
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand-to-metal charge transfer facilitates photocatalytic oxygen atom transfer (OAT) with cis-dioxo molybdenum(VI)-Schiff base complexes†
Thorsten
Dreher‡
 ,
Lukas
Geciauskas‡
,
Samuel
Steinfeld
,
Barbara
Procacci
,
Adrian C.
Whitwood
,
Jason M.
Lynam
,
Richard E.
Douthwaite
* and
Anne-K.
Duhme-Klair
*
,
Lukas
Geciauskas‡
,
Samuel
Steinfeld
,
Barbara
Procacci
,
Adrian C.
Whitwood
,
Jason M.
Lynam
,
Richard E.
Douthwaite
* and
Anne-K.
Duhme-Klair
*
Department of Chemistry, University of York, Heslington, YO10 5DD, York, UK. E-mail: richard.douthwaite@york.ac.uk; anne.duhme-klair@york.ac.uk
First published on 6th September 2024
Abstract
Systems incorporating the cis-Mo(O)2 motif catalyse a range of important thermal homogeneous and heterogeneous oxygen atom transfer (OAT) reactions spanning biological oxidations to platform chemical synthesis. Analogous light-driven processes could offer a more sustainable approach. The cis-Mo(O)2 complexes reported here photocatalyse OAT under visible light irradiation, and operate via a non-emissive excited state with substantial ligand-to-metal charge-transfer (LMCT) character, in which a Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O π*-orbital is populated via transfer of electron density from a chromophoric salicylidene-aminophenol (SAP) ligand. SAP ligands can be prepared from affordable commercially-available precursors. The respective cis-Mo(O)2-SAP catalysts are air stable, function in the presence of water, and do not require additional photosensitisers or redox mediators. Benchmark OAT between phosphines and sulfoxides shows that electron withdrawing groups (e.g. C(O)OMe, CF3) are necessary for photocatalytic activity. The photocatalytic system described here is mechanistically distinct from both thermally catalysed OAT by the cis-Mo(O)2 motif, as well as typical photoredox systems that operate by outer sphere electron transfer mediated by long-lived emissive states. Both photoactivated and thermally activated OAT steps are coupled to establish a catalytic cycle, offering new opportunities for the development of photocatalytic atom transfer based on readily-available, high-valent metals, such as molybdenum.
O π*-orbital is populated via transfer of electron density from a chromophoric salicylidene-aminophenol (SAP) ligand. SAP ligands can be prepared from affordable commercially-available precursors. The respective cis-Mo(O)2-SAP catalysts are air stable, function in the presence of water, and do not require additional photosensitisers or redox mediators. Benchmark OAT between phosphines and sulfoxides shows that electron withdrawing groups (e.g. C(O)OMe, CF3) are necessary for photocatalytic activity. The photocatalytic system described here is mechanistically distinct from both thermally catalysed OAT by the cis-Mo(O)2 motif, as well as typical photoredox systems that operate by outer sphere electron transfer mediated by long-lived emissive states. Both photoactivated and thermally activated OAT steps are coupled to establish a catalytic cycle, offering new opportunities for the development of photocatalytic atom transfer based on readily-available, high-valent metals, such as molybdenum.
Introduction
It is well recognised that light could help meet energy needs for driving chemical reactions,1 and the last decade has seen intense interest in photocatalysis for synthesis, environmental remediation, and energy storage.2 In particular, metal-based photocatalysts have been heavily investigated because of their favourable photophysical properties and opportunities for tailored design.3 Mechanistically, most homogeneous photoredox chemistry can be characterised as arising from outer-sphere single-electron transfer (SET) between a photoexcited catalyst and substrate to generate radicals, which undergo subsequent reactions (Scheme 1a).4–6 These photocatalytic reactions are typically mediated by emissive metal-to-ligand charge transfer (MLCT) excited states with lifetimes in the μs regime, sufficient to support bimolecular reactivity, as exemplified by precious metal polypyridyl complexes.7–10 Other established metal-based photo-reactions suitable for incorporation into catalytic cycles are ligand dissociation and reductive elimination from short-lived (<ns) excited states of organometallic complexes.11,12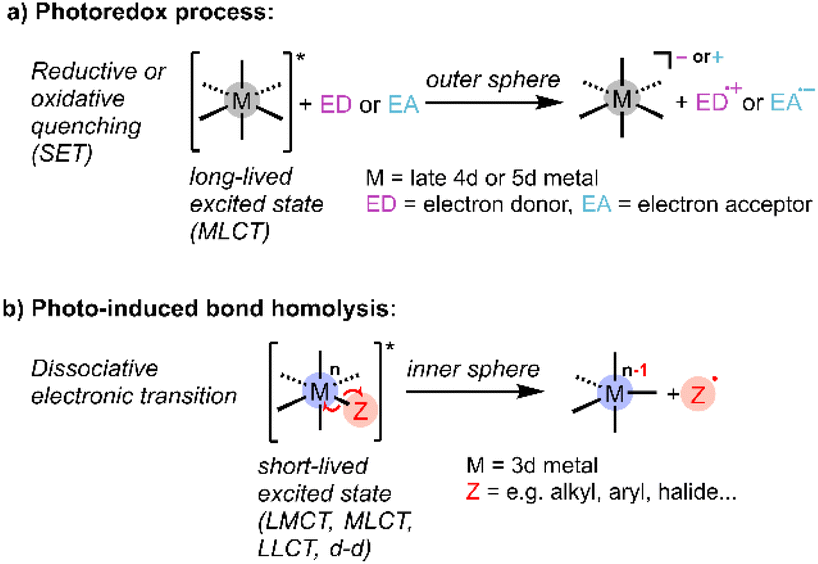 | ||
| Scheme 1 Schematic representation of conventional (a) photoredox and (b) bond homolysis processes used in homogeneous photocatalysis (SET: single-electron transfer; CT: charge transfer; M: metal, L: ligand). | ||
Recently there has been much interest in developing Earth-abundant metal-containing photocatalysts that operate via ligand-to-metal charge transfer (LMCT).11,13,14 These photocatalysts are typically high-valent complexes where LMCT induces radical reactivity via inner-sphere electron transfer, in some cases overcoming the thermodynamic limits of the more common outer-sphere SET via MLCT.15,16 Known reactivity of LMCT excited states include radical ligand dissociation, for example of halides (Scheme 1b), which can then exhibit catalytic reactivity,17 and atom abstraction, for example hydrogen atom transfer (HAT) from hydrocarbons observed for metal oxo complexes.18
The focus of the work presented here is the LMCT-induced reactivity of molybdenum oxo complexes to enable photocatalytic oxygen atom transfer (OAT) reactions. Molybdenum oxo and dioxo systems are found in molybdoenzymes, bio-inspired metal complexes, and metal molybdates,19–22 and are critical for supporting a wide range of thermally activated homogeneous and heterogeneous catalytic reactions found in biology, chemical synthesis, and industrial processes.23–25 These include a family of mononuclear molybdoenzymes containing an oxo ligand and pyranopterin dithiolene cofactor which acts as a non-innocent ligand, supporting OAT via two sequential one-electron transfer steps.23,26–29 Many classes of bio-inspired Mo-oxo and cis-Mo-dioxo complexes incorporating multidentate ligands have also been investigated for OAT reactions such as epoxidations.30–34 Heterogeneous metal molybdates catalyse a range of industrially-important heterogenous reactions such as propene oxidation, where surface Mo-oxo ligands are also implicated in concerted OAT-type reactivity.30,35 Mechanistically, all these reactions typically cycle through Mo(VI–IV) oxidation states via SET or concerted OAT.21 In addition to these thermally active Mo-catalysts, a number of photoactive systems incorporating the Mo(VI)-oxo motif are known (Scheme 2). Early work described [Mo(O)Cl4(H2O)]− (I) and [cis-Mo(O)2Br2(tBu-bipy)] (II) complexes for stoichiometric OAT to phosphines and alkenes under UV irradiation.36,37 More recently, a related [cis-Mo(O)2Cl2(tBu-bipy)] (III) system was shown to stoichiometrically activate C–H bonds via HAT.38,39 A characteristic limiting feature of these systems is the formation of a [(O)MoV–O–MoV(O)] oxo-bridged complex40–44 that prevents catalytic turnover.38 Partly to overcome this limitation, the C(O)OH-substituted system [cis-Mo(O)2X2((C(O)OH)2-bipy)] (X = Cl(IV), Br(V)) was supported on UV photoactive TiO2 nanoparticles.45–49 These composite systems can be regenerated in a separate dark oxidation step, mimicking a catalytic cycle. Photoactivated OAT to phosphines has been achieved catalytically using a dyad (VII) comprising a [RuII(bipy)3]2+ photosensitiser covalently-linked to a cis-MoVI(O)2 cocatalyst which operates under visible light but does not incorporate direct photoactivation of the cis-MoVI(O)2 moiety.50 This bio-inspired system requires methyl viologen as a redox mediator and significant catalyst degradation occurs. Of wider relevance, cis-WVI(O)2 complexes of highly-conjugated macrocyclic salen ligands (e.g.VI) have μs emissive states that mediate a number of photocatalytic transformations, including C–H activation and C–C coupling reactions, via SET-induced radical pathways.51 Collectively, these seminal reports suggest that there is an opportunity to exploit LMCT into a MoO π* orbital to support photocatalytic OAT that could expand the extensive thermal chemistry of this motif. We hypothesised that LMCT transitions that lead to the population of antibonding MoO orbitals can promote the labilisation of oxo ligands by decreasing their bond order, thereby facilitating photocatalytic OAT. We were motivated to explore d0 Mo-dioxo complexes that incorporate ligand systems that support charge-transfer transitions and enable catalytic turnover that is unrestricted by the formation of kinetically-inert [(O)MoV–O–MoV(O)] complexes under the chosen experimental conditions. Here we report a series of Mo-dioxo complexes of Schiff base ligands that are air stable, support LMCT under visible light irradiation and mediate photocatalytic OAT in the presence of water without photocatalyst degradation.
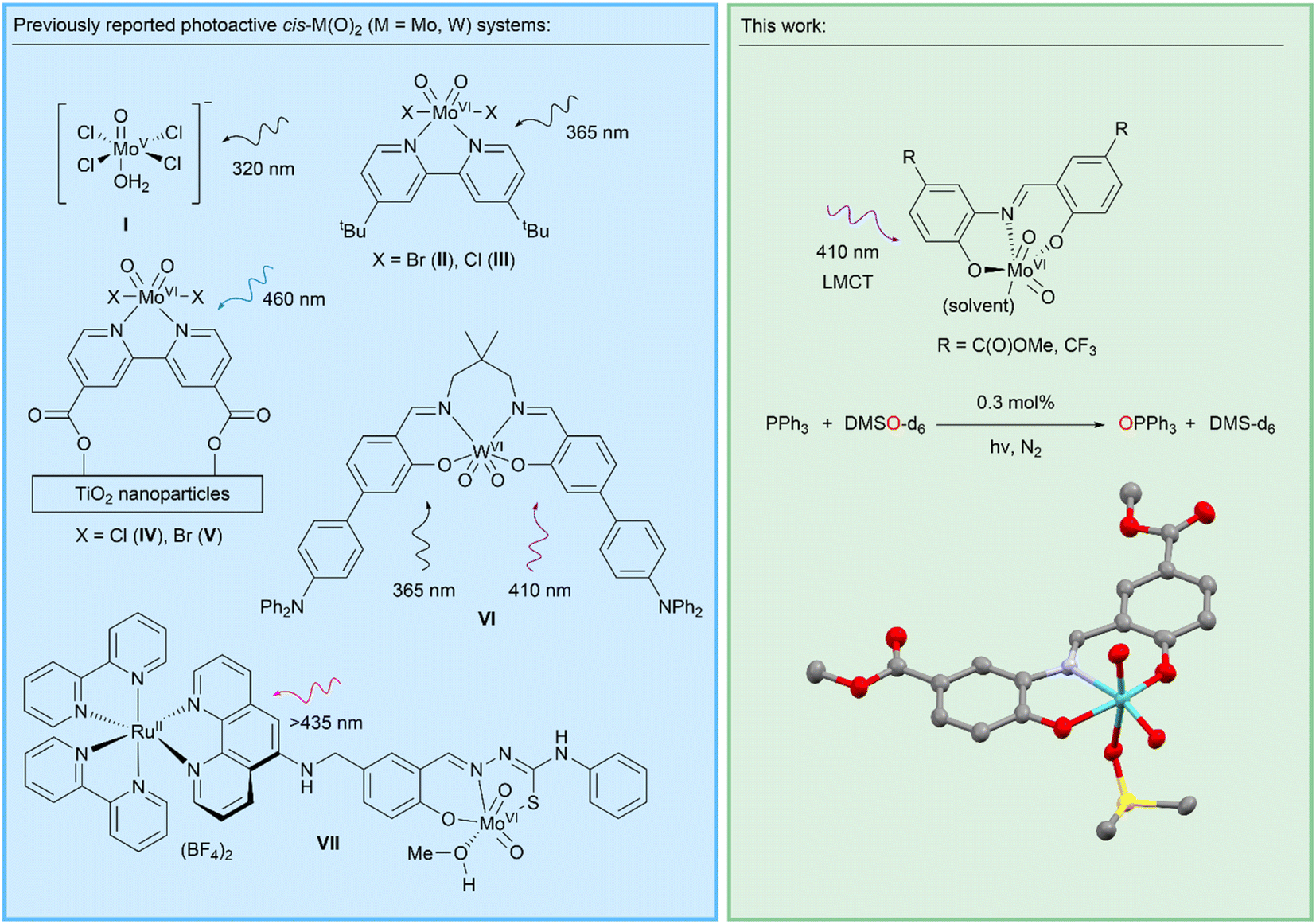 | ||
| Scheme 2 Previously-reported photoactive cis-Mo(O)2 and cis-W(O)2 complexes, and an exemplary photocatalytic system developed in this work, including a crystal structure of the [cis-MoVI(O)2(κ3-SAPC(O)OMe)(Me2SO)] complex (3C(O)OMeDMSO) reported here (ORTEP plot (50% probability); H atoms omitted for clarity; C, grey; N, lavender; O, red; S, yellow; Mo, cyan). | ||
Results and discussion
Synthesis and characterisation of ligands and complexes
The ligand system used in this work is based on a tridentate meridionally-coordinating salicylidene-aminophenol (SAP) motif, which is known to support visible-light induced LMCT transitions if coordinated to cis-MoVI(O)2.52,53 Since SAP-type ligands can be prepared in a single step from commercially available precursors (Scheme 3), substituent effects could be used to vary their spectroscopic and electronic properties systematically and potentially limit oxo-bridged bimetallic complex formation.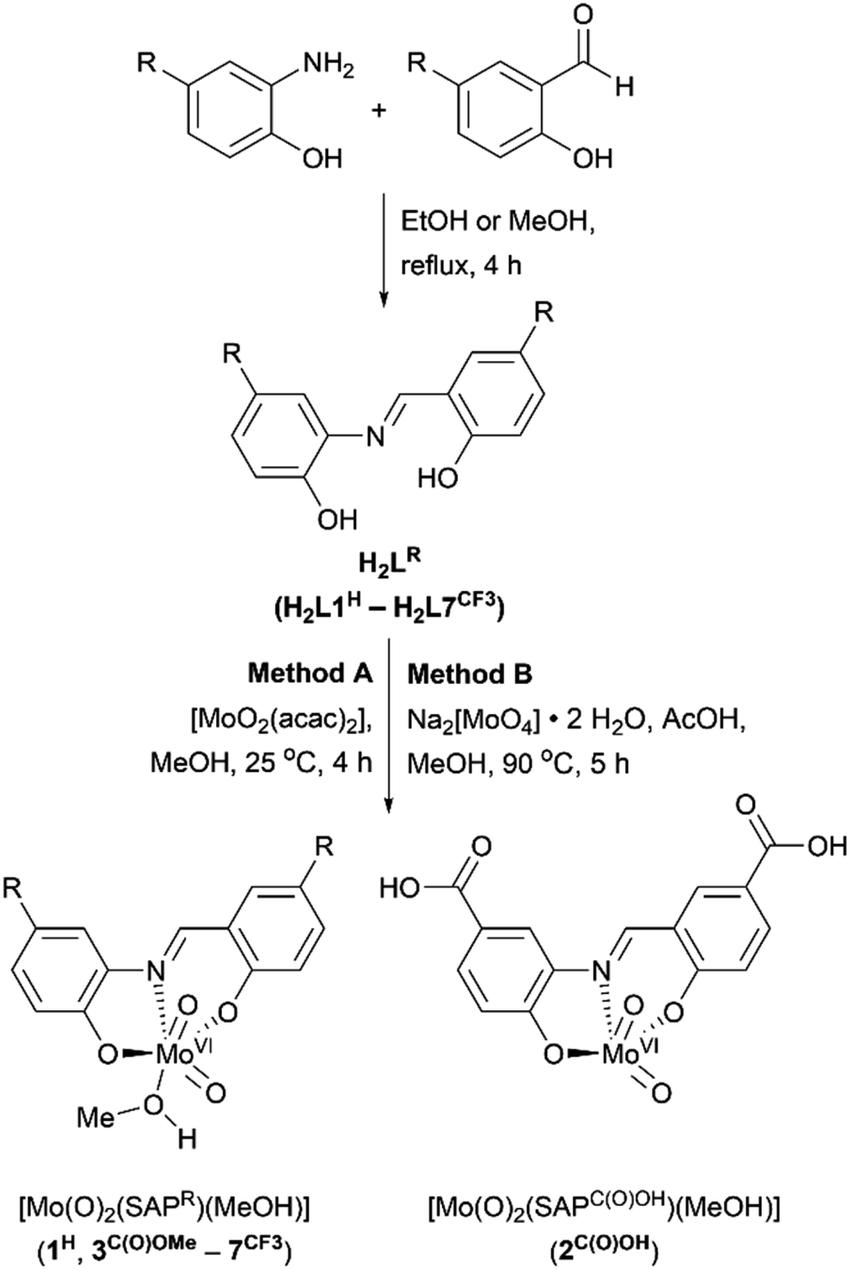 | ||
| Scheme 3 Synthesis of SAP-type ligands (H2LR) and cis-MoVI(O)2 complexes (1H–7CF3). Where R = H (H2L1H and 1H); C(O)OH (H2L2C(O)OH and 2C(O)OH); C(O)OMe (H2L3C(O)OMe and 3C(O)OMe); F (H2L4F and 4F); tBu (H2L5tBu and 5tBu); OMe (H2L6OMe and 6OMe); CF3 (H2L7CF3 and 7CF3). | ||
Reactions between SAP ligands and [MoO2(acac)2] or Na2[MoO4]·2H2O in methanol give the target complexes 1H–7CF3, comprising meridional SAP coordination to a cis-MoVI(O)2 moiety, with additional methanol coordination for most complexes giving [cis-MoVI(O)2(κ3-SAPR)(MeOH)] with pseudo-octahedral geometry. The only exception was complex 2C(O)OH which does not exhibit solvent coordination in the solid state. The proposed compositions were confirmed by 1H and 13C NMR, ATR-IR, and UV-vis spectroscopies, mass spectrometry, elemental analysis, and X-ray crystallography (see ESI†). Complexes 1H–7CF3 are pale yellow to dark red, and are air stable as solids and in solution, with good solubility in DMSO and DMF. All metrical data are consistent with similar cis-MoVI(O)2 complexes of tridentate ligands.44,54–59
Photocatalytic testing
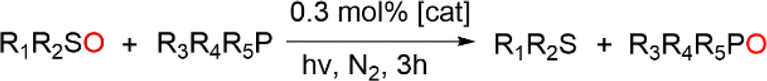
Previously, cis-MoVI(O)2-containing catalysts have been extensively studied for conventional thermal OAT between phosphines and sulfoxides as oxygen atom acceptors and donors, respectively, to mimic biological processes and investigate thermodynamic and kinetic features.21,40,41 In these studies, Hammett correlations are found for various ligand derivatives and substrates, and rates are reflective of the thermodynamic driving force between phosphine and sulfoxide.41,42,50,60 For comparison, we therefore initially examined the prototypical OAT reaction between PPh3 and DMSO-d6. Photoreactions were performed by irradiating at 410 nm, based on the UV-vis spectra of 1H–3C(O)OMe (see Fig. S31, ESI†) which show very similar features, including a broad absorption band centered around 420 nm, assigned to a transition with substantial LMCT character based on TD-DFT simulations (see below).Photoreactions with complexes 1H–3C(O)OMe in excess DMSO-d6 and 300 equivalents of PPh3 over 3 hours showed that complex 1H is essentially photoinactive. However, complexes 2C(O)OH and 3C(O)OMe exhibited 35% and 78% conversions of PPh3 to OPPh3, respectively, with essentially quantitative selectivity as determined by 1H and 31P NMR spectroscopy (Table 1, entries 1–3). In the absence of irradiation, complex 3C(O)OMe is inactive (Table 1, entry 4). Irradiation causes heating of the reaction mixture to 45 °C, however in the absence of light, no reaction occurs on heating to 45 °C, confirming that OAT is induced by light (Table 1, entry 5). Further control experiments showed that no appreciable activity is observed in the absence of catalyst 3C(O)OMe or using the SAP ligand precursor H2L3C(O)OMe (Table 1, entries 6–7). The reaction was also monitored by 1H NMR over 3 hours, requiring periodic cessation of irradiation.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | PPh3 conversion/%b | TON | TOF/h−1 |
| a General conditions: catalyst (1 mM in DMSO-d6), 300 equivalents PPh3, 410 nm, 3 h, under N2. b Conversion determined by 1H NMR spectroscopy. c In the dark. d In the dark and at 45 °C. e Added water (300 mM). f Under air. | ||||
| 1 | 1H | 3 | 9 | 3 |
| 2 | 2C(O)OH | 35 | 105 | 35 |
| 3 | 3C(O)OMe | 78 | 234 | 78 |
| 4c | 3C(O)OMe | 0 | 0 | 0 |
| 5d | 3C(O)OMe | 0 | 0 | 0 |
| 6 | None | 0 | 0 | 0 |
| 7 | H2L3C(O)OMe | 3 | 9 | 3 |
| 8e | 3C(O)OMe | 73 | 219 | 73 |
| 9f | 3C(O)OMe | 83 | 249 | 83 |
| 10 | 4F | 2 | 6 | 2 |
| 11 | 5tBu | 0 | 0 | 0 |
| 12 | 6OMe | 1 | 3 | 1 |
| 13 | 7CF3 | 97 | 291 | 97 |
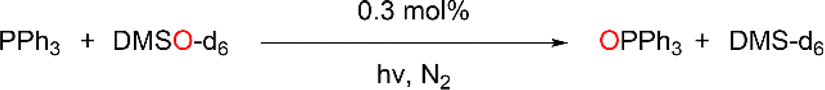
There is no apparent loss in activity for 3C(O)OMe over the monitored time (Fig. 1). The 1H NMR resonances of complexes 2C(O)OH and 3C(O)OMe remain unchanged after photoreaction, confirming photocatalyst stability for these complexes (see Fig. S45 and S46, ESI†). Furthermore, reactions using complex 3C(O)OMe tolerate exposure to both air and water, again with no apparent catalyst degradation (Table 1, entries 8–9). The observation of the remarkable photocatalytic activity and stability for 2C(O)OH and 3C(O)OMe prompted the investigation of complexes 4F–7CF3 containing a range of electron withdrawing (EWG) and donating (EDG) groups, respectively (Table 1, entries 10–13). Hammett plots (see Fig. S48, ESI†) show that there is a threshold for photocatalytic activity: compounds with EDGs are mostly inactive whilst the EWGs studied lead to increased activity, with [MoVI(O)2(SAPCF3)(MeOH)] (7CF3) approaching quantitative conversion after 3 hours of irradiation (Fig. 1). Conventional ‘thermal’ OAT catalysts typically show a linear Hammett correlation.41,42,50,60 For the active catalysts described here linearity is not observed indicating that the reaction rate is not driven by a single thermodynamic parameter.
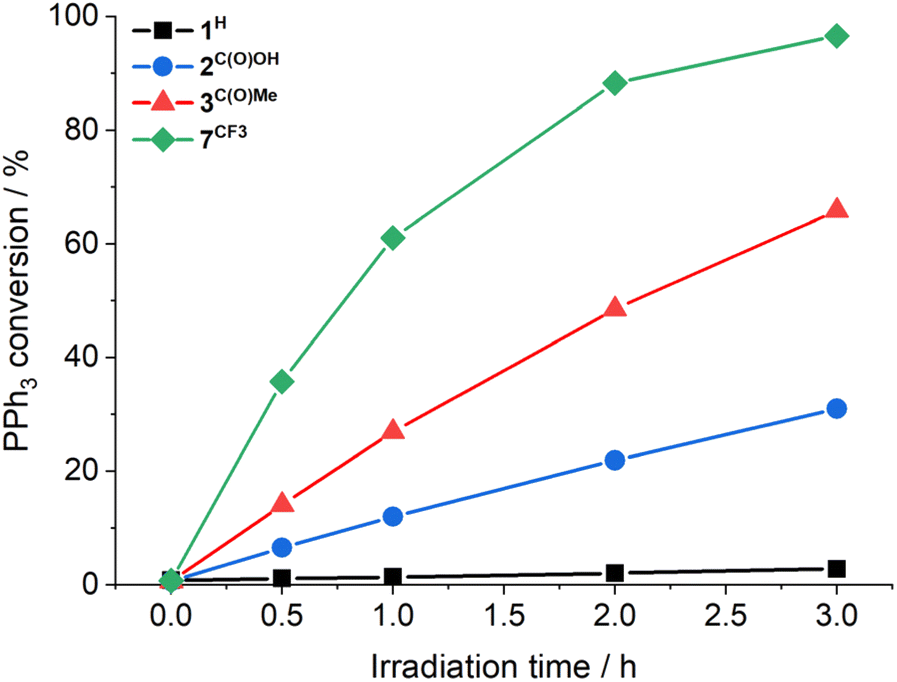 | ||
| Fig. 1 Photocatalytic OAT from DMSO-d6 to PPh3 as a function of time using 1H (black squares), 2C(O)OH (blue circles), 3C(O)OMe (red triangles), and 7CF3 (green diamonds) (1 mM in DMSO-d6), 300 equivalents PPh3, 410 nm, 3 h, under N2; PPh3 conversion was determined by 1H NMR spectroscopy. Note: the lines are added to guide the eye, and do not represent data fitting. | ||
Finally, we tested three different phosphines, using increasing equivalents (100, 200 and 400), in combination with catalysts 3C(O)OMe and 7CF3 and in all cases, the reactions were found to be pseudo-first order in phosphine (see Table S4, Fig. S49 and S50, ESI†), as expected based on literature reports.41,61–63 PPh3 and (P(p-F-Ph)3 gave rise to similar kobs values, in line with the respective Hammett parameters64 (p-F, σp = 0.06 and H, σp = 0). In comparison, P(p-CF3-Ph)3), the phosphine with the most electron-withdrawing substituent (p-CF3,σp = 0.53), gave rise to significantly lower pseudo-first-order rate constants. P(p-CF3-Ph)3 is not only the poorest nucleophile but also the most difficult phosphine to oxidise. Similar observations have previously been reported for thermally-activated catalysts.65
TD-DFT calculations and electronic spectroscopy
To assign the optical transitions that support photocatalysis, TD-DFT calculations were performed on complexes 1H and 3C(O)OMe, as representative photoinactive and photoactive complexes, respectively. The calculated transitions are in good agreement with the corresponding experimental UV-vis spectra (Fig. 2a) and show that the dominant orbital contribution to the lowest energy transition is HOMO → LUMO (>90% for both complexes). Examination of the orbital contributions to the HOMO demonstrate that it is localised on the SAP ligand, including the imine π-bond. The LUMO contains a ∼30% contribution from the Mo-based orbitals as well as oxo π*-orbital character (Fig. 2b). Therefore, the lowest energy excitation is probably best considered to be an LMCT §.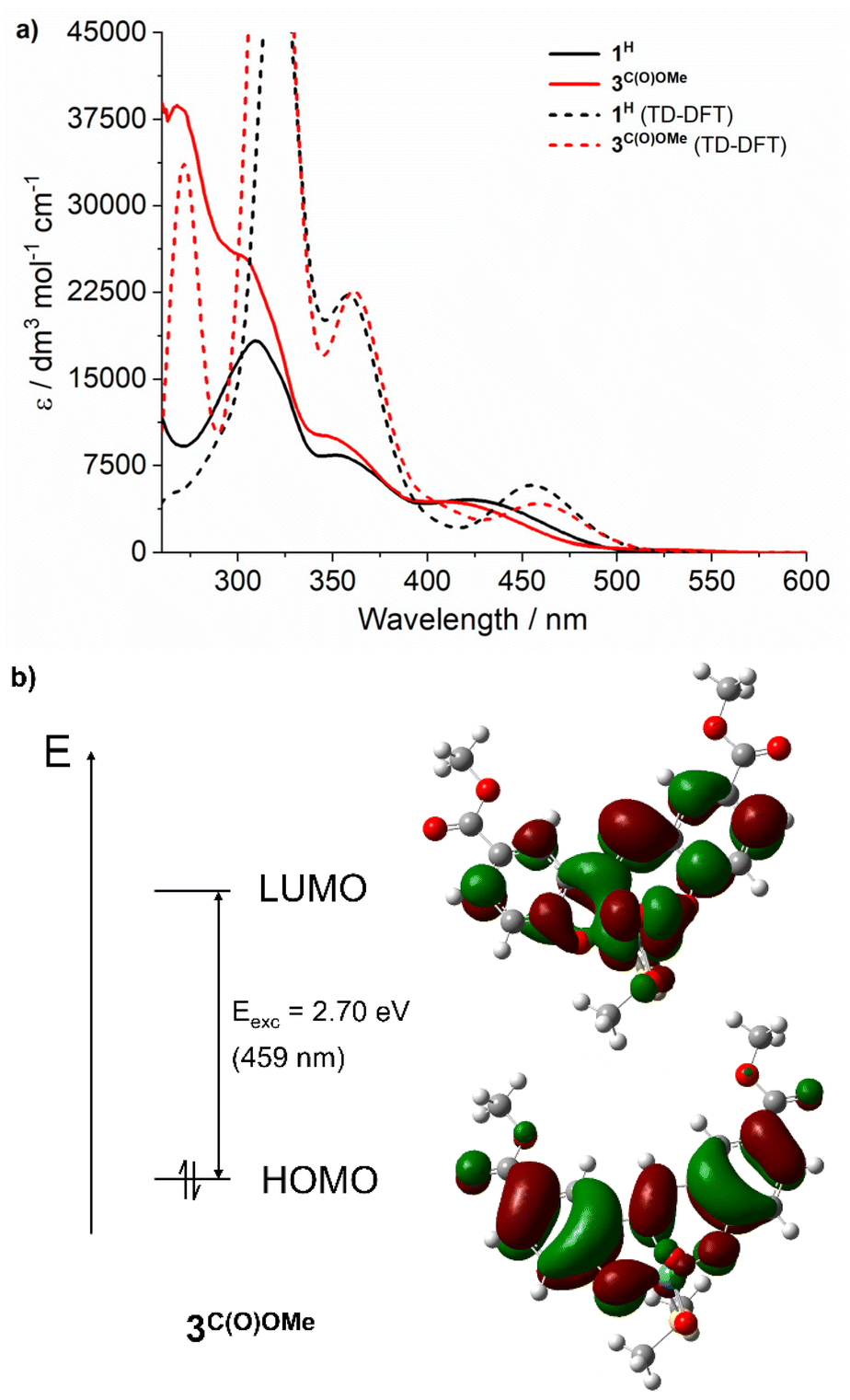 | ||
| Fig. 2 (a) Electronic absorption spectra of 1H and 3C(O)OMe at 0.05 mM in DMSO (solid lines), and spectra calculated at the (PBE0/DEF2-TZVPP//BP86/DEF2-SVPP; SCRF in DMSO) level of theory using TD-DFT simulations (dashed lines); Gaussian functions were fitted onto DFT determined transitions to obtain the calculated spectra. (b) Calculated ground state HOMO (iso-surface level 121) and LUMO (iso-surface level 122) orbitals for 3C(O)OMe at the PBE0/DEF2-TZVPP level. The calculated S0 → S1 transition at 459 nm is shown and is 92% HOMO → LUMO. | ||
Notably, SAP ligand substituent orbitals contribute little to either the HOMO or LUMO of 1H and 3C(O)OMe and do not induce significant changes to the electron density of metal-imine and metal-oxo orbitals. Photocatalysis was also investigated at 365 and 460 nm for complexes 2C(O)OH and 3C(O)OMe and showed little variation between OPPh3 conversion and irradiation wavelength across the LMCT band (Fig. 3). This is in accordance with Kasha's rule,66 since the observed photoactivity of the complexes results from the lowest-lying excited state and is independent of the irradiation wavelength.
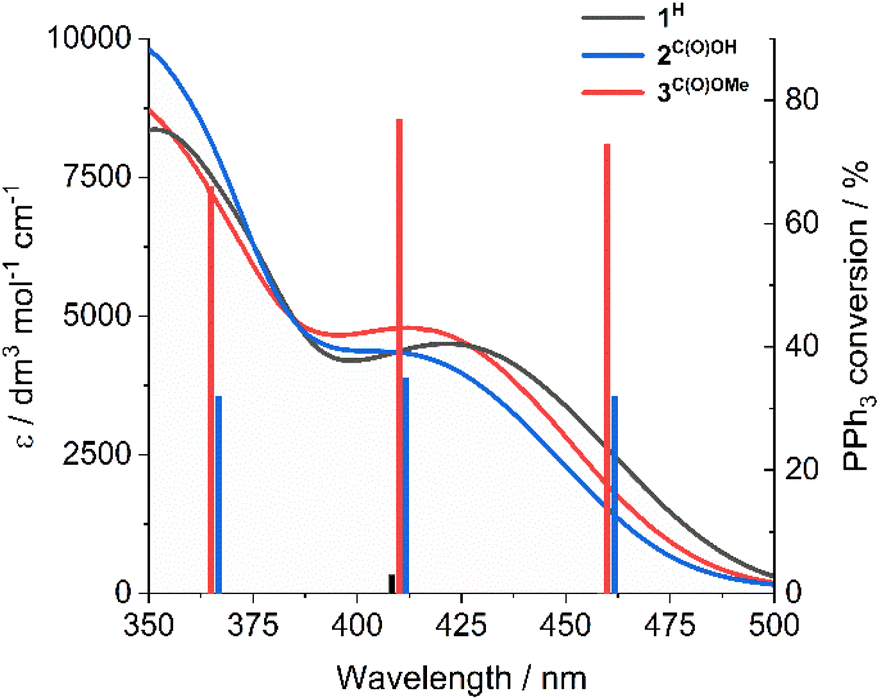 | ||
| Fig. 3 UV-vis spectra of 1H, 2C(O)OH, and 3C(O)OMe and PPh3 conversion as a function of irradiation wavelength. Photocatalytic conditions: catalyst (1 mM in DMSO-d6), 300 equivalents PPh3, 3 h, under N2. | ||
Steady-state and time-resolved fluorescence spectroscopies were used to compare the photoexcited states of 1H, 3C(O)OMe and SAP ligands H2L1H and H2L3C(O)OMe, respectively. The steady state emission spectra of the complexes and ligands both show a weak emission band at 525 nm with solutions of 1H and 3C(O)OMe exhibiting a lower emission by over an order of magnitude compared to H2L1H and H2L3C(O)OMe, respectively (see Fig. S52, ESI†). Time-resolved photoluminescence showed these low intensity emission bands of complexes and ligands also have similar lifetimes of 3–5 ns (see Fig. S53 and Table S17, ESI†) which, for 1H and 3C(O)OMe are also unaffected by the addition of PPh3 (see Fig. S54, ESI†). We cannot exclude that for 1H and 3C(O)OMe emission could be due to residual dissociated ligand. Regardless, it is clear that 1H and 3C(O)OMe are essentially non-emissive and the excited state lifetimes of the emissive states are short relative to intermolecular reaction rates. Collectively, the TD-DFT, UV-vis, and fluorescence spectroscopies, and wavelength independence of photocatalysis, indicate that initial photon absorption does not account for the marked difference in reactivity of complexes 1H and 3C(O)OMe and that OAT is not attributable to an emissive excited state.
Half-catalytic cycle OAT reactivity studies
Oxidation of PPh3 was also investigated under non-catalytic, stoichiometric conditions in the absence of DMSO-d6 as the oxygen atom donor. Reaction between 3C(O)OMe and 5 equivalents of PPh3 in DMF-d7 (a solvent unreactive under these conditions) was monitored using 1H and 31P NMR spectroscopy. The addition of PPh3 to 3C(O)OMe does not affect 1H and 31P NMR signals of either 3C(O)OMe or PPh3, indicating that no significant interaction is detectable in the ground state before irradiation and was further supported by 1H and 31P NMR spectra of 3C(O)OMe obtained in the presence of increasing equivalents of PPh3 (see Fig. S55, ESI†). On irradiation, the samples change colour from yellow to dark green within minutes (see Fig. S58, ESI†), with gradual loss of 1H NMR signals associated with PPh3 and 3C(O)OMe, and growth of signals due to OPPh3 and broader peaks upfield of 3C(O)OMe associated with a new SAP-containing species (see Fig. S59b, ESI†). No signals indicating coordination of PPh3 or OPPh3 to the Mo-complex are observed. In comparison, catalytic reactions in DMSO-d6 as the solvent (Table 1), remain yellow throughout the course of the reaction, presumably because the large excess of DMSO-d6 leads to fast re-oxidation of the dark green SAP-containing intermediate, thereby preventing its accumulation.Closure of the catalytic cycle, requiring reoxidation of the catalyst by DMSO-d6, was subsequently investigated by first irradiating a mixture of 3C(O)OMe and 5 equivalents of PPh3 in DMF-d7 for 21 h, and then adding 10 equivalents of DMSO-d6 in the dark at 45 °C which is the reaction temperature during irradiation. After 2 h at 45 °C, ca 75% of 3C(O)OMe was regenerated indicating that the reoxidation of the catalytic intermediate is a thermally driven process (see Fig. S60b, ESI†). An analogous experiment performed under irradiation gave 50% regeneration of 3C(O)OMe (see Fig. S59b, ESI†) which is lower due to continued photocatalytic turnover in the presence of excess PPh3.
Complementary IR spectroscopic studies with 1H and 3C(O)OMe in solution with 10 equivalents of PPh3 and 50 equivalents of DMSO-d6 in DMF allowed monitoring of the characteristic symmetric and asymmetric stretch vibrations at ca 910 and 930 cm−1, attributable to cis-Mo(O)2,57,60 and a band at 1196 cm−1 assigned to OP stretch vibration of the free OPPh3 product,67,68 respectively. On irradiation at 410 nm, the generation of a new species occurs with a single band at 948 cm−1 characteristic of a Mo(O) species,69 with concomitant generation of OPPh3 (Fig. 4), confirming OAT has occurred. On standing in the dark for 20 hours under a constant flow of nitrogen, significant regeneration of 3C(O)OMe is observed (see Fig. S65, ESI†).
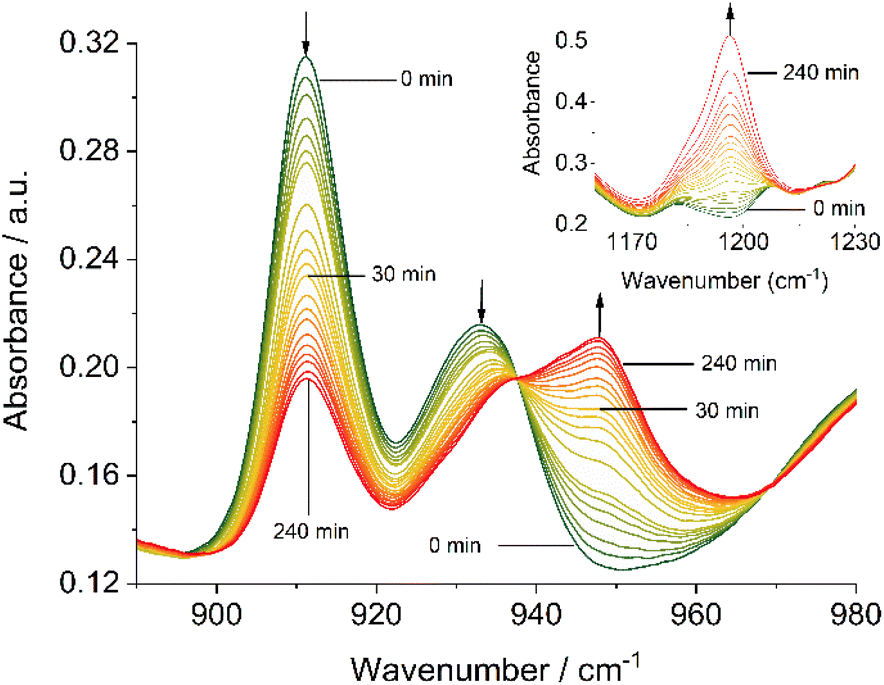 | ||
| Fig. 4 Solution-IR spectra in the region of the MoO stretching bands (890–900 cm−1) of 3C(O)OMe (50 mM in DMF), 10 equivalents PPh3 and 50 equivalents DMSO-d6 in DMF irradiated at 410 nm as a function of time; insert–OP stretching band (1160–1230 cm−1) of the same sample. | ||
Both, the formation of the Mo(O) intermediate and the regeneration of cis-MoVI(O)2 (3C(O)OMe) give rise to clear isosbestic points. For conventional ‘thermal’ OAT catalysed by cis-MoVI(O)2 complexes it has been reported previously that the putative MoIV(O) intermediates can comproportionate with remaining cis-MoVI(O)2 complexes to give stable, and often catalytically inert [(O)MoV–O–MoV(O)] oxo-bridged complexes.40–44,70 Here, after photo-activation of 3C(O)OMe, the presence of a single band at 948 cm−1 and isosbestic points are consistent with formation of a monomeric MoIV(O) intermediate or rapid comproportionation to a [(O)MoV–O–MoV(O)] complex in equilibrium with MoIV(O), as illustrated in Scheme 4, that reacts too rapidly to be observed.
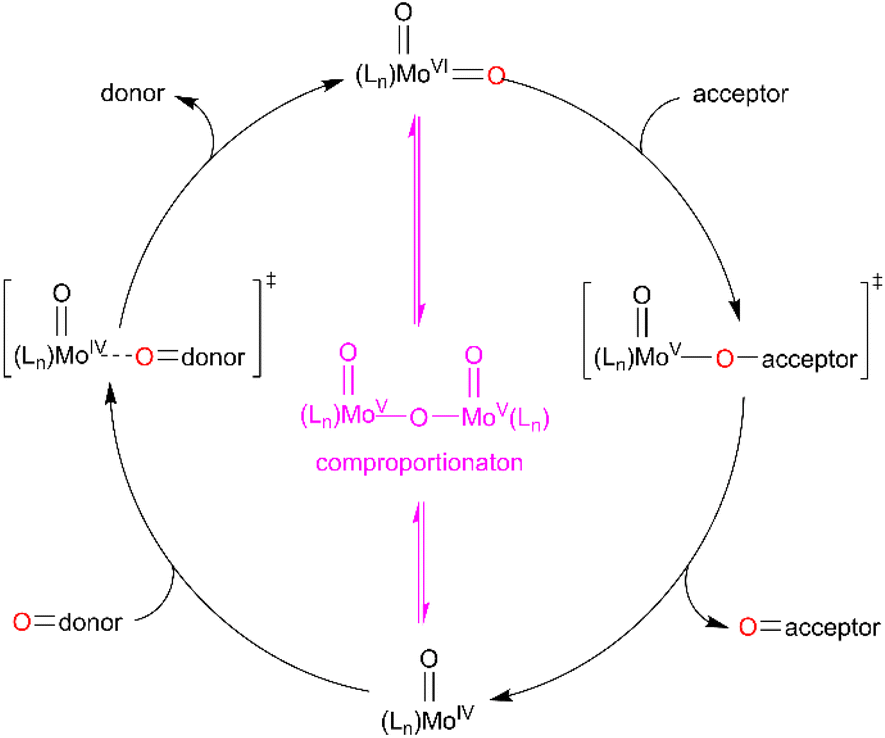 | ||
| Scheme 4 General OAT mechanism between an oxygen-atom acceptor and an oxygen-atom donor via MoVI/MoV/MoIV redox cycling. | ||
The IR-solution spectrum of 1H showed no change on irradiation, confirming that OAT from 1H to PPh3 does not occur (see Fig. S67 and S68, ESI†). Collectively, the 1H, 31P and IR-solution data show that OAT occurs under irradiation to give a Mo(O) complex intermediate and subsequent regeneration of cis-Mo(O)2 occurs thermally. Furthermore, the lack of catalytic turnover for 1H is not due to stoichiometric OAT and subsequent formation of a catalytically inactive [(O)MoV–O–MoV(O)] oxo-bridged bimetallic complex.71
Phosphine and sulfoxide scope and equimolar catalytic reactions
There is a clear opportunity to develop synthetically useful OAT chemistry using these systems and, as an example, the deoxygenation of sulfoxides is considered of synthetic utility for sulfide formation.72–74 For this purpose, the reaction solvent was changed from DMSO, a reactive solvent, to acetone, which is unreactive under these conditions. Photocatalytic OAT also occurs between equimolar ratios of a range of other phosphines and sulfoxides (Tables 2, S18 and S19, ESI†). Aryl, alkyl and substituted derivatives are tolerated, and the studied sulfoxides and phosphines all exhibited OAT with generation of the respective phosphine oxide and sulfide, showing complete selectivity for the OAT reaction. For example, using 3C(O)OMe and 300 equivalents of PPh3 and 300 equivalents of p-tolyl sulfoxide in acetone gives 97% conversion in 48 hours.|
|
|||
|---|---|---|---|
| Entry | Sulfoxide | Phosphine | Conversionb %c, (%)d |
| a General condition: 1 mM of 3C(O)OMe in acetone-d6, 300 mM sulfoxide and 300 mM phosphine, 410 nm, 3 h, under N2. b Conversions determined by 1H NMR spectroscopy. c Conversion of sulfoxide to sulfide. d Conversion of phosphine to phosphine oxide in brackets. e Determination of phosphine conversion prevented by overlapping signals. | |||
| 1 | R1 = R2 = Me | R3 = R4 = R5 = Ph | 14, (20) |
| 2 | R1 = R2 = CH2Ph | R3 = R4 = R5 = Ph | 21d |
| 3 | R1 = R2 = 4-Me-Ph | R3 = R4 = R5 = Ph | 46, (46) |
| 4 | R1 = Me; R2 = Ph | R3 = R4 = R5 = Ph | 35e |
| 5 | R1 = Me; R2 = 4-Me-Ph | R3 = R4 = R5 = Ph | 44, (42) |
| 6 | R1 = Me; R2 = 4-Br-Ph | R3 = R4 = R5 = Ph | 36, (39) |
| 7 | R1 = Me; R2 = 4-MeO-Ph | R3 = R4 = R5 = Ph | 46, (45) |
| 8 | R1 = R2 = Me | R3 = R4 = Me; R5 = Ph | 30, (36) |
| 9 | R1 = R2 = Me | R3 = Me; R4 = R5 = Ph | 35, (42) |
| 10 | R1 = Me; R2 = 4-Me-Ph | R3 = R4 = R5 = 4-F-Ph | 14e |
| 11 | R1 = Me; R2 = 4-Me-Ph | R3 = R4 = R5 = 4-MeO-Ph | 32, (47) |
| 12 | R1 = Me; R2 = 4-Me-Ph | R3 = R4 = R5 = 4-CF3-Ph | 40e |
Mechanism of the photocatalytic OAT reaction
The foregoing observations can be used to propose a catalytic mechanism and identify some notable features. Conventional thermal OAT using bio-inspired cis-Mo(O)2 complexes75 generally occurs via the nucleophilic attack of oxygen atom acceptor on a Mo-oxo π* orbital (Scheme 4), which results in concomitant weakening of the Mo-oxo and strengthening of the acceptor-oxo bond. In these reported systems Hammett relationships for catalyst ligands indicate that EWGs reduce localisation of negative charge in the transition state, thereby increasing the rate of the reaction.41,42,50,60 In comparison, complexes 1H–7CF3 show similar, but non-linear trends (Table 1, Fig. S48†) with H, F, OMe, and tBu ligand substitution giving inactive catalysts, whereas the EWGs, C(O)OMe, C(O)OH and CF3 support a broad range of OAT activity.The similarity of the electronic absorption spectra (see Fig. S31, ESI†) and absorption coefficients at 410 nm for 1H–7CF3 indicate that initial photon absorption does not account for the differences in the observed activity. In addition, similar emission data for ligands and complexes in the presence and absence of substrate (see Table S17, Fig. S52–S54, ESI†) suggest that the emissive excited states that are detectable do not mediate OAT. Nevertheless, 1H, 31P NMR and IR spectroscopies clearly show that, for inactive complexes, catalytic OAT fails because initial OAT to phosphine does not occur. There is no evidence for an inert intermediate after initial OAT that prevents catalytic turn over. Indeed, IR-solution spectroscopy clearly shows that SAP ligands with sufficiently electron-withdrawing substituents support the generation of a Mo(O) intermediate under irradiation which undergoes thermal regenerative OAT.
Based on the above observations, a mechanism is proposed (Scheme 5) where the initial photon absorption leads to the generation of a non-emissive excited state (i). Subsequently OAT occurs via either (ii) SET followed by a radical rebound pathway via caged-radicals, as proposed for some metal-oxyl systems,76–78 or alternatively, a concerted reaction to give (iii) via the nucleophilic addition of phosphine. Both pathways would be promoted by EWGs on the SAP ligands, with the SET pathway potentially less sensitive to phosphine steric features. Subsequently, elimination of OPPh3 from (iii) gives Mo(O) (iv) which undergoes thermal regenerative OAT from a sulfoxide donor. In comparison, recently reported photoactive W-dioxo complexes are proposed to photocatalyse via photo-induced SET pathways from an emissive state,51 similar to the well-established Ru- and Ir-bipyridyl photoredox catalysts,8,72 where substrate binding to the metal centre is not required, and radical-based chemical reactions are initiated via outer sphere electron transfer. Irradiation of 3C(O)OMe in DMSO in the presence of potential sacrificial electron donors,79 such as triethylamine (TEA), N,N-dimethylaniline (DMA), and 1, 3-methyl-2-phenylbenzimidazoline (BIH) showed a decrease in the amount of catalyst and a complex array of products (Fig. S86†). These observations are indicative of electron transfer and suggest that complex photoredox chemistry is occurring that leads to the decomposition of the catalyst. To try and differentiate between concerted and SET pathways, a radical scavenging experiment was undertaken. Since TEMPO is unsuitable as a radical scavenger because it reacts directly with catalyst 3C(O)Me under irradiation (see Fig. S87 and S88, ESI†), we selected 2,4,6-tri-tert-butylphenol (TTBP)80,81 as an alternative. A control experiment showed that on irradiation of a mixture of 3C(O)Me and TTBP in DMSO no change was observed in the 1H NMR spectrum (see Fig. S89, ESI†). On addition of PPh3 catalysis occurs giving 57% conversion compared to 69% for a parallel control reaction in the absence of TTBP (see Fig. S90, ESI†). The addition of electrolyte (250 mM [nBu4N][PF6]) to facilitate cage escape of any radicals formed via SET decreased the conversion further to 53% (see Fig. S96 and S97, ESI†). These observations could be suggestive of a SET mechanism in which the putative [PR3]˙+ radical does not significantly dissociate from the catalyst. The observed decrease in conversion, however, is not pronounced enough to allow a conclusive differentiation between the concerted and radical-rebound (caged-radical) pathways shown in Scheme 5. Similarly, catalysis in the presence of styrene and cyclohexene as putative radical traps also did not affect conversion of PPh3 to OPPh3 (see Fig. S98–S101†). In addition, we note the insensitivity of catalysis to dioxygen (Table 1, entry 9) suggesting that free [PR3]˙+ is not present.
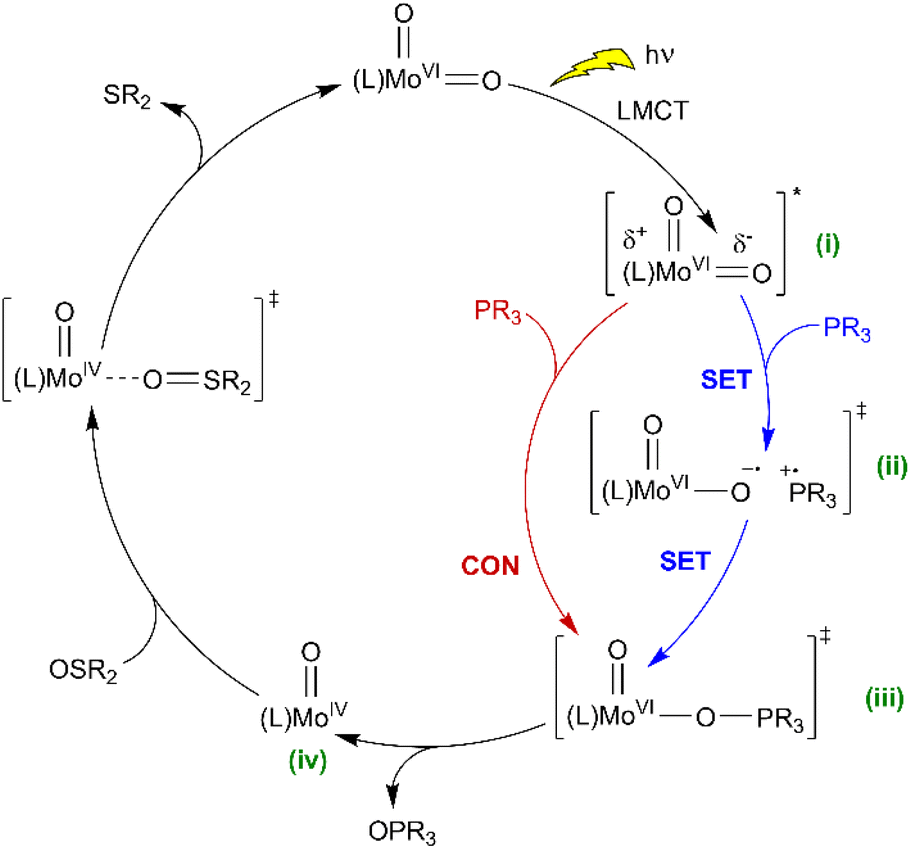 | ||
| Scheme 5 Proposed catalytic mechanisms for light-activated OAT between phosphine and sulfoxide substrates, catalysed by photoactive cis-MoVI(O)2 complexes (SET = single electron transfer, CON = concerted reaction, comproportionation as shown in Scheme 4 also conceivable). | ||
Conceivably, prior coordination of PR3 could be followed by intramolecular migration of PR3cis to MoV(O) which has been proposed for related stoichiometric reactions37 although in these cases spectral changes are observed on mixing PR3 with MoV which we do not observe here. Collectively, these observations suggest that the putative [PR3]˙+ radical does not significantly dissociate from (ii) but the data do not conclusively differentiate between the concerted and radical rebound (caged-radical) pathways shown in Scheme 5.
Of perhaps more direct relevance are recent hydrogen atom transfer (HAT) reactions to [cis-Mo(O)2X2(tBu-bipy)] (where X = F, Cl, Br) complexes where a LMCT excited state is proposed to mediate stoichiometric HAT from a hydrocarbon substrate to an oxo ligand via a step-wise SET-proton transfer or concerted pathways.38 In contrast LMCT here promotes bimolecular radical oxo-ligand transfer to a phosphine substrate. This reactivity is analogous to unimolecular radical dissociation of ligands with a single M–L bond such as halides via inner sphere electron transfer (Scheme 1).17
Conclusions
A novel class of photoactive Mo-based OAT catalysts was synthesised in two steps from inexpensive, commercially available precursors. The catalysts are tolerant to both water and oxygen. Compared to the established Mo-oxo catalytic chemistry of biological, bio-inspired, and heterogeneous systems, there are clear mechanistic differences for the photocatalytic cycle proposed here, which arise from photoactivation via LMCT. In contrast to most homogeneous photocatalytic redox systems, photo-induced reactivity does not arise from a long-lived emissive state, but rather from a non-emissive LMCT excited state. In common with thermal OAT chemistry mediated with cis-Mo(O)2 complexes, EWGs promote activity indicating the accumulation of negative charge in the rate-limiting transition state. A further important feature of the SAP complexes studied here is that under the chosen experimental conditions the formation of inert [(O)MoV–O–MoV(O)] complexes is not restricting catalytic turnover, which is sustained with remarkable catalyst stability, allowing equimolar OAT reactions with very low catalyst loading compared to many photoredox reactions. A unique feature of the work described here is the coupling of photoactivated and thermally activated OAT steps to establish a catalytic cycle. There is considerable scope to further understand mechanistic details of these LMCT-induced reactions that could guide future atom transfer catalyst developments to expand the reactivity beyond that described here, which is the focus of current studies.Data availability
All supporting data has been uploaded as part of the ESI.†§Author contributions
R. E. D. and A.-K. D.-K. directed the project. T. D., L. G., and S. S. carried out compound synthesis, characterisation, and respective data analysis. A. C. W. collected, solved, and refined the crystal data. T. D. and L. G. carried out photocatalytic testing of the reported complexes. L. G. and J. M. L. carried out computational calculations. L. G. and B. P. carried out FTIR-monitoring of non-catalytic reactions and analysed the respective data. T. D. tested the substrate scope. L. G. carried out sacrificial electron donor and radical scavenging experiments, catalysis with electrolyte and kinetic experiments with different phosphines. T. D., L. G., R. E. D. and A.-K. D.-K. co-wrote the original draft. All authors contributed to the editing of the manuscript. R. E. D. and A.-K. D.-K. were responsible for funding acquisition and project administration.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank EPSRC (EP/T518025/1) (L. G.) and (EP/T007338/1) (A.-K. D.-K.), the Wild Fund (T. D.), the Erasmus+ programme of the European Union (S. S.) and The University of York for financial support. J. M. L. is supported by an Industrial Fellowship from the Royal Society, (INF\R1\221057). The authors are grateful for computational support from the University of York High Performance Computing service, Viking, and the Research Computing team. The authors also thank Prof. Neil Hunt for useful discussions, use of FTIR equipment, and Prof. Martin L. Kirk for helpful discussions.Notes and references
- G. Ciamician, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- D. D. Dionysiou, G. Li Puma, J. Ye, J. Schneider and D. Bahnemann, ed. Photocatalysis: Applications, The Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- C. R. J. Stephenson, T. P. Yoon and D. W. C. MacMillan, Visible Light Photocatalysis in Organic Chemistry, Wiley, Weinheim, 2018 Search PubMed.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed.
- J. P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 130, 10188–10228 CrossRef.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- D. Banerjea and M. K. Bharty, in Mechanisms of Reactions of Metal Complexes in Solution, The Royal Society of Chemistry, Cambridge, 2022, ch. 9. pp. 497–521 Search PubMed.
- S. M. Treacy and T. Rovis, Synthesis, 2024, 56, 1967–1978 CrossRef CAS PubMed.
- N. R. East, R. Naumann, C. Förster, C. Ramanan, G. Diezemann and K. Heinze, Nat. Chem., 2024, 16, 827–834 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed.
- L. Chang, S. Wang, Q. An, L. Liu, H. Wang, Y. Li, K. Feng and Z. Zuo, Chem. Sci., 2023, 14, 6841–6859 RSC.
- Z. Xiao, C. G. Young, J. H. Enemark and A. G. Wedd, J. Am. Chem. Soc., 1992, 114, 9194–9195 CrossRef CAS.
- J. H. Enemark, J. J. A. Cooney, J. J. Wang and R. H. Holm, Chem. Rev., 2004, 104, 1175–1200 CrossRef CAS PubMed.
- R. H. Holm, Coord. Chem. Rev., 1990, 100, 183–221 CrossRef CAS.
- A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC.
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- J. Thrane, U. V. Mentzel, M. Thorhauge, M. Høj and A. D. Jensen, Catalysts, 2021, 11, 1329 CrossRef CAS.
- A. Stanislaus, A. Marafi and M. S. Rana, Catal. Today, 2010, 153, 1–68 CrossRef CAS.
- S. J. Nieter Burgmayer and M. L. Kirk, in Metallocofactors that Activate Small Molecules. Structure and Bonding, ed. M. W. Ribbe, Springer, Cham, 2019, vol. 179, pp. 101–152 Search PubMed.
- C. Gates, H. Varnum, C. Getty, N. Loui, J. Chen, M. L. Kirk, J. Yang and S. J. N. Burgmayer, Inorg. Chem., 2022, 61, 13728–13742 CrossRef CAS PubMed.
- K. Heinze, Coord. Chem. Rev., 2015, 300, 121–141 CrossRef CAS.
- S. Leimkühler and C. Iobbi-Nivol, FEMS Microbiol. Rev., 2015, 40, 1–18 CrossRef PubMed.
- Y. Shen, P. Jiang, P. T. Wai, Q. Gu and W. Zhang, Catalysts, 2019, 9, 31 CrossRef.
- W. Yan, M. Liu, J. Wang, J. Shen, S. Zhang, X. Xu, S. Wang, J. Ding and X. Jin, Chem. Rec., 2020, 20, 230–251 CrossRef CAS PubMed.
- P. Salonen, J. A. Schachner, A. Peuronen, M. Lahtinen, F. Belaj, N. C. Mösch-Zanetti and A. Lehtonen, Mol. Catal., 2023, 540, 113034 CrossRef CAS.
- M. K. Hossain, J. A. Schachner, M. Haukka, A. Lehtonen, N. C. Mösch-Zanetti and E. Nordlander, Polyhedron, 2017, 134, 275–281 CrossRef CAS.
- N. Zwettler, J. A. Schachner, F. Belaj and N. C. Mösch-Zanetti, Mol. Catal., 2017, 443, 209–219 CrossRef CAS.
- D. Chen, X. Zhang, H. Jiang and X. Yuan, Res. Chem. Intermed., 2020, 46, 4705–4721 CrossRef CAS.
- H. Arzoumanian, G. Agrifoglio, H. Krentzien and M. Capparelli, J. Chem. Soc., Chem. Commun., 1995, 655–656 RSC.
- R. A. Isovitsch, J. G. May, F. R. Fronczek and A. W. Maverick, Polyhedron, 2000, 19, 1437–1446 CrossRef CAS.
- S. Fosshat, S. D. M. Siddhiaratchi, C. L. Baumberger, V. R. Ortiz, F. R. Fronczek and M. B. Chambers, J. Am. Chem. Soc., 2022, 144, 20472–20483 CrossRef CAS PubMed.
- S. Fosshat, F. R. Fronczek and M. B. Chambers, Chem.–Eur. J., 2023, 29, e202300486 CrossRef CAS PubMed.
- M. S. Reynolds, J. M. Berg and R. H. Holm, Inorg. Chem., 1984, 23, 3057–3063 CrossRef CAS.
- J. Topich and J. T. Lyon, Polyhedron, 1984, 3, 61–65 CrossRef CAS.
- J. Topich and J. T. Lyon, Inorg. Chem., 1984, 23, 3202–3206 CrossRef CAS.
- J. M. Berg and R. H. Holm, J. Am. Chem. Soc., 1985, 107, 925–932 CrossRef CAS.
- S. A. Julia Craig, E. W. Harlan, B. S. Snyder, M. A. Whitener and R. H. Holm, Inorg. Chem., 1989, 28, 2082–2091 CrossRef.
- C. A. Páez, O. Lozada, N. J. Castellanos, F. O. Martínez, F. Ziarelli, G. Agrifoglio, E. A. Páez-Mozo and H. Arzoumanian, J. Mol. Catal. A: Chem., 2009, 299, 53–59 CrossRef.
- H. Martinez Q, Á. A. Amaya, E. A. Paez-Mozo, F. Martinez O and S. Valange, Catal. Today, 2021, 375, 441–457 CrossRef CAS.
- N. J. Castellanos, H. Martínez Q, F. Martínez O, K. Leus and P. Van Der Voort, Res. Chem. Intermed., 2021, 47, 4227–4244 CrossRef CAS.
- N. J. Castellanos, F. Martínez, F. Lynen, S. Biswas, P. Van Der Voort and H. Arzoumanian, Transition Met. Chem., 2013, 38, 119–127 CrossRef CAS.
- C. A. Páez, N. J. Castellanos, F. Martínez O., F. Ziarelli, G. Agrifoglio, E. A. Páez-Mozo and H. Arzoumanian, Catal. Today, 2008, 133–135, 619–624 CrossRef.
- A. B. Ducrot, B. A. Coulson, R. N. Perutz and A. K. Duhme-Klair, Inorg. Chem., 2016, 55, 12583–12594 CrossRef CAS PubMed.
- D. Yu, W. P. To, G. S. M. Tong, L. L. Wu, K. T. Chan, L. Du, D. L. Phillips, Y. Liu and C. M. Che, Chem. Sci., 2020, 11, 6370–6382 RSC.
- T. I. Robertson and P. N. Nelson, J. Mol. Struct., 2022, 1249, 131625 CrossRef CAS.
- S. Y. Ebrahimipour, M. Khosravan, J. White and S. Fekri, Appl. Organomet. Chem., 2018, 32, e4233 CrossRef.
- W. Wang, T. Guerrero, S. R. Merecias, H. García-Ortega, R. Santillan, J. C. Daran, N. Farfán, D. Agustin and R. Poli, Inorg. Chim. Acta, 2015, 431, 176–183 CrossRef CAS.
- O. A. Rajan and A. Chakravorty, Inorg. Chem., 1981, 20, 660–664 CrossRef CAS.
- M. Minelli, F. Namuswe, D. Jeffrey, A. L. Morrow, I. A. Guzei, D. Swenson, E. Bothe and T. Weyhermu, Inorg. Chem., 2006, 45, 5455–5464 CrossRef CAS PubMed.
- M. Cindrić, G. Pavlović, R. Katava and D. Agustin, New J. Chem., 2017, 41, 594–602 RSC.
- M. Cindrić, N. Strukan, V. Vrdoljak and B. Kamenar, Z. Anorg. Allg. Chem., 2004, 630, 585–590 CrossRef.
- J. Topich, Inorg. Chim. Acta, 1980, 46, 37–79 CrossRef.
- A. Rezaeifard, M. Jafarpour, H. Raissi, M. Alipour and H. Stoeckli-Evans, Z. Anorg. Allg. Chem., 2012, 638, 1023–1030 CrossRef CAS.
- R. Mayilmurugan, B. N. Harum, M. Volpe, A. F. Sax, M. Palaniandavar and N. C. Mösch-Zanetti, Chem.–Eur. J., 2011, 17, 704–713 CrossRef CAS PubMed.
- C. J. Whiteoak, G. J. P. Britovsek, V. C. Gibson and A. J. P. White, Dalton Trans., 2009, 2337–2344 RSC.
- A. Ducrot, B. Scattergood, B. Coulson, R. N. Perutz and A. K. Duhme-Klair, Eur. J. Inorg. Chem., 2015, 2015, 3562–3571 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- C. Lorber, M. R. Plutino, L. I. Elding and E. Nordlander, J. Chem. Soc., Dalton Trans., 1997, 3997–4003 RSC.
- N. J. Turro, V. Ramamurthy, W. Cherry and W. Farneth, Chem. Rev., 1978, 78, 125–145 CrossRef CAS.
- I. Bors, J. Kaizer and G. Speier, RSC Adv., 2014, 4, 16928–16930 RSC.
- C. Ercolani, M. Gardini, G. Pennesi and G. Rossi, J. Chem. Soc., Chem. Commun., 1983, 549–550 RSC.
- O. E. Brian Schultz, S. F. Gheller, M. C. Muetterties, M. J. Scott and R. H. Holm, J. Am. Chem. Soc., 1993, 115, 2714–2722 CrossRef.
- R. H. Holm, Chem. Rev., 1987, 87, 1401–1449 CrossRef CAS.
- X. Ma, C. Schulzke, Z. Yang, A. Ringe and J. Magull, Polyhedron, 2007, 26, 5497–5505 CrossRef CAS.
- A. K. Clarke, A. Parkin, R. J. K. Taylor, W. P. Unsworth and J. A. Rossi-Ashton, ACS Catal., 2020, 10, 5814–5820 CrossRef CAS PubMed.
- L. Shiri and M. Kazemi, Res. Chem. Intermed., 2017, 43, 6007–6041 CrossRef CAS.
- A. S. Surur, L. Schulig and A. Link, Arch. Pharm. (Weinheim, Ger.), 2019, 352, 1800248 Search PubMed.
- G. C. Tucci, J. P. Donahue and R. H. Holm, Inorg. Chem., 1998, 37, 1602–1608 CrossRef CAS.
- X. Huang and J. T. Groves, JBIC, J. Biol. Inorg. Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- Y. Shimoyama and T. Kojima, Inorg. Chem., 2019, 58, 9517–9542 CrossRef CAS PubMed.
- C. Limberg, Angew. Chem., Int. Ed., 2003, 42, 5932–5954 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- C. D. Cook, D. A. Kuhn and P. Fianu, J. Am. Chem. Soc., 1956, 78, 2002–2005 CrossRef CAS.
- A. Juneau, T. O. Hope, J. Malenfant, M. Mesko, J. McNeill and M. Frenette, ACS Catal., 2022, 12, 2348–2356 CrossRef CAS.
- Agilent. CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- Oxford Diffraction Ltd, Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm within, CrysAlisPro software, 2009 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- V. Béreau, V. Jubéra, P. Arnaud, A. Kaiba, P. Guionneau and J. P. Sutter, J. Chem. Soc., Dalton Trans., 2010, 39, 2070–2077 RSC.
- R. M. Irfan, D. Jiang, Z. Sun, D. Lu and P. Du, Dalton Trans., 2016, 45, 12897–12905 RSC.
- M. Livendahl, J. Jamroskovic, M. Hedenström, T. Görlich, N. Sabouri and E. Chorell, Org. Biomol. Chem., 2017, 15, 3265–3275 RSC.
- Akanksha and D. Maiti, Green Chem., 2012, 14, 2314–2320 RSC.
- P. Jaikhan, B. Buranrat, Y. Itoh, J. Chotitumnavee, T. Kurohara and T. Suzuki, Bioorg. Med. Chem. Lett., 2019, 29, 1173–1176 CrossRef CAS PubMed.
- H. Gehrke and J. Veal, Inorg. Chim. Acta, 1969, 3, 623–627 CrossRef CAS.
- M. M. Folkendt, B. E. Weiss-Lopez, J. P. Chauvel and N. S. True, J. Phys. Chem., 1985, 89, 3347–3352 CrossRef CAS.
- M. Bagherzadeh, S. Ataie, H. Mahmoudi and J. Janczak, Inorg. Chem. Commun., 2017, 84, 63–67 CrossRef CAS.
- X. Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 6280–6283 CrossRef CAS PubMed.
- A. Guha, R. Sanyal, T. Chattopadhyay, Y. Han, T. K. Mondal and D. Das, J. Mol. Struct., 2013, 1042, 104–111 CrossRef CAS.
- S. Kim, J. Y. Noh, K. Y. Kim, J. H. Kim, H. K. Kang, S. W. Nam, S. H. Kim, S. Park, C. Kim and J. Kim, Inorg. Chem., 2012, 51, 3597–3602 CrossRef CAS PubMed.
- B. Das, A. Chakraborty and S. Chakraborty, Spectrochim. Acta, Part A, 2020, 225, 117443 CrossRef CAS PubMed.
- R. Sanz, J. Escribano, Y. Fernández, R. Aguado, M. R. Pedrosa and F. J. Arnáiz, Synlett, 2005, 9, 1389–1392 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: The authors have cited additional references in the ESI.82–109. CCDC 2312692, 2312691, 2312695, 2312693 and 2312694. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02784a |
| ‡ These authors have contributed equally to this work. |
| § A triplet state lying ca 75 kJ mol−1 lower in energy that the S1 state was also located and we cannot rule out a situation in which there is rapid intersystem crossing (<5 ns) and it is actually the triplet manifold that is responsible for the observed chemistry. Our calculations indicate that there is still significant Mo character in the highest occupied SOMO in a similar vein to the S1 state so is consistent with the proposed mechanism which has light-induced reduction of the metal centre as a key step. |
| This journal is © The Royal Society of Chemistry 2024 |