
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in block copolymer-phospholipid hybrid vesicles: from physical–chemical properties to applications
Edit
Brodszkij
 * and
Brigitte
Städler
* and
Brigitte
Städler
Interdisciplinary Nanoscience Center (iNANO) Aarhus University, Gustave Wieds Vej 14, 8000 Aarhus C, Denmark. E-mail: ebk@inano.au.dk
First published on 24th June 2024
Abstract
Hybrid vesicles, made of lipids and amphiphilic block copolymers, have become increasingly popular thanks to their versatile properties that enable the construction of intricate membranes mimicking cellular structures. This tutorial review gives an overview over the different hybrid vesicle designs, and provides a detailed analysis of their properties, including their composition, membrane fluidity, membrane homogeneity, permeability, stability. The review puts emphasis on the application of these hybrid vesicles in bottom-up synthetic biology and aims to offer an overview of design guidelines, particularly focusing on composition, to eventually realize the intended applications of these hybrid vesicles.
1 Introduction
Over the past 20 years since the first reports on hybrid vesicles (HVs),1–4 ongoing research has illustrated that the incorporation of amphiphilic block copolymers into lipid bilayers changes the properties of the vesicles, e.g., the stability is enhanced in comparison to liposomes. The combination of block copolymers with lipids not only provides greater design flexibility but also preserves the self-assembly capabilities of the vesicles. Consequently, these HVs are gaining popularity as versatile platforms for drug delivery applications and synthetic biology.5 Applications of HVs in synthetic biology have been summarized in several reviews focusing on the obtained artificial systems.6–9 In particular, HVs hold great promise as an alternative for assembling plasma membranes imitates, especially when precise control over the membrane properties is essential for the development of synthetic life-like systems.10–14 Recently, Reimhult et al.15 reviewed the use of HVs for drug delivery applications comparing selected properties. Further, HVs appeared in many reviews focusing on lipid-based drug delivery vehicles, where they are highlighted as game changing prospects owing to their versatility.16–21 However, HVs in a descriptive manner were last time summarized by Schulz et al.22,23 and Le Meins et al.24 almost 10 years ago and only a selection of assemblies was mentioned by Go et al.25The aim of this tutorial review is to provide a detailed overview of the fundamental aspects as well as the recent advances and achievements in the area of HV assembly. We focus on various aspects of the vesicle and membrane properties. We aim to conduct a comprehensive comparison of numerous assemblies, with the intention of deriving conclusions that can provide inform for design criteria for an optimal synthetic cell membrane. Throughout this tutorial review, HV assemblies are referred to by their composition to highlight the used polymer & lipid.
2 Assembly and composition
To date, various polymers have been combined with phospholipids to assemble HVs and many have demonstrated long-term stability. Table 1 provides a summary of these assemblies including the type and ratio (mol% or w/w%) of their building blocks and the references to the publications. The choice of lipids typically depends on the study's objectives or application. Fully saturated lipid such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) or unsaturated zwitterionic lipids such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) or L-α-phosphatidylcholine (Soy) (Soy PC) are often the first choices. The most commonly used polymer for the hydrophilic block is poly(ethylene glycol) (PEG) or referred to poly(ethyl oxide) (PEO) in some publications. In contrast, the polymers used as the hydrophobic block are more diverse including poly(1,2-butadiene) (PBd), poly(isobutylene) (PiB), poly(dimethyl siloxane) (PDMS) and poly(cholesterol methacrylate) (PCMA). The chemical structures of the lipids and the polymers are summarized in Scheme 1.| Polymer | Lipid | Polymer content | Ref. |
|---|---|---|---|
| a Abbreviations: Lipids: DPPC: 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; DMPC: 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DOPC: (1,2-; dioleoyl-sn-glycero-3-phosphocholine); DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt); Soy PC (L-α-phosphatidylcholine (Soy)); Soy PS: anionic soy PS; POPC: 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine; EcCL: E. coli lipid extract and cholesterol; HSPC: L-α-phosphatidylcholine, hydrogenated (soy); DPhPC: 1,2- diphytanoyl-sn-glycero-3-phosphocholine; DOPS: 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt); DOPE: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine; Diyne PC(DC8,9PC): 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phosphocholine; egg SM: sphingomyelin (egg, chicken); EPC: L-α-phosphatidylcholine (egg, chicken) polymer: PMOXA: polymethyloxazoline; PDMS: poly(dimethyl siloxane); PEG: poly(ethylene glycol); PBd: polybutadiene; PIB: poly(isobutylene); PI: polyisoprene; PCL: poly(ε-caprolactone); PLA: poly(lactic acid); APOEGA: poly(oligoethylene glycol acrylate); PLauA: poly(lauryl acrylate); PIPPAm or PNIPAM: poly(N-isopropyl amide); PLMA: poly(lauryl methacrylate); PCMA: poly(cholesterol methacrylate); DMAAm or PDMAEMA: poly(dimethyl aminoethyl methacrylate); PCEA: poly(2-carboxyethyl acrylate); PHEMA: poly(2-hyroxyethyl mathacrylate); BuMA: poly(i-butyl methacrylate); PMPC: poly(2-methacryloyloxyethyl phosphorylcholine); PPO: poly(propylene oxide). | |||
| PMOXA21-b-PDMS73-b-PMOXA21 | DPPC | 10–60 mol% | 1 |
| PDMS60-b-PMOXA21 | DMPC | 90 mol% | 26 |
| PMOXA15-PDMS67-PMOXA15 PMOXA6-PDMS33-PMOXA6 | DOPC | 27 | |
| PDMS-g-PEG2 | DPPC, DOPC | 28 | |
| PDMS22-g-(PEG16)2 | Soy PC | 60–100 mol% | 29–34 |
| DOPC | |||
| DPPC DOTAP | |||
| Soy PS | |||
| PDMS16-g-(PEG12)2) | DPPC | 0–100 w/w% | 35–37 |
| PEG8-b-PDMS22-b-PEG8 | POPC | ||
| PEG12-b-PDMS43-b-PEG12 | |||
| PEG17-b-PDMS67-b-PEG17 | |||
| PDMS36-b-PEG23 | DOPC | 50, 80 w/w% | 38 |
| PDMS23-b-PEG13 | POPC | 75–90 w/w% | 39–42 |
| PDMS27-b-PEG17 | |||
| PDMS36-b-PEG23 | |||
| PEG8-b-PDMS22-b-PEG8 | POPC | 41 | |
| PEG12-b-PDMS43-b-PEG12 | |||
| PEG3-b-PDMS15-b-PEG3 | DOPC | 0.05–0.18 wt% | 43 |
| PEG14-b-PDMS15-b-PEG14 | |||
| PEG28-b-PDMS15-b-PEG28 | |||
| PBd22-b-PEG14 | HSPC | 75, 90 mol% | 44 |
| DOTAP | 75 mol% | 45 | |
| POPC, EcCl | 5–75 mol% | 46–53 | |
| Soy PC | 50 mol% | 54 | |
| PBd12-b-PEG11 | POPC | 25–75 mol% | 51–53 |
| PBd46-b-PEG30 | POPC | 20–90, mol% | 55 |
| PBd12-b-PEG9 | DOPS | 50% | 56 |
| PBd32-b-PEG21 | DOPC | ||
| PBd37-b-PEG22 | DOPC | 10–50 mol% | 58 |
| PBd23-b-PEG13 | DOTAP, DOPS, DOPC | 20–80% | 59 and 60 |
| PBd33-b-PEG22 | |||
| PBd37-b-PEG55 | |||
| PBd73-b-PEG44 | |||
| PBd22-b-PEG23 | DPPC, POPC | 70 w/w% | 61 and 62 |
| PBd11-b-PEG8 | DPPC, POPC egg SM | 63–65 | |
| PBd-b-PEG | POPC | Simulation | 66 |
| PBd12-b-PEG10 | DOPC, DOPS | 50–99 mol% | 67 |
| DOPE | |||
| PBd43-b-PEG20 | DPPC | 35, 65% | 68 |
| PIB87-b-PEG17 | DPPC | 50 mol% | 69–71 |
| PIB37-b-PEG48 | DOPC | ||
| PI-b-PEG (877 Da) | DPhPC | 90 mol% | 72 |
| PI-b-PEG (1530 Da) | |||
| PI-b-PEG (3188 Da) | |||
| PEG113-b-PCL46 | DPPC | 5, 10 mol% | 73 and 74 |
| PEG113-PCL50 | |||
| PEG113-b-PCL23 | |||
| PCL12-PEG45-PCL12 | DPPC | 0.001–2 mol% | 75 |
| PCL16-PEG104-PCL16 | |||
| PEG12-b-PCL9 | POPC | 50 mol% | 76 |
| PEG12-b-PCL6 | |||
| PEG45-b-PCL44 | DPPC | 20 mol% | 77 |
| PEG15-b-PLA25 | POPC | 50 mol% | 78 |
| PEG45-b-PLA54 | |||
| PEG12-b-PCL25 | |||
| PEG45-b-PCL25 | |||
| PEG45-b-PCL42 | |||
| PEG114-b-PCL88 | DPPC | 10−3 g L−1, 0.2 g L−1 | 79 |
| PEG114-b-P(CL42-co-LA85) | |||
| PEG114-b-P(CL14-co-LA138) | |||
| PEG114-b-PLA328 | |||
| P(IPPAm-co-DMAAm)-b-PLA | Diyne PC(DC8,9 PC) | 27 w/w% | 80 |
| POEGA-b-PLauA | HSPC | 81 | |
| PNIPAM106-b-PLauA25 | DPPC | 0.2–10 mol% | 82 |
| PNIPAM28-b-PLauA13 | |||
| PDMAEMA33-b-PLMA15 | HSPC | 83 | |
| PDMAEMA32-b-PLMA23 | |||
| PDMAEMA31-b-PLMA8 | EPC | 10 mol% | 84 |
| PDMAEMA58-b-PLMA23 | |||
| PEG113-P(8C-Chol)m m = 1.4, 2.2, 2,8, 3.3, 3.7, 4,8 | DPPC | 5 mol% | 85 |
| PCMA-b-PDMAEMA | POEPC, POPS, POPE, POPC | 90 w/w% | 86 and 87 |
| PCMA11-b-PEG113 | POPC/POPS | 88 | |
| PCMA11-b-P(METMA7-co-CEA104) | DOPC | 27 w/w% | 89 |
| PCMA10-b-PCEA170 | DOPC, DOPC, DOPE | 10 w/w% | 90–92 |
| P(CMA9-co-BuMA9)-b-PCEA186 | Soy PC | 1–90 w/w% | 93 |
| P(CMA8-co-HEMA3)-b-PCEA186 | |||
| PBuMA50-b-PCEA175 | |||
| PHEMA58-b-PCEA150 | |||
| PCMA-b-PMPC | DOPS, DOPC, DOPE | 94 | |
| Oligo(aspartic acid)4-b-PPO43 | DOPC | 10 mol% | 95 |
| Maltopentaose-b-PPO43 | DOPC | 10–90 mol% | 96 |
 | ||
| Scheme 1 Chemical structures of the lipids (top row) and the hydrophilic and hydrophobic polymers used in the block copolymers. PDMAEMA is also used as a hydrophilic block. For abbreviations meaning see Table 1. | ||
The block copolymer molecular mass typically falls within the range of 1.2 to 20 kDa. Block copolymers with low molecular mass share a more comparable size to the lipid counterpart. They were early on assumed to construct vesicles with a similar curvature, and hence being more suitable to form HVs than high molecular weight block copolymers. Even though it seems that an apparent size mismatch may lead to higher degree of phase separation between the lipids and the block copolymers (see later for details), using low molecular weight block copolymer is not a requirement. However, the properties of the HV are most likely to depend on the length of the block copolymer chains and their ability to mix in various ratios (Scheme 2).
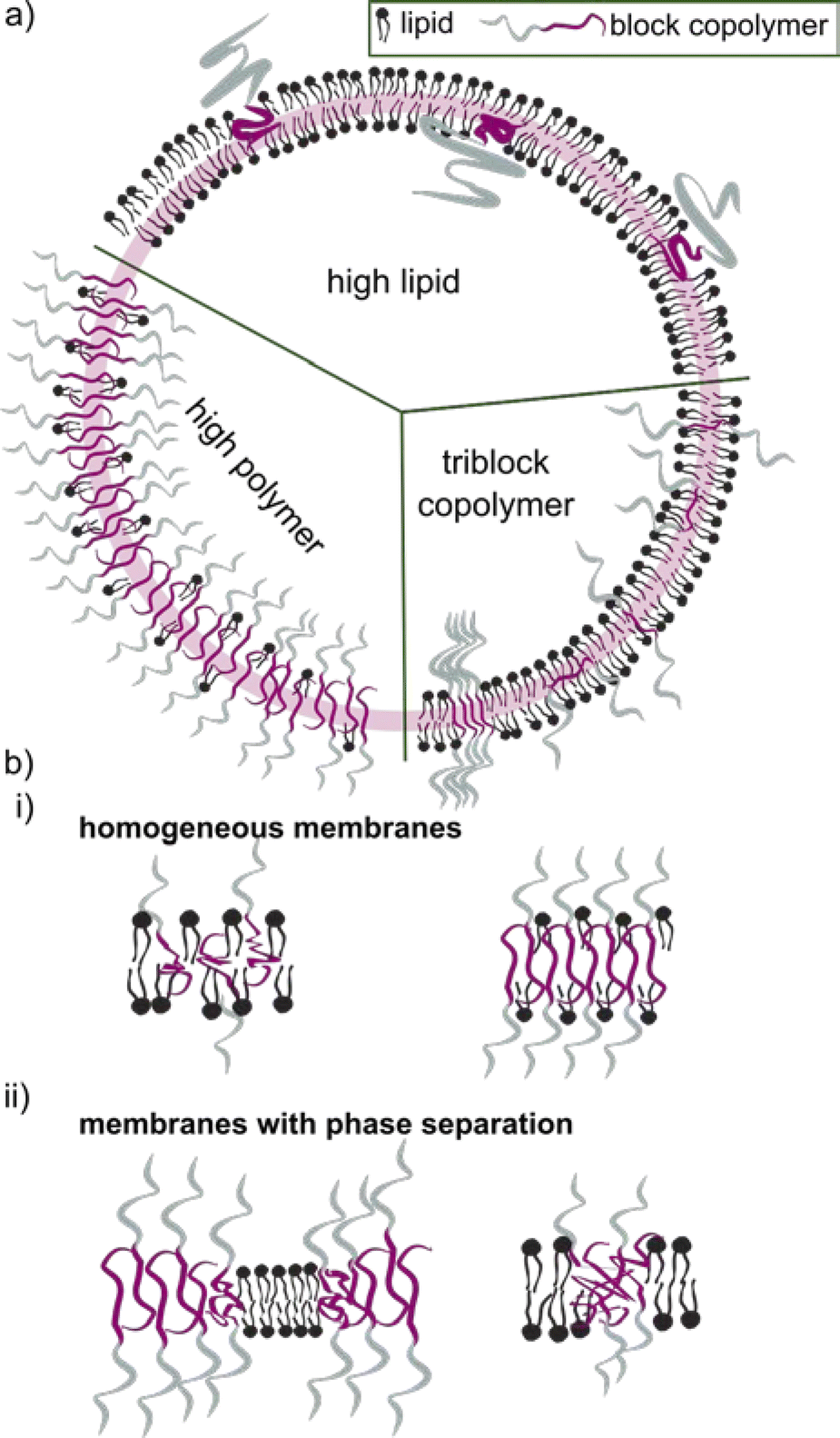 | ||
| Scheme 2 (a) Schematic representation of hybrid vesicles composed of lipids and block copolymers with various lipid block copolymer ratios. (b) Cartoon of the distribution of lipids and block copolymers in either a homogeneous membrane (i) or a membrane that shows phase separation (ii). | ||
Although small HVs and large HVs are spherical assemblies with sizes below 100 nm and 100 nm and below 1 μm, respectively, this review will refer to both these sizes of vesicles as smaller HV (sHV). Often authors tend to use small and giant as a description to distinguish between the reported vesicle types, that is misleading nomenclatura. sHVs are typically assembled using the thin film hydration method, which is well established for liposomes and polymersoms formation.5 Briefly, the block copolymer and lipid components are mixed in an organic solvent followed by solvent evaporation either under vacuum on a rotary evaporator or with a stream of nitrogen while rotating the flask, to create a thin lipid-polymer film that is left to dry under vacuum for 4–12 h. The film is rehydrated with aqueous solution (pure water, buffer, cell media etc.) that can contain cargo for encapsulation into the void of the vesicles. The assemblies are then subjected to either sonication or extrusion through polycarbonate membranes to form unilamellar sHVs in the range of 50–150 nm with narrow polydispersity. sHVs are often passed through a size exclusion column to remove micelles or non-encapsulated cargo. Complementarily, giant unilamellar HVs (gHVs) (sizes above 1 μm) are usually assembled via electroformation where a thin polymer lipid film is deposited on the surface of an electrode, usually an ITO coated glass slide. An electric current is applied during the rehydration step to induce the controlled formation of gHVs. Aqueous media with low conductivity (e.g., distilled water) can only be used, therefore, sucrose or glucose solutions are often considered to fill the gHVs in order to allow for their subsequent exposure to physiological buffers without rupturing due to osmotic imbalance between the gHVs void and the environment.97 Emulsion-based methods,98 which are often used for giant liposome assembly, are not common for gHVs formation. However, this method was used to form asymmetrical vesicles.99
The precise process by which block copolymers insert into the lipid bilayer remains debated. It is generally assumed that the components simultaneously detach from the surface during the rehydration phase and form a vesicular structure due to the hydrophobic effect.100 Nonetheless, evidence suggests that block copolymers can incorporate into pre-existing liposome membranes. For example, Mineart et al.85 demonstrated that the block copolymer PEG113-b-P(8C-Chol) could be incorporated into the lipid bilayer post-assembly by adding a micelle solution to pre-made liposomes. Similarly, PCMA-b-PCEA-GALA dissolved in DMSO was inserted into pre-made DOPC liposomes.91 Henderson et al.57 showed that PBd-b-PEG polymersomes could fuse with DOPC liposomes when the mixture was subjected to high salt concentration and mechanical stimuli.
The successful formation of HVs is typically confirmed through various methods including assessing Förster Resonance Energy Transfer (FRET) effects, measuring changes in zeta potential,60,88 determining membrane thickness in recorded cryogenic electron microscopy (cryo-EM) images, utilizing small angle X-ray scattering (SAXS) and monitoring alterations in various membrane properties.
Determining alterations from the theoretical expected membrane composition is important when the effect of the building blocks on the resulting membranes properties is investigated. However, confirming the exact constitution is challenging. The composition of the assemblies after rehydration are commonly reported to be the same as the ratio of the polymer and the lipid in the deposited thin films. However, this is not necessarily the case as it was pointed out by Dao et al. when studying the effect of the polymer's molecular weight on PEG-b-PDMS-b-PEG & DPPC-based assemblies.36 Cryo-EM images revealed the coexistence of various types of structures such as HVs, micelles, liposomes, and polymersomes. They argued that this finding was the result of the high line tension originating from the differences in thickness of the polymer-rich vs. lipid-rich areas. Similar observations were made for PBD-b-PEG & DPPC-based68 and poly(oligoethylene glycol acrylate)-b-poly(lauryl acrylate) POEGA-b-PLauA & hydrogenated L-α-phosphatidylcholine (HSPC)-based83 assemblies. We also showed recently that assemblies made of P(CMA-co-butyl methacrylate (BuMA))-b-PCEA or P(CMA-co-hydroxyethyl methacrylate (HEMA))-b-PCEA & soy PC93 and PCMA-b-poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC)) & DOPC94 had a micellar fraction present when more than ∼1 mol% block copolymer was used. However, we could only speculate if the micellar fraction contained lipids or consisted of block copolymers only. Furthermore, cryo-EM analysis of the vesicles indicated the presence of multiple populations (liposomes, HVs and polymersomes) in several other studies based on membrane thickness analysis.30,36,40
3 Size and composition of smaller HVs (sHVs)
The sizes of sHVs are determined usually by dynamic light scattering (DLS) and reported as hydrodynamic diameter (Dh) and polydispersity (PDI), which ranges, typically, between 40–400 nm with PDI of 0.1–0.3. Several studies have been carried out to determine the influence of the composition on the size of the HVs by changing the molecular weight of the block copolymer, by varying the block copolymer to lipid ratios, or by incorporating charged lipids among others. Determining the size of the sHVs is a commonly performed first characterization effort, which gives an indication of successful HV formation. PDI values above 0.3 are usually considered too polydisperse, suggesting assembly of non spherical vesicles or the presence of multiple populations. The curvature of the liposomes depends on the length of the lipid molecules, i.e., longer chains produce liposomes with larger diameters (Dh). Similarly, longer chain block copolymers form polymersomes with larger diameters. Therefore, the hydrophobic chain length is expected to influence the diameter of the sHVs due to the size mismatch between the lipids and block copolymers.The amount of the block copolymer content also affects the sHV size. For instance, increasing the PBd37-b-PEG22 content in PBd37-b-PEG22 & DOPC from 10 to 50 mol% increased the Dh from 150 to 200 nm.58 Similarly, the incorporation of higher amounts of poly(ε-caprolactone) (PCL)n-b-PEGm-b-PCLn into DPPC vesicles increased the Dh from 112 to 243 nm.75 In addition, the Dh of PB22-PEG14 & POPC-based HVs with 25, 50 or 75% block copolymer content were 60–80% smaller than the respective liposomes and polymersomes.46
However, there are also examples where the changes in Dh due to variations in composition were very small. For instance, the Dh slightly decreased with increasing the block copolymer content from 10 to 25 w/w% for PCMA11-b-PEG113 & POPS-based HVs.88 Similarly, Dh of PBd22-b-PEG14 & E. coli lipid extract-based HVs only changed a few nm when the block copolymer content was increase from 25 to 75 mol%.48 In another study, increasing the amount of PMOXA6-PDMS33-PMOXA6 from 10 to 20 mol% did not significantly affect the Dh of the resulting HVs. However, increasing the molecular weight of this block copolymer to PMOXA15-b-PDMS67-b-PMOXA15 led to an increase in Dh from 80 to 120 nm, which further increased by doubling the amount of block copolymer.27 This trend implies that the longer block copolymer chain prefers to form a vesicle with higher curvature. However, a contradictory example reported no change in Dh of approximately 100 nm for sHVs made of POPC and either PEG15-b-poly(lactic acid)PLA25 or PEG45-b-PLA54.78 It should be noted that low block copolymer content might not impact the vesicles curvature. In the same study, HVs made of either PEG45-b-PCL25 or PEG45-b-PCL42 & POPC had a very broad range of Dh with a main peak around 20 nm indicating the presence of a large micellar population.
The impact of the HV composition on the size is likely also associated with the interactions occurring between the block copolymer chains and the lipids. For instance, the size of the HVs was affected by the charge of the phospholipids (POPC vs. POPS vs. POEPC) when using PCMA11-b-poly(dimethyl aminoethyl methacrylate)77 (PDMAEMA), i.e., HVs containing POPC and POPS were 10–25% smaller than the corresponding liposomes, while POEPC containing HVs were ∼16% larger.86 This observation was presumably due to the attraction or repulsion between the slightly positively charged PDMAEMA, and the charge of the phospholipids. In contrast, when a block copolymer with a zwitterionic hydrophilic block (PCMA-b-PSMA) was used for HV assembly, their size did not change significantly independent whether 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), DOPC or a mixture of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)/DOPC was employed.94 In another example, by employing PCMA and its copolymers with BuMA or HEMA in the hydrophobic block, the size of the resulting HVs together with soy PC decreased from 178 nm to 150 nm upon the introduction of more hydrophilic monomers.93 This reduction reached ∼116 nm with an increasing block copolymer content. Here, we observed a micellar population, which might have influenced the measured Dh. Transmission electron microscopy (TEM) and cryo-EM images can also be used to gain information of the size and morphology of the HVs (Fig. 1). Recently, the effect of the polymer's hydrophilic length on the morphology of the vesicles was studied. Cryo-EM images showed that when the hydrophilic block length increased from 0.12 to 1.2 kDa in PEG-b-PDMS-b-PEG, the resulting HVs with DOPC became more spherical from distorted sphere. However the PDI also increased.43
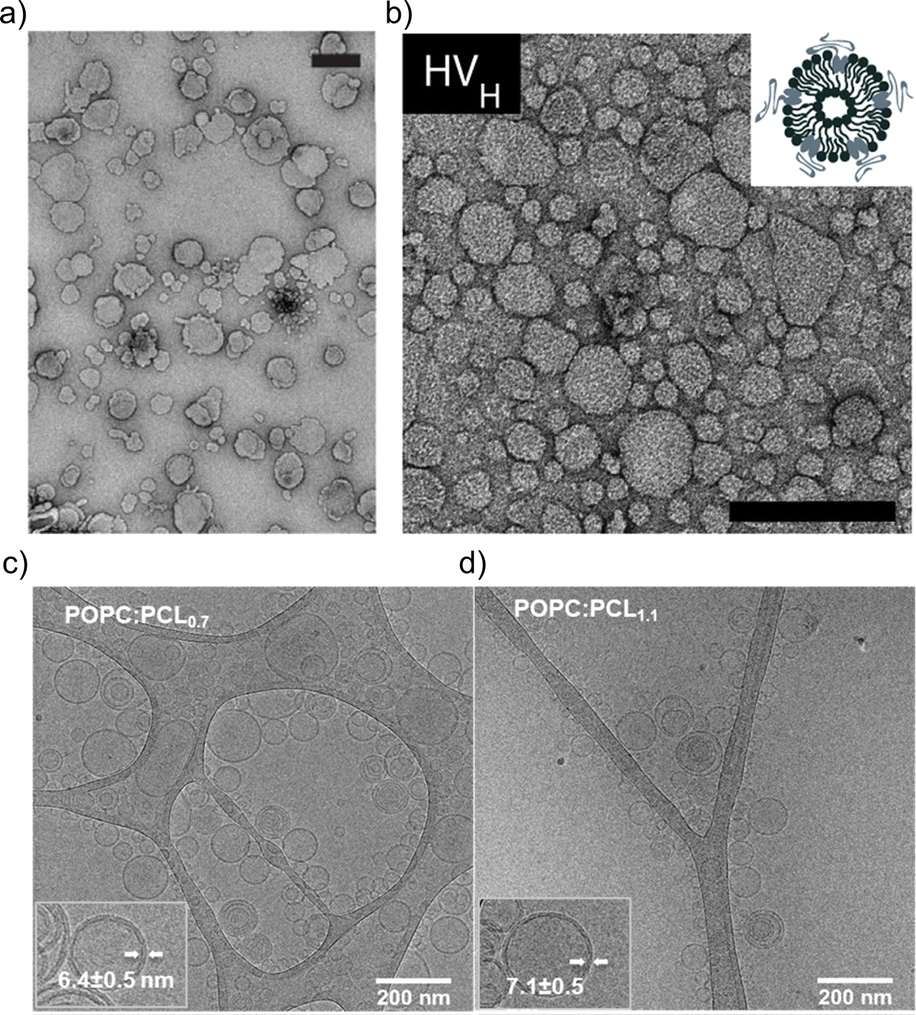 | ||
| Fig. 1 Typical electron microscopy images of HVs. TEM images of (a) PCMA11-b-P(METMA7-co-CEA104) & DOPC (Reprinted from ref. 89, Copyright 2024, with permission from Wiley) and (b) PCMA11-b-PEG113 & POPC (Reprinted from ref. 88, Copyright 2024, with permission from Elsevier). Cryo-EM images of (c) PEG12-b-PCL6 & POPC and (d) PEG12-b-PCL9 & POPC.76 Used under CC BY 3.0. https://www.mdpi.com/2073-4360/12/4/979. | ||
4 Lipid-polymer phase separation
The incorporation of block copolymers and lipids into a mixed membrane is typically envisioned via the alignment of the hydrophobic tails of the lipids and the block copolymers. The hydrophilic segment of diblock copolymers either faces into the void of the HV or towards the environment. Triblock copolymers are expected to have one hydrophilic blocks facing the external environment, while the other is positioned in the internal void (Scheme 2b). Dissipative particle dynamics simulation showed that the arrangement of block copolymers in the lipid bilayer can be characterized as bridge- and loop-shapes in the membrane, resulting in symmetrical and asymmetrical inner and outer membrane leaflets. The arrangement was highly influenced by the hydrophilic length of the block copolymer as well as repulsive energies between the hydrophobic block and the lipid tails.101 Moreover, the resulting hybrid membrane can have the building blocks either homogenously distributed or phase separated. Both types of membranes can be beneficial. Homogenous hybrid membranes have better stability and reproducibility, while (lipid) phase separation and raft formation are essential in membranes of living organisms, and are therefore anticipated to contribute with unique functions to man-made membranes.102Different factors influence if a specific block copolymer and lipid combination results in a homogenous or phase separated membrane. First, the fluidity of the lipid phase strongly affects the homogeneity of the hybrid membrane, i.e. lipids with melting temperature (Tm) below room temperature are more likely to facilitate homogenous hybrid membranes. Second, the ratio between the block copolymer and lipids is an important parameter, and homogeneous membrane often require very specific ratios. An example in this context are gHVs made of the triblock copolymer PDMS22-g-(PEG16)2 & POPC that had homogenous membranes with high block copolymer content (75 mol%), while block copolymer amounts below 50 mol% resulted in macrodomain formation that quickly evolved to fission through budding of the vesicle.33 In contrast, the same block copolymer & DPPC showed stable HVs with macrodomain formation for up to 75 mol% block copolymer content, and homogeneous membranes above 85 mol% block copolymer content. The difference was explained by the curvature energy and the line tension between the polymer-rich and the lipid-rich domains that caused budding of the vesicles. Furthermore, the HVs with DPPC were stable for longer time, due to the lower mobility of the DPPC lipids in the gel phase compared to POPC in the fluid state that allows faster movements of the components. Similarly, gHVs made of maltopentaose-b-poly(propylene oxide) (PPO)43 & DOPC had a homogeneous membrane up to 50 mol% block copolymer content, while above domain formation was observed.96
Hu et al. used dissipative particle dynamics simulation to model membrane formation with a block copolymer and saturated or non-saturated lipids. They found that phase separation is driven by a weak energy incompatibility between the lipids and the block copolymers. Above a critical lipid concentration, a mixing–demixing transition in the polymer-rich membrane can be observed, until a thermal condition in the hydrophobic layer is satisfied, with low thickness mismatch. This quality depends on the type of lipids and their Tm. Furthermore, significant thickness mismatch also drives phase separation.103
FRET measurements are an interesting opportunity to characterize hybrid membranes. The existence of a FRET signal not only gives an indication that the two components are in close proximity to each other, i.e. being part of the same assembly,1,32,34–36,57,64,89–95 but also the presence of domains can be determined by calculating the FRET efficiency. It is important to note that even though FRET is used very often, it is a qualitative rather than a quantitative measure for domain formation as smaller domains might not be detected, and FRET can always occur at the lipid–polymer interface. FRET measurements supported the existence of lipid domains by using NBD-labelled block copolymers and rhodamine-labelled phospholipids in PDMS-g-PEG2 & DPPC-based or PDMS-g-PEG2 & DOPC-based HVs.34 In a follow up study, the length of the blocks were systematically varied in the PEGn-b-PDMSm-b-PEGn (n = 8, 12 or 17 and m = 22, 43 or 67) triblock copolymer, and the effect on the hybrid membrane when assembled into sHV with DPPC was assessed.36 Phase separation was found for all tested compositions illustrated by a combination of small angle neutron scattering (SANS) and FRET measurements. Further, the sample contained liposomes, polymersomes and even worm-like micelles in addition to the sHVs with higher hydrophobic size mismatch of the hydrophobic block and the phospholipids,36 highlighting the impact of the lipid fluidity on the phase separation (Fig. 2a). Membrane homogeneity for gHVs is often determined visually by confocal laser scanning microscopy (CLSM). In contrast, gHVs made of POPC & PEG8-b-PDMS22-b-PEG8 had homogenous hybrid membranes below 10 w/w% lipid content, while gHVs consisting of POPC and higher molecular weight PEG12-b-PDMS43-b-PEG12 showed homogenous hybrid membranes up to 40 w/w% lipids as observed by CLSM (Fig. 2b).35 The decrease of the overall curvature (sHV vs. gHV) allowed for a better mixing of the building block. Macrodomain formation was observed for gHVs assembled from PIB87-b-PEG17 & DPPC or PIB37-b-PEG48 & DPPC. In the latter case, the DiDC18 dye was incorporated in the hybrid membrane that showed a preference for the less ordered (polymer) phase causing the highly ordered lipid domains to appear dark in the CLSM images. Furthermore, gHVs made of PIb37-b-PEG48 & DPPC had an interesting, ragged surface at temperatures below the Tm of DPPC. In contrast, gHVs made of PIb87-b-PEG17 & DPPC showed defects and ragged membranes for low block copolymer content (10%), while smooth spherical gHVs were observed at higher block copolymer content.70
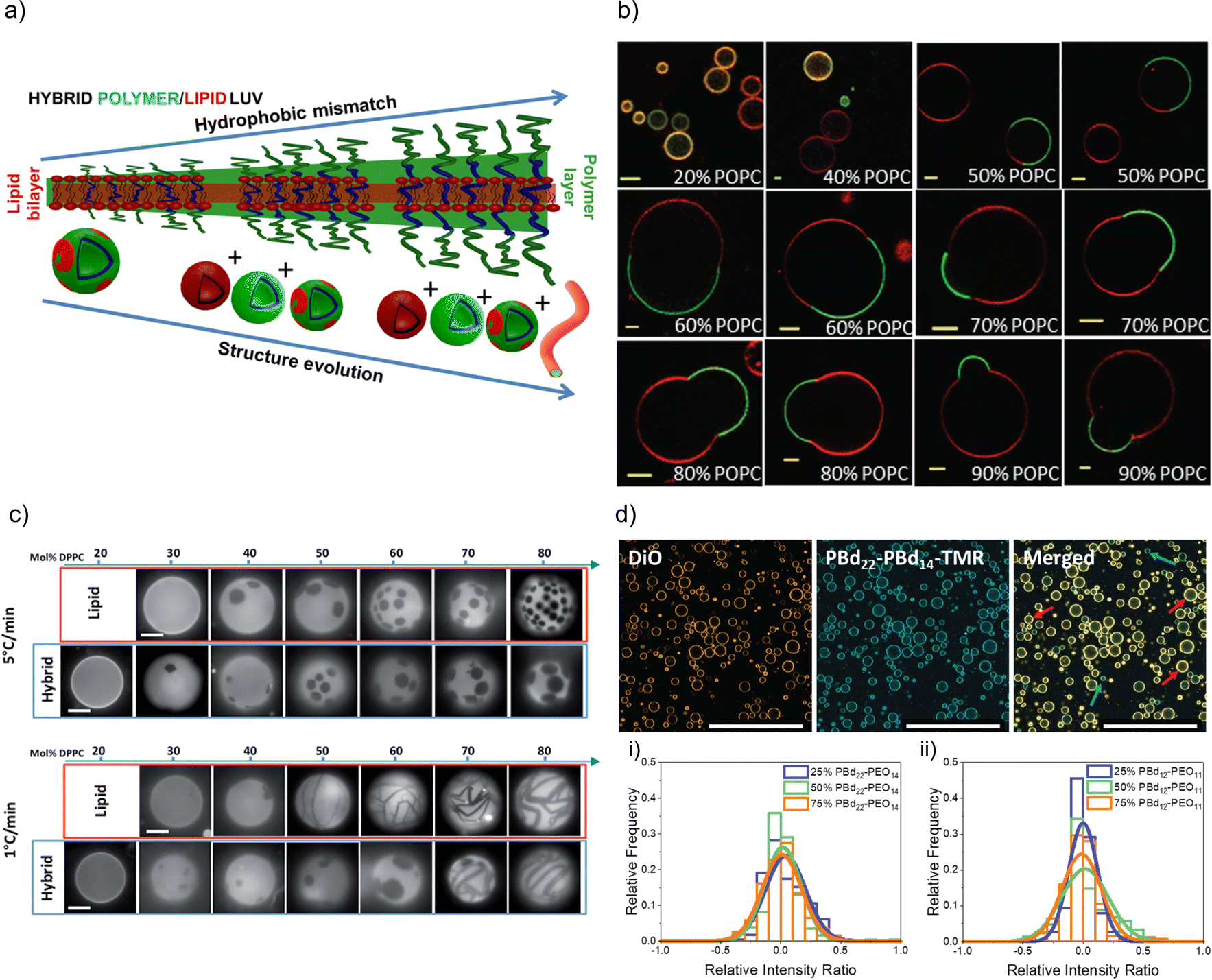 | ||
| Fig. 2 Lipid and polymer distribution in HVs. (a) Schematics of observed structures with increasing length of polymer including the presence of polymersomes, liposomes, HVs and worm like micelles, using PDMS-g-PEG &DPPC.36 Reprinted with permission from ref. 36. Copyright 2024 American Chemical Society. (b) Equatorial slices of hybrid 3K/POPC GHVs labelled with PDMS26-g-(PEG12)2-FITC (green channel) and DOPE-Rhod (red channel) visualized by CLSM at room temperature. Scale bar: 5 μm. Reprinted from ref. 35. Copyright 2024 the Royal Society of Chemistry. (c) Appearance of PDMS-g-PEG & DPPC (“hybrid”) vesicles compared with DOPC/DPPC (“lipid”) vesicles for different compositions and two cooling rates from the one-phase region. Scale bars: 10 μm. DOPE-Rhod is employed as the tracer. Reprinted from ref. 28. Copyright 2024 the Royal Society of Chemistry. (d) Images of intensity contributions from DiO and, PBd22-PEG14-TMR dyes individually in 25 mol% PBd12-PG11-based gHVs, and the merged channels. Red arrows indicate vesicles with greater DiO contribution, while blue arrows indicate vesicles with greater PBd22-PEG14-TMR contribution to the overall fluorescence intensity of the gHV. The histograms show the relative intensity contributions from DiO and PBd22-PEG14-TMR in (a) PBd22-PEG14/POPC and (b) PBd12-PEG11 & POPC-based HVs. An intensity ratio of 0 indicates equal relative fluorescence intensity contributions in HVs from both PBd22-PEG14-TMR and DiO. Scale bars: 200 mm.52 Used under CC BY 3.0. https://rsc.66557.net/en/content/articlelanding/2022/sm/d1sm01591e. | ||
Domain formation in HVs can be a temperature dependent effect. In particular, if the assembly temperature is raised above the Tm of saturated lipids for HV formation to allow for sufficient mobility required for assembly, followed by cooling to room temperature. The extent of domain formation was observed to be influenced by the cooling rate after electroformation. Phase-separated gHVs made of PDMS-g-PEG2 and DPPC at 53 °C via electroformation had distinct membrane tensions depending on the cooling rates after electroformation, i.e., cooling rates 1 and 5 °C min−1 resulted in low and high membrane tension respectively (Fig. 2c).28 Similarly, applying slower cooling rates after gHVs formation using PBd46-b-PEG30 & DPPC resulted in larger domains.104 In the case of slower cooling rates, the components have increased mobility, allowing them to attain a more energetically favourable arrangement before reaching the bilayer's gelation temperature, at which point the mobility of the components is substantially reduced.
Alternatively, domain formation can be induced post-assembly, e.g., domain formation was induced using the affinity of biotin to neutravidin. The otherwise homogeneous HVs made of PBd46-b-PEG30 & POPC with incorporated biotinylated polymer were driven to form macrodomains in gHVs upon crosslinking with neutravidin.55 However, when 5 mol% DSPE-PEG-biotin as a lipid was incorporated instead, no domain formation was induced. This difference was attributed to the fact that the hybrid system was not solely driven by the preferential biotin-protein interplay but also by the phospholipid-block copolymer interaction within the hybrid membrane. Furthermore, the addition of cholesterol can induced phase separation in homogeneous HVs,86 or it can have a fluidizing effect to overcome nanodomain formation.76 Interestingly, the introduction of cholesterol into a hybrid membrane composed of PBd46-b-PEG30 & DPPC induced the creation of domains exhibiting quasi-circular shapes, as opposed to the irregular patches observed in the absence of cholesterol.104 This occurrence is likely attributed to cholesterol's impact on lipid packing and the associated transitions in lipid phases.
In an elegant study, Schulz et al. showed the advantage of using PIB87-b-PEG17 & DPPC-based HVs in creating biomimetic models to understand receptor-ligand recognition.71 Ganglioside GM1 modified lipids were incorporated in the HVs, and the lateral diffusion was monitored depending on the amount of block copolymer used during assembly. The binding of cholera toxin B to GM1 caused phase separation in the initially uniform membrane for block copolymer contents up to 30%. However, at block copolymer concentrations exceeding 40%, no binding occurred, presumably due to the steric hindrance caused by the block copolymer chains.
Nonetheless, there are several examples of phospholipid and block copolymer combinations where phase separation was not observed.26,29,30,39,44,61,72,76,89 In many of these cases, the study's objective did not focus on the identification of composition boundaries related to phase separation. Consequently, no general conclusions could be drawn regarding whether these components consistently form homogeneous membranes irrespective of the environmental conditions. Often, homogeneity of the hybrid membranes were determined based on the analysis of CLSM images when fluorescently labelled components were used for assembly. A recent observation highlighted that, despite having homogeneous membranes, the quantity of lipid and block copolymer varied among different gHVs. This conclusion was drawn based on CLSM images captured from a large number of gHVs within the same population. gHVs made of PBd-b-PEG & POPC52 and either P(CMA-co-BuMA)-PCEA or P(CMA-co-HEMA)-b-PCEA & Soy PC93 showed different levels of fluorescent signal originating from the fluorescently labelled building blocks. This observation indicated that the quality of mixing of the components prior to drying of the lipid-block copolymer film on the electrode surface influenced the composition of the resulting gHVs. Further, there was no (or only very limited) mixing after the gHVs detached from the surface during electroformation (Fig. 2d).
5 Membrane properties
The plasma membrane of living cells has a diversity of properties, including its selective permeability, dynamic fluidity, integral role in cellular communication, and active participation in maintaining cell structure and homeostasis. This fluidity allows the membrane to undergo dynamic movements, such as lateral diffusion, flexion, and fusion with other membranes. The lateral mobility of the lipid and protein components are essential to maintain the above mentioned cellular processes. Consequently, several important properties need to be considered when assembling artificial membranes that will collectively imitate important aspects of the plasma membrane.5.1 Lateral mobility
The phospholipids in the bilayer of liposomes, which are often considered a very minimalistic model of the cell membrane, have free lateral diffusion depending on the type of phospholipids and/or the temperature. Consequently, phospholipids in hybrid membranes also exhibit lateral motion that not only affected by the type of phospholipid and temperature, but also by the amount and type of block copolymer. Lateral mobility can be measured by fluorescence recovery after photobleaching (FRAP), where the reappearance of the fluorescent signal is followed after photobleaching of a selected area. FRAP is typically used for gHVs. Fluorescence correlation spectroscopy (FCS) is typically used for sHVs, where the diffusion of fluorophore containing molecule is detected. The fluorophores can be chemically attached to a lipid or the block copolymer, or inserted into the bilayer, hence its movement is related to the diffusion of the lipids and the block copolymers.105 For instance, DPPC lipids in liposomes have limited lateral mobility at room temperature since the membrane is in the gel phase. However, when PIB87-b-PEG17 was incorporated in a DPPC membrane, the DPPC diffusion coefficient increased with increasing polymer content, indicating that the incorporation of the block copolymer affected the lipid organization, resulting in a more mobile system.71 In the hybrid membrane with 40 mol% block copolymer DPPC had 25% higher lateral mobility compared to membranes with 18 mol% polymer. In contrast, POPC lipids in liposomes have high lateral mobility at room temperature (D ∼9.8 ± 1.7 μm2 s−1) since the lipid bilayers is in the liquid phase. A ∼45× decrease in probe's diffusion coefficient was found when increasing amounts of PBd46-b-PEG30 were incorporated, to 0.22 ± 0.06 μm2 s−1.55 Hybrid membranes not only have lateral diffusion of the phospholipids but the incorporated block copolymer also exhibit mobility. For instance, as the block copolymer content decreased from 90 to 50 mol% in membranes composed of PEG8-b-PDMS22-b-PEG8 & POPC, the diffusion coefficient of a polymer-based probe decreased by a factor of 2 (from 3.55 ± 0.79 to 1.70 ± 0.32 μm2 s−1).37 While when the longer PEG12-b-PDMS43-b-PEG12 was incorporated with POPC into a hybrid membrane, the diffusion coefficient did not significantly change and was found between 1.41 ± 0.20 and 2.05 ± 0.41 μm2 s−1. The lipids diffused about 2.5× faster than the block copolymer in the same hybrid membrane.5.2 Phase transition (melting) temperature (Tm)
The fundamental relevance of the phase transition temperature (Tm) in lipid membranes as well as the glass transition (Tg) and the crystallization temperature (also called melting temperature and referred as Tm) in block copolymers defines the properties of the hybrid membrane, potentially influencing the HVs behavior during storage at 4 °C and during application at room temperature or 37 °C. Therefore, understanding the influence of the block copolymer insertion into the lipid bilayer and its effect on the packing of the lipids is an important question to address. Tm is often determined by measuring the melting enthalpy using differential scanning calorimetry (DSC), but other methods can also be employed (see below in the section about general polarization). For instance, the lipids' main transition peak, occurring at around 40 °C, was diminished and a second peak appeared when considering sHV made of PMOXA21-b-PDMS73-b-PMOXA21 & DPPC1 (Fig. 3). This observation was attributed to a change in the block copolymer's conformation as it was inserted into the membrane. The second peak became more distinct from the actual main transition peak of DPPC when the block copolymer content increased up to 40 mol%, while the temperatures of both peaks remained unaffected by the increase in block copolymer concentration. Tm remained unchanged with respect to the lipid-polymer ratio for gHVs made of PDMS-g-PEG & POPC and PDMS-g-PEG & DPPC for up to 70 mol% block copolymer content. However, above this threshold, the lipids were unable to meet and nucleate the gel phase, causing the melting transition of the lipids to disappear. Additionally, the melting enthalpy decreased as the block copolymer content increased, from 30 J g−1 to 15 J g−1 and from 52 J g−1 to 20 J g−1 for gHVs of PDMS-g-PEG & POPC and PDMS-g-PEG & DPPC, respectively.33 The melting temperature of sHVs made of either PMOXA15-PDMS67-PMOXA15 or PMOXA6-b-PDMS33-b-PMOXA6 & DOPC decreased compared to pure DOPC liposomes with increasing amount of block copolymer up to 10 mol% from −17.2 to −19.2 °C.27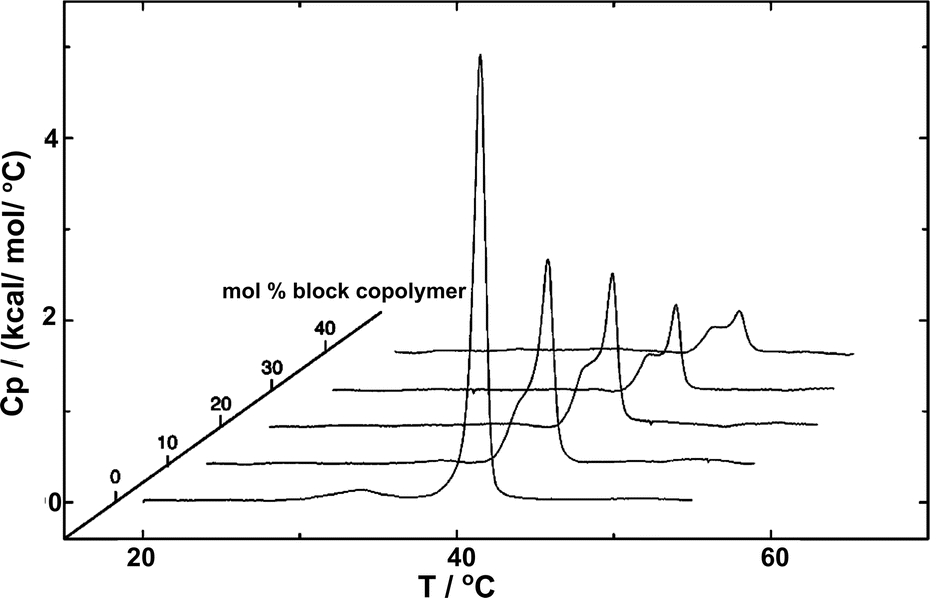 | ||
| Fig. 3 DSC experiments showing the endotherms of HVs made of DPPC with increasing PMOXA21-b-PDMS73-b-PMOXA21 content. Reprinted with permission from ref. 1. Copyright 2024 American Chemical Society. | ||
Some polymers such as PLA and PCL have a glass transition temperature and a crystallization temperature in the range of interest for biomedical applications. The typical transition behavior of these pure polymers was also observed in HVs made of PEG12-b-PCL10 & POPC, indicating that these phase transitions are not significantly affected by the presence of the lipids. The authors argued that the lack of change in Tm is due to phase separation of the components. sHVs with higher molecular weight block copolymers showed subtle features consistent with their melting transition, and the double transition peak maximums were shifted for PEG45-b-PCL42 from 36 °C and 46 °C to 32 °C and 44 °C, and for PEG45-b-PCL25 from 34 °C to 50 °C. In the same study, the Tm of the block copolymer in PEGn-b-PLAm & POPC was not observed.78 Similarly, using PEGm-b-PCLn & DPPC in the sHVs the presence of the polymers led to a significant increase of the main transition temperature (from 41 °C to 54 °C).74 The main transition enthalpy decreased significantly by the incorporation of polymer into DPPC liposomes. In sHVs composed of either PCL12-b-PEG45-b-PCL12 or PCL16-b-PEG104-b-PCL16 & DPPC no change in Tm at low block copolymer content (up to ∼24 w/w%) was observed. However, Tm was reduced by 5 °C for a ∼38 w/w% block copolymer content in the HVs.75 The latter observation indicated that the concentration of the block copolymer was essential to have an effect on the packing of the DPPC lipids. In another case, where HVs made of POEGA-b-PLauA & soy (HSPC) (a saturated lipid) were investigated, the pre-transition peak broadened and the transition enthalpy decreased with increased block copolymer concentration.81 The main peak position did not change significantly.
An interesting type of HVs considered the integration of thermo-responsive PNIPAM, in particular PNIPAM106-b-PlauA*25/PNIPAM28-b-PLauA13 & DPPC.82 DSC heating curves revealed differences in the 1st and 2nd heating cycles, with the Tm of the lipid shifting to higher values (above 41 °C), indicating structural rearrangements. However, the contribution of the thermo-responsive phase transition of PNIPAM at 32 °C was unfortunately not detectable during the heat cycles, presumably due to its low concentration.
5.3 Membrane fluidity
The fluidity of cell membranes is crucial for various cellular functions, including signal transduction, membrane trafficking, and the activity of membrane-bound enzymes and receptors. For example, temperature, pH, and the presence of certain ions can influence the membrane fluidity. Laurdan is often used to report on membrane packing since Laurdan's fluorescent emission properties are sensitive to the polarity of its environment (hydration of the membrane), leading to a red shift in the Laurdan emission peak. Fluidity of the membrane can be expressed as the general polarization (GP), which is a relation between Laurdan's emission peaks at 440 nm and 490 nm. GP values close to 1 indicate a highly ordered environment, while values close to −1 are related to a hydrated, disordered environment.106 Overall, measuring the GP is a valuable tool for understanding the biophysical properties of cell membranes as GP provides a quantitative measure of the fluidity or rigidity of lipid membranes. Changes in GP can be indicative of environmental factors affecting lipid vesicles.GP values for HVs made of PDMS-g-(PEG)2 & soy PC were found to be −0.43 compared to −0.28 for liposomes, indicating a more fluid membrane in the former case.30 The GP of PDMS-g-(PEG)2 & 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), PDMS-g-(PEG)2 & anionic soy PS (soy PS) and PDMS-g-(PEG)2 & DOPC also decreased compared to their respective liposomes. Furthermore, the amount of block copolymer in the assemblies influenced the degree of decrease in the measured GP.32
In contrast, the addition of PBd-b-PEG did not change the GP of POPC-based gHVs neither with increasing molecular weight nor with increasing block copolymer content, showing similar hydration and membrane ordering for the different gHVs.52 The insertion of cholesterol increases ordering in unsaturated lipid membranes, and the effect of polymerized cholesterol was studied for comparison. No change in the GP was found for HVs made of PCMA-b-PMPC and various lipids (DOPC, DOPS or DOPS/DOPE) compared to the corresponding liposomes.94 In contrast, PCMA-b-PCEA at lower concentration reduced the GP in HVs made with DOPC from ∼−0.25 to −0.3, while the GP increased to −0.10 at higher block copolymer content.90 The same effect was found for P(CMA-co-BuMA)-b-PCEA & soy PC and P(CMA-co-HEMA)-b-PCEA & soy PC, indicating that the cholesterol units homogenously distributed in the membrane of the vesicles.93 At the biological relevant temperature, the addition of PEG114-b-P(CL-co-LA)m in DPPC with various hydrophobic block lengths slightly increased the GP compared to liposomes.79 Similarly, the incorporation of PCL-b-PEG-b-PCL with DPPC into vesicles had only a small effect on the GP values at low polymer concentrations, but the GP decreased from ∼0.55 to 0.45 at a high polymer concentration.75 Increasing polymer content in HVs made of PBd11-b-PEG8 & DPPC resulted in decreased GP values from 0.6 to 0.38 (ref. 64) (Fig. 4). As mentioned above, GP values for DPPC lipid containing vesicles are often reported over a range of temperatures and are used for indicating the lipid melting transition temperature. The GP values upon heating reflect the phase transition, and the inflection point of the GP values measured with increasing temperature indicates the Tm. HVs made of PEG114-b-P(CL-co-LA)m with various hydrophobic block lengths and DPPC had a 3–4 °C higher Tm compared to DPPC liposomes.79 The overall change in GP values of DPPC liposomes was between 0.6 to −0.15 upon heating from 20 to 65 °C with a sharp change at the Tm (41 °C, GP = O). The addition of increasing amounts of PBd-b-PEG decreased the change in GP values upon heating, accompanied by a broader transition temperature.64 Furthermore, the Tm decreased more than 20 °C at the highest block copolymer concentrations. Recently, a multipeak analysis of Laurdan emission spectra has been employed to provide deeper insights into the phase behavior of hybrid membranes in several publications. This approach reveiled that alterations in both pH and salt concentration induced a shift in the transitional phase of sHVs composed of carboxylated PEG-b-PBd & egg sphingomyelin (egg-SM), while variations in molecular weight had no effect.65 The application of multipeak analysis to gHVs consisting of the same components (PEG-b-PBd & egg-SM and cholesterol) provided insights into the mechanism governing phase separation during the cooling of gHVs.63 The analysis suggested either spinodal decomposition or nucleation and growth as the underlying processes. Furthermore, the analysis indicated that cholesterol was more likely to be partitioned in the block copolymer-rich domains than in the lipid-rich domains.
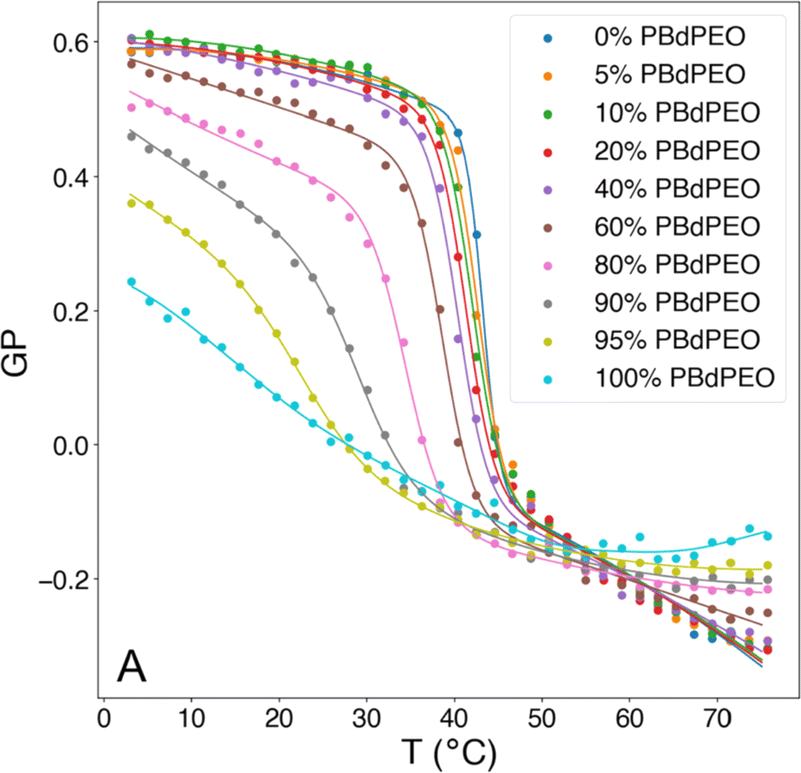 | ||
| Fig. 4 Generalized polarization (GP) of Laurdan, corresponding to membrane polarity, as a function of temperature and composition for HVs made of Bd11-b-PEG8 & DPPC. Reprinted from ref. 64, Copyright 2024, with permission from Elsevier. | ||
5.4 Viscoelastic properties
The viscoelastic properties of lipid bilayers determine their ability to change shape and deform in response to various mechanical stresses. This is crucial for the functioning of biological membranes, as cells are constantly subjected to mechanical forces. For example, during cell movement, the lipid bilayer needs to be flexible to allow cell shape changes and migration.Micropipette aspiration is the most used technique to study the bilayer's mechanical properties. During micropipette aspiration, the membranes of the gHVs are subjected to a mechanical (stretching) force caused by the air suction. The subsequent deformation of the membrane is recorded from where the lysis strain, the lysis stress and the area expension (stretching) modulus are calculated. For example, PEG14-b-PBD22 & HSPC with 75 mol% block copolymer content had an intermediate area expansion modulus of Ka = 112 dyn cm−1 between polymersomes (72 dyn cm−1) and liposomes (206 dyn cm−1).44 However, in the majority of cases, gHVs were reported to have lower lysis strain than their respective liposomes and polymersomes. For example, the area expansion modulus (Ka) in phase separated PDMS-g-PEG & DPPC gHVs was calculated to be lower than for the pure vesicles.28 Although these gHVs and the control multi-lipid liposomes (DOPC/DPPC) showed phase separation, the gHVs remained intact under the tested strain conditions, unlike the liposomes that ruptured. In another case, Ka of HVs made of PBd46-b-PEG30 & POPC increased up to 17% (from 112 ± 10 to 131 ± 15 mN m−1) with increasing lipid concentration (up to 30 mol%), while the critical lysis stress and strain decreased by 17% (from 15.3 to 12.7 mN m−1) and 40% (from 0.15 to 0.09), respectively, compared to polymersomes.55 The authors argued that despite a reduction in lysis stress and lysis strain, the higher stretching modulus, due to the integration of block copolymers and lipids into the hybrid membrane compensated for the toughness. This compensation was manifested in the greater applied tension needed to obtain a given strain.
In this context, the most studied block copolymer–lipid combination is PEG and PDMS in various architectures such as diblock copolymer, triblock copolymer or grafted copolymer by the Le Mains group. The stretching moduli increased for gHVs made of PEG12-b-PDMS43-b-PEG12 & POPC with increasing lipid content (0–20 w/w%) without phase separation observed on the macroscale.37 However, a decrease in lysis strain was found even for very low lipid content (5%), which was lower than for POPC liposomes. The authors suggested that, most likely due to the formation of nanodomains, lysis occurred at the borders of these domains. The elastic modulus exhibited an intermediate value between that of polymersomes and liposomes, but with high dispersity values, ranging from 80 to 155 mN m−1.1 Interestingly, intermediate mechanical properties were observed for gHVs made of PDMS27-b-PEG17 & POPC (with 10% lipid) compared to liposomes and polymersomes. Specifically, the lysis strain was found to be 7 ± 2%, compared to 3 ± 2% of POPC liposomes and 16 ± 4% for polymersomes. The lysis stress was the same as for POPC liposomes (8 ± 2 mN m−1) while the stretching moduli was comparable to the polymersomes (123 ± 12 mN m−1).39 In a follow up report, gHVs made of POPC lipids and PDMS-b-PEG with various block lengths were extensively studied to have a better understanding of the change in mechanical properties upon varying the molecular weight of the block copolymer and the block copolymer – lipid composition (up to 20% lipid) (Fig. 5). The area expansion moduli of these gHVs remained almost constant independently of the lipid composition (100 and 130 mN m−1). However, the lysis strain and stress decreased with the decreasing lipid fraction and increased with the molecular weight of the hydrophobic block. The lysis strain remained higher than that of POPC liposomes, except for gHVs made with the lowest molecular weight block copolymer. The lysis stress was more composition dependent, and lower values that the POPC liposomes were measured in some cases. Consequently, the calculated toughness of the HVs was highly dependent on the molecular weight of the block copolymer. In particular, gHVs assembled with the highest molecular weight PDMS-b-PEG had the most resistant hybrid membranes, with a tenacity always higher than POPC liposomes. The authors pointed out that in their previous efforts, gHVs formed with triblock copolymer PEG12-b-PDMS43-b-PEG12 & POPC had a membrane toughness lower than that of POPC liposomes even at a low lipid fraction (5 wt% POPC).41 The line tensions between macrodomains of gHVs made of the above mentioned polymers and POPC were calculated to further elaborate on the membrane properties. The compositions of the gHVs were chosen to allow for the assembly of stable budded vesicles with each hemisphere being formed by either the polymer phase or the lipid phase. Similar line tension values (0.32–0.82 pN) were obtained for PDMS23-b-PEG13, PDMS27-b-PEG17, or PEG8-b-PDMS22-b-PEG8 & POPC with 50% lipid content.42 gHVs made of PDMS-g-PEG & soy PC exhibited twice lower rigidity than soy PC liposomes.30 These gHVs exhibited lower bending rigidity than liposomes, comparable to that of polymersomes (11.6 ± 2.4 kBT) rather than showing intermediate softening. This result indicated that this property was largely determined by the prevailing membrane component.
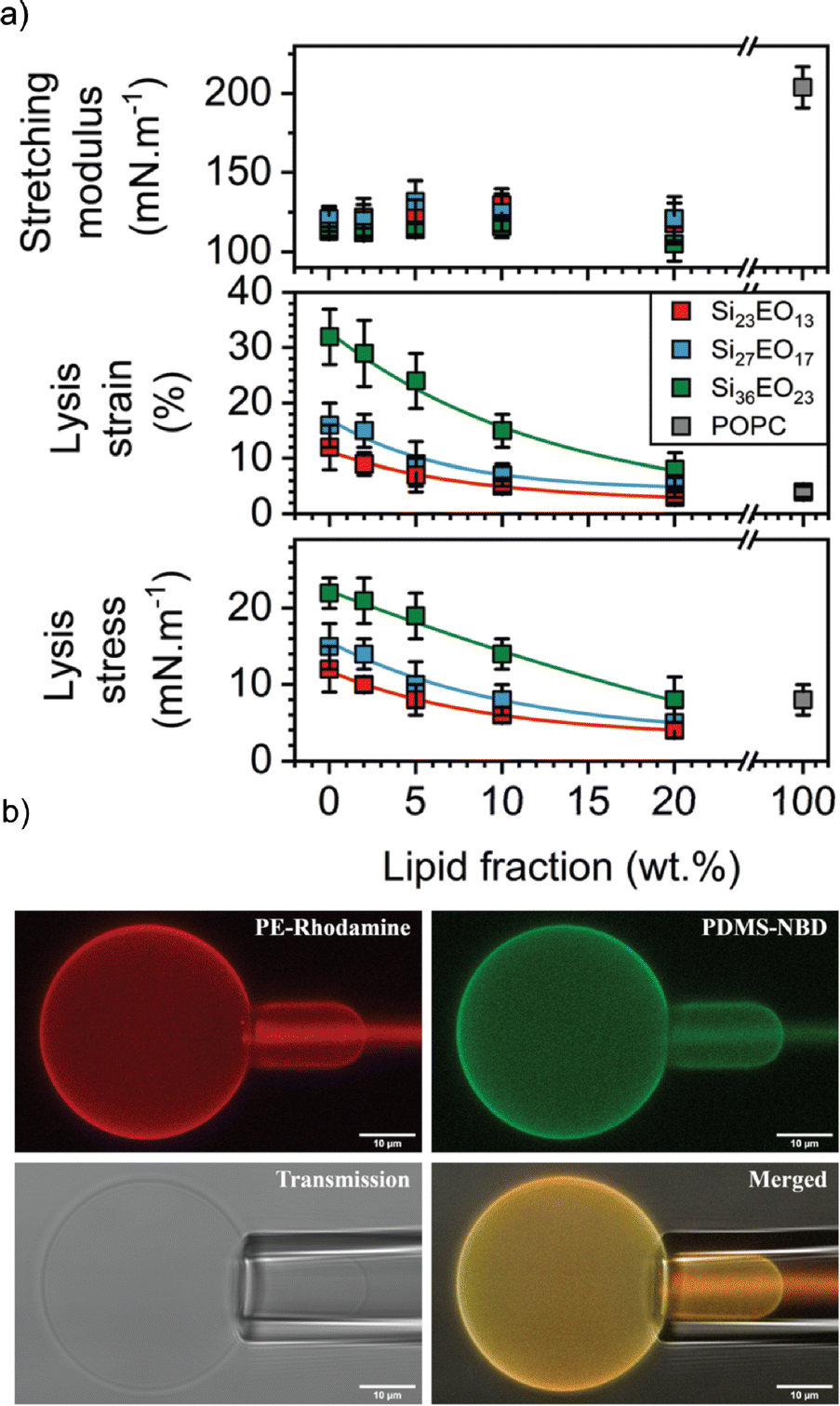 | ||
| Fig. 5 Membrane properties: (a) mechanical properties determined from the stress–strain curves: stretching modulus (top panel), lysis strain (middle panel) and lysis stress (bottom panel). (b) CLSM images of micropipette experiment in the different observation modes: PE rhodamine channel, PDMS NBD channel, PE rhodamine and PDMS NBD channel merged, and transmission. Scale bars: 10 μm. Reprinted from ref. 41. Copyright 2024 the Royal Society of Chemistry. | ||
In addition to micropipette aspiration, other methods were also utilized to measure the mechanical properties of membranes. For instance, Winzen et al. applied atomic force microscopy to characterize the viscoelastic behavior of gHVs.26 They compressed gHVs made of PDMS60-b-PMOXA21 & 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and obtained a Young's modulus of 15 ± 4 MPa that was similar to the corresponding polymersome stiffness. Progressive deformation on PBD-b-PEG & DPPC-based gHVs was studied with a microfluidic setup, where the gHVs were passed through a narrow, short channel by controlling the pressure of the flow.68 In this case, gHVs with 35% lipid content showed similar longitudinal deformation as the polymersomes despite domain formation. However, gHVs with 65% lipid content might feature both liposome-like transverse flattening and polymersome-like deformation, while slipping through the channel. The variations in stretching techniques employed in pipette aspiration, atomic force microscopy, and microfluidic conditions make it challenging to directly correlate their outcomes. Nevertheless, insights into alterations in membrane properties can be obtained through comparison of HVs to liposomes and/or polymersomes.
5.5 Membrane thickness
The membrane thickness has an effect on the mechanical properties of the membranes as well as on the permeability of the vesicles. Liposomes have a membrane thickness of around 3–6 nm, depending to largely on the length of the lipid tails and environmental factors, such as temperature or ionic strength. The membrane thicknesses can be obtained experimentally by various techniques including SANS, SAXS and cryo-EM. The latter has the advantage that individual vesicle can be observed (to e.g., identify phase separation) and measured and thus, gives information about variations with in the population.36The addition of copolymers has a substantial influence on the membrane thickness due to the higher molecular weight of the copolymer chains compared to the lipids, which therefore occupy a larger volume. For instance, molecular simulations showed that with increasing block copolymer content in PEG-b-PBd & DOPC-based HVs, the membrane thickness increased.66 Although there is theoretically a variation in membrane thickness based on the lipid-to-polymer ratio, in many instances, this difference may not be significant. For example, the average membrane thickness of HVs made of PDMS60-b-PMOXA21 & DMPC did not statistically differ from the thickness of polymersome membranes (17 ± 2 nm) independent on the amount of added lipids (10 or 50%) and were homogenous in all cases.26
Several studies aimed at elucidating the relationship between molecular weight and composition distribution in HVs by analyzing the membrane thickness. For instance, sHVs comprising of PCMA-b-pDMAEMA & POPC, POPE or POPS lipids had membrane thicknesses between 5.8 and 6.7 nm, which was thicker than the membrane of a POPC lipid bilayer (5.4 ± 0.5 nm).86 Moreover, sHVs made with low molecular weight PCMA-b-pDMAEMA had, on average, thinner membranes compared to sHVs assembled with high molecular weight PCMA-b-pDMAEMA. Furthermore, the presence of a bilayer structure was observable in all samples, indicating a homogeneous insertion of the block copolymers into the hybrid membrane. Cryo-EM analysis of sHVs assembled from PDMS-g-PEG & soy PC indicated that the membrane thickness slightly increased with increasing amount of block copolymer (4.40 ± 0.16 nm for liposomes, 4.86 ± 0.17 nm for sHVs and 5.25 ± 0.17 nm for polymersomes).30 The liposomes had a bilayer structure with sharp outer boundaries, while the block copolymer containing membrane appeared as fuzzy monolayers. Similar assemblies made of PDMS-g-PEG & soy PC had an increased membrane thickness when measured in potassium chloride-supplemented buffer compared to sucrose (6.1 ± 0.3 nm compared to 5.3 ± 0.2 nm) due to the ion association in the hydrophilic PEG layer.31 Similarly to the potassium chloride containing solution, the presence of the polymer in sHVs made of either PDMS26-g-(PEG12)2 & anionic soy PS or PDMS26-g-(PEG12)2 & cationic DOTAP allowed for strong association of the ions with the vesicle surfaces in tris(hydroxymethyl)aminomethane buffer, i.e., their membrane thickness increased from 5.8 ± 0.4 nm to 6.5 ± 0.4 nm for the anionic sHVs and from 5.8 ± 0.3 nm to 6.4 ± 0.3 nm for the cationic sHVs.32
Vesicles with inhomogeneous bilayer thicknesses can indicate nanodomain formation. For example, sHVs made of maltopentaose-b-PPO & DOPC had a variation within a single sHV between ca. 2.5 nm and ca. 6.5–7 nm, which corresponded to the average membrane thicknesses in pure vesicles and polymersomes, pointing to lipid and polymer-rich domains.96
Cryo-EM is often used as a complementary technique to SANS and SAXS with the aim to visually support the scattering data. Neutron scattering curves can give information about the shape and size of the scattering objects in solution by fitting various types of models to the scattering data. In SANS, scattering of neutron beams depends on the scattering length density (SLD) of the scattering nucleus. There is a difference in SLD between hydrogen and deuterium, thus, by mixing H2O and D2O in different proportions, the SLD of the solvent can be systematically varied and matched to the SLD of one of the components of the hybrid vesicle. This technique known as contrast matching, and it is possible to render a phospholipid invisible.107 For instance, sHVs assembled from PDMS-g-PEG2 & DPPC were measured with or without deuterated DPPC by SANS.34 The obtained shape factor was related only to the copolymer, i.e., the polymer contrast conditions, in the presence of deuterated lipids. In this condition, the obtained curves showed a disk-like shape, indicating polymer domains in the sHVs instead of homogeneous distribution when using the Krakty-Porod fit. In contrast, the assembly had a vesicular shape with an average bilayer thickness of 5.8 nm at 20 °C when non deuterated lipids were used. Cry-EM images of these sHVs showed either rounded spherical or faceted sHVs. The latter was ascribed to the gel state of the DPPC lipids. In another study, SANS curves obtained from sHV composed of PEGn-b-PDMSm & POPC with increasing overall molecular weight of the block copolymer in polymer contrast and lipid contrast conditions were fitted with the same Kratky-Porod model.40 The membrane thickness decreased with increasing lipid fraction when observing the polymer phase, suggesting interactions between the lipid chains and the block copolymer chains in the hybrid membrane. The higher molecular weight of the block copolymer did not influence the membrane thickness significantly (6.4–6.8 nm). In lipid contrast mode, sHVs with shorter block copolymer chains exhibited comparable membrane thicknesses to liposomes, while the sHVs with the longest block copolymer chain displayed a more pronounced deviation. The authors argued that this observation might be explainable by fission. However, in this case, it would be expected that the thickness of the second population should also remain unchanged compared to the polymersome, which was not observed.40 Perera et al. assembled sHVs made of PEGn-b-PDMS15-b-PEGn & DOPC where PDMS length was kept constant and the PEG was varied systematically.43 They used a polydisperse core-multishell model where the SLDs were taken into account, allowing to estimate the thickness of the water-rich hydrophilic shell and the denser PDSM and lipid-rich core. The SANS data revealed that the hydrophilic mass fraction of an amphiphilic polymer affected the hybrid membrane thickness and the sHV size distribution (Fig. 6a), which was in good agreement with the obtained cryo-EM images.
 | ||
| Fig. 6 (a) Change of the total membrane thickness of PEGn-b-PDMS15-PEGn (n = 3, 14, 28) & DOPC-based HVs with the increasing polymer concentration. Reprinted with permission from ref. 43. Copyright 2024 American Chemical Society. (b) Cryo-EM images of the membrane of vesicles made of (i) PBd22-b-PEG14 (50 mol%) & POPC, (ii) population 1, (iii) population 2 (thin and thick, respectively), and (iv) PBd22-b-PEG14 only (polymersomes).50 Used under CC BY 4.0. https://onlinelibrary.wiley.com/doi/full/10.1002/smll.202206267. | ||
The scattering profile in SAXS measurements is related to the electron density in the bilayer. Modeled SAXS data can give information about the shape and the size of the vesicle as well as the packing of the components in the bilayer. This technique is often used for studying membrane proteins and their configuration in the bilayer.108 SAXS was used to assess the effect of the chemical nature of the hydrophobic block on sHVs made of either BuMA-b-PCEA & soy PC or (PCMAn-co-BuMAm)-b-PCEA & soy PC with various molecular weights and polymer to lipid ratios.93 The SAXS data were fitted with a model, which took the presence of micelles in addition to the sHVs in the solution into account. The results showed that the sHVs had a higher membrane thickness between 40.4 ± 0.6 to 44.8 ± 1.2 Å compared to the soy PC liposomes (39 ± 0.2 Å), which slightly increased with increasing block copolymer content. However, the type of block copolymer did not influence the membrane thickness of the sHVs. In a related case, sHVs made of PCMA-b-PMPC & DOPC or DOPS had comparable membrane thicknesses to liposomes (∼4.6 nm) as determined by SAXS measurements.94 However, the scale of the Gaussians describing the head group regions were in general slightly larger for the sHVs than for the liposomes, suggesting the presence of the block copolymer in the membranes. In a recent publication by Seneviratne et al., a detailed high resolution cryo-EM and SAXS investigation of PBd22-b-PEG14 & POPC-based sHVs with increasing polymer content was presented.50 Overall, they found that the membrane thickness increased (between 5–10 nm) with increasing block copolymer fraction. However, two population of vesicles with different membrane thickness were observed at higher polymer content (>50%). The membranes did not show domain formation and atomic resolution cryo-EM images revealed the detailed membrane structure, showing similarly organized bilayer structures with different thickness (Fig. 6b).
In general, all three techniques give information about the membrane thickness. CryoEM images contain detailed structural information of individual vesicles, while, SAXS and SANS give average structural information in solution. Consequently, using these techniques in a complementary manner provides the most comprehensive understanding.
6 Stability
The limited stability is probably the major shortcoming of liposomes. This issue also includes their rather short shelf life, i.e., the liposomal colloidal stability is affected by membrane fusion within 48 h, at inappropriate storage conditions.5 In contrast, HVs often show exceptional stability without detectable changes in Dh and PDI over time. Several examples are reported with stability up to four weeks at 4 °C,26,46,78 and in a recent study even over 500 days.40 An aspect that affects the HV stability is the length of the polymer chains, e.g., HVs assembled from PEG12-b-PCL10 & POPC, PEG45-b-PCL25 & POPC or PEG45-b-PCL42 & POPC had lower stability for the shorter hydrophobic blocks (4 days vs. 3 weeks) (Fig. 7a).78 Conversely, HVs consisting of PEG113-P(8C-Chol) & DPPC exhibited higher stability for shorter hydrophobic blocks (up to 245 days), while high molecular hydrophobic segments were insufficiently incorporated leading to precipitation after 2 weeks.85 In another example, HVs made of PMOXA15-b-PDMS67-b-PMOXA15 & DOPC were stable for up to 3 weeks when 10 mol% triblock copolymer was used, while 20 mol% triblock copolymer reduced the stability of the HVs to 5 days.26 HVs assembled from PBd-b-PEG & POPC were subjected to different types of sterilization methods that can be required for biological applications such as drug delivery.51 Not surprisingly, only HVs that were below 400 nm remained intact after filtration through a 0.44 μm filter. Furthermore, freeze–thaw–vortex cycles also showed substantial destruction of the HVs, assessed by the substantial release of their cargo, pointing out that other ways of sterilization methods need to be tested.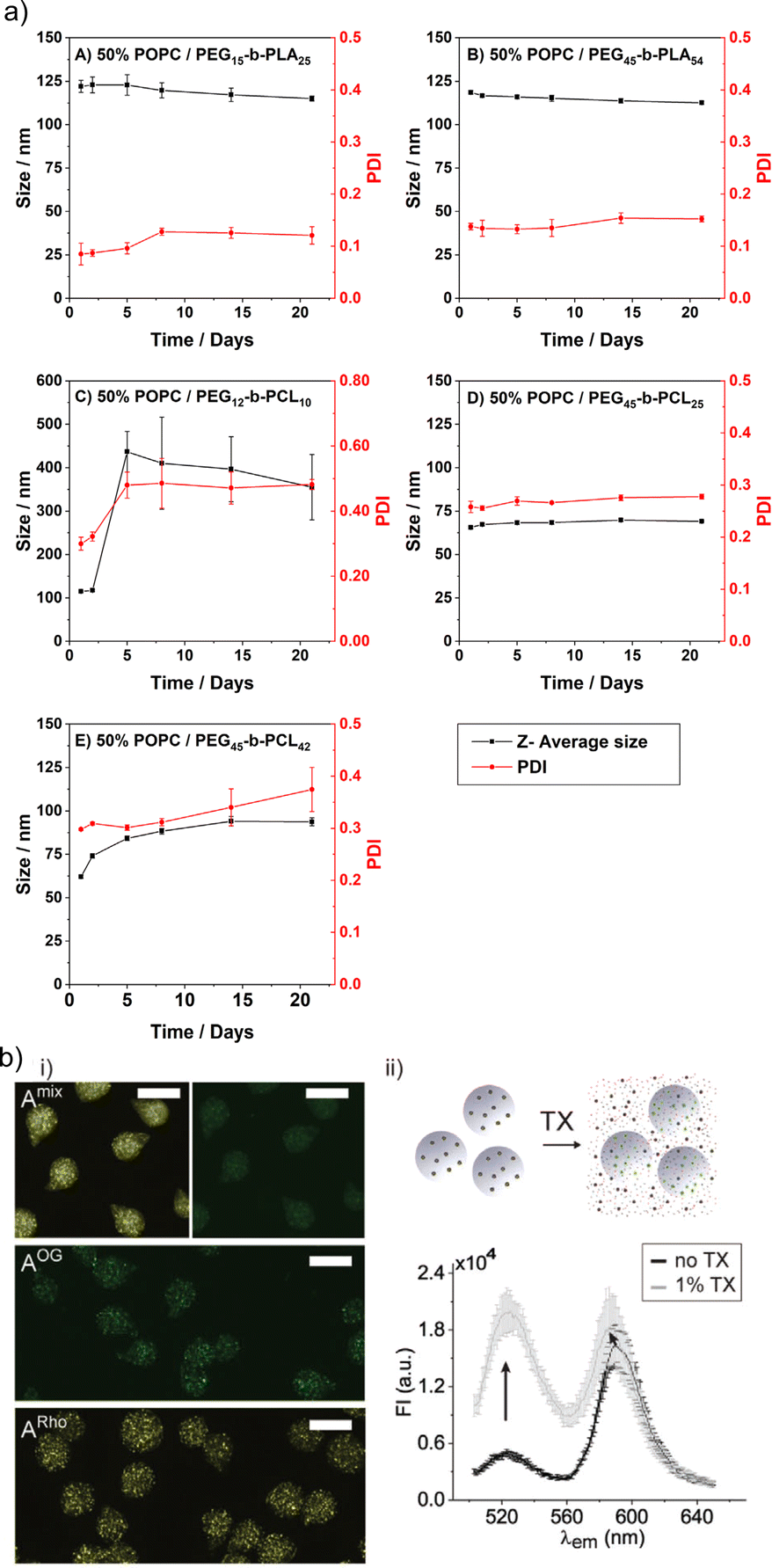 | ||
| Fig. 7 Stability of HVs. (a) Hybrid vesicle size stability at 4 °C. Samples are monitored by DLS over a period of 3 weeks: (A) PEG15-b-PLA25; (B) PEG45-b-PLA54; (C) PEG12-b-PCL10; (D) PEG45-b- PCL25 &POPC; and (E) PEG45-b-PCL42 &POPC. Reprinted from ref. 78, Copyright 2024, with permission from Elsevier. (b) (i) Representative fluorescent microscopy images of HVs encapsulated in alginate beads (A) with FRET pair labelled polymer and lipid (Amix) or only one component (AOG, and ARho). Scale bars: 100 μm. (ii) Illustration of sHVs encapsulated in alginate beads and their disassembly upon addition of Triton-X (top). Fluorescent emission spectra of Amix before (black line) and after (gray line) exposure to 1% Triton-X (bottom). Reprinted from ref. 89, Copyright 2024, with permission from Wiley. | ||
Exposure to surfactants such as Triton-X or bile as well as lipases is often used to determine the inherent stability of HVs. Lipases are a group of enzymes that catalyse the hydrolysis of lipids that play a crucial role in the digestion of lipids in the digestive tract. As expected, the stability of HVs towards lipases depends on the building blocks. For instance, sHVs made of PEG12-b-PCL6 & POPC, PEG12-b-PCL9 & POPC or PEG16-b-PLA38 & POPC were structurally affected by phospholipase A2 (PLA2) as monitored by calcein release and significant changes in their Dh and PDI.76 In another study, the hydrophobic block consisted of a copolymers of PCL and PLA with PEG as a hydrophilic segment. The structural integrity of the sHVs containing a rather large polymer PEG114–PLA328 was best preserved, delaying calcein release up to 5 min after PLA2 addition.79 In contrast, gHVs made of PBd46-b-PEG30 & DPPC & cholesterol (50 mol% block copolymer) remained topographically intact and spherical upon PLA2 addition.104 The gHVs lost their cholesterol-rich lipid domains when subjected to PLA2, but they showed self-healing, i.e., recovering the full circular structure after the lipid loss as visualized by CLSM. These gHVs remained structurally intact despite hole formation that allowed for the release of encapsulated cargo as big as proteinase K (28 kDa). In another study, gHVs made of PBd19-b-PEG27 & DPPC, exhibited a burst release of the cargo when exposed to lipases, although the overall structural integrity of the gHVs was preserved.61 In contrast, gHVs assembled from PBd19-b-PEG27 & POPC showed first a continuous release of the encapsulated cargo upon addition of PLA2 followed by buckling and release of the remaining cargo. The fact that the lipase could access the lipid molecules showed that the incorporated block copolymer did not provide a protecting stealth layer around the HVs, as it could be expected from PEG. This example illustrated that the HVs, even when containing high amounts of block copolymer, had large lipid domains, even for the non-saturated, more fluid membranes. Destabilization of HVs by surfactants depends on their concentration. Surfactants in low concentrations are often employed for the insertion of membrane proteins (see below). Typically, the HVs undergo complete structural disintegration when the surfactant concentrations reach a certain threshold. A common approach to exploit this fact is to use Triton-X at concentrations ranging from 0.03–1% to release the cargo and evaluate the encapsulation efficiency.1,46,51,61,75,76,78,79,93
The stability of the HVs is an important aspect for their use in drug formulation, e.g., for encapsulation of HVs into hydrogels as potential drug deposits. To this end, sHVs made of PCMA11-b-P(METMA7-co-PCEA104) & DOPC,89 PCMA11-b-PEG113 & POPC/POPS,88 or PCMA-PMPC & DOPS or DOPC94 were encapsulated into alginate microbeads. The preservation of the FRET effect due the fluorescently labeled block copolymer and the fluorescently labeled lipids suggested that the building blocks remained closely associated even after the sHVs were trapped in the hydrogel. Yet, the lipids leak out after addition of Triton-X or incubation in simulated gastric fluid, indicating the loss of structural integrity of the sHVs (Fig. 7b).
7 Permeability
The cell membrane's permeability is controlled in various ways and plays a significant role in keeping cellular homeostasis. Being able to influence permeability is a crucial characteristic for achieving cell membrane-like adaptability. Liposomes and especially polymersomes have very low inherent permeability,109 and recent progresses in this context is discussed in several comprehensive reviews.110–112 Often, HVs show improved permeability towards small molecules compared to their polymersome or liposome counterpart. The inherent permeability is usually determined/compared by monitoring the release of encapsulated (small hydrophilic) fluorescent molecules such as calcein or (carboxy)fluorescein. However, the main drawback is the low solubility of these dyes in aqueous media, and to achieve self-quenching concentrations (>20 mM), high pH is required, which can interfere with the assembly process of the vesicles. Further, the measured encapsulation efficiency in vesicles made by the rehydration method is inherently very low, generally below 10% as it is a passive encapsulation process. Nonetheless, this release assay remains the most viable option to address permeability of small vesicles.Most of the permeability studies are performed on sHVs that are extruded through a 100 nm membrane to obtain unilamellar vesicles. In general, the release of the dyes occurred within 5 days. Multilamellar vesicles likely do not release the encapsulated molecules completely.93 Studies have explored various methods to influence the permeability of HVs, including the lipid to copolymer ratio, molecular weight of the copolymer, and the use of charged lipids or polymer chains. For example, research has shown that increasing the PEG14-b-PB22 to POPC ratio in HVs could result in delayed release of encapsulated carboxyfluorescein at room temperature.46 The effect of the block copolymer molecular weight was explored by systematically changing the length of the blocks in PEGn-b-PLAm.78 The carboxyfluorescein release rates were influenced by the overall properties of the block copolymer, but there was no clear trend on the dependence of the release profile on the length of the various blocks. However, the release of carboxyfluorescein was slower at 4 °C than at 37 °C. In another example, the molecular weight of the block copolymer was found to affect the permeability of HVs made of PDMS-b-PEG & POPC, but not the lipid to block copolymer ratio.40 Specifically, assemblies made with the shortest polymer (PDMS23-b-PEG13) showed higher permeability, releasing fluorescein more efficiently than less permeable liposomes. HVs made with POPC and the longer block copolymers exhibited slightly lower permeability, but not significantly different from the corresponding polymersomes.
The chemistry of the hydrophobic block, which inserts into the bilayer, can be altered, by synthesizing block copolymers, which, in turn, can modify the permeability of the HVs. The calcein release from sHVs made of POE114-b-P(CLx-co-LAy) & DPPC, with different compositions of PCL and PLA, increased with increasing PLA content.79 When comparing block copolymers of PCMA with BuMA or HEMA as the hydrophobic block, it was observed that increasing the block copolymer content from 1–5 w/w% resulted in a rise in carboxyfluorescein release from the gHVs. gHVs made of soy PC and block copolymers with a PCMA homopolymer hydrophobic block or a copolymer of CMA and BuMA hydrophobic block did not cause any change to the gHVs' permeability. However, when using HEMA instead of BuMA in the copolymer, a significant increase in permeability of RhoX was observed.93 sHVs made of the aforementioned building blocks exhibited a faster release of carboxyfluorescein within 24 h as compared to soy PC liposomes, consistent with the trend observed in gHVs. In another study, when PCMA-b-PMPC was used in HVs with DOPC, DOPS or DOPS/DOPE, the encapsulated carboxyfluorescein was released at a similar rate within 24 h, indicating that the lipid type did not influence the permeability.94 The molecular weight of the hydrophilic block can also influence the permeability of HVs. Further, HVs made of maltopentaose-b-PPO43 & DOPC had lower permeability towards PEG-FITC (550 Da) than HVs made of maltopentaose-b-PPO26 & DOPC.96
pH-sensitive chemical groups give the possibility to modulate the flow of charged molecules across the membrane. For instance, negatively charged calcein was released within minutes from gHVs made of cationic lipids (CLS) & PDMS36-b-PEG23 upon acidification of the environment, i.e., protonation of the lipid heads, while calcein was retained in gHVs containing only zwitterionic POPC and non-chargeable lipids.38 Similarly, gHVs made of oligo(aspartic acid)4-b-PPO & DOPC were permeable to positively charged rhodamine 6G and non-charged PEG-FITC (550 Da) after immersing the assemblies into the fluorophore solution for 4 h. In contrast, negatively charged fluorescein was only able to pass through the hybrid membrane at pH 4 where the polymer side chains were uncharged.95 In addition, these sHVs showed higher permeability towards acetylthiocholine than DOPC liposomes probably due to the attraction forces between the negatively charged polymer chains and the positively charged molecule. The pH responsive nature of PDMAEMA allowed to fully release the encapsulated drug below its pKa (pH 6.5) from PDMAEMA-b-poly(lauryl methacrylate) & egg L-α-phosphatidylcholine (EPC)-based HVs, while at pH = 7.4 60% of the drug was retained.84
Inonophors passively transport specific type of ions across the cell membranes, and they are explored in synthetic cell membranes to establish more acidic or basic conditions in the vesicle's void. The ion channels valinomycin, nigericin and gramicidin A were incorporated into HVs made of PBd37-b-PEG22 & POPC to increase the permeability towards potassium ions(Fig. 8).58 The H+ and OH− permeability can be assessed by encapsulating environmental sensitive dyes such pyranine or trisodium 8-hydroxypyrene-1,3,6-trisulfonate to monitor the pH equalization rates inside of the vesicles. Kleineberg et al. showed that the rate of ion permeability was higher for both their HV system compared to liposomes. However HVs made of PBd22-b-PEG14 & soy PC showed lower permeability compared to PDMS12-g-(PEG32)2 & soy PC, assessed by valinomycin incorporation. The difference in permeability was probably due to the polymer architecture (linear vs. grafted) that likely inserted differently into the hybrid bilayer, forming a denser bilayer when PBd22-b-PEG14 was used.54
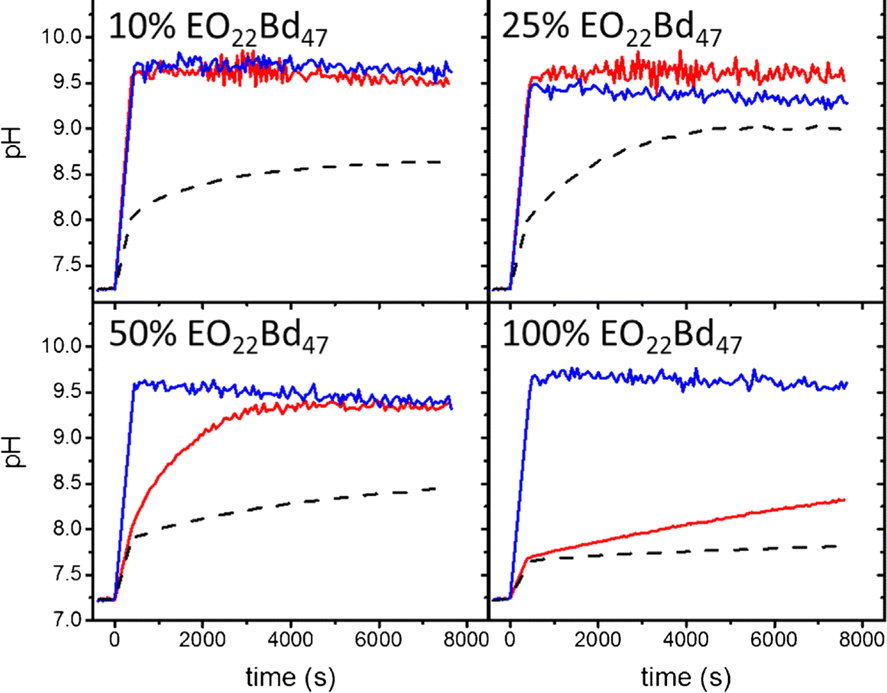 | ||
| Fig. 8 Permeability of HVs detected via monitoring the internal vesicle pH. HVs made of PBd37-b-PEG22 & POPC using 10%, 25%, and 50% and 100% block copolymer without any added ionophore (dashes) and after incubating with valinomycin (red tracing) and nigericin (blue tracing). Reprinted from ref. 58, Copyright 2024, with permission from Elsevier. | ||
8 Applications in bottom-up synthetic biology
The prospect of constructing artificial vesicular systems through the bottom-up assembly of biological and chemical components presents exciting opportunities for addressing previously unmet needs. By strategically combining modular scaffolds and functional building blocks, it becomes possible to engineer intricate systems featuring biomimetic or novel functionalities not found in nature.113–115The majority of the publications discussing HVs focus on physical–chemical properties of the assemblies but increasing numbers of reports push the concept towards an application. HVs were utilized as drug carriers,15,84 or mucopenetrating subunits,88 but the predominant envisioned purpose of HVs are their use as a scaffold for protein reconstruction, as nanoreactors/artificial organelles or artificial cells. Membrane-bound vesicles in the cellular environment contribute to the complexity, efficiency and specializations of cellular functions.
Membrane fusion is essential as it allows for the merging of membrane-bound organelles and vesicles, facilitating the exchange of materials and enabling cellular processes such as secretion, nutrient uptake, and communication. Controlled membrane fusion was achieved by charge-mediated fusion between positively charged HVs made of PDMS-g--(PEG)2 & DOTAP and negatively charged HVs made of PDMS-g--(PEG)2 & Soy PS for both, sHVs and gHVs. The authors found that the mixing of the semisynthetic membranes were depended on, but did not scale with, the charged lipid content. In gHVs, the fused vesicles even appeared as domains in the newly formed gHV in some cases, which may be a mean to control raft formation in synthetic membranes.32
8.1 Protein reconstruction
The plasma membrane contains membrane proteins fulfil a variety of crucial functions essential for the cell's survival and proper functioning. These membrane proteins are integral components of the lipid bilayer and contribute to the dynamic nature of the plasma membrane. The improved properties of HVs are anticipated to outweigh their lower resemblance to pristine phospholipid bilayers, e.g., the better hybrid membrane stability and elasticity116 are expected to maintain the functional integrity of the membrane proteins for longer compared to liposomes. The proteins are reconstituted into the hybrid membrane using established detergent-mediated reconstitution protocols used for proteoliposome assembly such as Triton-X,29,47,48,53,54 octyl-β-D-glucopyranoside,29,56,67 or dodecyl-maltoside.48 Various types of enzymes have been reconstructed into HVs, verifying the hypothesis that these vesicles can provide a suitable environment for the proteins as well as that the presence of the polymer did not impact the enzymatic function. Examples include the reconstitution of OmpF into HVs made of polyisopren(PI)-b-PEG & 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DPhPC),72 Cyt bo3 into HVs assembled with PBd22-b-PEG14 & POPC,47,53 or the efflux Pumps NaAtm1 and P-glycoprotein were successfully reconstructed into HVs made of PBd22-b-PEG14 & POPC and PBd22-b-PEG14 & E. coli lipid extract (EcCL), respectively.48 The enzyme pair bacteriorhodopsin and F1F0ATP synthase were successfully incorporated into HVs made of either PBd22-b-PEG14 & POPC or PDMS12-g-(PEG32)2 & Soy PC.29,30,54 In the latter case, Marusic et al. pointed out that the reason for the successful protein insertion and retained activity of the reconstructed enzyme was due to the suitable choice of copolymer, which resulted in a soft and fluid hybrid membrane and allowed for unhindered conformational changes and lateral diffusion. In a follow up study, the enzyme pair was inserted into HVs made of PDMS12-g-(PEG32)2 & Soy PS and PDMS12-g-(PEG32)2 & DOTAP. The enzyme activity was reduced in the presence of negatively charged lipids showing its sensitivity towards the environment.32 Recently, Catania et al. showed that the Cyt bo3 enzyme could be inserted into HVs made of PBd22-b-PEG14 & EcCL HVs without the use of typical surfactants but with the aid of styrene-maleic acid-based copolymer with a 74% efficiency.49 The role of surfactants in membrane proteine reconstitution is to destabilize the bilayer by swelling up the bilayer allowing an easy insertion of the protein. This procedure is tedious and requires the removal of the surfactant. Therefore, this finding represents a significant advancement. It not only enhances our comprehension of the bilayer structure of HVs but also provides a more accessible platform for studying membrane-bound enzymes in a more convenient manner.A recent example illustrated the incorporation of fusiogenic SNARE into sHVs made of PDMS12-g-(PEG32)2 & soy PC with remarkable amount of outward facing proteins and very high incorporation efficiency.31 The SNARE-containing sHVs were able to fuse together shown by mixing of their fluorescent content. The bending rigidity and pore edge tension was determined to be the key parameters for fusion. The authors proposed a plausible progression of fusion through cryo-EM snapshots. In addition, such synthetic materials were proposed as an amazing new tool for understanding dynamic membrane phenomenon and for the assembly of synthetic protocells (Fig. 9a).
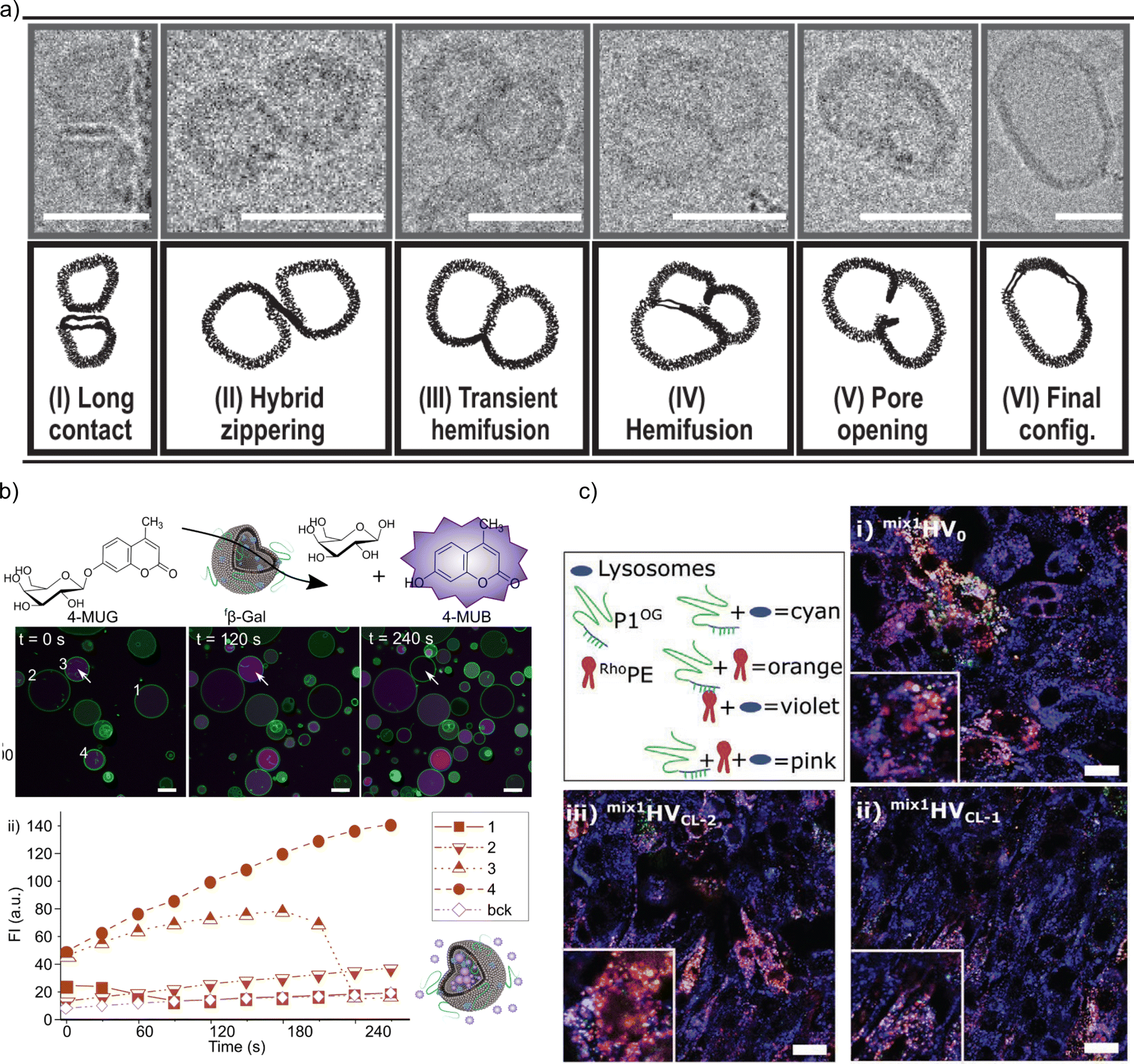 | ||
| Fig. 9 Applications of HVs as synthetic protocell and as nanoreactors. (a) Proposed fusion intermediates of SNARE-mediated fusion of HVs made of PDMS-b-PEG & soy PC. Scale bars: 30 nm.31 Used under CC BY 4.0. https://www.nature.com/articles/s41467-021-25294-z. (b) (i) Representative time-lapse CLSM images of β-Gal associated with HVs made of P(CMA-co-BuMa)-b-PCEA & soy PC immersed into 100 μM 4-MUG solution. The arrow indicates the sudden change in intensity (green: oregon green labelled polymer, magenta: 4-MUB). (ii) The increase of the fluorescent intensities of 4-MUB measured on CLSM image over time for selected 4 vesicles in comparison to the background (bck). Scale bars: 10 μm. Used under CC BY 4.0. https://www.sciencedirect.com/science/article/pii/S2352940722001846. (c) Representative CLSM images of cells incubated with HVs made of PCMA-b-PCEA & DOPE, PCMA-b-PCEA & DOPE with low cross linking, and PCMA-b-PCEA & DOPC with high crosslinking for 24 h (blue: LysoTracker deep red stained lysosomes; green: block copolymer; red: RhoPE). Scale bars: 20 μm.92 Used under CC BY 4.0. https://onlinelibrary.wiley.com/doi/10.1002/adbi.202200209z. | ||
8.2 Nanoreactors and artificial organelles
Nanoreactors are typically single compartment nanosized carriers that can perform encapsulated biocatalytic reactions due to entrapped (artificial) enzymes as recently reviewed in detail.6 These carriers can be based on (porous) silica nanoparticles, polymer capsules, liposomes, polymersomes or HVs among others. An essential property of the vesicular nanoreactors is their selective permeability, i.e., the substrate and product molecules can easily cross the membrane while the encapsulated enzymes are retrained. HVs offer unique opportunities in this context due to their hybrid nature. For instance, the polymer-rich domains in HVs made of oligo(aspartic acid)4-b-PPO43 & DOPC (10 mol% block copolymer) were shown to be able to serve as synthetic channels, allowing for the transfer of the small substrate molecule acetylthiocholine chloride to be hydrolyzed by the encapsulate enzyme acetylcholine esterase (AchE)s.95 The product was then released from the HVs to form a colored compound in a subsequent reaction for detection. Otrin et al. utilized HVs made of PDSM-g-PEG & soy PC to incorporate the chemical energy driven proton pump through a consecutive reaction of Cyt bo3 oxidase, which was used to drive ATP synthesis through F1F0ATP synthase.29 The composition of the hybrid membrane had no effect on the activity of Cyt bo3 oxidase alone. However, the ATP production rates dropped by 50% when HVs were used instead of liposomes. We utilized β-galactosidase to construct simple nanoreactors to evaluate the effect of the chemical changes in the used block copolymer. β-Galactosidase was found to be in the HVs membrane, but nonetheless, the fluorescent methylumbelliferone (4-MUB) production from β-D-galactopyranoside was observed in HVs made of P(CMA9-co-BuMA9)-b-PCEA186 & Soy PC as well as in HVs assembled from P(CMA8-co-HEMA3)-b-PCEA186. The enzymatic reaction was followed by CLSM imaging using gHVs (Fig. 9b). The produced fluorescent dye built up in the interior of the gHV until it was instantaneously released without any obvious damage of the structural integrity of the gHVs. The various hydrophobic blocks used had a significant influence on the lifetime of the associated enzyme. While using P(CMA9-co-BuMA9)-b-PCEA186 the enzymatic reaction was proceeding for up to 7 minutes. However in HVs made of P(CMA8-co-HEMA3)-b-PCEA186 & Soy PC the fluorescent content was released after 1 minute.93Nanoreactors that exhibit catalytic activity inside of mammalian cells are often referred to as artificial organelles. The design of a carrier system that can act as artificial organelles has many challenges including the requirement that they should remain intact upon cellular uptake and placement in the cytosol. We have observed that HVs made of PCMA11-b-P(METMA7-co-PCEA104) & DOPC lose their integrity when taken up by HepG2 cells. The block copolymer remained in the cells and the lipids were eliminated after 16 h.89 The fusiogenic GALA peptide was covalently attached to the block copolymer for the assembly of PCMA-b-PCEAGALA & DOPC-based HVs in addition to HVs made of PCMA-b-PCEA & DOPE to enhance their endosomal escape ability. Although both types of HVs could escape the lysosomes, the intracellular retention of the building blocks in RAW 264.7 mouse macrophages was only a few hours.91 In an attempt to address this issue, the functional groups on the block copolymer and the lipid head were cross-linked in HVs made of PCMA-b-PCEA & DOPE to improve their structural integrity. These HVs could withstand low surfactant concentrations. C8-D1A astrocytes showed lower uptake when incubated with the cross-linked HVs compared to the non-cross-linked HVs (Fig. 9c).92 In another effort, we demonstrated that HVs consisting of PCMA-b-PDMAEMA & POPC were able to produce H2O2 and nitric oxide when loaded with glucose oxidase and β-galactosidase, respectively, in RAW264.7 mouse macrophages.87 Furthermore, human macrophages differentiated from donor human peripheral blood mononuclear cells had higher intracellular nitric oxide levels when treated with β-galactosidase-containing nanoreactors in the presence of the substrate β-Nonoate compared to the controls.
9 Conclusions & outlook
Since the early days of assembling cell membrane-like systems, phospholipids are a popular choice because of their chemical nature and their ability to self-assemble into vesicles with tuneable sizes. Even though lipid vesicle-based artificial cells are essential in bottom-up synthetic biology, using only lipids is not sufficient to create artificial membranes, as they do not reflect the structural and functional complexity of cell membranes. Consequently, the addition of polymers to these lipid-based membranes is a step towards a more advanced design. The properties of polymers can be easily tuned and adjusted as the monomer of choice largely influences the physical–chemical properties of the resulting polymer chain. HVs therefore combine the best of the two worlds, the self-assembly ability of lipids and the chemical diversity of polymers. The fundamental properties including the homogenous distribution of the building blocks vs. their phase separation, the resulting vesicle sizes and membrane properties. In the latter case, different aspects interplay to give the overall hybrid membrane properties including the membrane thickness, the fluidity and viscoelastic properties. The improved stability and better control over the permeability are two core parameters that distinguishe HVs from liposomes. Although different fields benefit from HVs, they have proven to be particularly useful in bottom-up synthetic biology to improve protein reconstitution and to offer alternative scaffolds for nanoreactors and eventually artificial organelles and cells.The next step forward will require larger efforts to implement ‘smart’ polymers to obtain HVs with responsive properties. In addition, moving away from mostly synthetic polymers (and monomers) towards natural building blocks (e.g., peptides, sugars, oligonucleotides) is an underexplored opportunity. The biological evaluation of HVs is in the early days, and a better understanding of how cells, tissues and eventually living organisms respond to them is essential for their potential use in biomedicine.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Conceptualisation: E. B., B. S.; writing original draft E. B.; writing-review & editing E. B., B. S.; funding acquisition E. B., B. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 842640 (EB) and an ERC-Consolidator grant (grant agreement No. 818890).Notes and references
- T. Ruysschaert, A. F. P. Sonnen, T. Haefele, W. Meier, M. Winterhalter and D. Fournier, J. Am. Chem. Soc., 2005, 127, 6242–6247 CrossRef CAS PubMed.
- K. Kostarelos, P. F. Luckham and T. F. Tadros, J. Chem. Soc., Faraday Trans., 1998, 94, 2159–2168 RSC.
- K. Kostarelos, P. F. Luckham and T. F. Tadros, J. Colloid Interface Sci., 1997, 191, 341–348 CrossRef CAS PubMed.
- K. Kostarelos, T. F. Tadros and P. F. Luckham, Langmuir, 1999, 15, 369–376 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- X. Qian, I. Nymann Westensee, E. Brodszkij and B. Städler, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1683 CAS.
- C. Guindani, L. C. da Silva, S. Cao, T. Ivanov and K. Landfester, Angew. Chem., Int. Ed., 2022, 61, e202110855 CrossRef CAS PubMed.
- W. Jiang, Z. Wu, Z. Gao, M. Wan, M. Zhou, C. Mao and J. Shen, ACS Nano, 2022, 16, 15705–15733 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- C. Xu, S. Hu and X. Chen, Mater. Today, 2016, 19, 516–532 CrossRef CAS PubMed.
- W. M. Smigiel, P. Lefrançois and B. Poolman, Emerging Top. Life Sci., 2019, 3, 445–458 CrossRef CAS PubMed.
- W. Sato, T. Zajkowski, F. Moser and K. P. Adamala, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 1–21 Search PubMed.
- N. Amy Yewdall, A. F. Mason and J. C. M. Van Hest, Interface Focus, 2018, 8, 20180023 CrossRef PubMed.
- J. Shin, B. D. Cole, T. Shan and Y. Jang, Biomacromolecules, 2022, 23, 1505–1518 CrossRef CAS PubMed.
- E. Reimhult and M. M. Virk, J. Biomed. Res., 2021, 35, 301–309 CrossRef PubMed.
- M. Mohammadi, S. Taghavi, K. Abnous, S. M. Taghdisi, M. Ramezani and M. Alibolandi, Adv. Funct. Mater., 2018, 28, 1–18 CrossRef.
- C. E. Meyer, S. L. Abram, I. Craciun and C. G. Palivan, Phys. Chem. Chem. Phys., 2020, 22, 11197–11218 RSC.
- M. G. Gouveia, J. P. Wesseler, J. Ramaekers, C. Weder, P. B. V. Scholten and N. Bruns, Chem. Soc. Rev., 2023, 52, 728–778 RSC.
- V. De Leo, F. Milano, A. Agostiano and L. Catucci, Polymers, 2021, 13, 1027 CrossRef CAS PubMed.
- D. Liu, H. Sun, Y. Xiao, S. Chen, E. J. Cornel, Y. Zhu and J. Du, J. Controlled Release, 2020, 326, 365–386 CrossRef CAS PubMed.
- R. Tenchov, R. Bird, A. E. Curtze and Q. Zhou, ACS Nano, 2021, 15, 16982–17015 CrossRef CAS PubMed.
- M. Schulz and W. H. Binder, Macromol. Rapid Commun., 2015, 36, 2031–2041 CrossRef CAS PubMed.
- M. Schulz, A. Olubummo and W. H. Binder, Soft Matter, 2012, 8, 4849–4864 RSC.
- J. F. Le Meins, C. Schatz, S. Lecommandoux and O. Sandre, Mater. Today, 2013, 16, 397–402 CrossRef CAS.
- Y. K. Go and C. Leal, Chem. Rev., 2021, 121, 13996–14030 CrossRef CAS PubMed.
- S. Winzen, M. Bernhardt, D. Schaeffel, A. Koch, M. Kappl, K. Koynov, K. Landfester and A. Kroeger, Soft Matter, 2013, 9, 5883–5890 RSC.
- W. Shen, J. Hu and X. Hu, Chem. Phys. Lett., 2014, 600, 56–61 CrossRef CAS.
- D. Chen and M. M. Santore, Soft Matter, 2015, 11, 2617–2626 RSC.
- L. Otrin, N. Marušič, C. Bednarz, T. Vidaković-Koch, I. Lieberwirth, K. Landfester and K. Sundmacher, Nano Lett., 2017, 17, 6816–6821 CrossRef CAS PubMed.
- N. Marušič, L. Otrin, Z. Zhao, R. B. Lira, F. L. Kyrilis, F. Hamdi, P. L. Kastritis, T. Vidakovic-Koch, I. Ivanov, K. Sundmacher and R. Dimova, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15006–15017 CrossRef PubMed.
- L. Otrin, A. Witkowska, N. Marusic, Z. Zhao, R. B. Lira, F. L. Kyrilis, F. Hamdi, I. Ivanov, R. Lipowsky, P. L. Kastritis, R. Dimova, K. Sundmacher, R. Jahn and T. Vidakovic-Koch, Nat. Commun., 2021, 12, 4972 CrossRef CAS PubMed.
- N. Marušič, L. Otrin, J. Rauchhaus, Z. Zhao, F. L. Kyrilis, F. Hamdi, P. L. Kastritis, R. Dimova, I. Ivanov and K. Sundmacher, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2122468119 CrossRef PubMed.
- M. Chemin, P. M. Brun, S. Lecommandoux, O. Sandre and J. F. Le Meins, Soft Matter, 2012, 8, 2867–2874 RSC.
- T. P. T. Dao, F. Fernandes, M. Er-Rafik, R. Salva, M. Schmutz, A. Brûlet, M. Prieto, O. Sandre and J. F. Le Meins, ACS Macro Lett., 2015, 4, 182–186 CrossRef CAS PubMed.
- T. P. T. Dao, F. Fernandes, E. Ibarboure, K. Ferji, M. Prieto, O. Sandre and J. F. Le Meins, Soft Matter, 2017, 13, 627–637 RSC.
- T. P. T. Dao, A. Brûlet, F. Fernandes, M. Er-Rafik, K. Ferji, R. Schweins, J. P. Chapel, A. Fedorov, M. Schmutz, M. Prieto, O. Sandre and J. F. Le Meins, Langmuir, 2017, 33, 1705–1715 CrossRef CAS PubMed.
- T. P. T. Dao, F. Fernandes, M. Fauquignon, E. Ibarboure, M. Prieto and J. F. Le Meins, Soft Matter, 2018, 14, 6476–6484 RSC.
- V. Passos Gibson, M. Fauquignon, E. Ibarboure, J. Leblond Chain and J.-F. Le Meins, Polymers, 2020, 12, 637 CrossRef PubMed.
- M. Fauquignon, E. Ibarboure, S. Carlotti, A. Brûlet, M. Schmutz and J. F. Le Meins, Polymers, 2019, 11, 2013 CrossRef CAS PubMed.
- M. Fauquignon, E. Courtecuisse, R. Josselin, A. Mutschler, A. Brûlet, M. Schmutz and J. Le Meins, J. Colloid Interface Sci., 2021, 604, 575–583 CrossRef CAS PubMed.
- M. Fauquignon, E. Ibarboure and J. F. Le Meins, Soft Matter, 2021, 17, 83–89 RSC.
- M. Fauquignon, E. Ibarboure and J. F. Le Meins, Biophys. J., 2022, 121, 61–67 CrossRef CAS PubMed.
- R. M. Perera, S. Gupta, T. Li, C. J. Van Leeuwen, M. Bleuel, K. Hong and G. J. Schneider, ACS Appl. Polym. Mater., 2022, 4, 8858–8868 CrossRef CAS.
- Z. Cheng, D. R. Elias, N. P. Kamat, E. D. Johnston, A. Poloukhtine, V. Popik, D. A. Hammer and A. Tsourkas, Bioconjugate Chem., 2011, 22, 2021–2029 CrossRef CAS PubMed.
- X. Su, S. K. Mohamed Moinuddeen, L. Mori and M. Nallani, J. Mater. Chem. B, 2013, 1, 5751–5755 RSC.
- S. K. Lim, H. P. de Hoog, A. N. Parikh, M. Nallani and B. Liedberg, Polymers, 2013, 5, 1102–1114 CrossRef.
- S. Khan, M. Li, S. P. Muench, L. J. C. Jeuken and P. A. Beales, Chem. Commun., 2016, 52, 11020–11023 RSC.
- S. Rottet, S. Iqbal, P. A. Beales, A. Lin, J. Lee, M. Rug, C. Scott and R. Callaghan, Polymers, 2020, 12, 1049 CrossRef CAS PubMed.
- R. Catania, J. Machin, M. Rappolt, S. P. Muench, P. A. Beales and L. J. C. Jeuken, Macromolecules, 2022, 55, 3415–3422 CrossRef CAS PubMed.
- R. Seneviratne, G. Coates, Z. Xu, C. E. Cornell, R. F. Thompson, A. Sadeghpour, D. P. Maskell, L. J. C. Jeuken, M. Rappolt and P. A. Beales, Small, 2023, 2206267 CrossRef CAS PubMed.
- R. Seneviratne, L. J. C. Jeuken, M. Rappolt and P. A. Beales, Polymers, 2020, 12, 914 CrossRef CAS PubMed.
- R. Seneviratne, R. Catania, M. Rappolt, L. J. C. Jeuken and P. A. Beales, Soft Matter, 2022, 18, 1294–1301 RSC.
- R. Seneviratne, S. Khan, E. Moscrop, M. Rappolt, S. P. Muench, L. J. C. Jeuken and P. A. Beales, Methods, 2018, 147, 142–149 CrossRef CAS PubMed.
- C. Kleineberg, C. Wölfer, A. Abbasnia, D. Pischel, C. Bednarz, I. Ivanov, T. Heitkamp, M. Börsch, K. Sundmacher and T. Vidaković-Koch, ChemBioChem, 2020, 21, 2149–2160 CrossRef CAS PubMed.
- J. Nam, P. A. Beales and T. K. Vanderlick, Langmuir, 2011, 27, 1–6 CrossRef CAS PubMed.
- A. Mech-Dorosz, N. Bajraktari, C. Hélix-Nielsen, J. Emnéus and A. Heiskanen, Anal. Bioanal. Chem., 2020, 412, 6307 CrossRef CAS PubMed.
- I. M. Henderson and W. F. Paxton, Angew. Chem., Int. Ed., 2014, 53, 3372–3376 CrossRef CAS PubMed.
- W. F. Paxton, P. T. McAninch, K. E. Achyuthan, S. H. R. Shin and H. L. Monteith, Colloids Surf., B, 2017, 159, 268–276 CrossRef CAS PubMed.
- K. L. Willes, J. R. Genchev and W. F. Paxton, Polymers, 2020, 12, 745 CrossRef CAS PubMed.
- K. L. Willes, S. A. McFarland, T. E. Johnson, D. R. Hart and W. F. Paxton, ACS Appl. Nano Mater., 2022, 5, 13820–13828 CrossRef CAS.
- M. M. Virk and E. Reimhult, Langmuir, 2018, 34, 395–405 CrossRef PubMed.
- M. M. Virk, B. Hofmann and E. Reimhult, Langmuir, 2019, 35, 739–749 CrossRef PubMed.
- N. Hamada and M. L. Longo, Biochim. Biophys. Acta, Biomembr., 2022, 1864, 183887 CrossRef CAS PubMed.
- N. Hamada, S. Gakhar and M. L. Longo, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183552 CrossRef CAS PubMed.
- N. Hamada and M. L. Longo, Biochim. Biophys. Acta, Biomembr., 2022, 1864, 184026 CrossRef CAS PubMed.
- J. Steinkühler, M. L. Jacobs, M. A. Boyd, C. G. Villaseñor, S. M. Loverde and N. P. Kamat, Biomacromolecules, 2022, 23, 4756–4765 CrossRef PubMed.
- M. E. Palanco, N. Skovgaard, J. S. Hansen, K. Berg-Sorensen and C. Hélix-Nielsen, Bioinspiration Biomimetics, 2018, 13, 016005 CrossRef PubMed.
- C. Magnani, C. Montis, G. Mangiapia, A. F. Mingotaud, C. Mingotaud, C. Roux, P. Joseph, D. Berti and B. Lonetti, Colloids Surf., B, 2018, 168, 18–28 CrossRef CAS PubMed.
- A. Olubummo, M. Schulz, B. Lechner, P. Scholtysek, K. Bacia, A. Blume and W. H. Binder, ACS Nano, 2012, 6, 8713–8727 CrossRef CAS PubMed.
- M. Schulz, D. Glatte, A. Meister, P. Scholtysek, A. Kerth, A. Blume, K. Bacia and W. H. Binder, Soft Matter, 2011, 7, 8100–8110 RSC.
- M. Schulz, S. Werner, K. Bacia and W. H. Binder, Angew. Chem., Int. Ed., 2013, 52, 1829–1833 CrossRef CAS PubMed.
- M. Bieligmeyer, F. Artukovic, S. Nussberger, T. Hirth, T. Schiestel and M. Müller, Beilstein J. Nanotechnol., 2016, 7, 881–892 CrossRef CAS PubMed.
- N. Pippa, E. Kaditi, S. Pispas and C. Demetzos, Soft Matter, 2013, 9, 4073–4082 RSC.
- N. Pippa, D. R. Perinelli, S. Pispas, G. Bonacucina, C. Demetzos, A. Forys and B. Trzebicka, Colloids Surf., A, 2018, 555, 539–547 CrossRef CAS.
- M. A. Palominos, D. Vilches, E. Bossel and M. A. Soto-Arriaza, Colloids Surf., B, 2016, 148, 30–40 CrossRef CAS PubMed.
- A. K. Khan, J. C. S. Ho, S. Roy, B. Liedberg and M. Nallani, Polymers, 2020, 12, 979 CrossRef CAS PubMed.
- Y. K. Go, N. Kambar and C. Leal, Polymers, 2020, 12, 9–12 Search PubMed.
- S. Khan, J. McCabe, K. Hill and P. A. Beales, J. Colloid Interface Sci., 2020, 562, 418–428 CrossRef CAS PubMed.
- K. Flandez, S. Bonardd and M. Soto-Arriaza, Chem. Phys. Lipids, 2020, 230, 104927 CrossRef CAS PubMed.
- H. F. Li, C. Wu, M. Xia, H. Zhao, M. X. Zhao, J. Hou, R. Li, L. Wei and L. Zhang, RSC Adv., 2015, 5, 27630–27639 RSC.
- N. Pippa, D. Stellas, A. Skandalis, S. Pispas, C. Demetzos, M. Libera, A. Marcinkowski and B. Trzebicka, Eur. J. Pharm. Biopharm., 2016, 107, 295–309 CrossRef CAS PubMed.
- N. Naziris, A. Skandalis, A. Forys, B. Trzebicka, S. Pispas and C. Demetzos, J. Therm. Anal. Calorim., 2020, 141, 751–766 CrossRef CAS.
- N. Naziris, N. Pippa, V. Chrysostomou, S. Pispas, C. Demetzos, M. Libera and B. Trzebicka, J. Nanopart. Res., 2017, 19, 347 CrossRef.
- N. Naziris, N. Pippa, E. Sereti, V. Chrysostomou, M. Kędzierska, J. Kajdanek, M. Ionov, K. Miłowska, Ł. Balcerzak, S. Garofalo, C. Limatola, S. Pispas, K. Dimas, M. Bryszewska and C. Demetzos, Int. J. Mol. Sci., 2021, 22, 6271 CrossRef CAS PubMed.
- K. P. Mineart, S. Venkataraman, Y. Y. Yang, J. L. Hedrick and V. M. Prabhu, Macromolecules, 2018, 51, 3184–3192 CrossRef CAS PubMed.
- W. Zong, B. Thingholm, F. Itel, P. S. Schattling, E. Brodszkij, D. Mayer, S. Stenger, K. N. Goldie, X. Han and B. Städler, Langmuir, 2018, 34, 6874–6886 CrossRef CAS PubMed.
- Y. Zhang, N. Gal, F. Itel, I. N. Westensee, E. Brodszkij, D. Mayer, S. Stenger, M. Castellote-Borrell, T. Boesen, S. R. Tabaei, F. Höök and B. Städler, Nanoscale, 2019, 11, 11530–11541 RSC.
- E. Taipaleenmäki, G. Christensen, E. Brodszkij, S. A. Mouritzen, N. Gal, S. Madsen, M. S. Hedemann, T. A. Knudsen, H. M. Jensen, S. L. Christiansen, F. V. Sparsø and B. Städler, J. Controlled Release, 2020, 322, 470–485 CrossRef PubMed.
- E. Brodszkij, I. N. Westensee, M. Bertelsen, N. Gal, T. Boesen and B. Städler, Small, 2020, 16, 1906493 CrossRef CAS PubMed.
- P. De Dios Andres, I. N. Westensee, E. Brodszkij, M. A. Ramos-Docampo, N. Gal and B. Städler, Biomacromolecules, 2021, 22, 3860–3872 CrossRef CAS PubMed.
- C. Ade, X. Qian, E. Brodszkij, P. De Dios Andres, J. Spanjers, I. N. Westensee and B. Städler, Biomacromolecules, 2022, 23, 1052–1064 CrossRef CAS PubMed.
- C. A. Meyer, P. D. D. Andres, E. Brodszkij, I. N. Westensee, J. Lyons, S. H. Vaz and B. Städler, Adv. Biol., 2022, 2200209 Search PubMed.
- E. Brodszkij, I. N. Westensee, S. F. Holleufer, C. A. Ade, P. De Dios Andres, J. S. Pedersen, B. Stadler, P. D. D. Andres, J. S. Pedersen and B. Städler, Aplied Mater. Today, 2022, 29, 101549 CrossRef.
- J. M. Spanjers, E. Brodszkij, N. Gal and B. Stadler, Eur. Polym. J., 2022, 180, 111612 CrossRef CAS.
- T. Nishimura, S. Hirose, Y. Sasaki and K. Akiyoshi, J. Am. Chem. Soc., 2020, 142, 154–161 CrossRef CAS PubMed.
- N. Ozawa, S. Kosaka, S. Fujii and T. Nishimura, Polym. Chem., 2023, 14, 2198–2204 RSC.
- P. Walde, K. Cosentino, H. Engel and P. Stano, ChemBioChem, 2010, 11, 848–865 CrossRef CAS PubMed.
- P. A. Beales, B. Ciani and A. J. Cleasby, Phys. Chem. Chem. Phys., 2015, 17, 15489–15507 RSC.
- A. Peyret, E. Ibarboure, J. F. Le Meins and S. Lecommandoux, Adv. Sci., 2018, 5, 1–8 Search PubMed.
- D. Chandler, Nature, 2005, 437, 600–647 CrossRef PubMed.
- H.-C. Tsai, Y.-L. Yang, Y.-J. Sheng and H.-K. Tsao, Polymers, 2020, 12, 639 CrossRef CAS PubMed.
- T. Baumgart, S. T. Hess and W. W. Webb, Nature, 2003, 425, 821–824 CrossRef CAS PubMed.
- S.-W. Hu, C.-Y. Huang, H.-K. Tsao and Y.-J. Sheng, Phys. Rev. E, 2019, 99, 1–11 Search PubMed.
- J. Nam, T. K. Vanderlick and P. A. Beales, Soft Matter, 2012, 8, 7982–7988 Search PubMed.
- E. L. Elson, Biophys. J., 2011, 101, 2855–2870 CrossRef CAS PubMed.
- A. G. Jay and J. A. Hamilton, J. Fluoresc., 2017, 27, 243–249 CrossRef PubMed.
- J. E. Nielsen, T. K. Lind, A. Lone, Y. Gerelli, P. R. Hansen, H. Jenssen, M. Cárdenas and R. Lund, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 1355–1364 CrossRef CAS PubMed.
- J. S. Pedersen, C. Svaneborg, K. Almdal, I. W. Hamley and R. N. Young, Macromolecules, 2003, 36, 416–433 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967 CrossRef CAS PubMed.
- A. Larrañaga, M. Lomora, J. R. Sarasua, C. G. Palivan and A. Pandit, Prog. Mater. Sci., 2017, 90, 325–357 CrossRef.
- A. Belluati, I. Craciun, C. E. Meyer, S. Rigo and C. G. Palivan, Curr. Opin. Biotechnol., 2019, 60, 53–62 CrossRef CAS PubMed.
- H. Che and J. C. M. van Hest, ChemNanoMat, 2019, 5, 1092–1109 CrossRef CAS.
- S. Hirschi, T. R. Ward, W. P. Meier, D. J. Muller and D. Fotiadis, Chem. Rev., 2022, 122, 16294–16328 CrossRef CAS PubMed.
- W. Yuan, J. Piao and Y. Dong, Mater. Chem. Front., 2021, 5, 5233–5246 RSC.
- K. A. Podolsky and N. K. Devaraj, Nat. Rev. Chem, 2021, 5, 676–694 CrossRef CAS PubMed.
- M. L. Jacobs, M. A. Boyd and N. P. Kamat, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4031–4036 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |