
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetics of sulfur-transfer from titanocene (poly)sulfides to sulfenyl chlorides: rapid metal-assisted concerted substitution†‡
Pedro H.
Helou de Oliveira
a,
Patrick J.
Boaler
a,
Guoxiong
Hua
b,
Nathan M.
West
c,
Robert T.
Hembre
c,
Jonathan M.
Penney
d,
Malik H.
Al-Afyouni
d,
J. Derek
Woollins
e,
Andrés
García-Domínguez§
*a and
Guy C.
Lloyd-Jones
 *a
*a
aSchool of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: v1agarc9@ed.ac.uk; guy.lloyd-jones@ed.ac.uk
bSchool of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK
cEastman Chemical Company, 200 S Wilcox Dr, Kingsport, Tennessee 37660, USA
dFlexsys America L.P., 260 Springside Drive, Akron, OH 44333, USA
eDepartment of Chemistry, Khalifa University, Abu Dhabi, UAE
First published on 19th June 2024
Abstract
The kinetics of sulfur transfer from titanocene (poly)sulfides (RCp2TiS5, Cp2TiS4CMe2, Cp2Ti(SAr)2, Cp2TiCl(SAr)) to sulfenyl chlorides (S2Cl2, RSCl) have been investigated by a combination of stopped-flow UV-Vis/NMR reaction monitoring, titration assays, numerical kinetic modelling and KS-DFT calculations. The reactions are rapid, proceeding to completion over timescales of milliseconds to minutes, via a sequence of two S–S bond-forming steps (k1, k2). The archetypical polysulfides Cp2TiS5 (1a) and Cp2TiS4C(Me2) (2a) react with disulfur dichloride (S2Cl2) through rate-limiting intermolecular S–S bond formation (k1) followed by a rapid intramolecular cyclization (k2, with k2 ≫ k1 [RSCl]). The monofunctional sulfenyl chlorides (RSCl) studied herein react in two intermolecular S–S bond forming steps proceeding at similar rates (k1 ≈ k2). Reactions of titanocene bisthiophenolates, Cp2Ti(SAr)2 (5), with both mono- and di-functional sulfenyl chlorides result in rapid accumulation of the monothiophenolate, Cp2TiCl(SAr) (6) (k1 > k2). Across the range of reactants studied, the rates are relatively insensitive to changes in temperature and in the electronics of the sulfenyl chloride, moderately sensitive to the electronics of the titanocene (poly)sulfide (ρ(Ti–(SAr)) ≈ −2.0), and highly sensitive to the solvent polarity, with non-polar solvents (CS2, CCl4) leading to the slowest rates. The combined sensitivities are the result of a concerted, polarized and late transition state for the rate-limiting S–S bond forming step, accompanied by a large entropic penalty. Each substitution step {[Ti]–SR′ + Cl–SR → [Ti]–Cl + RS–SR′} proceeds via titanium-assisted Cl–S cleavage to generate a transient pentacoordinate complex, Cl–[Cp2TiX]–S(R′)–SR, which then undergoes rapid Ti–S dissociation.
Introduction
Titanocene pentasulfide, Cp2TiS5 (1a, Scheme 1), is a bench-stable and easily handled pentasulfide dianion surrogate.1,2 Pentasulfide 1a, and related polysulfides, (e.g. Cp2TiS4C(Me)2, 2a),2a allow the selective preparation of sulfur homocycles (Sn),3 heterocycles (E2Sn),4 and polysulfanes (R2Sn)5 from sulfenyl chlorides (SxCl2, RSxCl). Titanocene bis and mono-thiolates, Cp2Ti(SR)2 and Cp2TiCl(SR), are also efficient sulfide anion surrogates.3c,6,7 These S–S bond-forming reactions generally proceed quantitatively under mild conditions (≤25 °C) making pentasulfide 1a and analogues one of the central platforms for the synthesis of polysulfides.3d,8,9 Despite this widespread application, studies of the mechanism(s) of these sulfur transfer processes are scarce,2a,7a,10,11 and as far as we are aware the kinetics of the general process in Scheme 1 have not been investigated.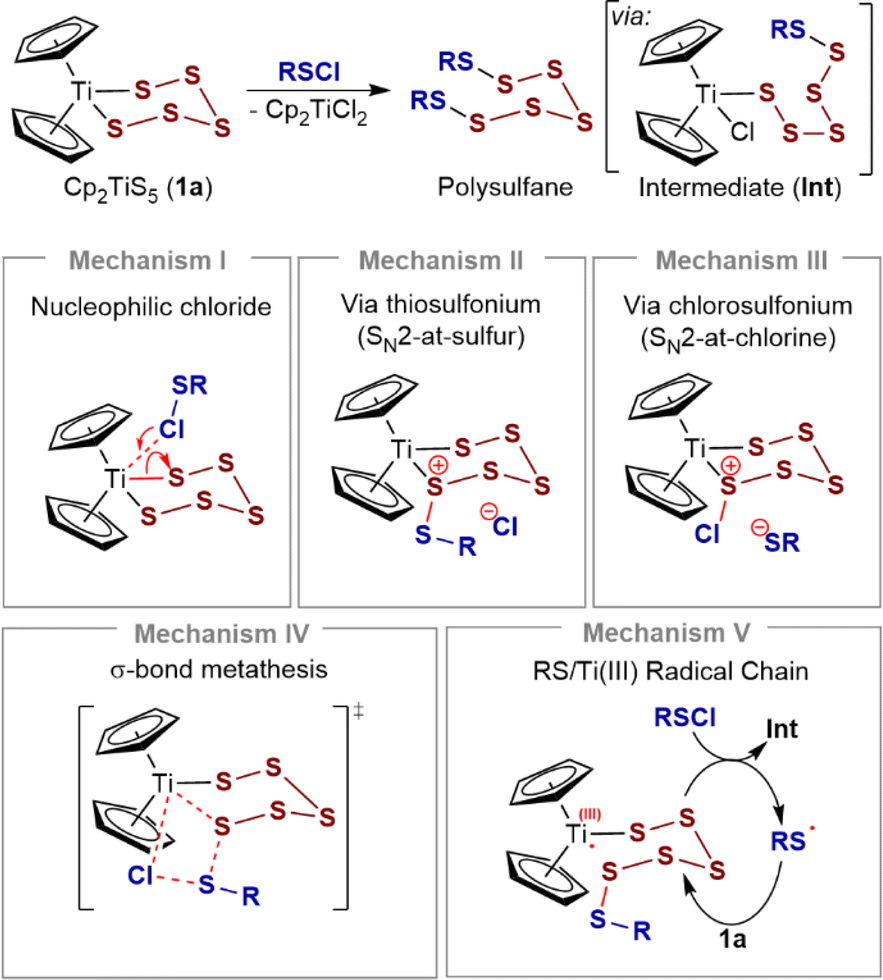 | ||
| Scheme 1 Reaction of titanocene pentasulfide (1a) with sulfur(II) chlorides: general features and potential reaction mechanisms (I–V). | ||
The reaction of pentasulfide 1a with RSCl species is generally accepted to proceed in two steps, via a chlorotitanocene intermediate (Int), Scheme 1. Support for this includes the isolation of intermediates from the slow reactions of the sterically hindered sulfenyl chloride Ph3CSCl with Cp2TiS4CMe2 (2a) and with Cp2TiS4(cyclo-C6H10).7a Münchow and Steudel proposed that nucleophilic attack by the chlorine atom in RSCl at the titanium atom opens the metallacycle (mechanism I).7a
However, sulfur compounds can react ambiphilically and several alternative mechanisms can also be considered for the S–S bond forming steps. For example, concerted or step-wise SN2-at-sulfur reactions are feasible,12–14 with 1a acting as the nucleophile (mechanism II). Alternatively, the S–S bond forming steps could involve formation of chlorosulfonium ions (mechanism III), concerted σ-bond metathesis (mechanism IV), or mechanisms involving sulfenyl radicals15 and Ti(III)16 chain carriers (mechanism V), or analogous chain processes. There is also the possibility that 1a acts as a reservoir to reactive ring-opened species.17 Depending on specific kinetic parameters, these last two possibilities are unlikely to result in simple bimolecular kinetics (see ESI, Section S9‡).
Results and discussion
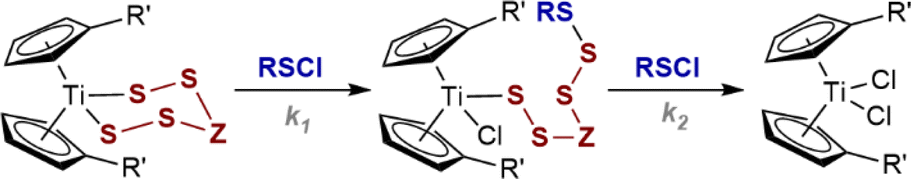
Herein, we report on the kinetics of sulfur transfer from titanacyles (1 and 2) to sulfenyl chlorides S2Cl2 and to RSCl reagents (3 and 4), including analysis of the effects of solvent and temperature on the rate. The experimental kinetic data are complemented with KS-DFT calculations to assess the feasibility of proposed reaction pathways and elucidate the key role of the titanocene in these nucleophilic substitutions. We also briefly report on the reactions of acyclic Ti-thiophenolates with S2Cl2. The overall insight to the kinetics and mechanism allows the identification of parameters that can be used to tune the rate and selectivity.Kinetics of the reaction of S2Cl2 with titanacycle 1a
We began by investigating of the reaction of S2Cl2 with Cp2TiS5 (1a). Preliminary analysis by 1H NMR spectroscopy confirmed quantitative conversion within seconds in CH2Cl2 solution, accompanied by a characteristic change in colour from deep-red (Cp2TiS51a, λmax 490 nm) to light-orange (Cp2TiCl2, λmax 393 nm).17b,c Stopped-flow UV-Vis analysis (SF-UV) of the reactions of 1a, at initial concentrations in the range 0.05–0.15 mM, with a large excess of S2Cl2 (for conditions, see the ESI, Section S2‡) were in agreement with pseudo first-order kinetics, with two clear isosbestic points (λ = 378, 422 nm, e.g.Fig. 1). The data correlated with the same exponent (−kobst) when the initial concentrations of the limiting reagent [1a]0 were doubled or halved (see Section S2 in the ESI‡) confirming the first-order18 kinetic dependency on the titanium complex 1a in the rate-limiting event. Extraction of kobs from exponential decays in absorption at 495 nm afforded linear correlations against [S2Cl2]0 (see Section S2 in the ESI‡) confirming first-order kinetic dependency on S2Cl2. The overall process {1a + S2Cl2} is thus governed by second order kinetics, first order in both components, i.e., −d[1a]/dt = k1[1a][S2Cl2], with the empirical19 bimolecular rate constant, k1 = 320 M−1 s−1 at 22 °C (Table 1, entry 1).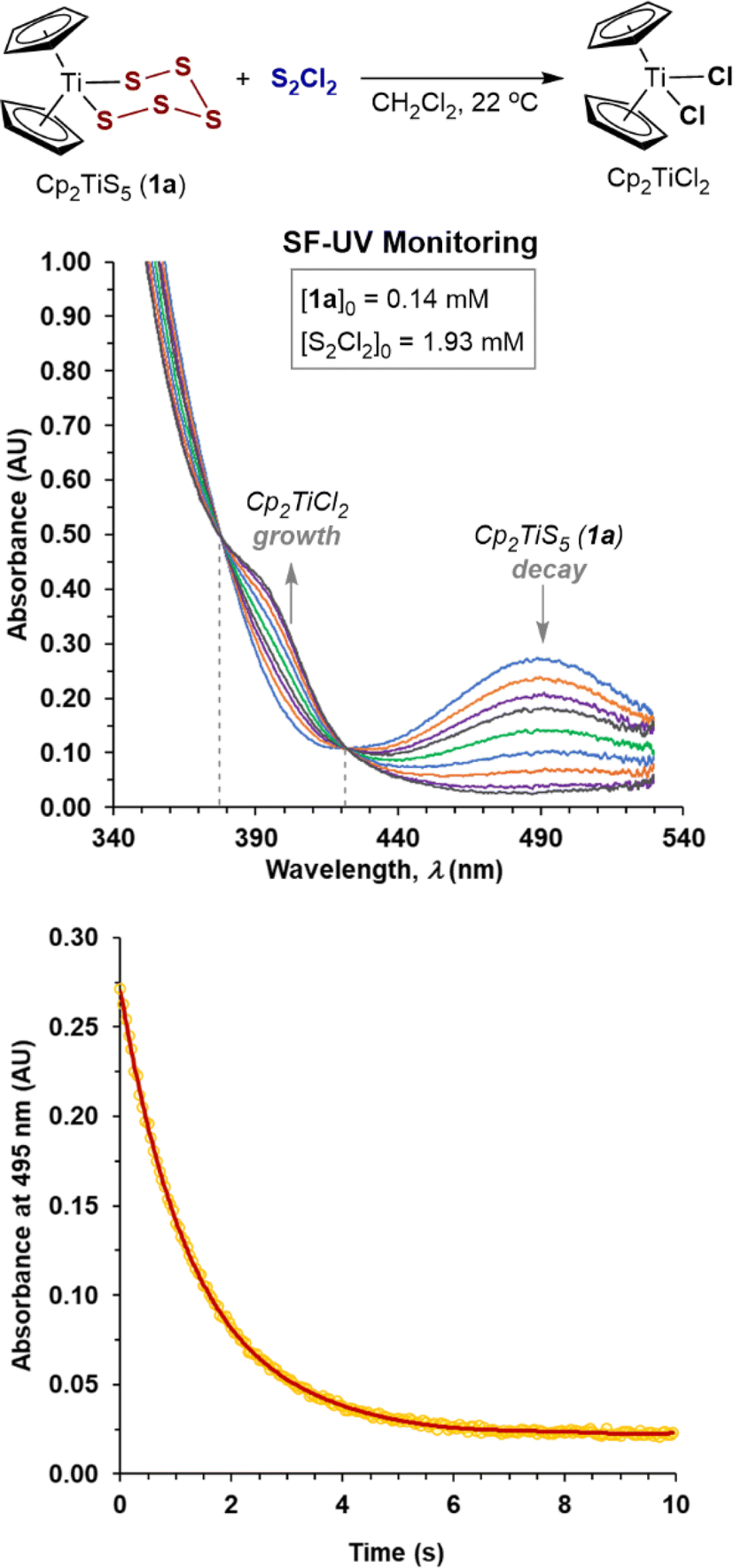 | ||
| Fig. 1 Stopped-flow UV-Vis temporal evolution of spectra and exponential decay in absorbance of 1a at 495 nm on reaction with excess S2Cl2 in CH2Cl2 at 22 °C. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Titanocene | RSCl | k 1 (M−1 s−1) | k 2 (M−1 s−1) |
| a No accumulation of an intermediate is observed in these processes, k2 > 102k1. | ||||
| 1 | 1a | S2Cl2 | 3.2 × 102 | —a |
| 2 | 1b | S2Cl2 | 2.4 × 102 | —a |
| 3 | 1c | S2Cl2 | 2.4 × 102 | —a |
| 4 | 1d | S2Cl2 | 2.0 × 102 | —a |
| 5 | 1e | S2Cl2 | 0.9 × 102 | —a |
| 6 | 2a | S2Cl2 | 3.4 × 104 | —a |
| 7 | 1a | 3a | 4.8 × 101 | 6.8 × 101 |
| 8 | 1a | 3b | 1.7 × 103 | 6.9 × 103 |
| 9 | 1a | 3c | 4.4 × 103 | 9.8 × 103 |
| 10 | 1a | 4a | 4.4 × 104 | 5.7 × 104 |
| 11 | 1a | 4b | 4.2 × 104 | 5.0 × 104 |
| 12 | 1a | 4c | 4.9 × 104 | 5.0 × 104 |
| 13 | 1a | 4d | 4.1 × 104 | 4.6 × 104 |
| 14 | 1a | 4e | 4.5 × 104 | 4.3 × 104 |
| 15 | 1a | 4f | 1.9 × 104 | 6.8 × 103 |
|
||||
Effects of solvent on the rate of reaction of S2Cl2 with 1a
The ability to conduct SF-UV kinetic assays at low concentrations of 1a allowed exploration of a range of solvents without issues relating to solubility of the reactants, intermediates, and titanocene dichloride and cycloheptasulfur products. Solvent polarity substantially affects the rate (k1, Fig. 2) and there is a reasonable correlation with Catalán's polarity parameters: log(k1) = 49.5{0.97 × SA + 0.03 × SdP} + 0.56; R2 = 0.98; see Section 2.3 in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 The three orders of magnitude range in rate across the series of solvents spanning from CS2 (slowest) to CHCl3 (fastest) is indicative of development in polar character on approach to the rate limiting transition state.
20 The three orders of magnitude range in rate across the series of solvents spanning from CS2 (slowest) to CHCl3 (fastest) is indicative of development in polar character on approach to the rate limiting transition state.
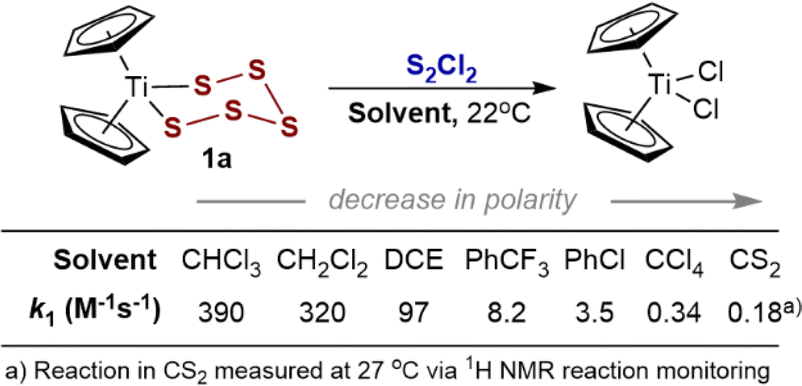 | ||
| Fig. 2 Solvent effects on the empirical19 rate constant (k1) values for the pseudo first-order reaction of 1a with excess S2Cl2 at 22 °C. | ||
The kinetics of the reaction in CS2 (ref. 3a) and in CCl4, were slow enough (k1 = 0.18 and 0.34 M−1 s−1, respectively) to allow in situ continuous 1H NMR spectroscopic reaction monitoring.21 The NMR profiles (see ESI, Section S5‡) showed no evidence for accumulation of intermediates, and remained consistent with bimolecular kinetics, i.e., −d[1a]/dt = k1[1a][S2Cl2], across a range of concentrations. Overall, the analyses eliminate radical chain (mechanism V) and slow irreversible Ti–S pre-dissociation mechanisms.
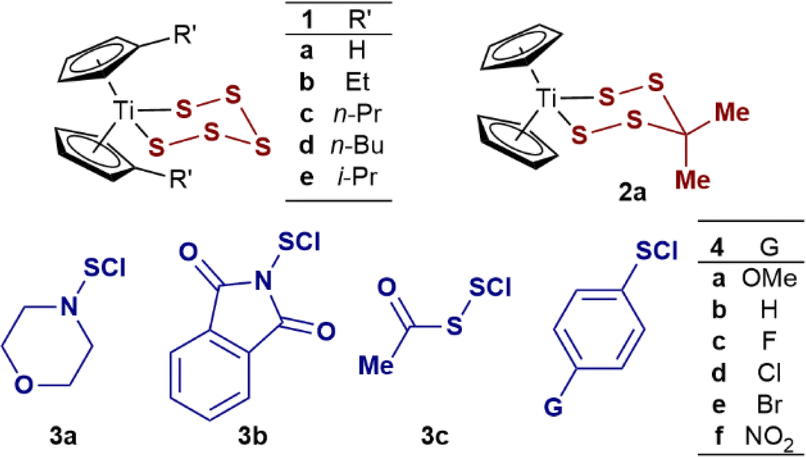
Kinetics of the reactions of S2Cl2 with related titanacycles 1b–e and 2a
Reactions of S2Cl2 in CH2Cl2 with structurally related titanacycles 1b–e and 2a also gave bimolecular kinetics, i.e., first order in both S2Cl2 and the titanacycle (Table 1, entries 2–6). Alkyl substitution of the cyclopentadienyl groups increases the solubility of titanocene (poly)sulfides in organic solvents.1a,2b The rates of reaction of the alkylated analogues 1b–e were all slightly slower than 1a (Table 1, entries 2–5) with the relative rates ranging from 0.8 (1b, Et) to 0.3 (1e, i-Pr).While compensating steric/electronic contributions cannot be discarded, it is reported that the donor effects of cyclopentadienyl substituents do not significantly affect the electron density on Ti in the titanapentasulfide group.22 The similar inductive effects of the alkyl substituents (e.g. Et, i-Pr, σp = −0.15)23 suggest these changes to result primarily from steric hindrance. However, attempts to correlate these small differences against reported steric parameters, such as angle-descriptions for η5-cyclopentadienide ligands,24 did not show a clear trend (see ESI, Section S2.4‡). In contrast to the negligible differences in reactivity of 1a–e, replacing the sulfur atom at position 4 in the pentasulfide ring had a profound effect, with the rate of Cp2TiS4C(Me)2 (2a) being two orders of magnitude faster (Table 1, entry 6) than with titanacycle 1a (Table 1, entry 1). The kinetics were again first order in both components, leading to the same overall bimolecular empirical rate relationship: –d[2a]/dt = k1[2a][S2Cl2].
Kinetics of the reactions of monofunctional sulfur chlorides (RSCl) 3a–c and 4a–f with titanacycle 1a
The SF-UV analyses of the reactions of monofunctional RSCl reagents 3a–c and 4a–f with 1a showed distinctly different temporal UV-Vis absorption profiles (Fig. 3) to those with S2Cl2 (Fig. 1). The absence of isosbestic points accompanied by an increase then decay in absorbance at 495 nm indicates accumulation of a UV-Vis absorbing reactive intermediate (Fig. 3). The difference between the reactions involving the monofunctional RSCl reagents compared to S2Cl2 is attributed to the Cp2TiCl(S7Cl) intermediate generated by the latter undergoing rapid intramolecular S–S bond formation, vide infra.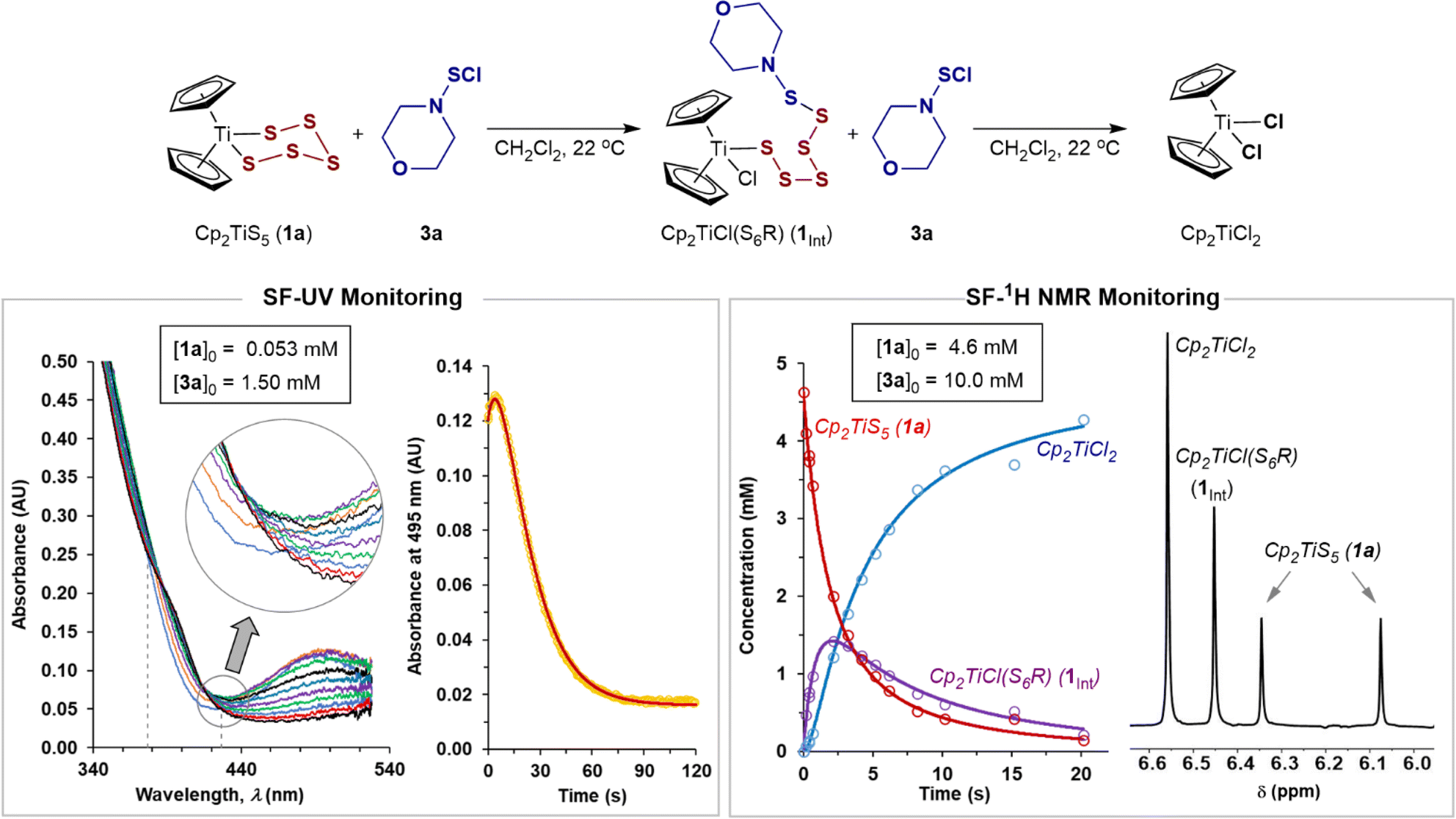 | ||
| Fig. 3 Stopped-flow UV and 1H NMR monitored evolution of the reaction of 1a with excess sulfenyl chloride 3a. Fitted models: solid lines; experimental data: circles. | ||
Stopped-flow 1H NMR spectroscopic (SF-NMR)25 analysis of the reaction of 1a with the least reactive monofunctional reagent, N-morpholinosulfenyl chloride 3a, confirmed the generation of a transient species. This was tentatively assigned as the polysulfanido intermediate Cp2TiCl(S6R), consistent with Münchow and Steudel's proposal.7a In this case the values for k1 and k2 (Table 1, entry 7) were independently determined by numerical methods fitting of both the NMR and SF-UV data (Fig. 3). The rate constants for the much more reactive RSCl reagents 3b, c and 4a–f (Table 1, entries 8–15) were extracted from numerical modelling of the UV-Vis data only, because the reactions were too fast to monitor by SF-NMR.
Each model employed multiple averaged data sets, varying [RSCl]0 (see, ESI, Section S3‡) with the net absorbance assumed to arise from Beer–Lambert additivity of all species. The temporal evolution of the SF-UV data then depends on the initial reactant concentrations, the rate constants k1 and k2, and the molar absorption coefficients (ε) of 1a, Cp2TiCl2, and Cp2TiCl(S6R). However, without being able to independently determine εint, we instead used the rate ratio (k2/k1) as a dimensionless constraint to find unique solutions to the data. These constraints were determined using analytical solutions to rapid-mixing NMR titration data26 (see, ESI Section S4.2‡).
The identity of the sulfur electrophile has a major impact on the rates of reaction of 1a with 3a–c and 4a–f, with k1 and k2 spanning three orders of magnitude (Table 1, entries 1, 7–15). The rate ratios (k2/k1) are also dependent on the identity of RSCl. For example 3b is significantly more reactive towards the corresponding Cp2TiCl(S6R) intermediate than the pentasulfide (1a), k2/k1 = 4. These results confirm that generation of the polysulfido intermediates in high yield from the reaction of titanacycles (1 and 2) with monofunctional sulfenyl chlorides is challenging.7a The aryl sulfenyl chlorides (ArSCl, 4a–f, Table 1, entries 10–15) showed the highest reactivity towards 1a, with rate constants indicative of millisecond reaction timescales at typical concentrations employed for synthetic procedures (0.05–0.1 M).3 However, within the series, which spans classic electron-donating (p-OMe, 5a) through electron-withdrawing (p-NO2, 5f) substituents, there is no meaningful correlation against Hammett substituent parameters (see ESI, Section S3.15‡). An analogous lack of correlation has previously been used to invoke concerted, SN2-like, mechanisms for nucleophilic substitutions at sulfur, rather than step-wise addition–elimination processes.27
Activation parameters for the reactions of S2Cl2 and 3a with titanacycles 1a and 2a in CH2Cl2
The kinetics of the first S–S bond forming step19 (k1) in the reaction of 1a, and of 2a, with S2Cl2, and both S–S bond forming steps (k1 and k2) in the reaction of 1a with N-morpholinosulfenyl chloride 3a, were investigated by variable temperature SF-UV kinetics. Overall, the rates are insensitive to temperature changes, showing small increases or decreases, or no change at all, depending on the identity of the titanacycle and sulfenyl chloride reagent (Fig. 4).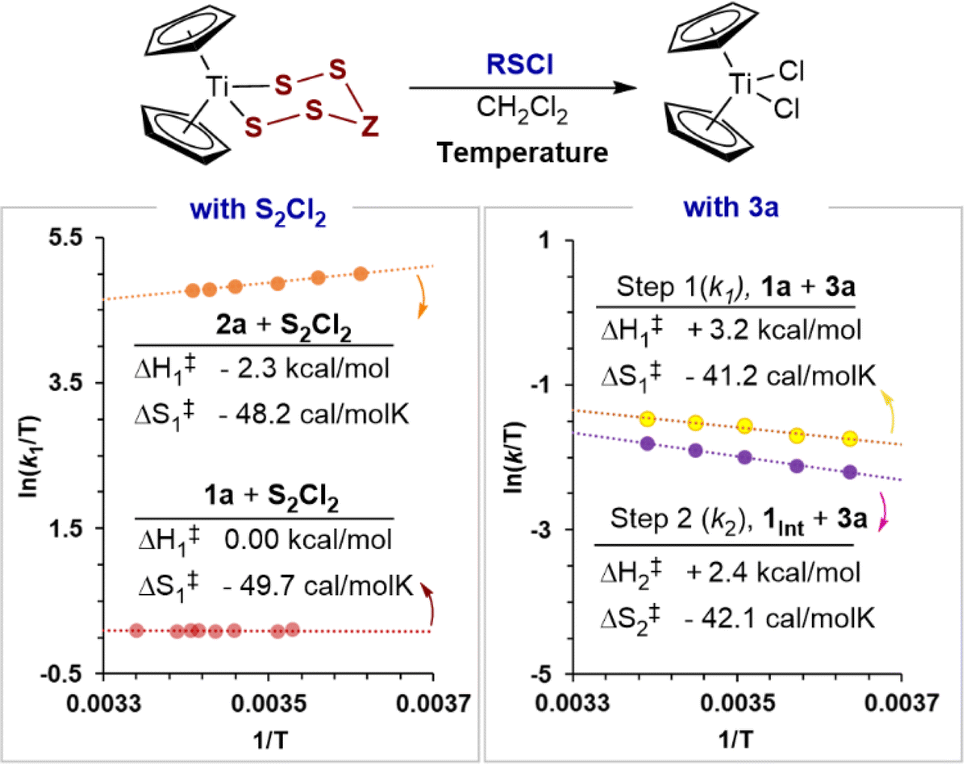 | ||
| Fig. 4 Eyring analyses using process rate constants for the reaction of titanacycles with chlorosulfanes. The calculated activation parameters correspond to the microkinetic S–S bond forming events.19 | ||
The rate constants for the reaction of 1a with S2Cl2 remain invariant, within experimental error, between 10 and 26 °C. The reaction of S2Cl2 with titanacycle 2a slightly increases in rate on cooling, (k277K ≈ 1.2k295K). Conversely, both of the empirical19 rate constants (k1, k2) for the reaction of 1a with 4a slightly decrease on cooling (k275K ≈ 0.8k295K). Eyring analyses (Fig. 4) showed all these effects arise from activation barriers that are dominated by large negative changes in entropy, with only minor enthalpic contributions (i.e., ΔG‡/RT ≈ −ΔS‡/R). Large negative entropies of activation can arise from significant solvent reorganisation around the transition state. However, this is more commonly observed in polar protic solvents and is unlikely to be significant in CH2Cl2. Overall, the results are indicative of highly organised associative transition states which result in large losses in translational degrees of freedom in the reactants, relative to the ground state.
Computational interrogation of the reaction of S2Cl2 with titanacycles 1a and 2a
The reaction of 1a with S2Cl2 was selected for initial assessment of various computational methods.28 Hybrid density functionals and DLPNO-CCSD(T) failed to qualitatively describe experimental trends, likely due to a significant multideterminant character in some calculated species (see ESI, Section S11.2‡). Based on these preliminary computational assays, the meta-GGA functional, r2SCAN,29 was chosen for further investigations.Free energy profiles of sulfur transfer reactions of S2Cl2 with 1a and 2a are shown in Fig. 5A. Both profiles qualitatively agree with experimental results in that the initial sulfur transfer is rate-limiting, and that titanacycle 2a is significantly more reactive than 1a. Sulfur transfer from the titanocene to the sulfur chloride were found to proceed through a four-membered transition state, similar to mechanism IV, but without accompanying dissociation of the Ti–S bond. The transfer initially leads to a coordinatively saturated 18-electron titanium complex (Fig. 5, Int1), prior to this opening into a 16-electron polysulfide intermediate (Fig. 5, Int2). Other pathways explored computationally (e.g. mechanism III) were much less favourable than the process discussed above (see ESI Section S11.3‡), and no appropriate local minimum could be found resulting from the attack of the chlorine atom of S2Cl2 at titanium (mechanism I).7a
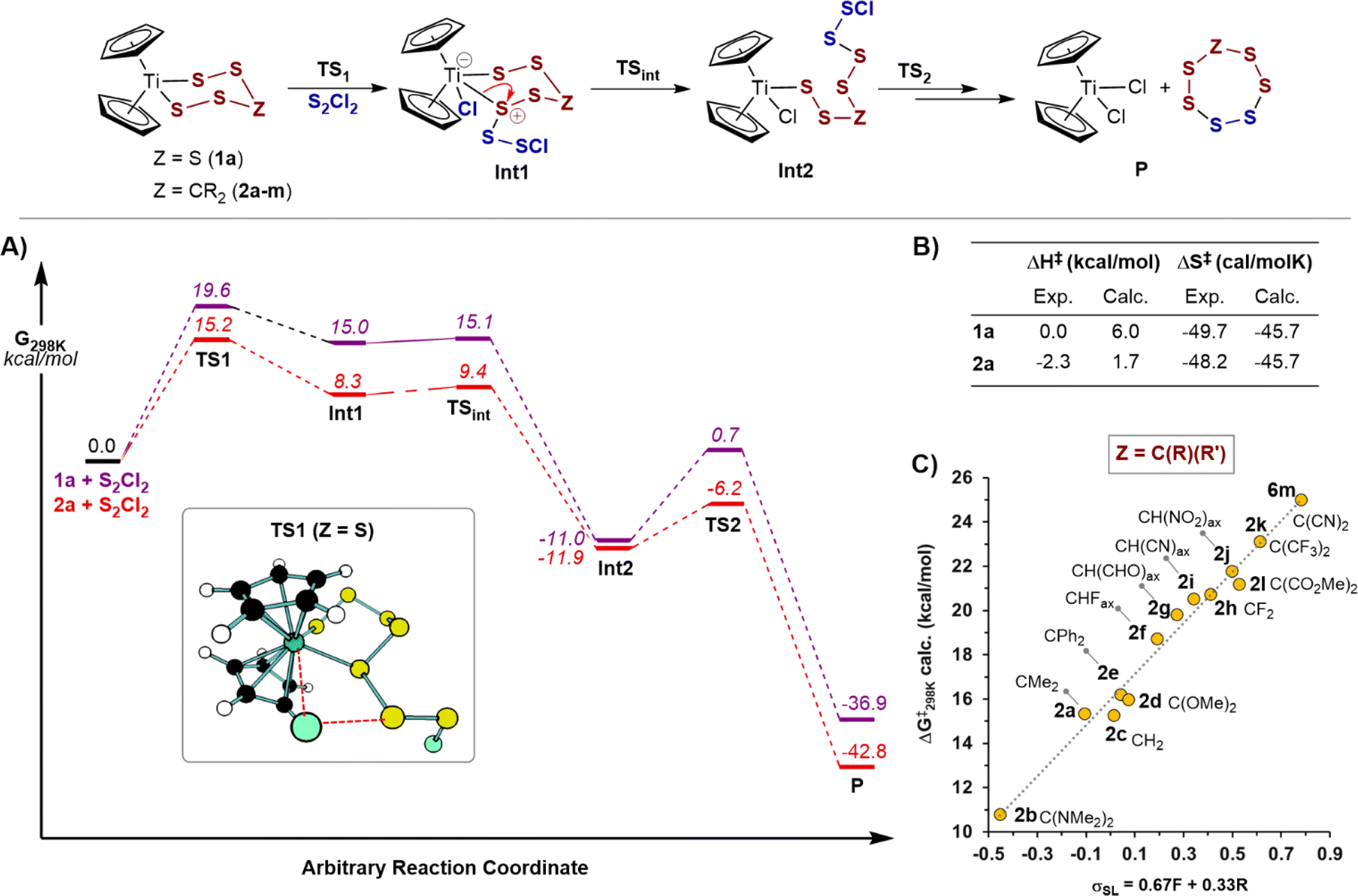 | ||
| Fig. 5 (A) Computed free energy reaction profile of 1a and 2a with S2Cl2 including transition state geometry TS1 for {1a + S2Cl2}. (B) Comparison of experimental (295 K) and computed values of activation parameters of the rate limiting transition state (TS1) for the reaction of 1a and 2a with S2Cl2. (C) Virtual reaction of other titanacycles Cp2TiS4Z (2b–m) with S2Cl2: correlation of microkinetic free energy barriers for TS1 against Swain–Lupton substituent constants. Computational calculations performed with: r2SCAN-D4-SMD(CH2Cl2)/ma-def2-QZVPP(Ti); ma-def2-TZVPP(C, H, S, Cl, O, N, F)//r2SCAN-3c, T = 298.15 K, p = 1.0 atm, qRRHO cutoff = 50 cm−1, c = 1.0 M. | ||
Comparison of the computational activation parameters for the rate controlling barriers with those determined experimentally are shown in Fig. 5B. Considering statistical corrections,19 the computational predictions are in reasonable agreement with results from experiment. The negligible changes in enthalpy and the large entropy of activation values for the reaction of 1a (|ΔS‡exp − ΔS‡calc| = 4.0 cal mol−1 K−1) and 2a (|ΔS‡exp − ΔS‡calc| = 2.5 cal mol−1 K−1) further support a concerted metathesis-like mechanism.30 These results also reinforce the conclusion that the large entropic barriers are not caused by solvent reorganisation.
The qualitative correlations found for trends in the reactions of other titanocenes (see ESI, Section S11.4‡) prompted us to further investigate the electronic effects caused by replacing the distal sulfur in 1a with carbon groups (2a and analogues 2b–m, Fig. 5C). The microkinetic free energies of activation correlated well with inductive-weighted Swain–Lupton parameters (ρSL = +11.5; R2 = 0.98),23,31 with electron-donating groups resulting in lower reaction barriers. That the correlation includes substituents of various sizes, and ground state geometries (2b, 2k and 2l have a twist-boat like geometry), suggests that steric factors, and other effects such as angle strain, only play a minor role on the relative reaction rates.
Kinetics of the reaction of S2Cl2 with titanoncene thiophenolates 5a–d and 6a–d
To expand the study to acyclic titanocene sulfide di-anion surrogates, we analysed the kinetics of the reactions of thiophenolato complexes, Cp2Ti(SAr)2 (5a–d) and Cp2TiCl(SAr) (6a–d) with S2Cl2, and with N-morpholinosulfenyl chloride 3a. 1H NMR spectroscopic analysis confirmed that complexes 5a and 6a are stable in CH2Cl2 solution at room temperature for at least 24 hours in the dark.Complexes 6a–d are however sensitive to ambient light, especially 6a (see ESI Section S8‡), and additional precautions were taken to avoid visible light exposure when preparing and handling their solutions.32 SF-UV analyses of the reactions of bis(thiophenolato) titanocenes 5a–d with excess of S2Cl2 showed this class of substrate to be exceptionally reactive. Indeed, 5a–d are converted into 6a–d within the deadtime (<10 ms) of the measurement method. This is then followed by exponential decay of the monothiolates 6a–d to Cp2TiCl2 (Fig. 6A). The same empirical rate constant (k2), within experimental error, was independently determined using an isolated sample of 6a. These features are indicative of a consecutive process for 5a–d, with the first step being significantly faster than the second (k1/k2 ≥ 100).
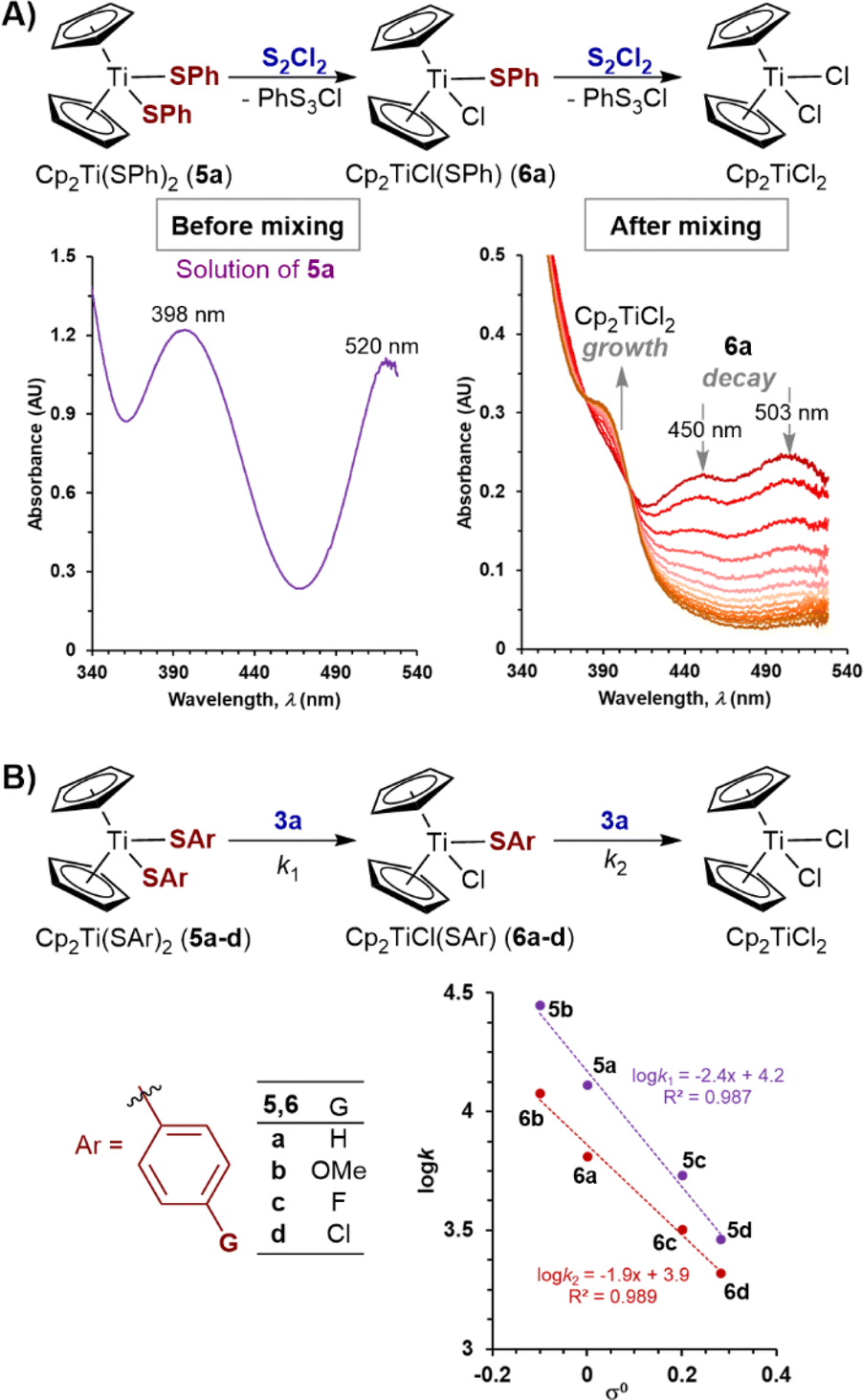 | ||
| Fig. 6 (A) SF-UV monitoring of the reaction of titanocene bisthiophenolate 5a with S2Cl2 at 22 °C. (B) Yukawa–Tsuno linear free energy correlation for thiophenolates 5a–d and 6a–d in reaction with 3a. | ||
The high reactivity of the bis(thiophenolato) titanocenes 5a–d is consistent with findings of Osakada and Yamamoto on the relatively slow kinetics of transmetallation of PtClMe(COD) with titanocene monothiolate 6a in THF.11b However, in contrast to the second order kinetics observed for the reactions with sulfenyl chlorides studied herein, Osakada and Yamamoto found that monothiolate 6a undergoes indirect reaction with PtClMe(COD) via5a generated through reversible disproportionation, resulting in overall third-order kinetics.11b
Direct comparison of the empirical rate constants for the reactions of titanacycles 1 and 2 with S2Cl2 (Table 1) against the same process for thiolate derivatives 6a–d is not possible because the sulfenyl chloride co-product (PhS3Cl) from the latter is highly susceptible to spontaneous desulfuration.33 This phenomenon results in the observed decay rates of 6a–d being the net effect of several simultaneous substitution processes. Nevertheless, similarly to titanacycles 1 and 2, the rates of reaction of 6a with S2Cl2 show a low sensitivity to temperature (k281K ≈ 1.1k297K). Qualitative assessment of effects of para-substitution in the aromatic ring in thiophenolate derivatives 6a–d, shows that electron-donating groups accelerate the substitution processes.34
To make quantitative comparisons, SF-UV analyses of the reactions of 5a–d with the less reactive N-morpholinosulfenyl chloride 3a were conducted. The spectra showed the distinctive features of a sequential reaction: lack of isosbestic points and initial increase then decay in absorbance. Deconvolution of data showed moderate selectivity of 3a for the bisthiolate complexes (k1/k2 = 1.4 to 2.5),19 with the greater the electron donating ability of the thiolate, the higher the selectivity. Linear free energy relationships using Hammett substituent constants23,30 showed moderate curvature (see ESI Section S3.15‡), for both experimental and computational data, despite the known correlation of nucleophilicity parameters for thiolates.35 Better correlations for both steps (k1, ρ = −2.4; k2, ρ = −1.9) were obtained using the method of Yukawa and Tsuno,36 (Fig. 6B). Both analyses indicate significant development of positive charge at the nucleophilic Ar–S–Ti sulfur atom during the transition state,37 consistent with that found in computational analysis of the reaction of S2Cl2 with 2a–m (Fig. 5C).
Conclusions
Using a combination of stopped-flow UV-Vis and stopped-flow NMR techniques, kinetic analyses, numerical modelling, and KS-DFT-computations, we have investigated the reactions of titanocene (poly)sulfides with a variety of sulfenyl chlorides to generate sulfur homocycles (Sn), heterocycles (E2Sn), and polysulfanes (R2Sn). Systematic variations in the structure of the reactants, reaction temperature, and solvent polarity have served to kinetically characterise key parameters influencing the phenomenological19 process rates. Collectively, the study provides data that supports a different mechanism to that previously proposed.7a The process has all of the characteristics of a nucleophilic substitution at R–S–Cl by the titanium-bound sulfur, in a concerted, late, and metathesis-like four-membered transition state, in which chloride departure is assisted by the metal (Fig. 5).Titanocene pentasulfide 1a is about two orders of magnitude less reactive than the isopropylidene analogue 2a, and about three orders of magnitude less reactive than monosulfide analogues 5a–d and 6a–d, to intermolecular attack of sulfenyl chlorides. In other words, the nucleophilicity of the titanium-bound sulfur is progressively attenuated by the increasing number of concatenated sulfur atoms, with the effect greatest in pentasulfide 1a. The results also rationalise the challenges in isolating titanocene polysulfido complexes in high yields using monofunctional sulfur chlorides as electrophiles, due to consecutive processes (k1, k2) having similar rates. Two key outcomes from these investigations inform practical protocols for sulfur transfer from titanocenes:
(1) The low sensitivity of the process rates to temperature makes cooling useful for the preparation of thermally sensitive organic (poly)sulfides without sacrificing reactivity. Solvent polarity can also be used to tune process rates, and incorporation of alkyl substituents on the titanocenes used to enhance solubility and thus process concentrations without a significant decrease in nucleophilicity.
(2) Titanocene thiolates undergo very rapid reactions with sulfenyl chlorides. They can be stored indefinitely as solids in the dark at −40 °C, are stable in solution for hours, and can generally be handled under air but do require protection from exposure to ambient light. Compared to the mono(thiolato)titanocenes, the bis(thiolates) are easier to access and more stable to visible light, but retain high nucleophilicity, thus making them the more practical sulfur transfer reagents.
Overall, the mechanistic features elucidated can be considered for other nucleophilic substitutions involving transition metal polysulfides, electrophiles or titanocene nucleophiles under non-aqueous conditions.
Data availability
Experimental kinetic data, NMR spectra, UV-Vis spectra, SF-methods, kinetic analyses, computational methodologies, energies, and further discussion and analysis can be found in the ESI.‡ Cartesian coordinates of optimised geometries are available in the OptimisedGeometries.xyz file.Author contributions
AGD and GCL-J directed the investigation. PHHO and AGD conducted all experimental work, except for the synthesis of 1b–1f which was carried out by GH. PHHO conducted the computational studies. PHHO, AGD, PJB and GCL-J analysed the data. All authors contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Eastman Chemical Company and Flexsys America L. P. for funding. We thank Dr Vera Münchow (Italy) for providing an optimised procedure for the purification of complex 2a. We thank Sumit Chakraborty and Javier Grajeda (Eastman) for valuable discussions. We thank the University of Edinburgh Compute and Data Facility (ECDF) for access to computing resources, the EPSRC Programme Grant “Boron: Beyond the Reagent” (EP/W007517), and Royal Society (URF\R1\221133 and RG\R2\232324) for support. AGD is a Royal Society University Research Fellow.Notes and references
- (a) A. Shaver, J. M. McCall, G. Marmolejo, F. Bottomley, E. C. Ferris and J. A. Gladysz, Inorg. Synth., 1990, 59–65 CrossRef CAS; (b) H. Köpf, B. Block and M. Schmidt, Chem. Ber., 1968, 101, 272–276 CrossRef; (c) J. Köpf and B. Block, Chem. Ber., 1969, 102, 1504–1508 CrossRef; (d) J. M. McCall and A. Shaver, J. Organomet. Chem., 1980, 193, C37–C39 CrossRef CAS , for alternative synthetic procedures, see: ; (e) E. Samuel, Bull. Soc. Chim. Fr., 1966, 11, 3548–3564 Search PubMed; (f) E. Samuel and C. Gianotti, J. Organomet. Chem., 1976, 113, C17–C18 CrossRef CAS.
- (a) D. M. Giolando and T. B. Rauchfuss, Organometallics, 1984, 3, 487–489 CrossRef CAS; (b) D. M. Giolando, T. B. Rauchfuss, A. L. Rheingold and S. R. Wilson, Organometallics, 1987, 6, 667–675 CrossRef CAS; (c) C. M. Bolinger, J. E. Hoots and T. B. Rauchfuss, Organometallics, 1982, 1, 223–225 CrossRef CAS; (d) C. M. Bolinger, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 1981, 103, 5620–5621 CrossRef CAS.
- (a) M. Schmidt, B. Block, H. D. Block, H. Köpf and E. Wilhelm, Angew Chem. Int. Ed. Engl., 1968, 7, 632–633 CrossRef CAS; (b) R. Steudel and R. Strauss, J. Chem. Soc., Dalton Trans., 1984, 1775–1777 RSC; (c) D. Dirican, N. Pfister, M. Wozniak and T. Braun, Chem.–Eur. J., 2020, 26, 6945–6963 CrossRef CAS PubMed; (d) R. Steudel and B. Eckert, Solid Sulfur Allotropes, in Elemental Sulfur and Sulfur-Rich Compounds I. Top. Current. Chem., ed. R. Steudel, Springer, Berlin, Heidelberg, 2003, vol. 230, pp. 1–79 CrossRef.
- For selected examples, see: (a) R. Steudel, S. Forster and J. Albertsen, Chem. Ber., 1991, 124, 2357–2359 CrossRef CAS; (b) A. Ishii and R. Yamashita, J. Sulfur Chem., 2008, 29, 303–308 CrossRef CAS; (c) R. Steudel, The Synthesis of Sulfur- and Selenium-Containing Organic and Inorganic Rings from Titanocene Precursors, in Studies in Inorganic Chemistry, ed. R. Steudel, Elsevier, 1992, vol. 14, ch. 13, pp. 223–253 Search PubMed.
- (a) R. Steudel, M. Pridohl, J. Buschmann and P. Luger, Chem. Ber., 1995, 128, 725–728 CrossRef CAS; (b) R. Steudel, K. Hassenberg, V. Münchow, O. Schumann and J. Pickardt, Eur. J. Inorg. Chem., 2000, 921–928 CrossRef CAS; (c) H. Schmidt and R. Steudel, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 557–558 CrossRef CAS.
- For selected examples of titanocene thiolate derivatives, see: (a) A. Shaver, S. Morris and A. Desjardins, Inorg. Chim. Acta, 1989, 161, 11–12 CrossRef CAS; (b) A. Shaver and S. Morris, Inorg. Chem., 1991, 30, 1926–1930 CrossRef CAS; (c) G. Fachinetti and C. Floriani, J. Chem. Soc., Dalton Trans., 1974, 2433–2436 RSC; (d) H. Köpf and H. Schmidt, Z. Anorg. Allg. Chem., 1965, 340, 139–145 CrossRef; (e) D. Taher, S. Klaib, T. Stein, M. Korb, G. Rheinwald, A. Ghazzy and H. Lang, Polyhedron, 2018, 148, 70–75 CrossRef CAS; (f) N. W. Alcock, H. J. Clase, D. J. Duncalf, S. L. Hart, A. McCamley, P. J. McCormack and P. C. Taylor, J. Organomet. Chem., 2000, 605, 45–54 CrossRef CAS; (g) See also ref. 11..
- (a) V. Münchow and R. Steudel, Eur. J. Inorg. Chem., 2004, 718–725 CrossRef; (b) R. Steudel, O. Schumann, J. Buschmann and P. Luger, Angew. Chem., Int. Ed., 1998, 37, 492–494 CrossRef CAS; (c) N. Takeda, N. Tokitoh and R. Okazaki, Polysulfido Complexes of Main Group and Transition Metals, in Elemental Sulfur and Sulfur-Rich Compounds II. Top. Current. Chem., ed. R. Steudel, Springer, Berlin, Heidelberg, 2003, vol. 231, pp. 153–202 Search PubMed.
- For a review on organic polysulfane chemistry, see: R. Steudel, Chem. Rev., 2002, 102, 3905–3946 CrossRef CAS PubMed.
- For examples of other reagents for the regulated synthesis of disulfides and polysulfides (n > 2) see: (a) X. Xiao, M. Feng and X. Jiang, Angew Chem. Int. Ed. Engl., 2016, 55, 14121–14125 CrossRef CAS PubMed; (b) X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191 CrossRef PubMed; (c) J. Xue and X. Jiang, Nat. Commun., 2020, 11, 4170 CrossRef CAS PubMed; (d) Q. Yu, L. Bai and X. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202314379 CrossRef CAS PubMed , and references therein..
- For a kinetic study of the relatively slow reaction of Cp2TiS5 with dimethyl acetylenedicarboxylate alkynes, see: (a) C. M. Bollinger and T. B. Rauchfuss, Inorg. Chem., 1982, 21, 3947–3954 CrossRef , for related processes with Zn(II)-reagents; (b) R. J. Pafford, J.-H. Chou and T. B. Rauchfuss, Inorg. Chem., 1999, 38, 3779–3786 CrossRef CAS; (c) M. Ballesteros and E. Y. Tsui, Dalton Trans., 2020, 49, 16305–16311 RSC.
- For kinetic studies on the the relatively slow transmetallation of sulfur from titanocenes into Pt(II) complexes, see: (a) K. Osakada, Y. Kawaguchi and T. Yamamoto, Organometallics, 1995, 14, 4542–4548 CrossRef CAS; (b) K. Osakada, T. Oshoda and T. Yamamoto, Bull. Chem. Soc. Jpn., 2000, 73, 923–930 CrossRef CAS.
- For examples of studies of substitutions with sulfur compounds at sufenyl-chlorides, see: C. G. Moore and M. Porter, J. Chem. Soc., 1958, 2888–2904 Search PubMed , note 584. (a) C. G. Moore and M. Porter, Tetrahedron, 1960, 9, 58–64 CrossRef CAS; (b) F. Pietra and D. Vitali, J. Chem. Soc. B, 1970, 623–625 RSC.
- (a) E. Ciuffarin and A. Fava, Nucleophilic Substitution at Sulfur, in Progress in Physical Organic Chemistry, ed. A. Steitwieser Jr and R. W. Taft, John Wiley and Sons, Inc., 1968, vol. 6, ch. 2, pp. 81–110 Search PubMed; (b) J. L. Kice, Nucleophilic Substitution at Different Oxidation States of Sulfur, in Progress in Inorganic Chemistry, ed. J. O. Edwards, John Wiley and Sons, Inc., 1972, vol. 17, pp. 148–206 Search PubMed; (c) S. Oae, in Organic Sulfur Chemistry: Structure and Mechanism, ed. J. T. Doi, CRC Press Taylor & Francis Group, Florida, 1st edn, 1991, vol. 1, ch. 4, pp. 119–181 Search PubMed.
- For a review on computational analysis of SN2-reactions, see: T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315–1330 CrossRef CAS PubMed.
- For selected examples of radical chain reactions using sulfenyl chlorides proceeding via thiyl radicals as carriers, see: (a) D. D. Tanner, N. Wada and B. G. Brownlee, Can. J. Chem., 1973, 51, 1870–1879 CrossRef CAS; (b) J. F. Harris Jr, J. Org. Chem., 1966, 31, 931–935 CrossRef; (c) R. G. Guy and I. Pearson, J. Chem. Bull. Soc. Jpn., 1977, 50, 541–542 CrossRef CAS.
- For selected examples on the role of titanocene(III) in halogen abstraction, see: (a) G. Hersant, M. B. S. Ferjani and S. M. Bennett, Tetrahedron Lett., 2004, 45, 8123–8126 CrossRef CAS; (b) Y. Qian, G. Li, X. Zheng and Y.-Z. Huang, Synlett, 1991, 489–490 CrossRef CAS; (c) X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836–14843 CrossRef CAS PubMed , titanocene(III) chloride can also react with disulfides delivering sulfur-centered radical species; (d) R. S. P. Coutts, J. R. Surtees, J. M. Swan and P. C. Wailes, Aust. J. Chem., 1966, 19, 1377–1380 CrossRef CAS.
- 1H NMR spectra of complex 1a show it exists as a mixture of two chair-conformers with inversion barriers reported to be ΔG298.15‡ = 76.3 kJ mol−1 (k = 1.65 s−1; t0.5 = 0.42 s) based on VT-NMR line-shape analyses: (a) E. W. Abel, M. Booth and K. G. Orrell, J. Organomet. Chem., 1978, 160, 75–79 CrossRef CAS , while the inversion has been justified by ‘the usual chair-to-chair’ interconversion, evidence against a ring-opening pathway has not been provided; (b) Upon UV-irradiation, a homolytic pathway is favoured over heterolytic Ti-S cleavage: A. E. Bruce, M. R. M. Bruce and D. R. Tyler, J. Am. Chem. Soc., 1984, 106, 6660–6664 CrossRef CAS , see also: ; (c) M. R. M. Bruce, A. Kenter and D. R. Tyler, J. Am. Chem. Soc., 1984, 106, 639–644 CrossRef CAS.
- This eliminates the possibility of a higher order dependency on 1a exhibiting pseudo first-order kinetics via self/auto-catalysis. The latter situation is detected by the exponent (–kt) to the first-order decay being dependent on the initial reactant concentration. For an example of this, see: P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed.
- In some cases, the possibility of degenerate pathways for one or more specific steps in the reaction mean that the phenomenological process rates lead to empirical constants (k1, k2) that differ from the microkinetic rate constants. For example, in the case of {1a + S2Cl2} the presence of two identical Ti–S bonds in 1a and two identical S-Cl bonds in S2Cl2 results in four degenerate pathways for the first S–S cleavage, and thus statistical corrections (in this specific case, k1/4) are required for calculation of activation parameters; see Section S.11.2 in the ESI‡ for further discussion. The statistical effects must also be considered when calculating selectivity for two microkinetic steps from the relative rate constants (k1/k2) for the two steps in the phenomenological process, and vice versa.
- (a) J. Catalán, J. Phys. Chem. B, 2009, 113, 5951–5960 CrossRef PubMed; (b) There was not a meaningful correlation of the process rate constants with the ET30 scale, dipole moment or dielectric constant of various solvents (see the ESI Section S2.3‡)..
- Y. Ben-Tal, P. J. Boaler, H. J. A. Dale, R. E. Dooley, N. A. Fohn, Y. Gao, A. Garcia-Dominguez, K. M. Grant, A. M. R. Hall, H. L. D. Hayes, M. M. Kucharski, R. Wei and G. C. Lloyd-Jones, Prog. Nucl. Magn. Reson. Spectrosc., 2022, 129, 28–106 CrossRef CAS PubMed.
- (a) A. Shaver and J. M. McCall, Organometallics, 1984, 12, 1823–1829 CrossRef; (b) G. Tainturier and B. Gautheron, Phosphorus Sulfur, 1988, 36, 11–14 CrossRef CAS; (c) The tert-butyl-Cp analogue of 1a was not explored but this substituent is anticipated to have a negligible inductive effect on the reactivity..
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- N. J. Coville, M. S. Loonat, D. White and L. Carlton, Organometallics, 1992, 11, 1082–1090 CrossRef CAS.
- (a) R. Wei, A. M. R. Hall, R. Behrens, M. S. Pritchard, E. J. King and G. C. Lloyd-Jones, Eur. J. Org Chem., 2021, 2331–2342 CrossRef CAS; (b) H. J. A. Dale, G. R. Hodges and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2023, 145, 18126–18140 CrossRef CAS PubMed.
- A competition of 1a and 2a for substoichoimetric quantities of S2Cl2 added by microsyringe to their solution in a 5 mm NMR tube gave rate constant ratios that differed by two orders of magnitude to those obtained using efficient rapid mixing by flow methods, see Section S7 in the ESI§.
- (a) C. Brown and D. R. Hogg, Chem. Commun., 1967, 38–39 RSC; (b) For a full discussion on concerted versus associative-dissociative pathways in SN2 at sulfur, see ref. 12b..
- Computations were performed using ORCA software, F. Nesse, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- (a) J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed; (b) S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- For examples of concerted σ-bond methatheses with similar activation parameters see: R. Waterman, Organometallics, 2013, 32, 7249–7263 CrossRef CAS.
- For the para-fluorine substituent we used the value of σp = 0.15, which is better parameterized for reactions in anhydrous organic solvents: (a) A. T. Shulgin and A. W. Baker, Nature, 1958, 182, 1299 CrossRef CAS; (b) T. J. A. Corrie, L. T. Ball, C. A. Russell and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 245–254 CrossRef CAS PubMed.
- For studies on the behaviour of titanocene (poly)sulfides on irradiation with UV-light, see: (a) H. Kunkely and A. Vogler, Z. Naturforsch., B: J. Chem. Sci., 1998, 53, 22 Search PubMed; (b) Ref. 17b, which reports on the higher sensitivity of these thiolate complexes to irradiation compared to Cp2TiS5..
- A previous report in polysulfane synthesis involving aryl polysulfane chlorides (ArSxCl) as intermediates stated that they ‘must be protected from moisture and pure starting materials must be used since the intermediates and end products were not purified by distillation due to their very considerable tendency to disproportionate: H. J. Langer and J. B. Hyne, Can. J. Chem., 1973, 51, 3403–3411 CrossRef CAS.
- The reactions of titanocene thiophenolate derivatives 5a–d and 6a–d with ArSCl reagents were too fast to measure using the SF-UV equipment available. This shows these titanocene thiophenolates are much more nucleophilic than the corresponding thiophenols, ArSH. For the kinetics of the latter, see: A. García-Domínguez, N. M. West, R. T. Hembre and G. C. Lloyd-Jones, Thiol Chlorination with N-Chlorosuccinimide: HCl-Catalyzed Release of Molecular Chlorine and the Dichotomous Effects of Water, ACS Catal., 2023, 13, 9487–9494 CrossRef.
- P. M. Jüstel, C. D. Pignot and A. R. Ofial, J. Org. Chem., 2021, 86, 5965–5972 CrossRef PubMed.
- (a) Y. Yukawa and Y. Tsuno, Bull. Chem. Soc. Jpn., 1959, 32, 971–981 CrossRef CAS; (b) Y. Yukawa, Y. Tsuno and M. Sawada, Bull. Chem. Soc. Jpn., 1966, 39, 2274–2286 CrossRef CAS.
- The mechanistic similarities of the reactions of 5a–d and 6a–d to the analogous reactions of titanacycles 1a and 2a, as well as reasonable correlations with computation (see ESI§) argues against a change in mechanism across the series.
Footnotes |
| † Dedicated to the memory of Prof. Dr Ralf Steudel (March 25, 1937–February 12, 2021) a life-long pioneer in the chemistry of sulfur. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02737j |
| § AGD is a Royal Society University research fellow. |
| This journal is © The Royal Society of Chemistry 2024 |