
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceManganese catalyzed chemo-selective synthesis of acyl cyclopentenes: a combined experimental and computational investigation†
Koushik
Sarkar
a,
Prativa
Behera
b,
Lisa
Roy
 *b and
Biplab
Maji
*a
*b and
Biplab
Maji
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, 741246, West Bengal, India. E-mail: bm@iiserkol.ac.in
bInstitute of Chemical Technology Mumbai, IOC Odisha Campus Bhubaneswar, Bhubaneswar, 751013, India. E-mail: l.roy@iocb.ictmumbai.edu.in
First published on 5th August 2024
Abstract
Cyclopentenes serve as foundational structures in numerous natural products and pharmaceuticals. Consequently, the pursuit of innovative synthetic approaches to complement existing protocols is of paramount importance. In this context, we present a novel synthesis route for acyl cyclopentenes through a cascade reaction involving an acceptorless-dehydrogenative coupling of cyclopropyl methanol with methyl ketone, followed by a radical-initiated ring expansion rearrangement of the in situ formed vinyl cyclopropenone intermediate. The reaction, catalyzed by an earth-abundant metal complex, occurs under milder conditions, generating water and hydrogen gas as byproducts. Rigorous control experiments and detailed computational studies were conducted to unravel the underlying mechanism. The observed selectivity is explained by entropy-driven alcohol-assisted hydrogen liberation from an Mn-hydride complex, prevailing over the hydrogenation of unsaturated cyclopentenes.
Introduction
Cyclopentenes are abundant in natural products (Scheme 1a) and are used as pharmaceuticals and medicinal compounds. Because of such importance, the synthesis of cyclopentenes seeks attention.1–5 Compared to the venerable cyclohexene synthesis via the Diels–Alder reaction, the preparation of cyclopentenes via [3 + 2] cycloaddition is limited in prominence.6 The synthesis of cyclopentenes via hydrogenation of cyclopentadiene suffers from harsh reaction conditions and poor selectivity due to the uncontrolled over-reduction.7–9 Other approaches to the synthesis of cyclopentenes via ring-closing metathesis needed the synthesis of pre-functionalized alkenes and precious metal catalysts.10,11 Vinyl cyclopropane rearrangement required multistep starting materials and harsh thermal,12–14 photochemical,15,16 or transition metal-catalyzed reaction conditions.12,16–18 Recently, carbonyl-alkene metathesis19 and photochemical annulation reactions6 have also been applied to the synthesis of cyclopentenes. However, the requirement of expensive synthesis of starting materials limits the usefulness of these methodologies. Undoubtedly, intermolecular divergent catalytic protocols utilizing abundant materials are underdeveloped and are in demand to render new retrosynthesis of these families of carbocycles.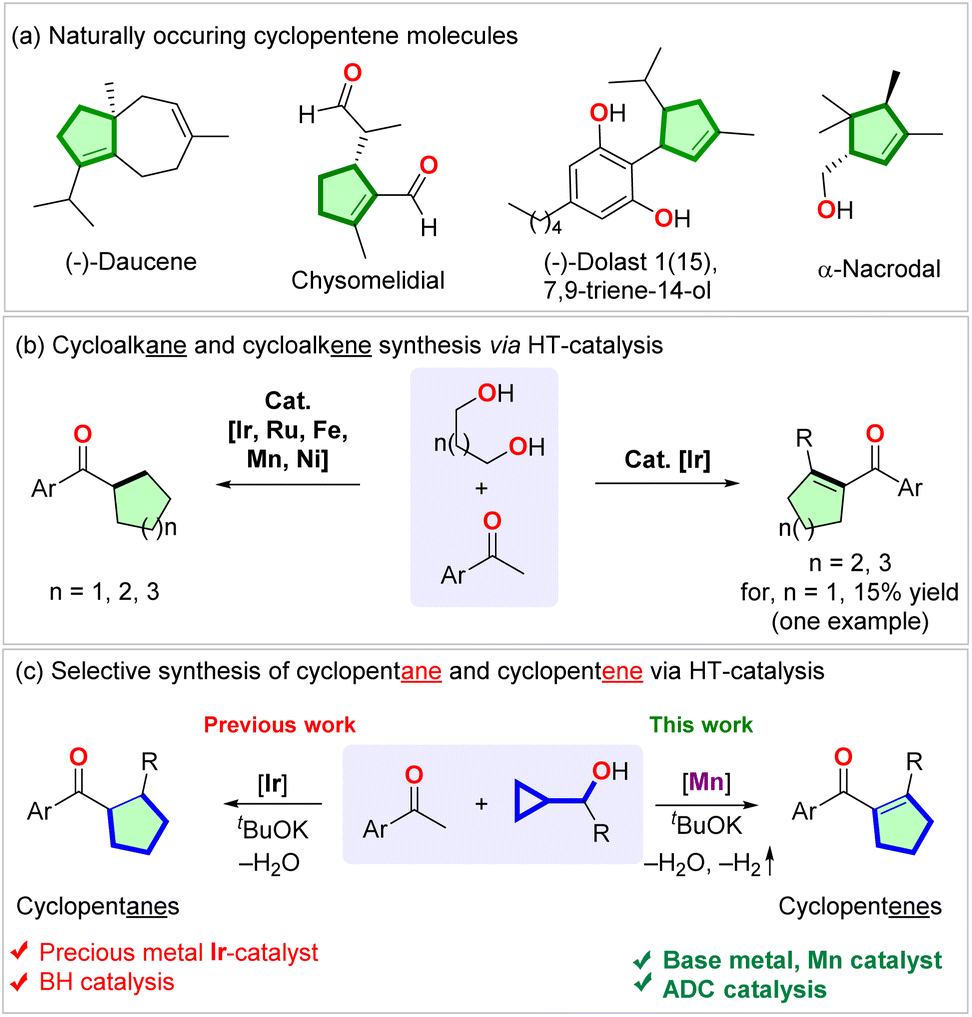 | ||
| Scheme 1 (a) Selected naturally occurring cyclopentene drug molecules; (b) previous report of the synthesis of cyclopentenes; (c) HT-mediated synthesis of cycloalkanes and cycloalkenes; (d) selective synthesis of cyclopentanes and cyclopentenes via HT-catalysis. | ||
Hydrogen transfer (HT) catalysis has become a versatile tool for waste-free redox transformations.20,21 Not long ago, Donohoe, Leitner, Maji, and Adhikary utilized the borrowing hydrogenation (BH) catalysis to synthesize cycloalkanes via the annulation of aryl ketones and diols using iridium,22–24 manganese,25,26 and nickel27 catalysts (Scheme 1b, left side). By tuning the ligand and the reaction conditions, the iridium-catalyzed reaction enabled the chemo-selective synthesis of cyclohexenes from pentamethylacetophenone and 1,5-diols (Scheme 1b, right side).24 However, the same reaction using 1,4-diol gave only 15% yield of one cyclopentene.24 Recently, Donohoe reported iridium-catalyzed preparation of saturated cyclopentanes (Scheme 1c, left side).28 However, to our knowledge, the applications of HT catalysis in synthesizing cyclopentenes have not been developed thus far (Scheme 1c, right side).
Employing an earth-abundant metal as a catalyst has an added advantage to sustainable chemical synthesis.21,29 Manganese complexes have recently been recognized as powerful catalysts for diverse (de)hydrogenation and hydroelementation reactions.30–32 Particularly, Mn(I)-complexes showed excellent activities in dehydrogenative coupling reactions without needing an acceptor for the liberated hydrogen.33,34 We are recently intrigued by the possibility of synthesizing multi-substituted acyl cyclopentenes via the Mn(I)-complex catalyzed HT-mediated coupling of cyclopropyl methanol with methyl ketone, followed by a single electron transfer (SET)-initiated ring expansion rearrangement of the in situ formed vinyl cyclopropenone intermediate (Scheme 1c, right side). We envisioned that by tailoring the catalyst and reaction conditions, it would be possible to facilitate the acceptorless liberation of a hydrogen molecule from a Mn(I)-hydride intermediate before the hydrogenation of the weakly polarized tetrasubstituted alkene could take place. It could thus enable the isolation of these unsaturated compounds, leading to the development of an unprecedented divergent synthesis of cyclopentenes with the generation of water and hydrogen as the byproducts. Herein, we report the realization of the above mechanistic hypothesis. Furthermore, controlled experiments and extensive DFT calculations were performed to understand the reaction mechanism.
Results and discussion
Reaction optimization
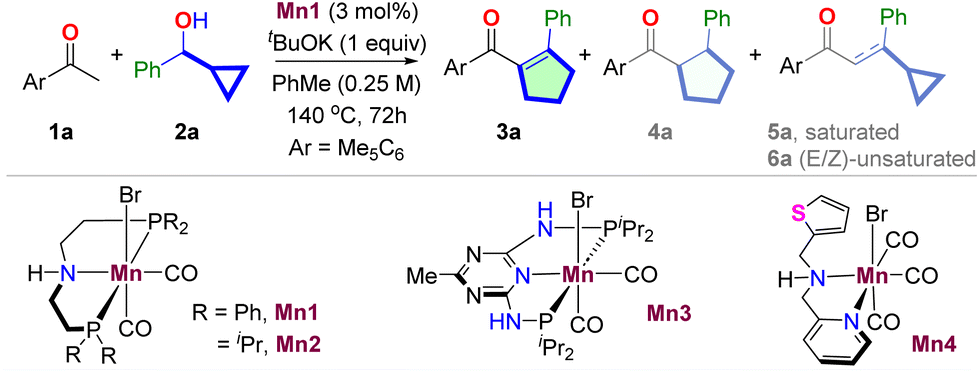
We began the project by inquiring about the reaction of pentamethyl acetophenone 1a and 1-phenyl cyclopropyl methanol 2a for synthesizing acyl cyclopentene 3a (Table 1, Section S4†). We realized the unique selectivity issue as the HT-mediated cascade of 1a and 2a could, together with the desired cyclopentene 3a, yield saturated cyclopentane 4a, BH product 5a, and unrearranged product 6a. Upon extensive optimization, we have found that the manganese complex Mn1 derived from the commercially available PhMACHO-ligand produced the desired cyclopentene 3a in 90% yield in the presence of tBuOK in toluene at 140 °C (entry 1). While 4a was undetected, trace amounts of 5a and 6a were observed by gas chromatography. The reaction was found to be sensitive to the bifunctional ligands. The Mn(I)-complexes Mn2,3 derived from iPrMACHO- and PN5P-ligands, respectively, provided inferior results (entries 2 and 3). The N,N-chelated Mn(I)-complexes Mn4 that we previously used for cycloalkane synthesis also gave poor results (entry 4).26 A significant amount of cyclopentane 4a was formed in the later case. The reaction was also sensitive to the bases used (entries 5–7). tBuOK performed better than other bases tested. Among the solvents, n-hexane gave a similar efficiency (entry 9), while others provided poor outcomes (entries 9–10). Control experiments suggested the necessity of each reaction component for successful product formation (entries 11–12).|
|
|||
|---|---|---|---|
| Entry | Deviation from the above | Selectivity 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a:4a:5a:6a :3a:4a:5a:6a |
% Yield of 3a |
a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Mn-catalyst (3 mol%), tBuOK (1 equiv.), PhMe (0.4 mL), 140 °C, 72 h. Selectivity and yields were calculated via gas chromatography using mesitylene as the internal standard. The poor mass balance in some cases is due to the complex, undetected product mixture. Ar![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C6Me5. C6Me5.
|
|||
| 1 | None | 2:90:0:5:2 |
90 (75) |
| 2 | Mn2 instead of Mn1 | 5:51:2:3:0 |
51 |
| 3 | Mn3 instead of Mn1 | 8:57:4:2:0 |
57 |
| 4 | Mn4 instead of Mn1 | 6:72:22:0:0 |
72 |
| 5 | NaOtBu instead of KOtBu | 44:45:0:0:0 |
45 |
| 6 | LiOtBu instead of KOtBu | 40:20:0:3:0 |
20 |
| 7 | KOH instead of KOtBu | 55:34:0:2:0 |
34 |
| 8 | Hexane instead of PhMe | 3:87:4:0:0 |
87 |
| 9 | Dioxane instead of PhMe | 4:34:4:0:0 |
34 |
| 10 | t AmOH instead of PhMe | 50:4:0:0 |
4 |
| 11 | Without Mn1 | 100:0:0:0:0 |
0 |
| 12 | Without KOtBu | 100:0:0:0:0 |
0 |
Tolerances of the method
With the set of optimized conditions, we explored the scope of Mn(I)-catalyzed cascade cyclopentene synthesis (Table 2). The reaction was minimally affected by the sterics, as o-, m-, and p-tolyl cyclopropylmethanol reacted with 1a at similar efficiencies, yielding the desired cyclopentenes 3b–3d in similar yields. The reaction tolerates halogens at different positions of the aryl ring, delivering 3e–3h in moderate yields. Aryl cyclopropylmethanol containing p-OMe, p-SMe, and p-iPr groups also reacted smoothly, providing the cyclopentenes 3i–3k in 50–60% yields. In particular, the reaction was equally efficient with the more challenging alkyl cyclopropylmethanols 3l–3s. 1-Cyclopropylpropan-1-ol provided 3l in 88% yield. While the increase in the chain length (3m) slightly lowered the yield, branching (3n) imparted minimal influence. A terminal alkene in 3o is retained under these hydrogen-liberating conditions. Similar observations were made with phenyl, ether, and thioether-containing substrates 3p–3r. Interestingly, dicyclopropylmethanol can also yield β-cyclopropyl cyclopentene 3s in a moderate 40% yield. Similarly, β-cyclobutyl cyclopentene 3t was synthesized in 50% yield. However, for the 1-phenyl cyclobutyl methanol, we obtained an exocyclic cylobutyl alkene product 3u, with 70% yield (Section S4.9†). The structures of 3a and 3u were confirmed via single-crystal X-ray crystallography.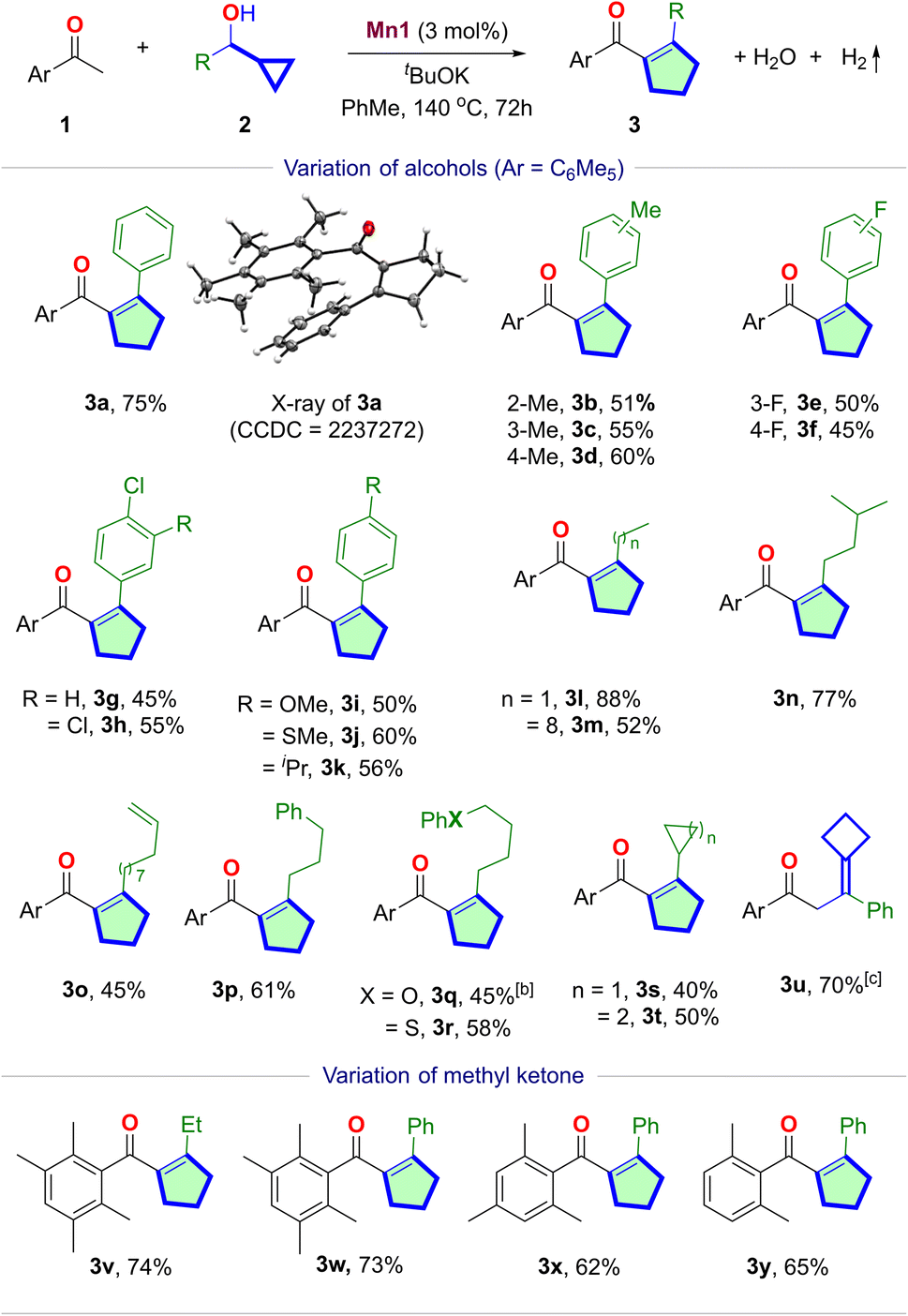
Diverse ortho-disubstituted acetophenones can be utilized as the C1-units yielding cyclopentenes 3v–3y in 62–74% yields. However, when 2a was treated with acetophenone under standard conditions, a complex mixture resulted, indicating the necessity of ortho-substituents that can sterically shield the carbonyl group to slow down reduction and aldol condensations. The advantageous effect of ortho-disubstituted methyl ketone has previously been demonstrated.22
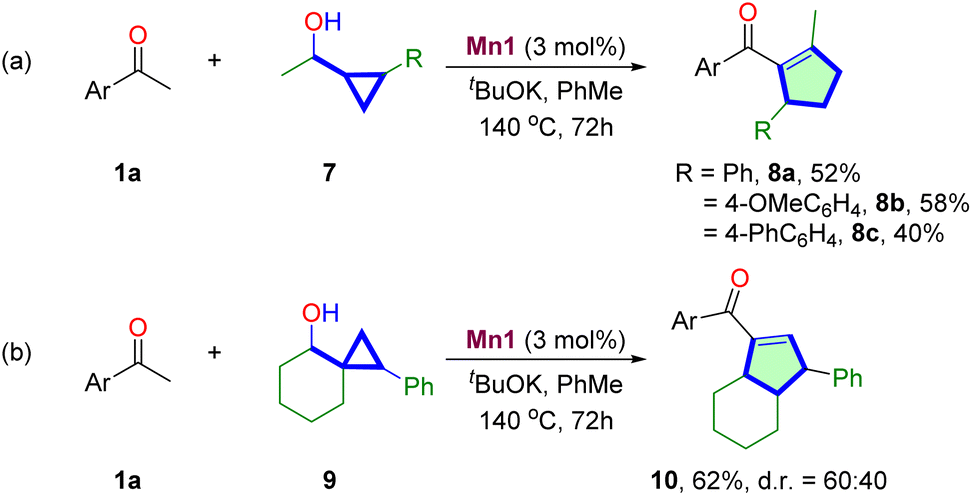
Then, we were prompted to explore the scope of substituted cyclopropane methanols for synthesizing multisubstituted cyclopentenes (Table 3). The reactions of 1,2-disubstituted cyclopropane methanols 7 with 1a proceed smoothly to produce 1,2,3-trisubstituted cyclopentenes 8a–8c in moderate 40–58% yields. The reaction of spiro-bicyclic cyclopropyl alcohol 9 was also investigated. The reaction proceeds at a similar efficiency, producing ring-expanded bicyclic product 10 in 62% yield, albeit in moderate 60:40 d.r.
|
3a Reaction conditions: 1a (0.1 mmol), 7 or 9 (0.2 mmol), Mn-catalyst (3 mol%), tBuOK (1 equiv.), PhMe (0.4 mL), 140 °C, 72 h. ArC6Me5.
|
|---|
|
Synthetic utility
To explore the synthetic utility of the Mn(I)-catalyzed cascade reaction, we have performed derivatization of isolated cyclopentene products (Scheme 2). The Pd/C catalyzed hydrogenation of 3a and produced the saturated carbocycle 11 in 70% yield. Corey–Chaykovsky cyclopropanation of 3l delivered bicyclo[3.1.0]hexan-1-yl core 12 in 62% yield and >99:1 d.r. We have also conducted the epoxidation of 3l. The reaction with tBuOOH/NaOH produced the tetra-substituted bicyclic epoxide 13 in a high 84% yield with >99:1 d.r. To further functionalize the product, we have performed late-stage γ-amination of 3avia bromine treatment in the presence of piperazine. Allyl amine 14 was isolated in 63% yield. The molecular structure of 14 was confirmed via single-crystal X-ray crystallography.35 Finally, the pentamethyl aryl group can easily be deprotected via retro-Friedel–Craft reactions. The free cyclopentene-1-carboxylic acid 15 was isolated in 75% yield.
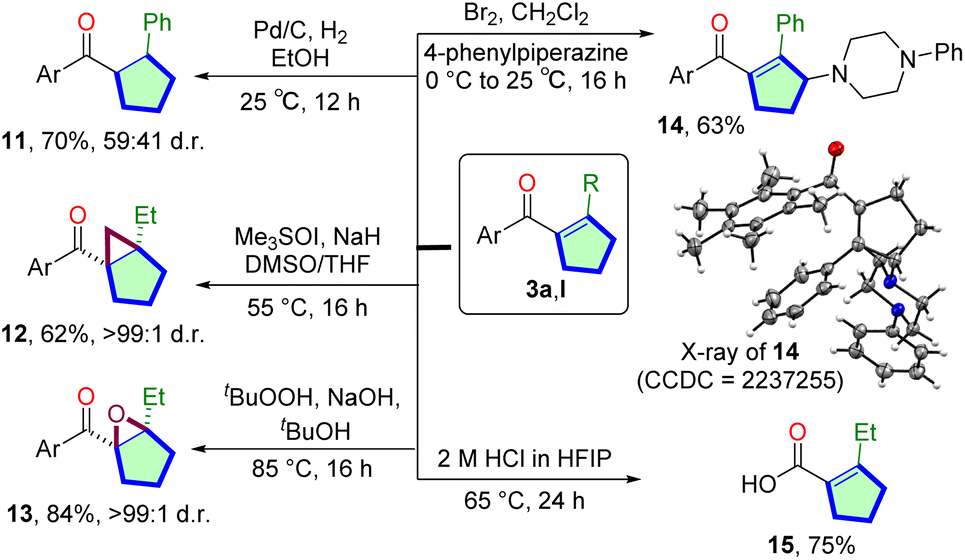 | ||
| Scheme 2 Derivatization of the product. | ||
Mechanistic studies and proposed mechanism
The Mn1-catalyzed dehydrogenation of 2a in the absence of 1a produced the corresponding ketone 16a in 80% yield (Scheme 3a, see Section S7.1†). The reaction proceeded without an acceptor, and hydrogen gas was detected upon GC analysis of the reaction headspace. The deuterium kinetic isotope effect experiment with 2a and 2a-d1 shows kH/kD = 1.67 (Scheme 3b, see Section S7.2†). It indicated that the dehydrogenation of secondary cyclopropyl alcohol could be the slowest step of the catalytic cycle. The reaction of 16a with 1a under the standard conditions gave 61% yield of product 3a (Scheme 3c, see Section S7.3†). The same reaction without Mn1 also gave a similar yield of 3a. However, no reaction occurred in the absence of tBuOK. It confirms the intermediacy of 16a and highlights the necessity of the base for the condensation and rearrangement steps.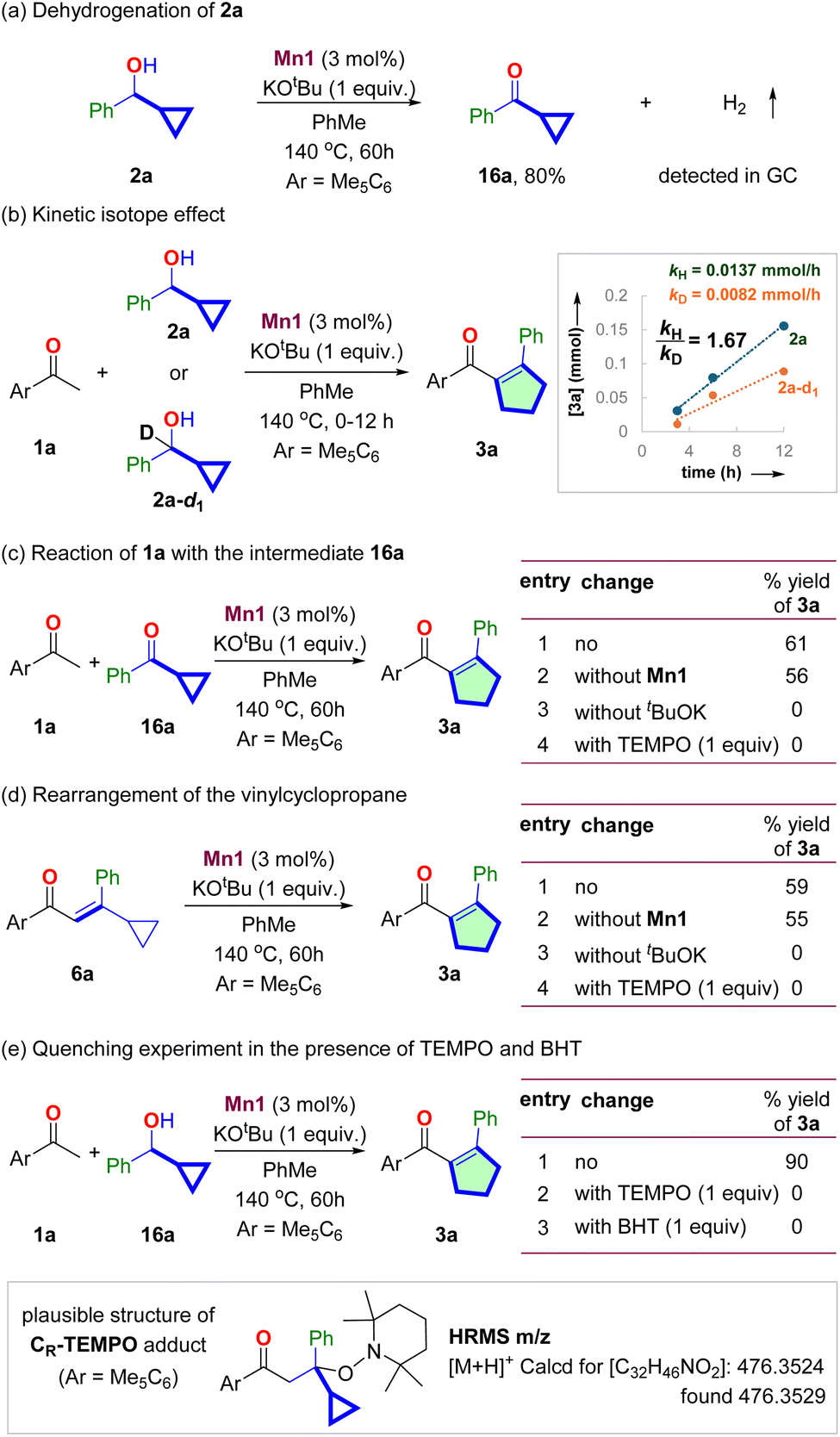 | ||
| Scheme 3 Mechanistic studies. (a) Dehydrogenation of 2a, (b) kinetic isotope effect study, (c) reaction of 1a with 16a, (d) probing the intermediacy of vinyl cyclopropane, (e) probing the involvement of radicals and plausible structure of the TEMPO adduct. | ||
We then synthesized the vinyl cyclopropane 6a and performed control experiments to probe its intermediacy (Scheme 3d, Section S7.4†). The subjection of 6a under standard conditions resulted in 59% yield of 3a (Scheme 3b, entry 1). The same reaction without Mn1 also provided 55% yield of 3a (entry 2). However, in the absence of tBuOK, 3a did not form, and 70% of 6a was recovered as an E/Z mixture (entry 3). These experiments suggested that (i) 6a is an intermediate for this reaction. (ii) Thermal vinyl-cyclopropane rearrangement did not occur at the reaction temperature. (iii) tBuOK mediates the rearrangement of 6a to 3a.
Notably, the isomerization of 6a to 3a stops in the presence of known radical quenchers, such as 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) (entry 4). The reaction of 1a with 2a in the presence of TEMPO and butylated hydroxytoluene (BHT) also did not produce 3a (Scheme 3e, see Section 7.5†). A similar observation was also made for the reaction of 16a with 1a (Scheme 3c, entry 4). High-resolution mass spectrometric analysis of the reaction mixture detected m/z = 476.3529, corresponding to a TEMPO adduct with the composition [C32H45NO2], suggesting the involvement of radical species during the rearrangement step (Scheme 3e).
The studied reaction holds promise for an intriguing ring expansion cascade. To provide in-depth insights into the reaction mechanism, we have further investigated the reaction using density functional theory at B3LYP-D3(BJ)/SMD(toluene)/def2-TZVPP (Fig. 1, 2, and Section S8†). For simplicity, we have bifurcated the reaction into (a) Mn-catalyzed dehydrogenation (Fig. 1a and b show the reaction mechanism and corresponding energy profile, respectively) and (b) the aldol condensation coupled to the SET cascade rearrangement (Fig. 2 shows the energy profile and structures of stationary states).
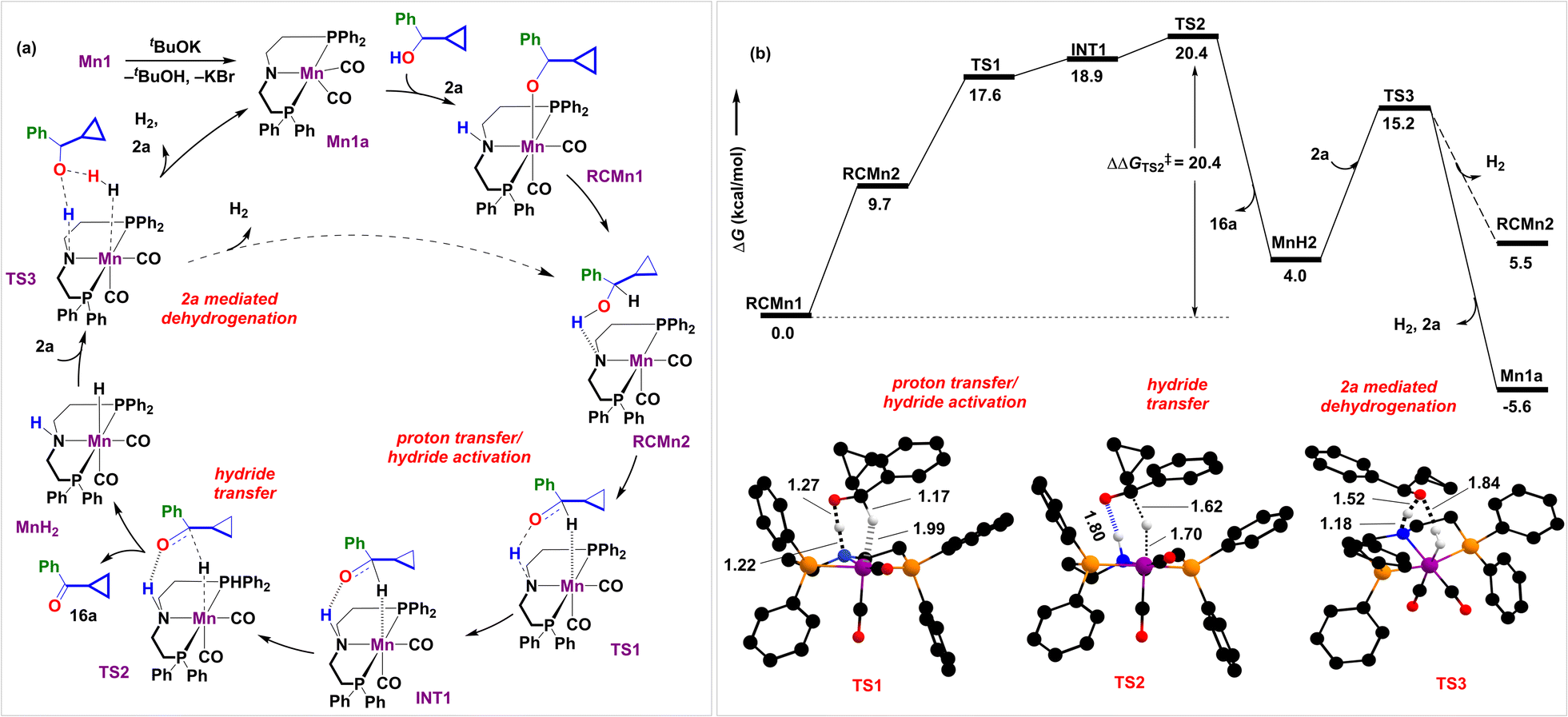 | ||
| Fig. 1 (a) Proposed mechanism for the Mn1-catalyzed dehydrogenation of 2a and (b) the corresponding reaction free energy profile at B3LYP-D3(BJ)/SMD(toluene)/def2-TZVPP (in kcal mol−1) for the Mn catalyzed dehydrogenation of alcohol. Color coding for optimized geometries (truncated) TS1, TS2, TS3: C(black), H(white), O(red), P(orange), N(blue), Mn(purple). Distances shown are in units of Å. Unimportant hydrogen atoms are not shown for clarity. | ||
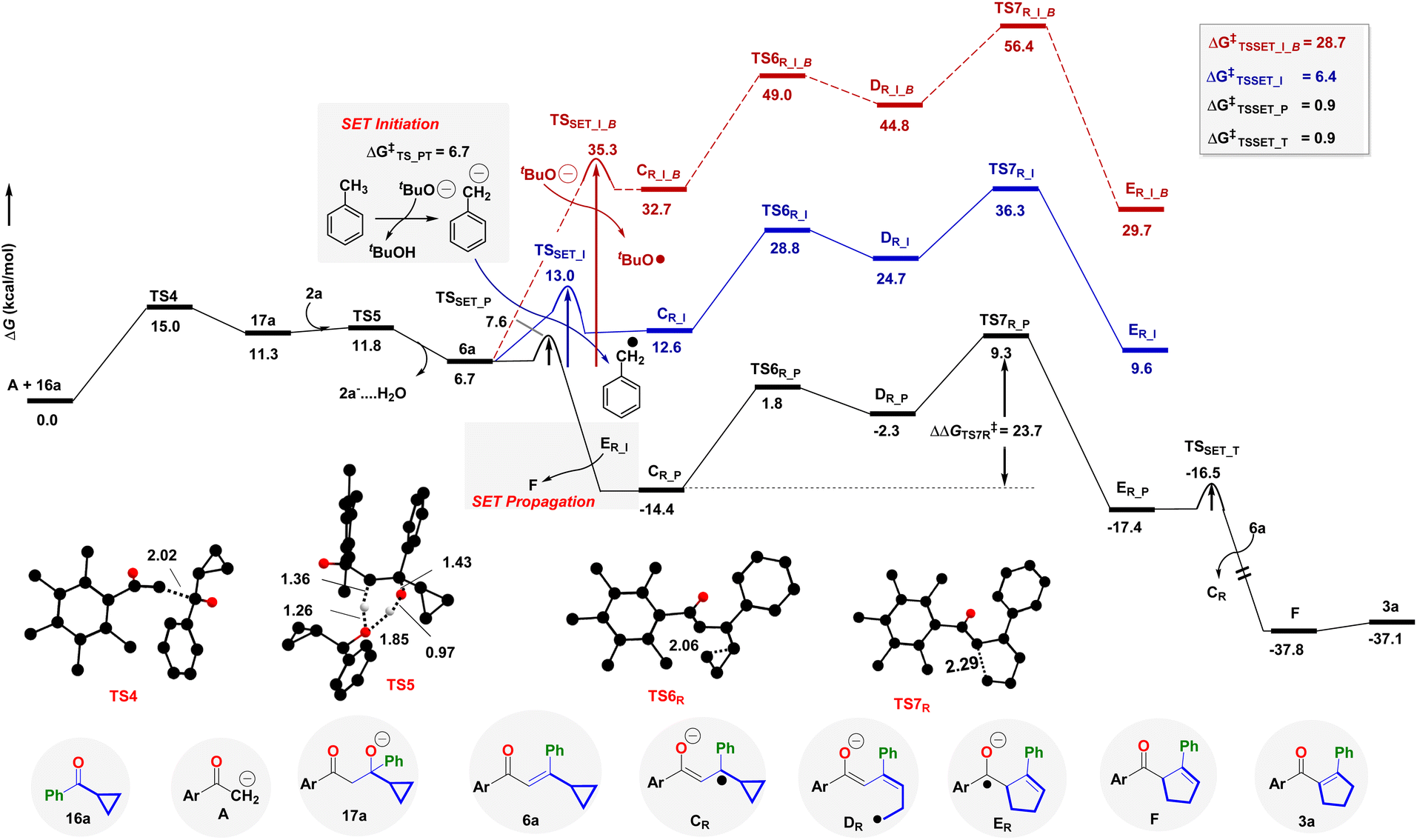 | ||
| Fig. 2 Reaction free energy profile at B3LYP-D3(BJ)/SMD(toluene)/def2-TZVPP (in kcal mol−1) for the synthesis of cyclopentene 3a. Color coding for optimized geometries (truncated) TS4, TS5, TS6R, TS7R: C(black), H(white), O(red), P(orange), N(blue), Mn(purple). Distances shown are in units of Å. Unimportant hydrogen atoms are not shown for clarity. | ||
We began by investigating the proposed catalytic cycle for the Mn1-catalyzed dehydrogenation of alcohol 2a to ketone 16a (Fig. 1a). As anticipated, our computational studies indicate that the high spin quintet (28.8 kcal mol−1) and the intermediate spin triplet (26.0 kcal mol−1) of the octahedral complex Mn1 are high in energy as compared to the singlet ground-state complex. Thus, we have considered all singlet state structures to compute the catalytic cycle. In the presence of an external base, the complex Mn1 undergoes dehydrobromination to give the catalytically active amido complex Mn1a (Fig. 1a), reported earlier as a penta-coordinate intermediate in analogous Mn complexes on treatment with tBuOK.36 We have recently characterized Mn1a.37 Similar pre-activation of the catalyst has been crucial to facilitate substrate binding, as observed by us and others.38,39 Indeed, adding alcohol 2a to Mn1a results in a computationally identified octahedral reactant complex, RCMn1 (ΔG = −9.9 kcal mol−1). Here, the alcoholic O–H bond is activated by the Mn–N framework, leading to the alkoxide coordination with the metal and protonation of the amido group. A similar resting state has been identified by Gauvin and others in acceptorless dehydrogenation of ethanol by the analogous Mn2 complex employing multinuclear NMR spectroscopy.40,41 In congruence with previous reports,40–44 we hypothesize that the catalytic cycle consists of three crucial steps: proton transfer/hydride activation, hydride transfer, and 2a alcohol mediated H2 release. Next to RCMn1, an adduct RCMn2 is hypothesized that shows strong N⋯H–O interaction and initiates metal–hydride interaction. This triggers a reversal proton transfer between the hydroxyl and the amido groups, complemented with hydride activation at 1.17 Å in TS1.40 In fact, this has been validated by intrinsic reaction coordinate scans (see Fig. S6†). This is a facile process with an intrinsic energetic cost of 7.9 kcal mol−1, generating INT1 in an endergonic manner. Next is the crucial β-hydride elimination through TS2 at a moderate barrier (ΔG‡ = 20.4 kcal mol−1). However, the tBuOK-mediated dehydrogenation of 2a required a 29 kcal per mol barrier. A similar dehydrogenation of alcohol is a rather difficult process by a Ni–phenanthroline complex, as observed by one of us,45,46 presumably due to the absence of strong O−⋯H–N hydrogen bonding in TS2, emphasizing on the crucial role of the N–H bifunctionality in the MACHO ligand of Mn1. In fact, Fu et al. have reported lowered reactivity of N-methyl substituted analogous Mn complexes in upgradation of ethanol.41TS2 is succeeded by releasing the ketone 16a and forming an Mn(I)-H intermediate MnH2. The process is slightly endergonic (∼4.0 kcal mol−1). We have previously identified similar Mn(I)-alkoxy (RCMn1) and Mn(I)-hydride (MnH2) complexes via NMR experiments.47 The ketone 16a further participates in a separate aldol condensation reaction (vide infra).
Experimentally, H2 gas evolution was confirmed through gas chromatographic analysis of the reaction headspace (see Sections S7.1 and S7.8†). The development of partial hydride and protic characters on hydrogen atoms over Mn and N in MnH2 is hypothesized to trigger the release of H2 (Fig. 1a). This can occur either through an inner-sphere self-dehydrogenation process46 when the N–H proton couples to the Mn–H hydride (Fig. 3 and S5†) or through an outer-sphere concerted proton-relay mechanism involving an alcohol 2a (Fig. 1 and 3).41,48 Out of the two possibilities, the former overcomes a higher intrinsic barrier (ΔΔG‡ = 24.3 kcal mol−1, viaTS3self, Fig. 3b) than the latter (ΔΔG‡ = 11.1 kcal mol−1, viaTS3, Fig. 3b) concerning the reference state, MnH2, suggesting that H2 evolution would be kinetically feasible on treatment with 2a. Interestingly, the stronger H–H interaction at 0.79 Å in TS3 as compared to 0.91 Å in TS3self might be responsible for the observed role of alcohol in facilitating the dehydrogenation process (see Section S8.4†).48
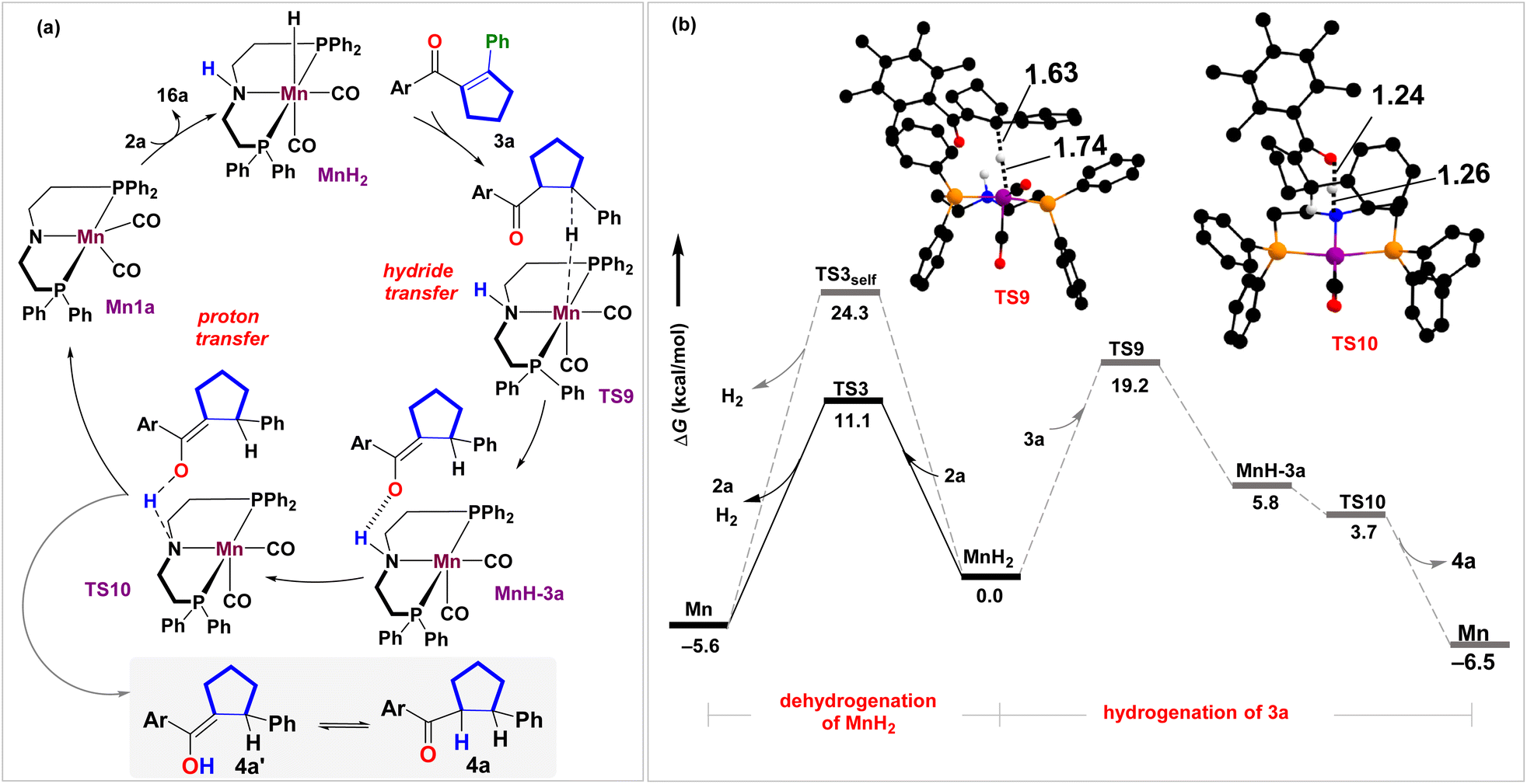 | ||
| Fig. 3 (a) Reaction mechanism and (b) corresponding free energy profile at B3LYP-D3(BJ)/SMD(toluene)/def2-TZVPP (in kcal mol−1) for the hydrogenation of cyclopentene 3a. Color coding for optimized geometries (truncated) TS9, TS10: C(black), H(white), O(red), P(orange), N(blue), Mn(purple). Distances shown are in units of Å. Unimportant hydrogen atoms are not shown for clarity. | ||
The aldol condensation of 1a and 16a to the enone 6a proceeds via the intermediacy of 17a (Fig. 2, Section S8†). The base tBuO− initiates the reaction through TS4 at 15.0 kcal mol−1 kinetic barrier; whereas alcohol 2a helps in the dehydration viaTS5 at ΔG‡ = 11.8 kcal mol−1. However, dehydration through the intermediacy of tBuOH is sluggish at ΔG‡ = 14.7 kcal mol−1. At this stage, we have explored different possibilities for rearranging 6a to 3a, either through radical mediation or nucleophilic attack (Fig. 2, see Section S8† for details). The tBuO− mediated SET initiation pathway required a 28.7 kcal mol−1 energetic requirement (TSSET_I_B,Fig. 2 and S4†). We identify that tBuO− could execute the deprotonation of an explicit solvent molecule (ΔG‡ = 6.7 kcal mol−1) that results in the generation of the tolyl anion that could facilitate the initiation process on 6a at a SET barrier of 6.4 kcal mol−1 (TSSET_I, Fig. 2). Thus, the SET transforms enone 6a to the enolate radical CR in an endoergic fashion. Once CR is formed, it undergoes a stepwise ring opening and ring closer rearrangement process through TS6R and TS7R, to deliver the enone radical anion ER, with the intermediacy of the transient radical species DR. Notably, the radical initiation with the tolyl anion affords the effective barriers of TS6R and TS7R as 28.8 and 36.3 kcal mol−1, respectively. This is totally in agreement with the high temperature (140 °C) requirement.
However, once ER is produced in situ, it significantly curtails down the kinetic barrier for SET to 0.9 kcal mol−1, as well as stabilizing the radical anion intermediate CR as referenced with 6a (Fig. 2). Hence, the radical chain propagation by an electron transfer from ER transforms 6a to CR and furnishes the required driving force to develop a β,γ-unsaturated 5-membered cyclopentene F during the radical chain termination. This is the reason the radical propagation steps, as shown by the black lines in Fig. 2, display a lower transition state barrier TS7R (23.7 kcal mol−1) and overall exothermicity. Thereby, isomerism of F leads to the targeted α,β-unsaturated cyclopentene product 3a. Our study also unfurls the unique role of catalytic electrons in novel cascade reactions and augments the chemical space with the already rich literature on efficient electron-catalyzed reactions.49
Once we reach product molecule 3a, one may fathom MnH2-mediated hydrogenation of the cyclopentene ring to the saturated product 4a (Fig. 3 and S5†), as observed previously with the [Ir(cod)Cl]2/cataCXiumA catalyst.28 Here, we propose a stepwise hydride/proton transfer to β-C and carbonyl O-centers, respectively, to generate the corresponding enol intermediate 4a′, which gets interconverted to 4a. To initiate the hydrogenation process, 3a binds to MnH2 through weak CO⋯H–N intermolecular H-bonding in TS9 (Fig. 3 and S5†), which catalyzes the initial Mn → C(β) hydride transfer at an energetic barrier of 19.2 kcal mol−1 followed by a relatively stable intermediate, MnH-3a. The consequent proton transfer step occurs (TS10) at a low energetic requirement to generate the target cyclopentane in an exoergic fashion. Essentially, under the experimental conditions, the transfer hydrogenation cascade mechanism is possible. However, a closer inspection reveals that the alcohol-mediated dehydrogenation of MnH2 is kinetically more facile (ΔΔG‡ = 11.1 kcal mol−1, viaTS3) than hydrogenation of 3a (ΔΔG‡ = 19.2 kcal mol−1, viaTS9). The competitive H2 release mechanism should be favorable entropically and thus rationalize the observation of an unsaturated cyclopentene product 3a (see Section S8.3†).
Conclusion
We reported the first example of the synthesis of acyl cyclopentenes via coupling cyclopropane methanol and aryl methyl ketones. A manganese complex derived from a commercially available ligand catalyzed the reaction, delivering the products in moderate to high yields with good selectivities. Water and hydrogen gas were produced as the byproducts. The detailed experimental and computational studies elucidated the reaction mechanism that involved multistep manganese-catalyzed acceptorless alcohol dehydrogenation, aldol condensation, and SET-mediated vinyl cyclopropane ring expansion. Facile alcohol-assisted hydrogen liberation over the hydrogenation of enones explains the selectivity for cyclopentene products. Further, we propose an uncharted mechanism of cooperativity between metal/single electron catalysis that we believe could inspire the design of new cascade reactions.Data availability
The ESI† includes all experimental details, including optimization of the synthetic method, synthesis, and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products, crystallography, and computation details are included as well.Author contributions
KS and BM conceived the project. KS performed experiments, analyzed products, and performed experimental mechanistic studies with input from BM. PB performed the DFT calculations with input from LR. KS and PB wrote the initial draft with input from BM and LR. BM and LR edited the manuscript. BM and LR acquired financial support for the development of this project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
BM thanks IISER Kolkata and CSIR 02(0405)/21/EMR-II for financial support. LR thanks SERB, India (SPG/2020/000754) for funding. KS and PB thank CSIR and ICT-IOCB, respectively, for PhD fellowships.References
- M. Ansell, Supplements to the 2nd Edition of Rodd's Chemistry of Carbon Compounds. A Modern Comprehensive Treatise, 2008 Search PubMed.
- J. Meinwald and T. H. Jones, J. Am. Chem. Soc., 1978, 100, 1883–1886 CrossRef CAS.
- K. M. Daane, G. Y. Yokota, V. M. Walton, B. N. Hogg, M. L. Cooper, W. J. Bentley and J. G. Millar, Insects, 2020, 11, 635 CrossRef PubMed.
- A. J. Ferreira and C. M. Beaudry, Tetrahedron, 2017, 73, 965–1084 CrossRef CAS.
- J.-B. Sortais, R. Buhaibeh and Y. Canac, in Manganese Catalysis in Organic Synthesis, ed. S. Jean-Baptiste, Wiley-VCH, Weinheim, 2021, pp. 39–66, DOI:10.1002/9783527826131.ch2.
- F. J. Sarabia, Q. Li and E. M. Ferreira, Angew. Chem., Int. Ed., 2018, 57, 11015–11019 CrossRef CAS PubMed.
- Y.-S. Feng, J. Hao, W.-W. Liu, Y.-J. Yao, Y. Cheng and H.-J. Xu, Chin. Chem. Lett., 2015, 26, 709–713 CrossRef CAS.
- H. Gao, Y. Xu, S. Liao, R. Liu, J. Liu, D. Li, D. Yu, Y. Zhao, Y. Fan and J. Memdr, Science, 1995, 106, 213–219 CAS.
- C. Liu, Y. Xu, S. Liao and D. Yu, J. Mol. Catal. A, 2000, 157, 253–259 CrossRef CAS.
- G. Domínguez and J. Pérez-Castells, Chem.–Eur. J., 2016, 22, 6720–6739 CrossRef PubMed.
- O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197–1212 CrossRef CAS PubMed.
- Z. Goldschmidt and B. Crammer, Chem. Soc. Rev., 1988, 17, 229–267 RSC.
- C. G. Overberger and A. E. Borchert, J. Am. Chem. Soc., 1960, 82, 1007–1008 CrossRef CAS.
- J. J. Li, in Name Reactions: A Collection of Detailed Reaction Mechanisms, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 606–607 Search PubMed.
- D. Armesto, A. Ramos, E. P. Mayoral, M. J. Ortiz and A. R. Agarrabeitia, Org. Lett., 2000, 2, 183–186 CrossRef CAS PubMed.
- R. I. Khusnutdinov and U. M. Dzhemilev, J. Organomet. Chem., 1994, 471, 1–18 CrossRef CAS.
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed.
- H. Albright, A. J. Davis, J. L. Gomez-Lopez, H. L. Vonesh, P. K. Quach, T. H. Lambert and C. S. Schindler, Chem. Rev., 2021, 121, 9359–9406 CrossRef CAS PubMed.
- J. M. Ketcham, I. Shin, T. P. Montgomery and M. J. Krische, Angew. Chem., Int. Ed., 2014, 53, 9142–9150 CrossRef CAS PubMed.
- K. Das, M. K. Barman and B. Maji, Chem. Commun., 2021, 57, 8534–8549 RSC.
- R. J. Armstrong, W. M. Akhtar, J. R. Frost, K. E. Christensen, N. G. Stevenson and T. J. Donohoe, Tetrahedron, 2019, 75, 130680 CrossRef.
- R. J. Armstrong and T. J. Donohoe, Tetrahedron Lett., 2021, 74, 153151 CrossRef CAS.
- L. B. Smith, R. J. Armstrong, D. Matheau-Raven and T. J. Donohoe, J. Am. Chem. Soc., 2020, 142, 2514–2523 CrossRef CAS PubMed.
- A. Kaithal, L.-L. Gracia, C. Camp, E. A. Quadrelli and W. Leitner, J. Am. Chem. Soc., 2019, 141, 17487–17492 CrossRef CAS PubMed.
- A. Jana, K. Das, A. Kundu, P. R. Thorve, D. Adhikari and B. Maji, ACS Catal., 2020, 10, 2615–2626 CrossRef CAS.
- A. K. Bains, A. Kundu, D. Maiti and D. Adhikari, Chem. Sci., 2021, 12, 14217–14223 RSC.
- S. Wübbolt, C. B. Cheong, J. R. Frost, K. E. Christensen and T. J. Donohoe, Angew. Chem., Int. Ed., 2020, 59, 11339–11344 CrossRef PubMed.
- Y. Cai, F. Li, Y.-Q. Li, W. Zhang, F.-H. Liu and S. L. J. T. L. Shi, Tetrahedron Lett., 2018, 59, 1073–1079 CrossRef CAS.
- K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CAS.
- S. Waiba and B. Maji, ChemCatChem, 2020, 12, 1891–1902 CrossRef CAS.
- M. K. Barman, S. Waiba and B. Maji, Angew. Chem., Int. Ed., 2018, 130, 9264 CrossRef.
- CCDC 2237272 (3a), 2237255 (14), 2237258 (6a), 2237269† (5a), and 2376543 (3u) contain the supplementary crystallographic data..
- Y. Wang, L. Zhu, Z. Shao, G. Li, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2019, 141, 17337–17349 CrossRef CAS PubMed.
- S. Waiba, K. Maji, M. Maiti and B. Maji, Angew. Chem., Int. Ed., 2023, 62, e202218329 CrossRef CAS PubMed.
- J. A. Luque-Urrutia, T. Pèlachs, M. Solà and A. Poater, ACS Catal., 2021, 11, 6155–6161 CrossRef CAS.
- K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- S. Fu, Z. Shao, Y. Wang and Q. Liu, J. Am. Chem. Soc., 2017, 139, 11941–11948 CrossRef CAS PubMed.
- J. A. Luque-Urrutia, T. Pèlachs, M. Solà and A. Poater, ACS Catal., 2021, 11, 6155–6161 CrossRef CAS.
- L. M. Azofra and L. Cavallo, Theor. Chem. Acc., 2019, 138, 64 Search PubMed.
- W. M. Dagnaw, Y. Lu, R. Zhao and Z.-X. Wang, Organometallics, 2019, 38, 3590–3601 CrossRef CAS.
- V. Arun, L. Roy and S. De Sarkar, Chem.–Eur. J., 2020, 26, 16649–16654 CrossRef CAS PubMed.
- L. Roy and A. Paul, Chem. Commun., 2015, 51, 10532–10535 RSC.
- S. Waiba, M. Maiti and B. Maji, ACS Catal., 2022, 12, 3995–4001 CrossRef CAS.
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2237272, 2237255, 2237258, 2237269 and 2376543. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02842b |
| This journal is © The Royal Society of Chemistry 2024 |