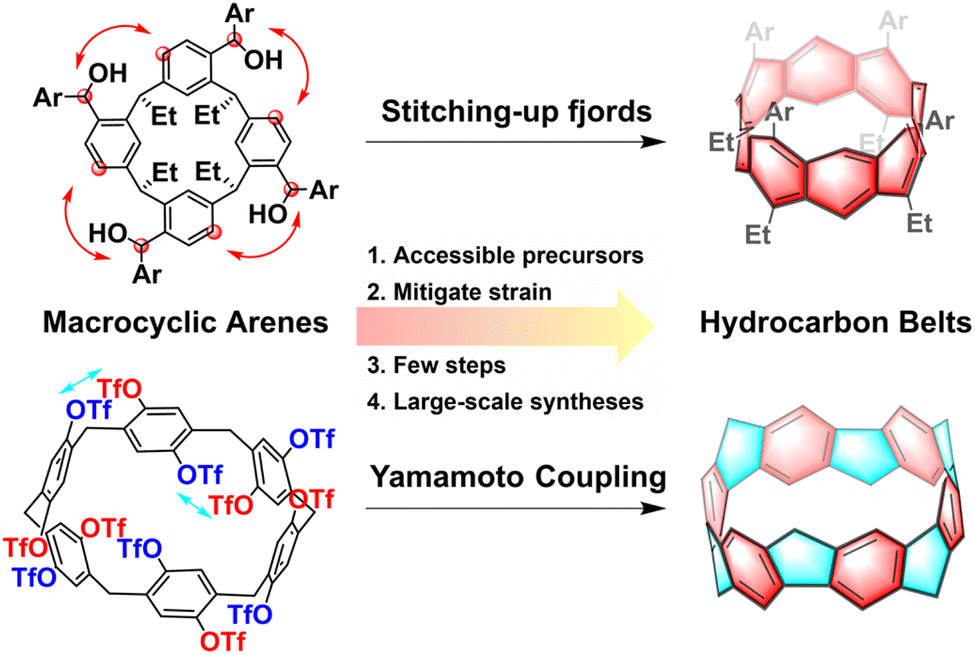 | ||
| Fig. 1 Strategies and advantages for the synthesis of hydrocarbon belts using traditional macrocyclic arenes. | ||
2. Hydrocarbon belts based on different macrocyclic arenes
2.1. Hydrocarbon belts based on resorcin[n]arenes
Resorcin[n]arenes, synthetic macrocyclic hosts in supramolecular chemistry, can be conveniently prepared with relatively high yields via acid-catalyzed condensations involving resorcinol and aldehydes.25,26 In 2018, Lucas first synthesized two hydrocarbon belts from resorcin[4]arenes through Scholl reactions. Subsequently, Wang made significant advancements by introducing a stitching-up-fjords strategy for synthesizing hydrocarbon belts from resorcin[n]arenes in 2020. The key feature of the method is to close the fjord regions of resorcin[n]arenes through multiple intramolecular alkylation reactions such as intramolecular Friedel–Crafts alkylation and acylation, intramolecular olefin metathesis, intramolecular nucleophilic aromatic substitution reaction (SNAr) and C–N cross-coupling.Lucas introduced a novel class of conjugated hydrocarbon belt 4, derived from resorcin[4]arenes derivatives through sequential Scholl reactions.27 The structure of belt 4 reveals that each appended aryl group is positioned adjacent to an available aryl site on the neighboring core ring, facilitating the formation of a new seven-membered ring through intramolecular ortho-annulation. Notably, the core structure of belt 4 shows near-perfect “vase” (C4v) symmetry. While the cavity of belt 4 is notably more constrained compared to conventional extended cavitands featuring tetraaryl and alkyl–ether groups, it can accommodate small neutral guests such as CH2Cl2 and ethylbenzene. The synthetic pathway for belt 4 is delineated in Fig. 2. Initially, the precursor tetratriflate macrocycle 1 was synthesized from a reduced symmetry resorcin[4]arene derivative in four steps. Subsequently, macrocycle 1 underwent a straightforward four-fold Suzuki–Miyaura cross-coupling with 3,4-dimethoxyphenylboronic acid to yield macrocycle 2. Following treatment with FeCl3 in CH3NO2/CH2Cl2, macrocycle 2 exclusively yielded the A/C-bis-annulated product 3 in 90% yield as the chiral P/M racemate. Belt 4 was then obtained in a 48% yield by treating macrocycle 3 with further oxidization with DDQ/TfOH in CH2Cl2. Overall, the concise synthetic route, coupled with the high yields of the products, underscores the efficiency of this approach for synthesizing homologous hydrocarbon belts.
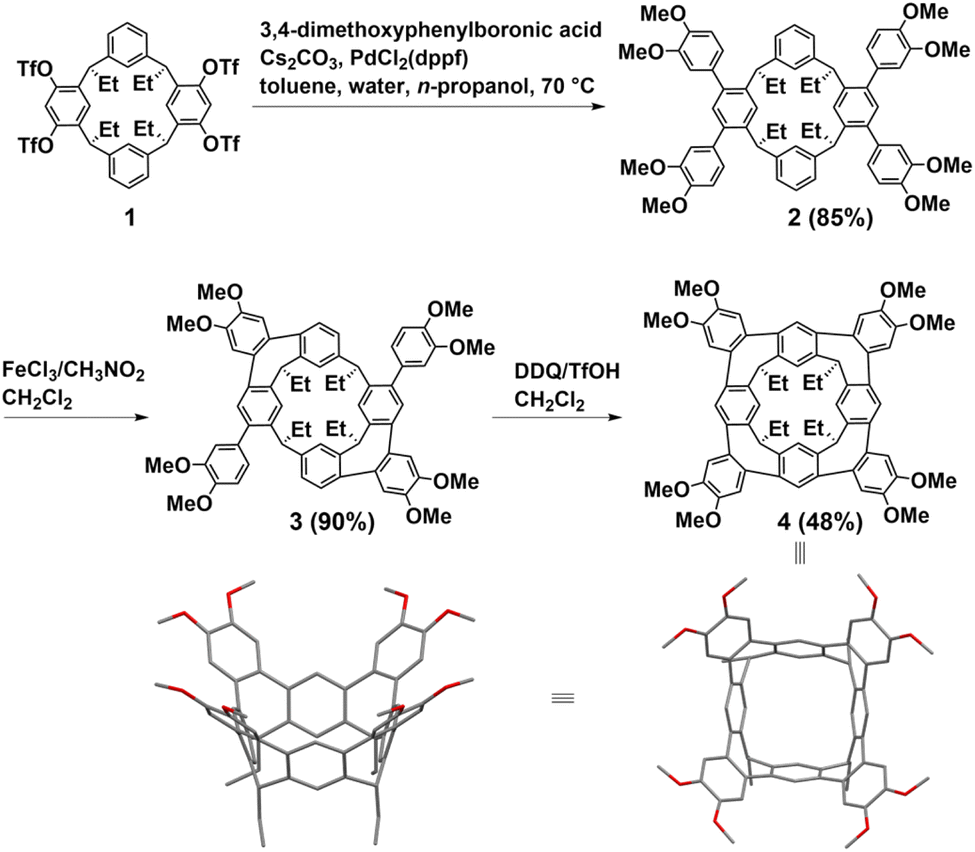 | ||
| Fig. 2 Synthetic route to hydrocarbon belt 4 by Scholl reactions. | ||
Wang employed a similar approach and proposed the “stitching-up fjords” strategies to synthesize fully conjugated and partially saturated belt[n]arenes. This method involved utilizing pre-organized and pre-functionalized constitutions of single-stranded resorcin[n]arenes. Both (partially) saturated belt[8]arene 8 and belt[8]arene 12 exhibit a consistent rigid, belt-shaped core structure of C4v symmetry and a cylindrical configuration, with the average spacing between two directly aligned benzene rings being 5.85 Å. In partially saturated belt[12]arene 20, six benzene rings and six 1,4-cyclohexadiene rings are sequentially fused in a linear fashion to form a nearly-equilateral hexagonal cylinder. The long diagonal averages approximately 1 nm in length, while the cylinder's height is roughly 0.3 nm. The synthetic routes to (partially) saturated belt[8]arene 8 and fully conjugated belt[8]arene 14 are shown in Fig. 3a. The synthesis of belt[8]arene initiated with an asymmetric yet readily accessible derivative of resorcin[4]arene. Through a series of transformations, resorcin[4]arene 5 was synthesized, with four triflate groups positioned at predetermined positions. Tetraisopropenyl-bearing resorcin[4]arenes 6 were obtained by a straightforward Suzuki coupling reaction with a boron reagent. By employing Tf2O, an efficient intramolecular Friedel–Crafts alkylation reaction occurred at room temperature, bridging two distal fjords to form product 7 with a satisfactory yield. Subsequent intramolecular Friedel–Crafts alkylation processes on 7 proceeded smoothly to close up the remaining two fjords, yielding nonconjugated belt[8]arene 8 in 16–19% yields. For conjugated belt[8]arene 14, the Stille coupling between 5 and vinylstannane led to a tetravinylated resorcin[4]arene 9. Ozonolysis of 9 resulted in 10, followed by Grignard reactions that yielded macrocyclic tetraol 11. Closing all four fjords in resorcin[4]arene 11 yielded the (partially) saturated belt[8]arene derivative 12 through an intramolecular Friedel–Crafts alkylation reaction, existing as pairs of crystalline stereoisomers, which were characterized by X-ray crystallography. Subsequent cascade reactions, involving quadruple aromatization and Diels–Alder addition reactions in a single step, resulted in the unstable but isolable belt[8]arene–DDQ adduct 13 by oxidative aromatization of a mixture of stereoisomers 12 with excess DDQ. The core structure of the belt[8]arene precursor 13 featured an alternating fusion of four phenyl rings and four 1,4-cyclohexadiene rings. After sequential retro-Diels–Alder reactions and laser irradiation (λ = 355 nm) of precursor 13, the belt[8]arene derivative 14 was detected in the MALDI mass spectrum.28 Using a similar intramolecular coupling strategy, the symmetric resorcin[6]arene derivative 15 was also utilized in the creation of a larger precursor to the belt[12]arene, known as hydrocarbon belts 20, a type of substituted collar[12]arene (Fig. 3b).29
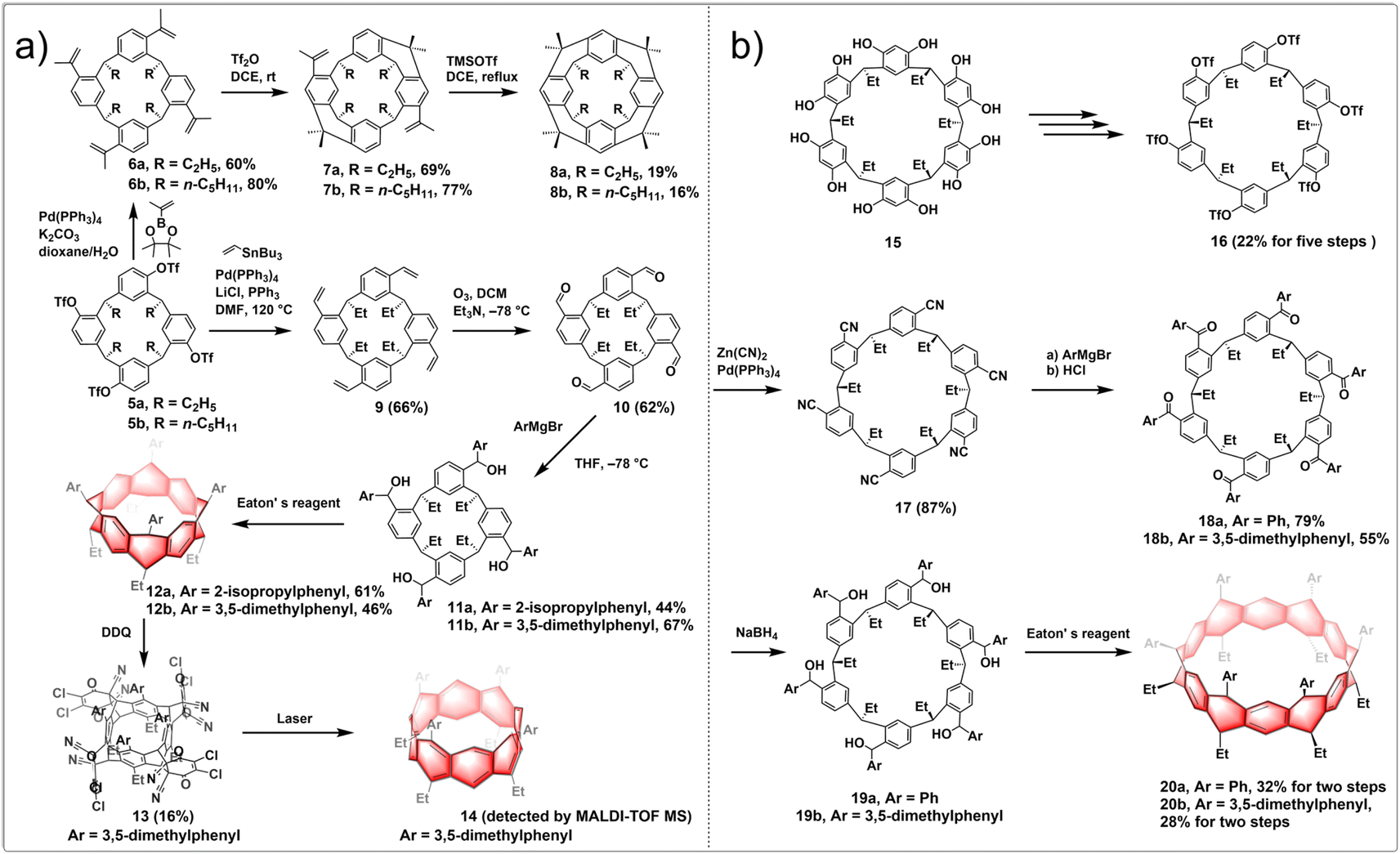 | ||
| Fig. 3 Synthetic routes to (a) belt[8]arene and (b) substituted collar[12]arene by intramolecular Friedel–Crafts alkylation. | ||
This synthetic approach effectively addresses the significant challenges associated with belt[n]arenes, including high strain energy and potential instability during synthesis.30 The “stitching-up fjords” strategy proves to be an effective method to overcome strain energies, while the incorporation of bulky aryl groups in the structure's rim can help stabilize the final product.31 More importantly, these works demonstrated the possibility of constructing fully conjugated zigzag-type hydrocarbon belts. The development of this innovative and pragmatic synthetic approach paves the way for producing belt-shaped molecules utilizing readily accessible macrocyclic arenes.
To investigate the versatility of the stitching-up fjords approach in crafting assorted novel and edge-functionalized zigzag hydrocarbon belts, Wang embarked on a study concerning the synthesis of carbonyl-embedded nanobelts. The research focused on the design of multiple intramolecular acylation reactions; the pivotal steps are sequential “stitching-up fjords” of a resorcin[4]arene derivative 22. This process ultimately yielded the carbonyl-embedded nanobelt 25, a crucial intermediate for the generation of a diverse array of functionalized molecular belts (Fig. 4a). The synthesized belt molecules exhibit a variety of geometric shapes, including the square prism configuration of belt 27, the truncated cone formations of belts 28 and 29, and the elliptical cylinder conformation of belt 30, depending on the functional groups present on the edges. The synthetic routes involved the conversion of resorcin[4]arene derivative 21 into compound 22 through a series of reactions including ozonolysis, oxidization and esterification. Heating 22 in a sealed tube with (CF3CO)2O/TfOH/MeOH led to compound 23 separated from a pair of isomers. Subsequent reduction of 23 using Et3SiH/TFA as a reducing agent afforded product 24 in an excellent yield. Molecular belt 25 was synthesized through the intramolecular Friedel–Crafts acylation reaction of the carboxylic acid of 24 under the influence of Eatons' reagent, albeit in a slightly lower yield (56%). Reducing the two carbonyl groups of 25 with NaBH4 led to diol-functionalized belt 26, while the treatment of 25 with n-butyllithium resulted in the formation of tertiary alcohol-containing belt 27. Besides, following the Baeyer–Villiger oxidation process, two isomeric belt molecules were produced, including the C2v-symmetric dilactone 28 and the meso-dilactone 29. Heating compound 25 with (PhSeO)2O in chlorobenzene also led to a dealkylative carbonylation reaction occurring preferentially at both methine positions, ultimately yielding molecular belt 30, which incorporated two 1,4-quinone subunits, in a satisfactory yield.32
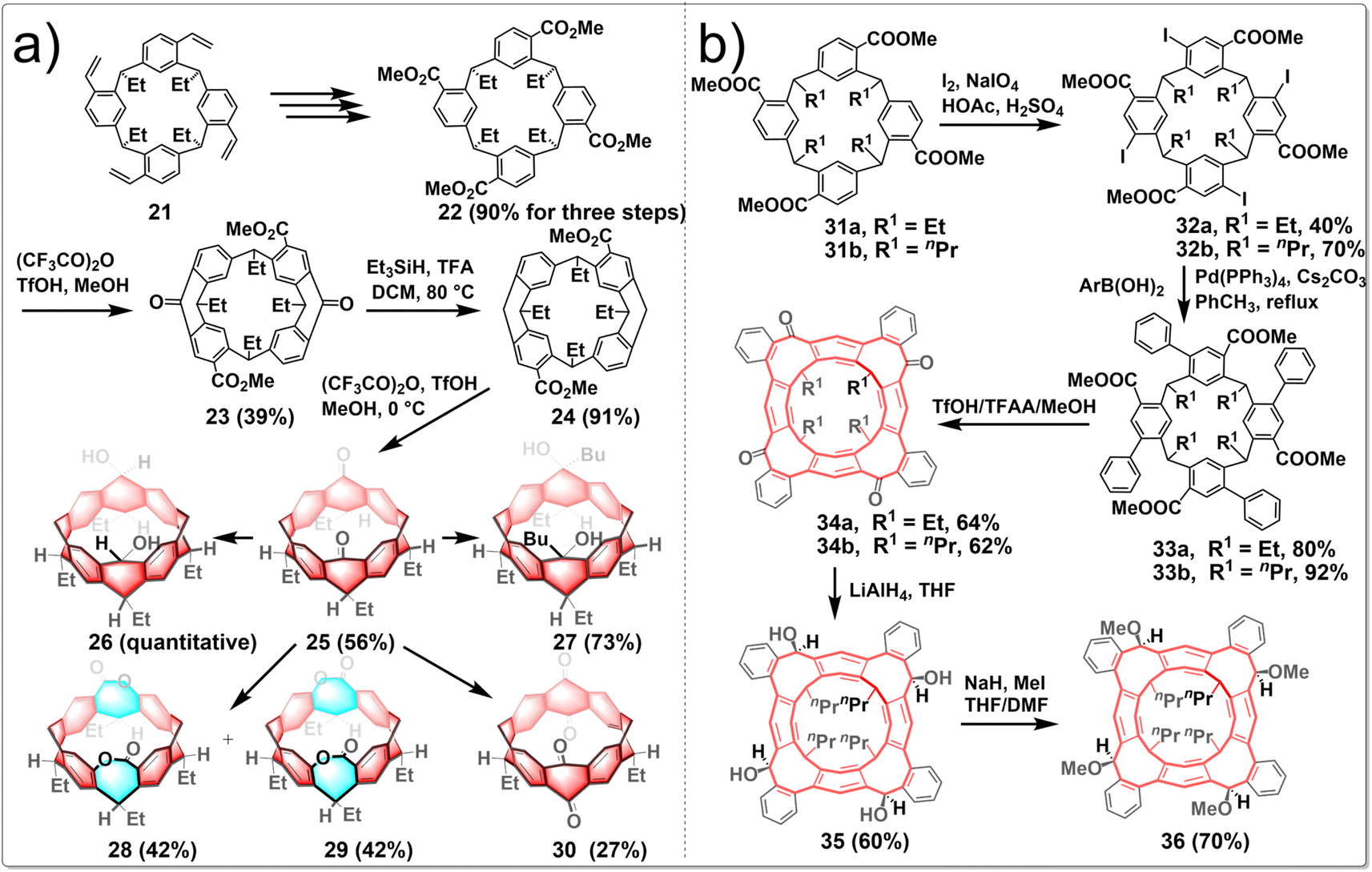 | ||
| Fig. 4 Synthetic routes to (a) biscarbonyl-functionalized hydrocarbon belts and (b) zigzag-type hydrocarbon belts containing functionalized eight-membered rings by intramolecular Friedel–Crafts acylation. | ||
Wang reported several unprecedented zigzag-type hydrocarbon belts featuring functionalized eight-membered rings. The resulting belt molecules 34, 35, and 36 exhibited subtly twisted belt structures with different truncated conical cavities, in which the interconnected eight-membered rings adopt a boat–boat configuration. The synthetic route is shown in Fig. 4b. The treatment of macrocycle 31 with a combination of iodine and NaIO4 in an acetic acid and sulfuric acid mixture resulted in the creation of iodoarene 32 with yields ranging from 40% to 70%. Subsequent fourfold Suzuki–Miyaura coupling reactions of iodoarene 32 with various arylboronic acids led to the production of compound 33. These compounds underwent multiple intramolecular Friedel–Crafts acylation reactions, facilitated by a mixture of CF3SO3H, (CF3CO)2O, and MeOH, to yield the nanobelt 34a with 64% yield. The ketone groups within these molecular belts were crucial sites for further chemical modifications, enabling the synthesis of other functionalized belts. Conversely, reduction of 34 with LiAlH4 yielded a mixture of stereoisomers, with compound 35 emerging as the predominant product at a 60% yield. The exhaustive methylation of 35 using methyl iodide in the presence of NaH produced a methoxylated belt 36. Furthermore, the functionalized belts were powerful hosts to form stable complexes with cesium ion. Additionally, enantiopure C4-symmetric molecular belts can be readily obtained and exhibited notable circularly polarized luminescence (CPL) activity.33 Generally, Friedel–Crafts acylation represents a potent strategy for constructing edge-functionalized hydrocarbon belts, as it facilitates the introduction of ketone moieties, which are readily amenable to further modification.
A stitching-up fjords strategy featuring exhaustive triflation of phenolic hydroxyl groups followed by the transition-metal-catalyzed vinylation and intramolecular olefin metathesis allowed for the efficient construction of belt[n]arene[n]tropilidenes (n = 4, 6). These belts adopted double-stranded structures with distinctive truncated cone cavities. The process began with cost-effective and readily available resorcin[n]arenes. Diverse hydrocarbon belts with a range of structural variations were synthesized through selective hydrogenation of olefin and phenyl subunits. Furthermore, the resultant molecular belts functioned as synthetic host materials for small compounds like p-xylene and nitromethane. The synthetic route is shown in Fig. 5a. In the presence of pyridine as an acid scavenger, the reaction of resorcin[4]arenes 37 with an excess of Tf2O at room temperature produced triflate intermediates 38 in nearly quantitative yields. The subsequent Stille cross-coupling reaction between 38 and tributyl(vinyl)stannane yielded vinyl-substituted resorcin[4]arenes 39. Then intramolecular olefin metathesis of 39 was successfully conducted in heated CH2Cl2 with Grubbs-II catalyst, leading to the synthesis of the targeted hydrocarbon belt molecules 40.34 The oxidation of 40 with benzeneseleninic anhydride selectively produced tetrakis(α-diketone)-functionalized belt intermediates. Then a subsequent condensation reaction afforded hydrocarbon belts 41 fused with quinoxaline moieties.35 In summary, this research established a concise and efficient synthetic method for creating functionalized hydrocarbon belts and showed that these belts are powerful macrocyclic hosts for molecular recognition and self-assembly.
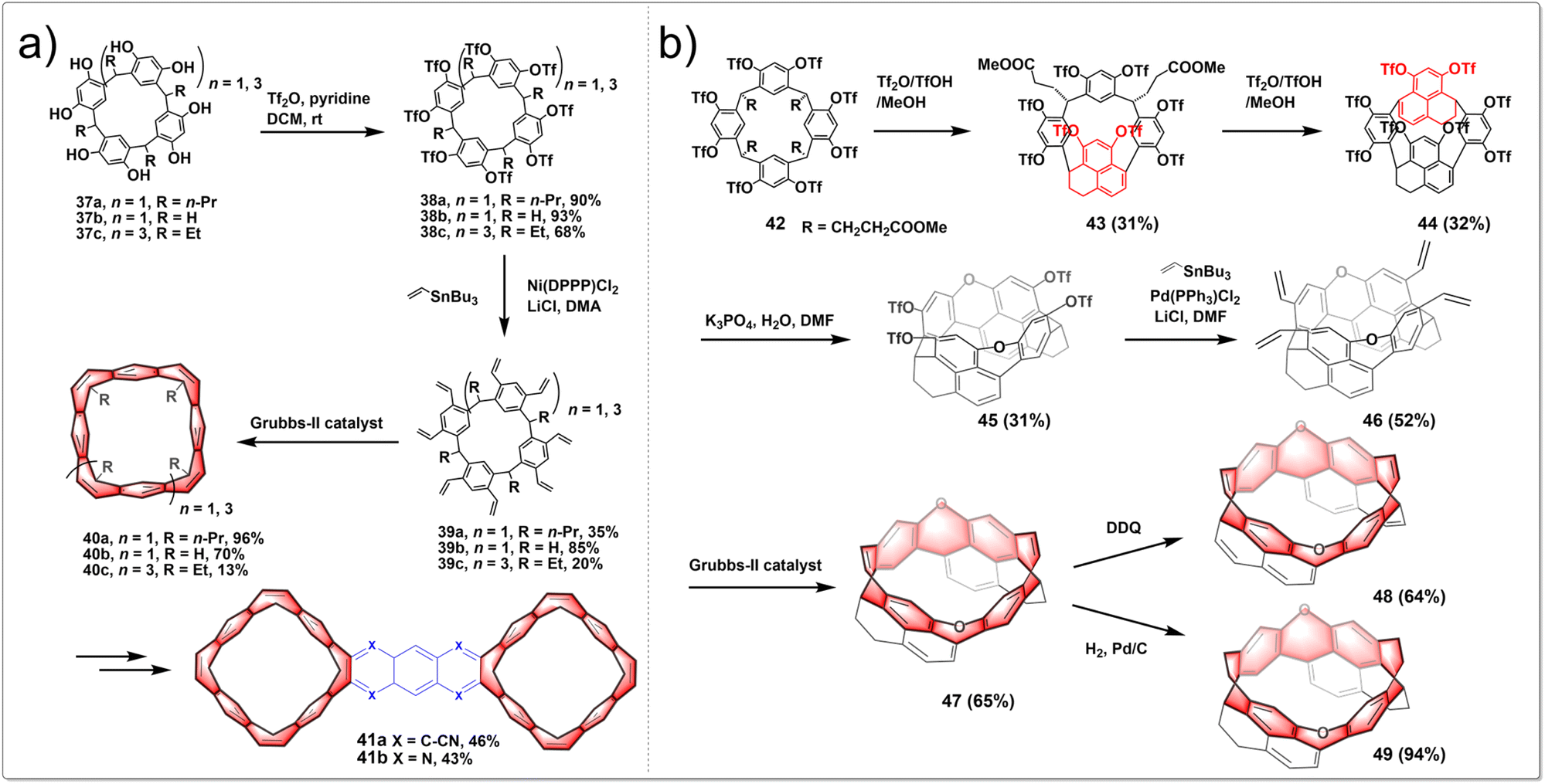 | ||
| Fig. 5 Synthetic routes to (a) belt[n]arene[n]tropilidenes and (b) oxygen-doped chiral hydrocarbon belts by intramolecular olefin metathesis. | ||
By employing intramolecular nucleophilic aromatic substitution and ring-closing olefin metathesis reactions, highly strained, depth-expanded oxygen-doped chiral molecular belts 47, 48 and 49 were successfully synthesized. These novel molecular belts have more rigid cavities and superior chiroptical properties compared to previously reported ones. It demonstrated that the chiral belts 47, 49 and 49 exhibit excellent CPL activities, with a maximum g luminescence dissymmetry factor (glum) reaching 0.022. The synthetic route is shown in Fig. 5b. Initially, a resorcinol derivative 42 was heated in a mixture of trifluoroacetic acid anhydride Tf2O, TfOH and MeOH, yielding two products, including 43 and its isomeric counterpart. Subsequent heating of compound 43 in a mixture of Tf2O, TfOH, and MeOH led to a separable mixture of macrocycle 44 and its isomer in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Treatment of a solution of 44 in DMF with K3PO4 and H2O yielded a dioxa-linked half-belt product 45via a double intramolecular nucleophilic aromatic substitution reaction. Efficient Stille cross-coupling of 45 with a vinyltin reagent produced a tetravinyl-substituted half-belt product 46. A dual olefin metathesis, catalyzed by Grubbs-II catalyst, yielded a dioxa-embedded zigzag-type molecular belt 47 in 65% yield. Oxidizing 47 with DDQ in refluxing toluene resulted in belt 48. Meanwhile, catalytic hydrogenation of 47 achieved the product 49 in almost quantitative yield.36 In conclusion, this research paves the way for the creation of chiral hydrocarbon molecular belts with expanded depth.
:1 ratio. Treatment of a solution of 44 in DMF with K3PO4 and H2O yielded a dioxa-linked half-belt product 45via a double intramolecular nucleophilic aromatic substitution reaction. Efficient Stille cross-coupling of 45 with a vinyltin reagent produced a tetravinyl-substituted half-belt product 46. A dual olefin metathesis, catalyzed by Grubbs-II catalyst, yielded a dioxa-embedded zigzag-type molecular belt 47 in 65% yield. Oxidizing 47 with DDQ in refluxing toluene resulted in belt 48. Meanwhile, catalytic hydrogenation of 47 achieved the product 49 in almost quantitative yield.36 In conclusion, this research paves the way for the creation of chiral hydrocarbon molecular belts with expanded depth.
The architectures of zigzag hydrocarbon belts and their heteroatom-embedded versions are not only aesthetically appealing, but also possess intriguing physical and chemical characteristics. Belts with embedded heteroatoms could serve as fundamental templates or seeds for the development of zigzag-type layered materials featuring heterostructures, holding promise for applications in optoelectronic. The synthesis of various oxygen/nitrogen-doped zigzag hydrocarbon belts has been achieved by intramolecular SNAr and intermolecular C–N cross-coupling processes from resorcin[4]arene derivatives. To construct heteroatom-linked octahydrobelt[8]arenes, cyclization reactions are facilitated by the preorganized conformations of mono-macrocyclic, half-belt, and quasi-belt compounds. The six-membered heterocyclic rings in the resulting hydrocarbon belts 51, 53, 55 exhibited a distinctive boat conformation with equatorially positioned alkyl groups, leading to the formation of strained square-prism-shaped belt structures. Furthermore, the photophysical and redox properties of the novel heteroatom-bearing belts differed from those of their octahydrobelt[8]arene counterparts. The synthetic route is shown in Fig. 6a. The one-pot reaction of 50 under basic conditions using K3PO4 yielded the target products 51. After optimizing the reaction conditions, this practically convenient, one-pot method improved the yield of 51 to 8–34%. The formation of belts 53 from the reaction of 52 and ArNH2 did not consistently yield high amounts due to the challenge posed by the accumulation of macrocyclic strain, making the formation of the second N-heterocycle difficult. To circumvent the problem, cross-coupling reaction between quasi-belt intermediates 54 and ArNH2 under the identical palladium-catalyzed conditions afforded belts 53, with greatly improved yields. When compounds 54 were treated with K3PO4 in hot DMF, the desired belt products 55 were generated as the sole belt products in 60–81% yields.37 These unique belts containing heteroatoms also showed distinct photophysical and redox characteristics compared to those of their octahydrobelt[8]arene counterparts. Subsequently, they conducted the efficient synthesis of a highly strained nitrogen-doped zigzag hydrocarbon belts, which involved an unconventional four-fold m-bromination, followed by Pd-catalyzed intramolecular C–N bond formation. Additionally, the oxidation of a p-methoxyphenyl-substituted belt[4]arene facilitated the formation of a singlet diradical dication nitrogen-doped zigzag belts.38
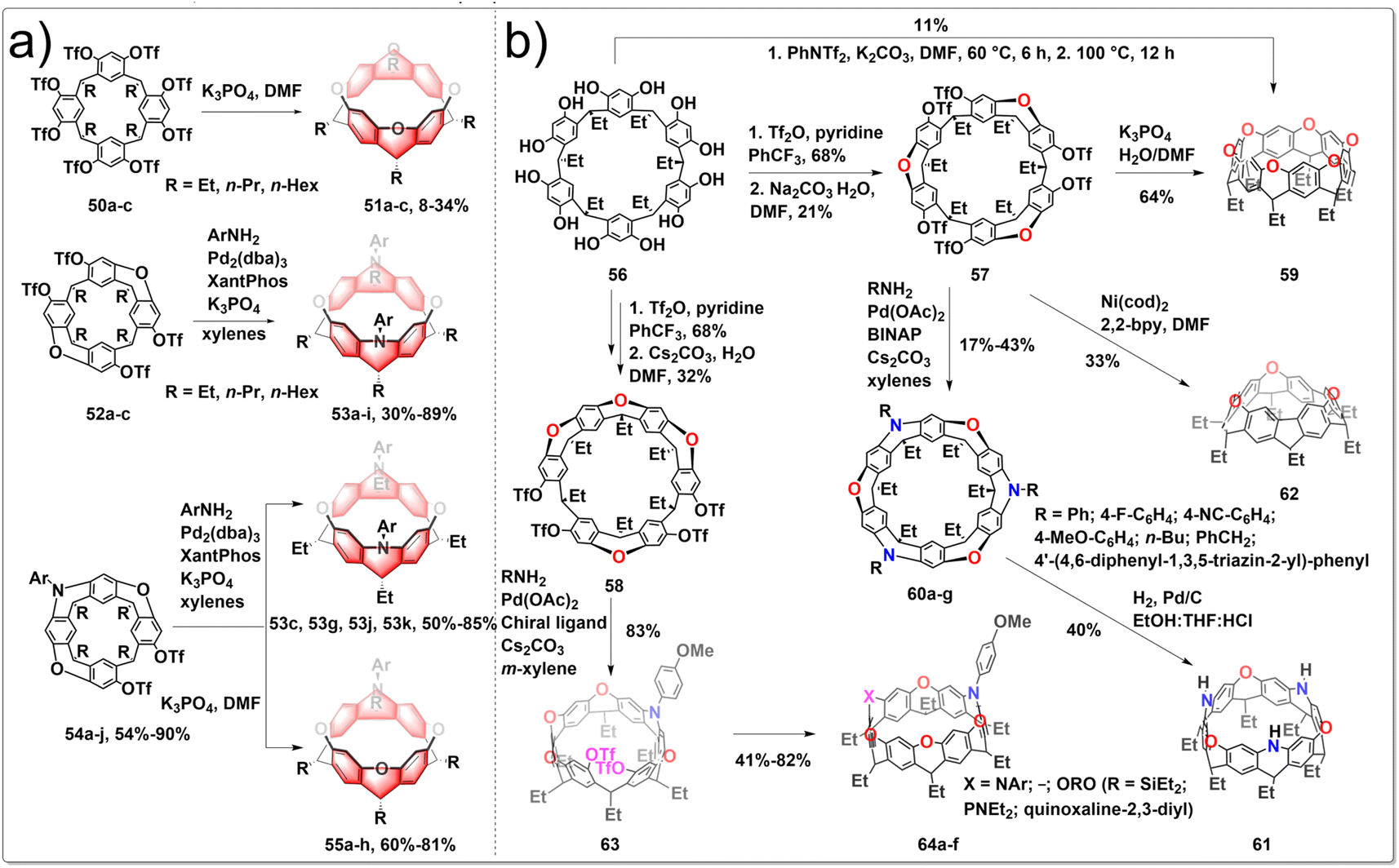 | ||
| Fig. 6 Synthetic routes to diverse (a) oxygen/nitrogen-doped hydrocarbon belts and (b) heteroatom-doped zigzag-type hydrocarbon belts by intramolecular SNAr and C–N cross-coupling. | ||
The incorporation of heteroatoms into zigzag-type hydrocarbon belts has been shown to give rise to intricate and symmetrically complex structures that are inherently chiral, exhibiting exceptional physical and chemical properties with potential applications. Additionally, the exploration of the chiral nature of heteroatom-incorporated zigzag belts represents a novel and significant research direction. Recently, Wang developed a series of heteroatom-doped zigzag-type hydrocarbon belts from readily available resorcin[6]arene.39 The synthetic route is shown in Fig. 6b. Resorcin[6]arene 56 was obtained conveniently as a by-product in the synthesis of resorcin[4]arene, serving as the starting materials. After carefully screening the reaction conditions, half belts 57 and 58 were prepared selectively in acceptable yields. In the presence of K3PO4, compound 57 underwent selective hydrolysis and the subsequent SNAr reactions produced belt 59 in good yields. Under the catalysis of Pd(OAc)2/BINAP, RNH2 reacted with 57 to form the corresponding triaza–trioxa-doped belts 60 with various aromatic substituents and chromophores decorating the belt edge. Catalytic hydrogenolysis of 60 led to the formation of NH-embedded belt 61, a valuable intermediate for the fabrication of diverse belt products simply based on the versatile reactivities of NH moieties. Mediated by Ni(cod)2/2,2-bipyridine, compound 57 underwent triple intramolecular Yamamoto coupling reactions to afford belt 62 in 33% yield. The high enantiopurity of intermediate 63 from the desymmetrizative C–N bond formation reaction of 58 with p-anisidine suggested the excellent enantioselectivity. After undergoing various chemical transformations, enantiopure chiral nanobelts 64 were obtained with ee values exceeding 99%.
2.2. Hydrocarbon belts based on pillar[n]arenes and prism[6]arene
Pillararenes are a class of macrocyclic molecules composed of 2,5-dialkoxyphenyl units connected by methylene (–CH2–) bridges.40,41 Among them, pillar[5]arene stands out as the most conformationally stable member, with higher homologues like pillar[6–15]arenes being synthesized through the expansion of pillar[5]arene rings.42 Meanwhile, prism[6]arenes, with a folded, cuboid-shaped conformation, are synthesized through methylene-bridging macrocyclization of 2,6-diethoxynaphthalene in high yield.43,44 Motivated by the pioneering concept by Vögtle to create cycloparaphenylenes by starting with already existing macrocycles, researchers were encouraged to explore the synthesis of a new type of non-alternant aromatic belts. In this unique structure, the cyclic paraphenylene chain is ladderized with methylene bridges, with functionalized pillararenes and prismarenes being considered as promising precursors for constructing new hydrocarbon belts.In 2020, Itami introduced a method for synthesizing methylene-bridged hydrocarbon belts based on pillar[6]arenes. This approach involved utilizing a triflate-functionalized pillar[6]arene as a macrocyclic precursor and employing a six-fold nickel-mediated Yamamoto coupling as the intramolecular cyclization process to generate non-alternant aromatic belts. The synthetic route is shown in Fig. 7a. Initially, perethylated pillar[6]arene 65 was efficiently prepared from 1,4-diethoxybenzene in satisfactory yield. Then, the fully hydroxylated form of pillar[6]arene 66 was achieved through de-ethylation with BBr3 in an 88% yield and 67 was obtained following triflation in 68% yield. Following these steps, a nickel-mediated intramolecular Yamamoto coupling reaction was conducted, leading to the isolation of the hydrocarbon belt product 68a in 18% yield. X-ray diffraction analysis validated the structural characteristics of the hydrocarbon belt 68a, revealing a slightly cylindrical elliptical deformation in the crystal lattice. Notably, the strain energy of the 68a falls between [6]cycloparaphenylenes and [6,6]nanobelt, up to 110.2 kcal mol−1.45 In 2023, Itami further reported the synthesis of size-dependent belts, based on pillar[8]arenes and pillar[10]arenes using the same strategy as for 68a. By optimizing the reaction conditions, they improved the overall yield of 68a to 1.7%. Additionally, belts 68b and 68c were obtained in overall yields of 0.04 and 0.1%, respectively. Furthermore, an interesting “paratrooper band” current along the backbone of 68a–c was found both experimentally and theoretically.46
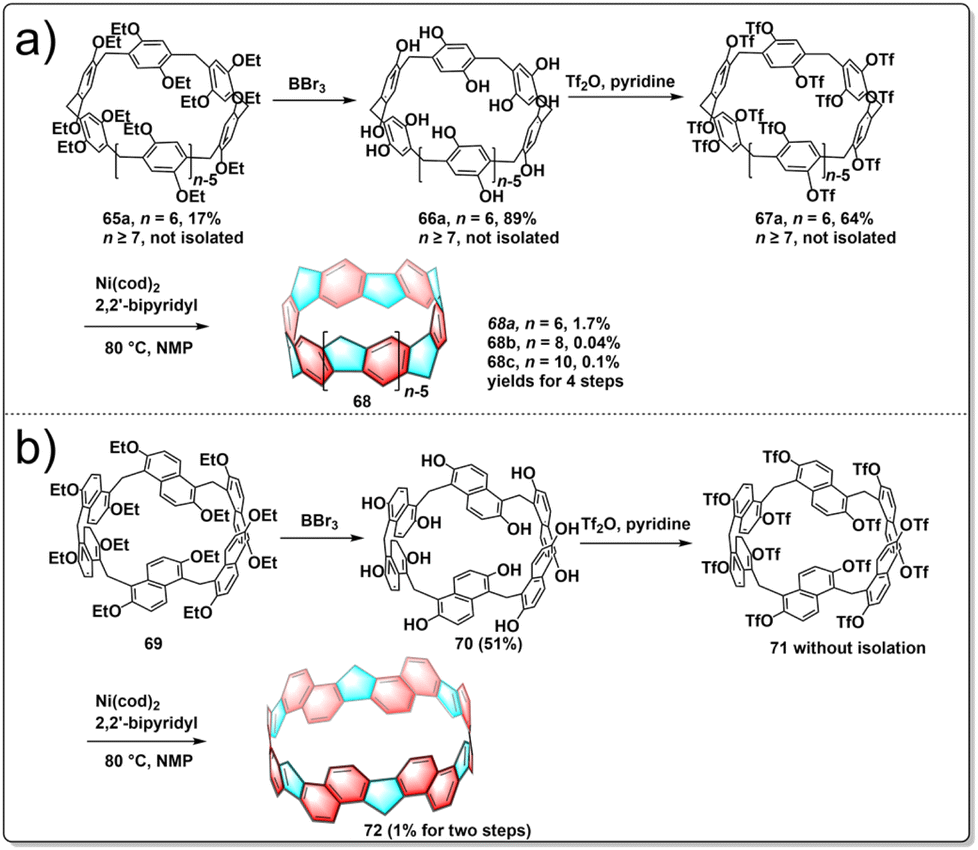 | ||
| Fig. 7 Synthetic routes to hydrocarbon belts from (a) pillar[n]arenes and (b) prism[6]arene. | ||
The production of alternative methylene bridged nanobelts is of tremendous interest. Encouraged by the results of previous experiments, in 2023, another novel structurally constrained hydrocarbon belt, methylene bridged [6]cyclo-2,6-naphthylene 72, was synthesized from triflated prism[6]arene 71.47 The polyhydroxylated prism[6]arene 69 was synthesized through methylene-bridging macrocyclization of 2,6-diethoxynaphthalene in 51% yield. The synthesis of 72 was achieved in three steps by the same strategy as used for belts 68a–c. After purification, 72 was successfully isolated in 1% yield over two steps from 70. It was found that 72 has a lower solubility and stability under air and light than do 68a–c (Fig. 7b). Belt 72 has a low strain energy (77.4 kcal mol−1) and displays bright fluorescence (Φ = 0.20). This result highlights the fact that the synthesis of size-dependent and extended conjugated hydrocarbon belts through intramolecular Yamamoto reactions is a general and effective approach.
2.3. Hydrocarbon belts based on fluoren[3]arenes and calix[3]carbazole
Although hydrocarbon belts have been successfully synthesized from pillararenes and prism[6]arene, their syntheses were inefficient due to the inward vertical rotation of the OTf groups, lowering their reactivities. Despite the wide range of macrocyclic arenes reported so far, the macrocyclic structures conducive to generating hydrocarbon belts remain limited. In 2021, Chen developed a new approach to synthesize conjugated hydrocarbon belts using two different new macrocyclic arenes, fluoren[3]arenes and calix[3]carbazoles. Fluoren[3]arenes are conveniently synthesized in 48–67% yield by BF3·OEt2-catalyzed condensation between 2,7-alkoxyl-substituted fluorenes and paraformaldehyde.48 Calix[3]carbazole is prepared in 73% yield by the FeCl3·6H2O-catalyzed one-pot condensation between 2,7-dimethoxy-9-phenylcarbazole and paraformaldehyde in dichloromethane.49 Followed by demethylation, triflation and intramolecular aryl–aryl coupling reactions, C(sp3)-bridged [6]cycloparaphenylenes and nitrogen-doped aromatic belts were then obtained in high yield, providing a general and highly efficient approach to the synthesis of hydrocarbon belts from these easily available macrocyclic arenes.The synthetic route to belts 76 is shown in Fig. 8a. Initially, catalyzed by BF3·OEt2, fluoren[3]arenes 73 were synthesized by one pot condensation of 2,7-dialkoxyl-substituted fluorenes and polyformaldehyde. After BBr3 demethylation and trifluoromethanesulfonic anhydride treatment, compounds 75 were obtained in yields up to 98%. Intramolecular aryl coupling was carried out through Ni(cod)2 to obtain aromatic belts 76, with yields up to 90%, significantly higher than previously reported methods. This new approach effectively addresses the long-standing issues of low yields in the synthesis of aromatic belts. Chen also conducted the synthesis of belt 78, characterized by a highly symmetrical rigid structure and strong blue fluorescence. The synthesis started from triflated 2,7-fluoren[3]arene 75, and 78 was conveniently obtained through transition-metal-catalyzed vinylation and intramolecular olefin metathesis in good yield. Subsequently, 78 was modified with acetyl chloride and AlCl3 to obtain 79 with a single acetyl group, achieving a yield of 56%. Following this, pure chiral isomers P-79 and M-79 were obtained by chiral separation. These enantiomeric forms of 79 not only exhibit strong green fluorescence and narrow energy gaps but also display CPL properties with a glum value of 0.002. This provides an effective method for the design and construction of inherently chiral conjugated macrocycles with strong fluorescence and CPL properties, which will facilitate their application in supramolecular chemistry and materials science.50
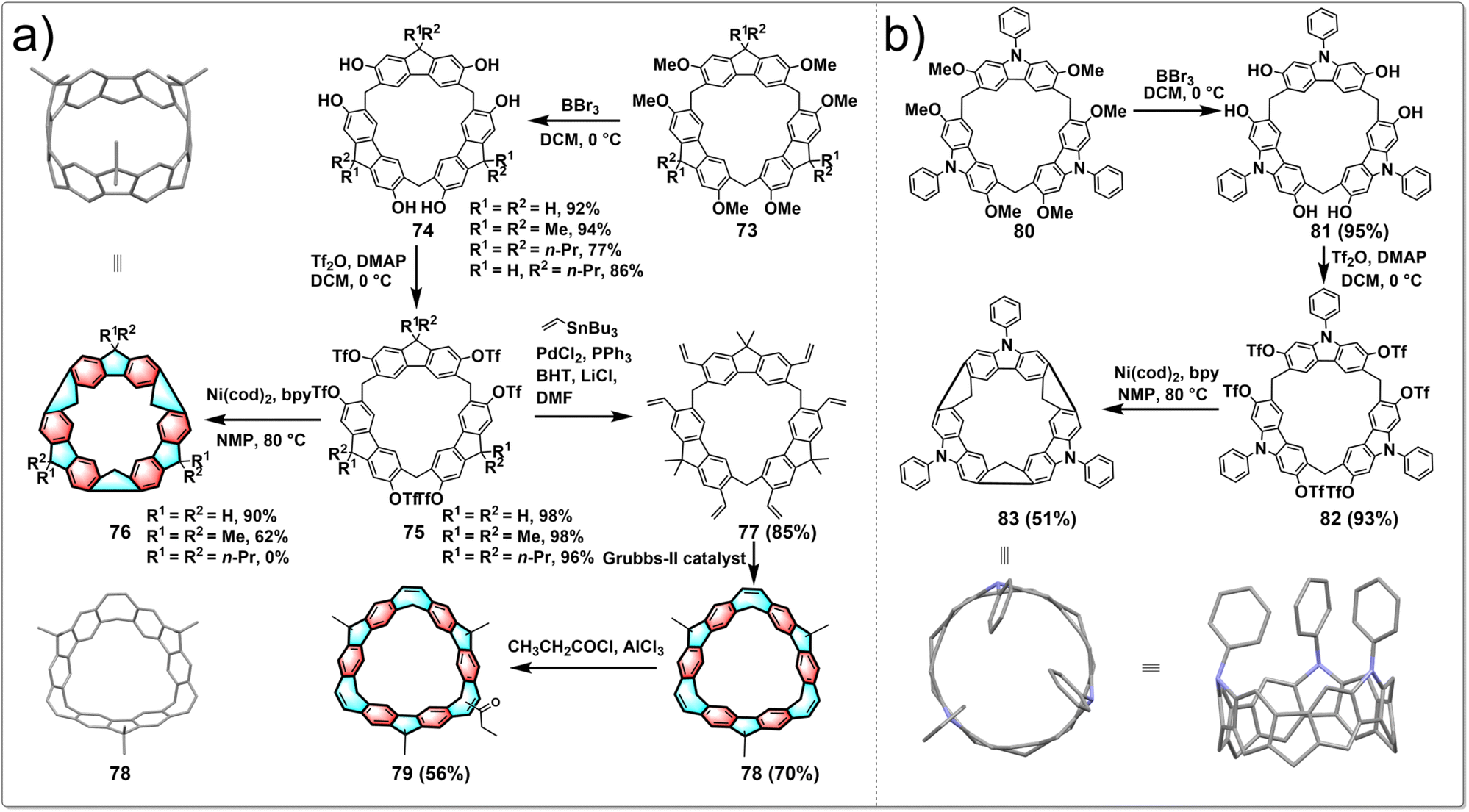 | ||
| Fig. 8 Synthetic routes to hydrocarbon belts from (a) fluoren[3]arenes and (b) calix[3]carbazoles. | ||
Inspired by the highly efficient synthesis of hydrocarbon belts 76 from fluoren[3]arenes and the excellent performance of nitrogen-doped carbon nanotubes, Chen reported the pioneering development of the first “nitrogen-doped” aromatic belt containing [6]cycloparaphenylenes skeleton, achieving a breakthrough in the field of aromatic belts. The synthetic strategy resembles that for 76, as shown in Fig. 8b. Target molecular belt 83 was obtained through demethylation, triflation and intramolecular aryl–aryl coupling reactions in high yield. The aromatic belt 83 features a rigid conjugated structure and deep cavity, capable of encapsulating a dichloromethane molecule in both solution and solid state. Remarkably, this belt displays intense green fluorescence and exhibits a narrow HOMO–LUMO energy gap. In addition, the introduction of the carbazole moiety renders the aromatic belt a promising organic donor for the design of new organic functional materials like thermally activated delayed fluorescence materials.51
3. Summary and outlook
In this review, we have outlined the synthetic methodologies utilized in the production of hydrocarbon belts derived from a range of readily available macrocyclic arenes, including resorcin[n]arenes, pillar[n]arenes, prism[6]arene, calix[3]carbazole, and fluoren[3]arenes. Our focus has been on the pivotal macrocyclization and belt-forming steps, involving the Scholl reaction, intramolecular Friedel–Crafts alkylation and acylation, intramolecular olefin metathesis, intramolecular SNAr and C–N cross-coupling, as well as Yamamoto coupling techniques. The “stitching-up fjords” approach, originating from resorcin[n]arenes and proceeding via multiple intramolecular alkylation processes, emerges as a promising strategy for the facile and effective synthesis of partially hydrogenated nanobelts.Although significant progresses have been achieved in the synthesis of hydrocarbon belts using diverse strategies, this field remains in its infancy, offering abundant prospects for further investigation. Areas yet to be explored include the synthesis of Vögtle[n]belts, [n]cyclophenacenes, and [n]cyclacenes.3 Furthermore, the advancement of effective ring-closure techniques for the formation of hydrocarbon belts is essential, and there should be an increased focus on employing novel macrocyclic arenes in the creation of hydrocarbon belts, such as acridane[n]arene, xanthene[n]arenes, biphen[n]arenes, helic[n]arenes and so on.16,24 Exploring new methodologies to overcome these limitations represents a promising avenue for future research. Lastly, a comprehensive exploration of the properties of hydrocarbon belts remains pending. Investigations into their host–guest properties, optical and electronic properties and chiral properties are still in their early stages. Understanding and harnessing these properties will unlock a myriad of applications across diverse fields.
Author contributions
G. F., Z. Z. and G. W. performed the literature search, analyzed the published results, and wrote the manuscript. G. F., Z. Z., G. W., L. S. prepared the review figures. B. H. and F. H. provided key advice and supervised the preparation of the text and revised the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
F. H. thanks the National Key Research and Development Program of China (2021YFA0910100), the National Natural Science Foundation of China (22035006, 22320102001, 22350007), Zhejiang Provincial Natural Science Foundation of China (LD21B020001), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006), and the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (2022R01005) for financial support. F. H. thanks The Chemistry Instrumentation Center of Zhejiang University for technical support. B. H. thanks the National Natural Science Foundation of China (22201252) and the Baima Lake Laboratory Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (LBMHZ24E020003).References
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 1–14 CrossRef.
- Y. Segawa, A. Yagi, K. Matsui and K. Itami, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed.
- Q. Guo, Y. Qiu, M. Wang and J. F. Stoddart, Nat. Chem., 2021, 13, 402–419 CrossRef CAS PubMed.
- T. Shi and M. Wang, CCS Chem., 2021, 3, 916–931 CrossRef CAS.
- K. Cheung, Y. Segawa and K. Itami, Chem.–Eur. J., 2020, 26, 14791–14801 CrossRef CAS PubMed.
- S. Nishigaki, Y. Shibata, A. Nakajima, H. Okajima, Y. Masumoto, T. Osawa, A. Muranaka, H. Sugiyama, A. Horikawa, H. Uekusa, H. Koshino, M. Uchiyama, A. Sakamoto and K. Tanaka, J. Am. Chem. Soc., 2019, 141, 14955–14960 CrossRef CAS PubMed.
- J. Nogami, Y. Nagashima, H. Sugiyama, K. Miyamoto, Y. Tanaka, H. Uekusa, A. Muranaka, M. Uchiyama and K. Tanaka, Angew. Chem., Int. Ed., 2022, 61, e202200800 CrossRef CAS PubMed.
- J. Nogami, D. Hashizume, Y. Nagashima, K. Miyamoto, M. Uchiyama and K. Tanaka, Nat. Synth., 2023, 2, 888–897 CrossRef.
- J. Zhu, Y. Han, Y. Ni, G. Li and J. Wu, J. Am. Chem. Soc., 2021, 143, 2716–2721 CrossRef CAS PubMed.
- J. Xie, X. Li, S. Wang, A. Li, L. Jiang and K. Zhu, Nat. Commun., 2020, 11, 3348 CrossRef CAS PubMed.
- J. Wang and Q. Miao, Org. Lett., 2019, 21, 10120–10124 CrossRef CAS PubMed.
- Y. Segawa, D. R. Levine and K. Itami, Acc. Chem. Res., 2019, 52, 2760–2767 CrossRef CAS PubMed.
- D. Eisenberg, R. Shenhar and M. Rabinovitz, Chem. Soc. Rev., 2010, 39, 2879–2890 RSC.
- R. Gleiter, B. Esser and S. Kornmayer, Acc. Mater. Res., 2009, 42, 1108–1116 CAS.
- H. Chen and Q. Miao, J. Phys. Org. Chem., 2020, 33, e4145 CrossRef CAS.
- X. Han, Y. Han and C. Chen, Chem. Soc. Rev., 2023, 52, 3265–3298 RSC.
- F. H. Kohnke, A. M. Z. Slawin, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 1987, 26, 892–894 CrossRef.
- P. R. Ashton, N. S. Isaacs, F. H. Kohnke, A. M. Z. Slawin, C. M. Spencer, J. F. Stoddart and D. J. Williams, Angew. Chem., Int. Ed., 2003, 27, 966–969 CrossRef.
- K. Y. Cheung, K. Watanabe, Y. Segawa and K. Itami, Nat. Chem., 2021, 13, 255–259 CrossRef CAS PubMed.
- Y. Han, S. Dong, J. Shao, W. Fan and C. Chi, Angew. Chem., Int. Ed., 2021, 60, 2658–2662 CrossRef CAS PubMed.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed.
- K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838–847 CAS.
- Z. Xia, S. Pun, H. Chen and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 10311–10318 CrossRef CAS.
- J. Pfeuffer-Rooschuz, L. Schmid, A. Prescimone and K. Tiefenbacher, JACS Au, 2021, 1, 1885–1891 CrossRef CAS.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- M. J. McIldowie, M. Mocerino, B. W. Skelton and A. H. White, Org. Lett., 2000, 2, 3869–3871 CrossRef CAS.
- J. N. Smith and N. T. Lucas, Chem. Commun., 2018, 54, 4716–4719 RSC.
- T. Shi, Q. Guo, S. Tong and M. Wang, J. Am. Chem. Soc., 2020, 142, 4576–4580 CrossRef CAS.
- T. Shi, S. Tong and M. Wang, Angew. Chem., Int. Ed., 2020, 59, 7700–7705 CrossRef CAS PubMed.
- Y. Segawa, A. Yagi, H. Ito and K. Itami, Org. Lett., 2016, 18, 1430–1433 CrossRef CAS PubMed.
- T. Shi, S. Tong, L. Jiao and M. Wang, Org. Mater., 2020, 2, 300–305 CrossRef CAS.
- Y. Zhang, S. Tong and M. Wang, Angew. Chem., Int. Ed., 2020, 59, 18151–18155 CrossRef CAS PubMed.
- Y. Peng, S. Tong, Y. Zhang and M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302646 CrossRef CAS PubMed.
- Q. Zhang, Y. Zhang, S. Tong and M. Wang, J. Am. Chem. Soc., 2020, 142, 1196–1199 CrossRef CAS PubMed.
- Y. Zhang, S. Tong and M. Wang, Org. Lett., 2021, 23, 7259–7263 CrossRef CAS.
- J. Chen, Z. Jiang, H. Xiao, S. Tong, T. Shi, J. Zhu and M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301782 CrossRef CAS PubMed.
- M. Tan, Q. Guo, X. Wang, T. Shi, Q. Zhang, S. Hou, S. Tong, J. You and M. Wang, Angew. Chem., Int. Ed., 2020, 59, 23649–23658 CrossRef CAS PubMed.
- M. Xie, S. Tong and M. Wang, CCS Chem., 2023, 5, 117–123 CrossRef CAS.
- X. Wang, X. Zhang, S. Tong, Q. Guo, M. Tan, C. Li and M. Wang, CCS Chem., 2024, 6, 1198–1210 CrossRef CAS.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- X. Hu, Z. Chen, L. Chen, L. Zhang, J. Hou and Z. Li, Chem. Commun., 2012, 48, 10999–11001 RSC.
- R. D. Regno, P. D. Sala, D. Picariello, C. Talotta, A. Spinella, P. Neri and C. Gaeta, Org. Lett., 2021, 23, 8143–8146 CrossRef.
- P. D. Sala, R. D. Regno, L. D. Marino, C. Calabrese, C. Palo, C. Talotta, S. Geremia, N. Hickey, A. Capobianco, P. Neri and C. Gaeta, Chem. Sci., 2021, 12, 9952–9961 Search PubMed.
- Y. Li, Y. Segawa, A. Yagi and K. Itami, J. Am. Chem. Soc., 2020, 142, 12850–12856 CrossRef CAS.
- H. Kono, Y. Li, R. Zanasi, G. Monaco, F. F. Summa, L. T. Scott, A. Yagi and K. Itami, J. Am. Chem. Soc., 2023, 145, 8939–8946 CrossRef CAS.
- K. Itami, A. Yagi, H. Kono and N. Kai, Synlett, 2023, 34, 1433–1436 Search PubMed.
- X. Du, D. Zhang, Y. Guo, J. Li, Y. Han and C. Chen, Angew. Chem., Int. Ed., 2021, 60, 13021–13028 CrossRef CAS PubMed.
- X. Zhou, G. Li, P. Yang, L. Zhao, T. Deng, H. Shen, Z. Yang, Z. Tian and Y. Chen, Sens. Actuators, B, 2017, 242, 56–62 CrossRef CAS.
- X. Du, X. Han, Y. Han and C. Chen, Chem. Commun., 2023, 59, 227–230 RSC.
- F. Zhang, X. Du, D. Zhang, Y. Wang, H. Lu and C. Chen, Angew. Chem., Int. Ed., 2021, 60, 15291–15295 CrossRef CAS PubMed.
Footnote |
| † G. F., Z. Z. and G. W. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |