
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOut-of-equilibrium dynamics of a grid-like Fe(II) spin crossover dimer triggered by a two-photon excitation†
Jose
de Jesus Velazquez-Garcia
 *a,
Krishnayan
Basuroy
a,
Joanne
Wong
b,
Serhiy
Demeshko
b,
Franc
Meyer
b,
Insik
Kim
c,
Robert
Henning
c,
Yannic U.
Staechelin
d,
Holger
Lange
ef and
Simone
Techert
*ag
*a,
Krishnayan
Basuroy
a,
Joanne
Wong
b,
Serhiy
Demeshko
b,
Franc
Meyer
b,
Insik
Kim
c,
Robert
Henning
c,
Yannic U.
Staechelin
d,
Holger
Lange
ef and
Simone
Techert
*ag
aDeutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany. E-mail: jose.velazquez@desy.de; simone.techert@desy.de
bInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, Göttingen, 37077, Germany
cCenter for Advanced Radiation Sources, The University of Chicago, Argonne National Laboratory, 9700 South Cass Ave, Lemont, Illinois 90439, USA
dInstitute of Physical Chemistry, Universität Hamburg, Martin-Luther-King-Platz 6, Hamburg, 20146, Germany
eThe Hamburg Center for Ultrafast Imaging, Universität Hamburg, 22761 Hamburg, Germany
fInstitute of Physics and Astronomy, Universität Potsdam, Karl-Liebknecht-Str. 24, 14476 Potsdam, Germany
gInstitut für Röntgenphysik, Georg-August-Universität Göttingen, Friedrich-Hund-Platz 1, Göttingen, 37077, Germany
First published on 25th July 2024
Abstract
The application of two-photon excitation (TPE) in the study of light-responsive materials holds immense potential due to its deeper penetration and reduced photodamage. Despite these benefits, TPE has been underutilised in the investigation of the photoinduced spin crossover (SCO) phenomenon. Here, we employ TPE to delve into the out-of-equilibrium dynamics of a SCO FeII dimer of the form [FeII(HL)2]2(BF4)4·2MeCN (HL = 3,5-bis{6-(2,2′-bipyridyl)}pyrazole). Optical transient absorption (OTA) spectroscopy in solution proves that the same dynamics take place under both one-photon excitation (OPE) and TPE. The results show the emergence of the photoinduced high spin state in less than 2 ps and with a lifetime of 147 ns. Time-resolved photocrystallography (TRXRD) reveals a single molecular reorganisation within the first 500 ps following TPE. Additionally, variable temperature single crystal X-ray diffraction (VTSCXRD) and magnetic susceptibility measurements confirm that the thermal transition is silenced by the solvent. While the results of the OTA and TRXRD utilising TPE are intriguing, the high pump fluencies required to excite enough metal centres to the high spin state may impair its practical application. Nonetheless, this study sheds light on the potential of TPE for the investigation of the out-of-equilibrium dynamics of SCO complexes.
Introduction
Spin crossover (SCO) complexes exemplify molecular bistability, transitioning between low spin (LS) and high spin (HS) states under various stimuli. This phenomenon involves a transition between electronic states with different numbers of unpaired electrons for octahedral complexes with a d4–d7 electron count, most typically for octahedral d6 systems going from LS singlet (S = 0, t62ge0g configuration) to HS quintet states (S = 2, t42ge2g configuration). This electronic change leads to alterations in molecular structure, volume, magnetic susceptibility and colouration, which drastically modify the physical properties of the systems depending on the spin state.1–5 Due to the significant structural differences between their HS and LS states, six-coordinated Fe(II) complexes of nitrogen-donor ligands are commonly employed in SCO research.6 Among the different stimuli used, temperature and light are probably the most commonly used.3,7,8Investigation of the dynamics of light-induced transition as well as the lifetimes and relaxation pathways are vital to provide insight into the fundamental process underlying SCO phenomena as well as to design new materials with potential application in optoelectronic devices,9 memory devices10 and electrical switches.11 Therefore, extensive research has been conducted on the photoswitching dynamics of SCO systems in solution and solid state. For solutions, the dynamics have been widely explored using ultrafast optical, X-ray or Raman spectroscopies. Consequently, comprehensive description of the subtle coupling between changes in electronic state and the molecular reorganisation has been provided by the groups of McCusker, McGarbey, Hendrickson and others.12–21 In solid state SCO systems, the out-of-equilibrium dynamics have been explored by using ultrafast optical spectroscopy and time-resolved photocrystallography. While the photoswitching dynamics of single crystals of mononuclear materials have been explored by Collet et al.,22–30 the structural reorganisation upon excitation in oligomeric SCO complexes has recently been reported by our groups.31–33
The out-of-equilibrium dynamics in SCO solids is a multistep process in which each step, namely, photoinduced, elastic and thermal, takes place at particular time scales, occurring in the ps, ns and μs regimes, respectively.22–30 In the first step, a laser pulse locally photoswitches a fraction of molecules from LS to HS. Here, the photoswitched fraction is proportional to the laser excitation density: one absorbed photon switches one metal centre. This photoswitching is accompanied by a single structural change at molecular scale in mononuclear complexes, while in oligonuclear complexes with strong elastic linking between metal centres, the spin transition is concomitant with a constant molecular reorganisation during 100s of ps.31–33 In the second step, the internal pressure due to HS swelling and lattice heating induces lattice expansion on the ns time scale. Here, an additional switching takes place towards the HS state, increasing the HS fraction during this step. Finally, in the third step, the heating of the lattice by the laser pulse leads to a thermal population of the HS state which occurs in the microsecond time scale.
It is important to note that previous studies have predominantly focused on the dynamics following one-photon excitation (OPE). The potential of two-photon excitation (TPE) for the study of the out-of-equilibrium dynamics remains largely unexplored. Curiously, the notion of using TPE for photoswitching has been introduced in literature,34,35 but it has barely been used.36,37 This gap leaves several questions without an answer such as:
Can TPE be used to explore the structural dynamics of SCO solids from fs to ms time scales?
Does TPE impact the lifetime of the photoinduced HS state in solid state?
What role does TPE play in each step of the out-of-equilibrium dynamics in solids?
What impact does TPE have in the photoconversion?
While this work will not provide all of the answers, our contribution aims to shed light on the out-of-equilibrium dynamics in SCO complexes using TPE. Our present investigation centres around the corner complex dimer [FeII(HLH)2]2(BF4)4·2MeCN (where HLH = 3,5-bis{6-(2,2′-bipyridyl)}pyrazole), here called FE2, previously reported by some of us.38 The complex has a [2 × 2] grid-like arrangement of four ligand strands with FeII ions at two opposite corners and two Npz–H⋯Npy (pz = pyrazole; py = pyridine) hydrogen bonds at each of the remaining two corners, as depicted in Fig. 1. The complex is part of a family of metallogrids based on the pyrazole-based ligand strands, the dynamics of which have been reported previously.31–33,39,40 Throughout this work, some properties of the FE2 grid will be compared with those of other members of this family, namely: [Fe4IIL4H](BF4)4·2MeCN (FE4H),41 [Fe4IIL4Me](BF4)4·2MeCN (FE4Me)42 and [Fe3IIL2H(HLH)2](BF4)4·4MeCN (FE3),43 where LR = 4-R-3,5-bis{6-(2,2′-bipyridyl)}pyrazole; R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, CH3. FE3 can be considered as a defect [2 × 2] grid structure with one metal ion missing, and FE2 as a double defect grid with two missing metal ions.
H, CH3. FE3 can be considered as a defect [2 × 2] grid structure with one metal ion missing, and FE2 as a double defect grid with two missing metal ions.
 | ||
| Fig. 1 (a) Pyrazole-bridged compartmental ligand HL. (b) Molecular representation of the [FeII(HLH)2]24+ grid. Fe(A)and Fe(B) ions are in the LS state. Counter ions, solvent molecules and hydrogen atoms are omitted for clarity. (c) Schematic representation of the FE2 grid. | ||
Our findings for FE2 in solution reveal that the transition from the LS to the HS state takes place at similar timescale whether induced by OPE (at 400 nm) or TPE (at 800 nm). We also report the molecular reorganisation upon two-photon excitation at 780 nm in solid state during the photoinduced step. Additionally, we demonstrate that the co-crystallising solvent in FE2 suppresses the thermal transition, emphasising that light serves as an alternative pathway to access the HS in spin crossover complexes.
Methods
Synthesis
The FE2 metallogrid and its ligands were synthesised following the procedure reported in the literature.38X-ray diffraction
Single-crystal X-ray diffraction experiments were performed from 100 K to 390 K. The latter is the maximum temperature possible before the decay of crystallinity became significant. The data were collected in increments of 10 K from 100 K to 290 K, and in increments of 20 K from 290 K to 390 K. The heating rate was maintained at 360 K h−1. The X-ray data were collected in phi scan mode on undulator synchrotron radiation with λ = 0.6199 Å at P11 beamline in PETRA III, DESY, Hamburg, Germany. Indexing of the X-ray diffraction pattern, unit cell refinement and spot integration were performed using the XDS program package.44 The crystal structures were solved with the SHELXT45 structure solution program within the Olex2 software package.46 The structures were refined against F2 isotropically, followed by full matrix anisotropic least squares refinement by SHELXL.47 For all structures, hydrogen atoms were fixed geometrically in idealised positions and allowed to ride with the respective C or N atoms, to which each was bonded, in the final cycle of refinement. Crystal data and structure refinement parameters for the 100 K dataset are provided in Table S1,† while the selected bond lengths and bond angles at the same thermal ellipsoid plot and packing of molecules down all crystallographic axes at 100 K are given in Fig. S1–S5.†Magnetic studies
Temperature-dependent magnetic susceptibility measurements were carried out with a Quantum-Design MPMS-XL-5 SQUID magnetometer equipped with a 5 Tesla magnet in the range from 10 K to 400 K and 400 K to 200 K at a magnetic field of 0.5T. The powdered sample was contained in a Teflon bucket and fixed in a non-magnetic sample holder. Each raw data file for the measured magnetic moment was corrected for the diamagnetic contribution of the sample holder and the Teflon bucket. The molar susceptibility data were corrected for the diamagnetic contribution. Temperature-independent paramagnetism (TIP) and a Curie-behaved paramagnetic impurity (PI) with spin S = 5/2 were included according to χcalc = (1 − PI)·χ + PI·χmono + TIP. Simulation of the experimental magnetic data was performed with the julX program.48Steady state spectroscopy
Room temperature UV-vis spectrum of FE2 was acquired on a Varian Cary-5E UV-vis Spectrometer, employing a scan rate of 0.2 nm s−1. The wavelength range covered 200–800 nm with a step size of 0.5 nm. The absorption spectrum was collected on 10 mm cuvettes, using a reference spectrum of acetonitrile, which was also used to dissolve the FE2 compound.Optical transient absorption spectroscopy
The femtosecond OTA measurements were performed using an amplified Ti-sapphire laser system (Spitfire-Ace, 800 nm, 1 kHz, 35 fs; Spectra Physics) in combination with a commercial TA setup (HELIOS, Ultrafast Systems). The amplifier output was split into two parts for the optical pump and probe beam. For the pump beam, the 800 nm laser fundamental and its second harmonic, generated in a BBO crystal, were used. The probe pulse passed a delay stage and was used to generate white light in a CaF2 crystal. The nanosecond to microsecond TA measurements were conducted employing a commercial setup (EOS, Ultrafast Systems) that uses an electronically triggered white light laser as a probe source. The FE2 was dissolved in acetonitrile to reach the concentration of 1 mM, and the solution was placed in a 2 mm-thick cuvette for the TA measurement.Time-resolved pink Laue crystallography
Time-resolved intensity data were collected at the BioCARS station at the Advanced Photon Source, Argonne National Laboratory, IL, USA. TRXRD data were collected at an undulator setting of 15 keV with a Rayonix MX340-HS detector. Pulses from a Ti:sapphire laser tuned to 780 nm wavelength and 2.0 (mJ mm−2)/pulse were used as a pump source and set perpendicular to the φ-rotation axis. Attempts to obtain high-quality datasets using a 390 nm wavelength excitation were unsuccessful due to the fast deterioration of the crystals. The time delay between the laser pump and the X-ray probe was set by varying the arrival times of the picosecond laser pulses to the synchrotron X-ray pulses. Delay times (dt) between the laser pump and the probe were set to 200 ps and 500 ps. An additional reference measurement was taken without any laser excitation, the so-called ‘static’ dataset.The recorded datasets for all time delays covered a 180° scan with a 1° step. Laser-OFF and laser-ON frames were collected in immediate succession to minimise the effect of long-range fluctuations in the beam's position or intensity. The OFF/ON cycle pump–probe cycle was repeated five times for each frame to allow for subsequent statistical background estimation and filtering of the intensities. A new crystal was used for each time delay to prevent the effects of degradation. Crystals used in this study had approximate dimensions of 200 × 120 × 100 μm3. The temperature was set to 100 K for all datasets.
In a previous work,31 photodifference maps for the photoinduced step were rather noisy. To avoid this, the LaueUtil tool kit49,50 was used to obtain the photodifference maps for both delay times: 200 ps and 500 ps. However, the data reduction to obtain the structural models was done with the software package Precognition/Epinorm,51 using variable elliptical integration for all time delays. Since the individual datasets at 200 ps and 500 ps were not enough to create an accurate model, after integration both datasets were combined during the wavelength normalisation and data reduction. During this step the laser-OFF and laser-ON datasets were scaled with the ‘static’ dataset, but the repetitive and symmetry-related reflections were merged separately. Then, the reduced datasets were used to obtain the structural models at laser-ON and laser-OFF conditions. For all datasets, initial models of the crystal structures were taken from those obtained from the monochromatic X-ray diffraction experiment at 100 K. Subsequently, the structures were refined using the SHELXL within the Olex2 software package. Thermal ellipsoid plots (50% of probability) for all time-resolved datasets are provided in ESI (Fig. S9 and S10†).
Results
Multitemperature analysis
A detailed structural description of FE2 at 100 K, 290 K and 390 K is provided in Table S3.† The structural analysis obtained at the above-mentioned temperatures shows that the compound maintains its space group (P21/n) throughout the investigated temperature range. Within the asymmetric unit, two crystallographic-symmetry-independent metal ions are observed. Each ion is bonded to two HL ligand stands forming a dimer with a grid-like shape, where two opposite corners are devoid of the metal ion but hold together by two Npz–H⋯Npy hydrogen bonds (Fig. 1). Upon closer examination of the {Fe–N} coordination environment, it is observed that FE2 adopts a 2LS configuration at 100 K, as evidenced by the 〈Fe–N〉 bond lengths, which show typical values for LS ions (∼1.94 Å). Additionally, the distortion of each {Fe–N} coordination sphere, measured by the trigonal distortion parameter (Θ, see ESI Fig. S6†), shows values of Θ between 276 and 292°. These values resemble those observed in the LS ions of the previously reported FE4Me and FE3 grids.31,33It is well known that the SCO phenomenon takes place on the metal ions, and it can propagate all along the solid material causing structural changes across different scales: from the metal site up to the macroscopic scale. At the metal site, the LS to HS transition has as consequences a reversible expansion of the 〈Fe–N〉 bond length (∼0.2 Å) and a deformation of the {Fe–N} polyhedron. VT-SCXRD studies allow for viewing the temperature dependence of the 〈Fe–N〉 bond lengths and the distortion of the coordination sphere for both crystallographic-symmetry-independent metal ions. Previous VT-SCXRD studies in FE4Me and FE3 showed that the temperature dependence of 〈Fe–N〉 and Θ of the transiting ions resembles the magnetic behaviour, while metal ions in materials with silenced thermal SCO show an almost linear increase of those values on heating the crystal. As shown in Fig. 2, the temperature dependence of 〈Fe–N〉 and Θ for both ions in FE2 reflects the magnetic behaviour, showing no significant changes. Below 350 K the average metal–ligand bond length expands by less than 0.01 Å, while the angular distortion increases by less than 10°. It is worth noting that above such temperature both ions exhibit a noticeable increase in both the 〈Fe–N〉 and Θ, which suggests the beginning of the LS → HS transition at both metal sites. However, similar to the previously reported FE3 grid, this change is attributed to the loss of the co-crystallising solvent, and it is not a feature of the solvated sample.
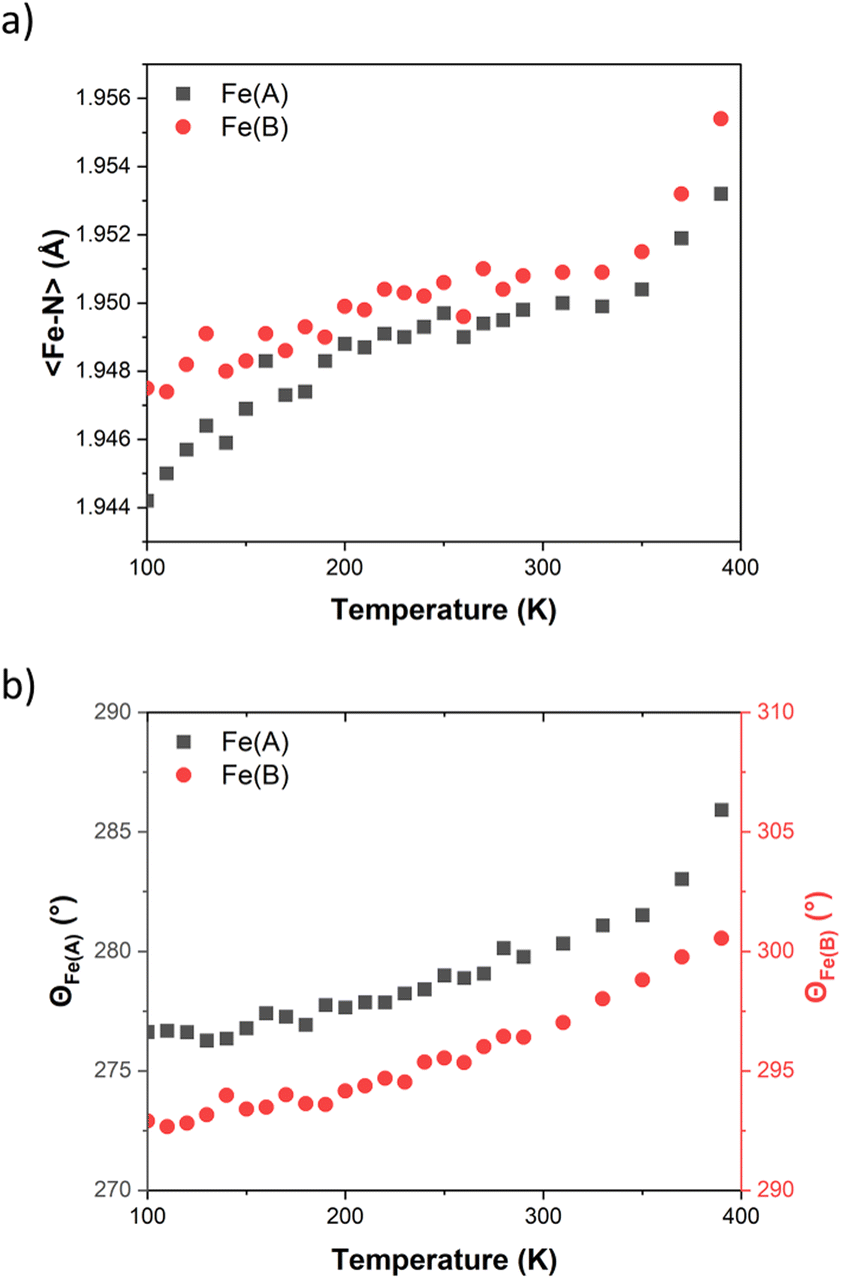 | ||
| Fig. 2 Temperature dependence of (a) 〈Fe–N〉 bond length and (b) angular distortions (Θ) in FE2. | ||
One of the key aspects to consider during the VT-SCXRD analysis is the expansion of the grid with the temperature. Fig. 3 shows a large change of the separation between the two metal centres of ∼0.1 Å with the increase of temperature. As previously observed in the defect FE3 grid, such a large change is primarily driven by the flexibility provided by the metal-devoid vertexes featuring hydrogen bonds, since systems with strongly linked metal centres tend to restrict the thermal expansion of the grid, as was demonstrated for FE4Me.
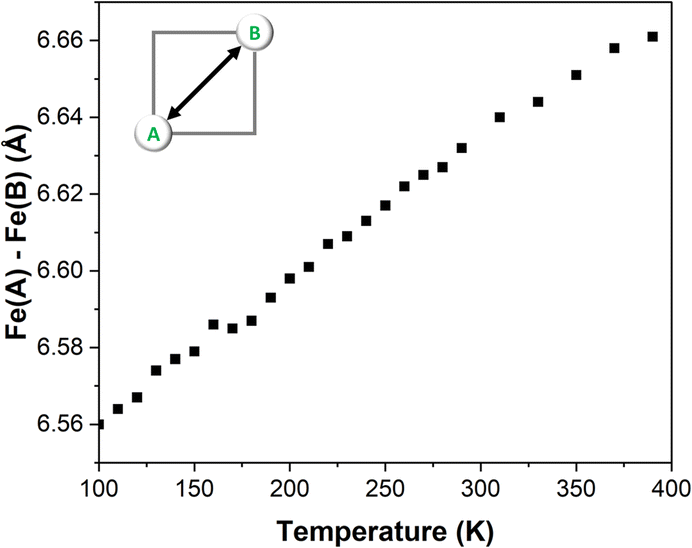 | ||
| Fig. 3 Temperature dependence of the Fe–Fe distances in FE2. | ||
Magnetic measurements
The magnetic measurement of FE2 (Fig. 4) indicates the diamagnetic [LS-LS] state between 10 and 300 K. At even higher temperature, there is a small slope of χMT, which corresponds to the formation of approximately 12.5% HS state of both Fe ions at 400 K, as suggested by the VT-SCXRD (see above). This effect is not completely reversible and is most likely associated with the loss of solvent molecules as observed in the VT-SCXRD analysis. After solvent loss, approximately 2.5% of the FE2 molecules are in HS-HS state at room temperature.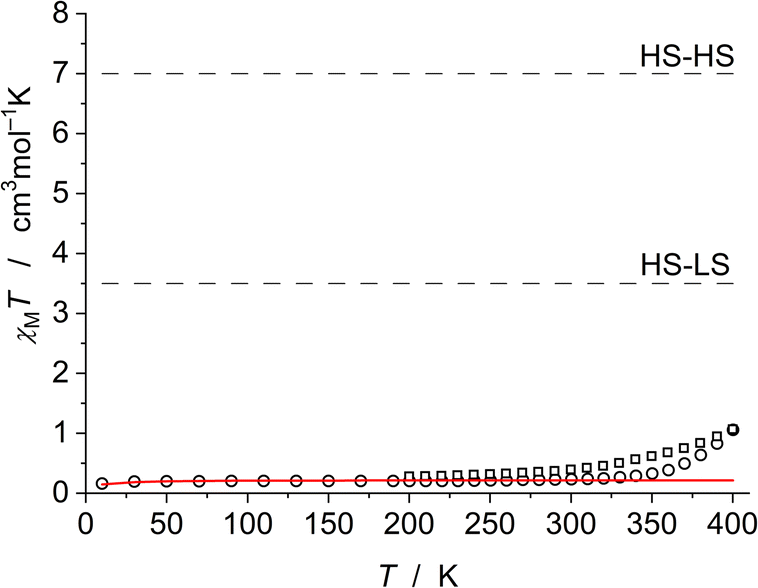 | ||
| Fig. 4 Magnetic data of FE2 between 10 and 400 K (open circles) and 400 and 200 K (open squares). The solid red line represents the simulation for the [LS-LS] case with the addition of the temperature-independent paramagnetism (TIP = 320 × 10−6 cm3 mol−1) and a Curie-behaved paramagnetic impurity (PI = 5%) with spin S = 5/2. Dashed lines show the expected χMT values for the [HS-HS] and [HS-LS] situations as a guide. | ||
The assumption that the crystals lose 2MeCN during the SQUID measurement is based on the fact that the sample is heated up to 400 K, the measurement takes place under reduced pressure (of approx. 3–5 mbar) and the magnetic moment is irreversibly slightly higher afterwards. In case of the VT-SCXRD, the crystals form different domains after 330 K and in rare cases where the temperature increase is significantly slower, it can lead to a fully desolvated crystal without significant losing crystallinity or formation of different domains. We will explore the desolvated sample in a future publication.
Dynamics in solution
The UV-visible spectrum of FE2 in MeCN at room temperature (Fig. 5) is similar to that previously reported for the FE4H, FE4Me and FE3 grids. The absorbance below 350 nm is attributed to π–π* transition in the ligand, while the band spanning the 500–650 nm range is ascribed to the metal-to-ligand charge-transfer (MLCT) transition of mixed singlet-triplet character. It is worth noting that no observable band is found in the 750–800 nm range, and hence no one-photon absorption should be expected in this range.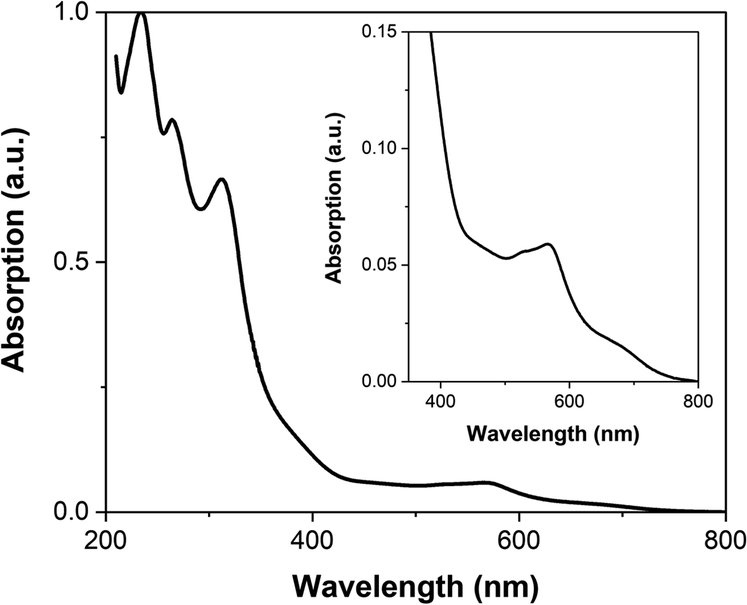 | ||
| Fig. 5 UV-vis spectrum of FE2 grid in acetonitrile. (inset) Enlargement of the 350–800 nm range. | ||
Fig. 6a shows the photoinduced dynamics following femtosecond excitation of FE2 at 400 nm in MeCN at room temperature for a fluence of 1.5 mJ cm−2. The spectra reveal the emergence of the ground state bleach (GSB) caused by the depletion of the ground state signal due to photoexcitation by the pump pulse. This GSB in FE2 is akin to what was observed in the FE4H and FE3 grids, indicating a LS to HS transition in less than 1 ps. The differential spectra at 10 ps delay time and fluences from 0.100 to 1.5 mJ cm−2 (Fig. 6b) exhibit an increase of the GSB with higher excitation fluences. As depicted in Fig. 6c, the differential absorption at 10 ps for the 565 nm probe demonstrates that the GSB signal exhibits a linear dependency with the rise in excitation fluence, which is typical for a one-photon absorption process.
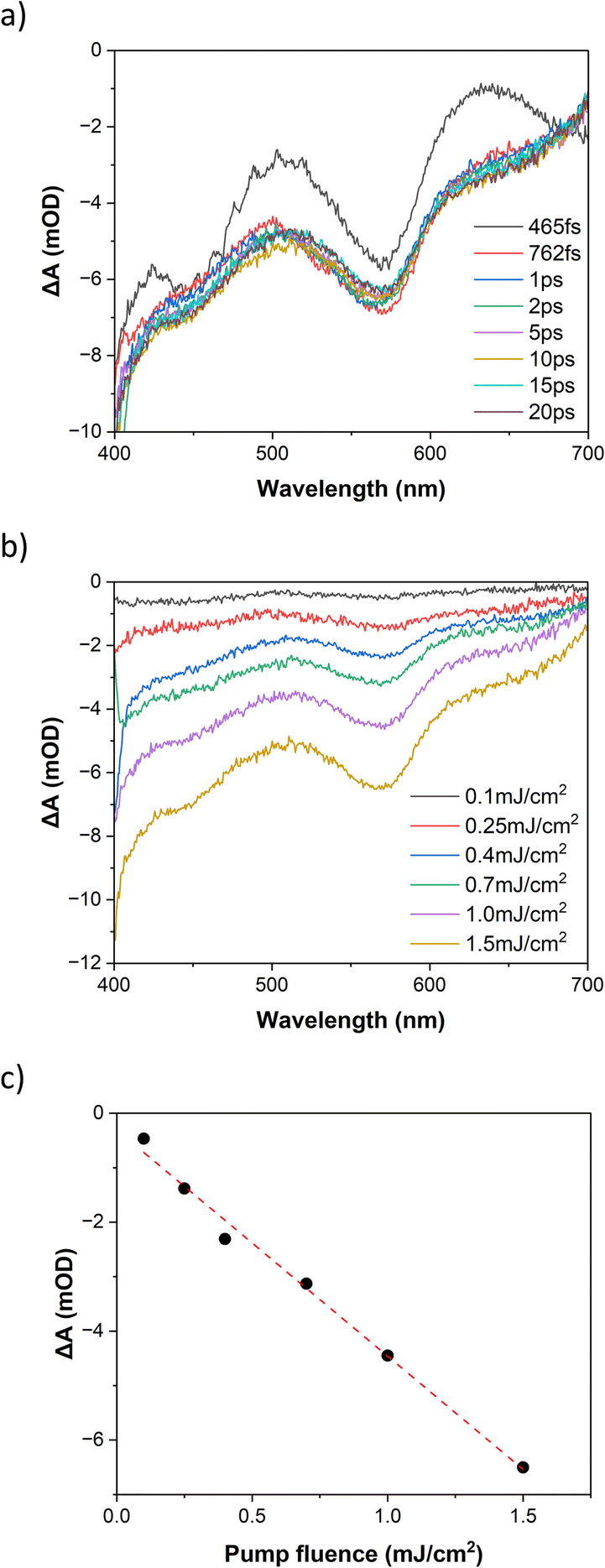 | ||
| Fig. 6 Transient optical spectra following 400 nm excitation of FE2 grid (a) at different delay times, pump fluence of 1.5 mJ cm−2 and (b) various pump fluences after 10 ps delay. (c) Differential absorption after 10 ps delay, probed at 565 nm, linear fit as guide to the eye. | ||
The photoinduced dynamics at 800 nm excitation of FE2 in MeCN at room temperature for fluence 17.3 mJ cm−2 is shown in Fig. 7a. Similar to the 400 nm excitation, a GSB is observed from 2 ps on after laser excitation. However, a coherent artifact impairs the visualisation before such delay time. Nevertheless, the dynamics at 800 nm excitation are expected to be similar to those at 400 nm excitation as has been seen in other metal complexes.36,52 The power dependency at 10 ps is given in Fig. 7b and c. The increase in excitation fluence results in a stronger GSB signal as observed in the 400 nm excitation. However, unlike 400 nm excitation, at 800 nm excitation, the GSB signal exhibits a quadratic dependency with the rise on the excitation fluence. Such quadratic power dependence is characteristic of the TPE and it has been widely reported in literature.53–57 A linear dependence of the GSB with pump fluence would be expected in the case of even a small amount of OPE. These results confirm that although there is no absorption band at 800 nm in the UV-vis spectrum, the LS to HS transition at 800 nm is achieved via a two-photon absorption process.
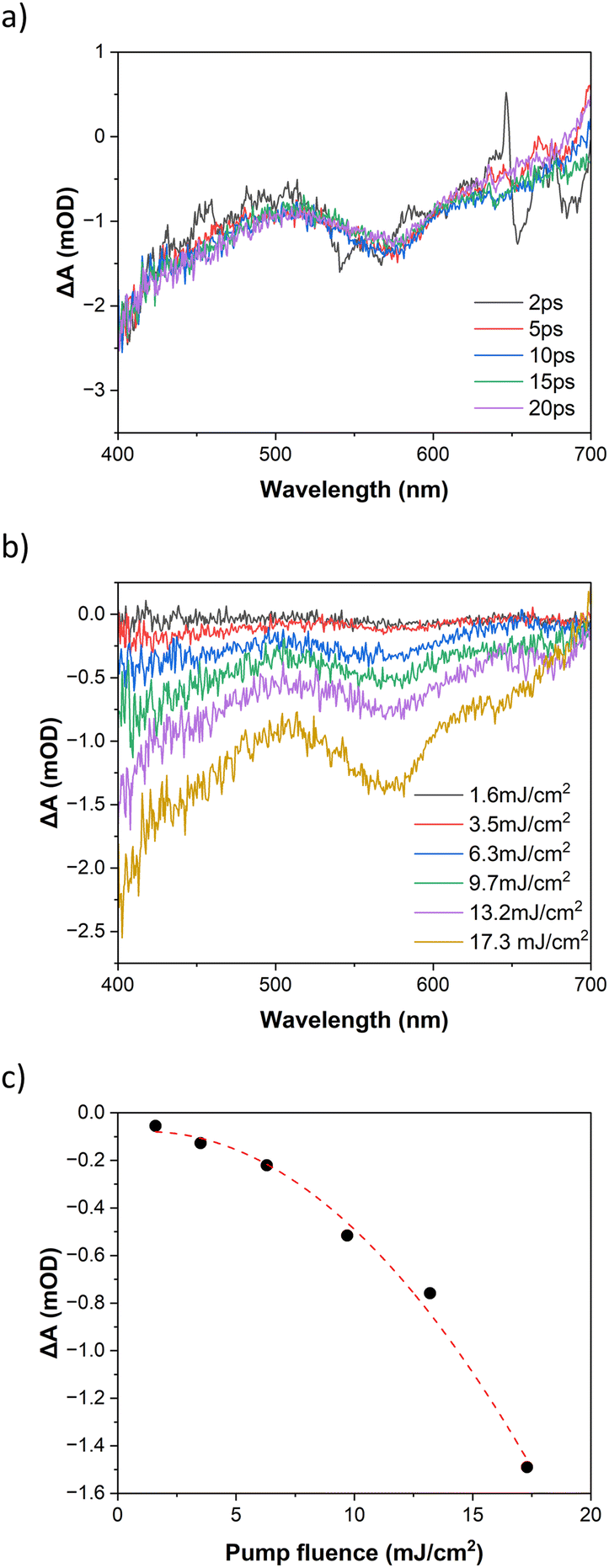 | ||
| Fig. 7 Transient optical spectra following 800 nm excitation of FE2 grid (a) at different delay times, pump fluence of 17.3 mJ cm−2 and (b) various pump fluences after 10 ps delay. (c) Differential absorption after 10 ps delay, probed at 565 nm, quadratic fit as guide to the eye. | ||
The bleach recovery dynamics for both 400 nm and 800 nm excitation wavelengths using a 565 nm probe are presented in Fig. S8.† The figure demonstrates that regardless of using one-photon or two-photon absorption, the recovery dynamics will be the same. The exponential fitting yields a single-exponential decay with a time constant of (147 ± 2)ns. This shows that the lifetime of the photoinduced HS state will be longer in the FE2 than in the FE3 grid (123 ± 4)ns (ref. 39) but shorter than in the FE4H grid (210 ± 5)ns.40
Molecular reorganisation in solid state
The light-induced structural changes in solid state can be illustrated by photodifference maps58–61 (Fig. 8). The maps have the form FONobs − FOFFobs and are generated from all independent reflections with I/σ(I) > 3 (see ESI† for more information). The isosurfaces are drawn at 0.095e Å−3 for 200 ps and 0.087e Å−3 for 500 ps datasets to highlight the displacement of the Fe ions. The molecular response on excitation is observed by the shift of the electron density of the Fe(A) and Fe(B) ions. Positive electron density peaks (green) on the maps pinpoint where Fe(II) ions have shifted upon excitation, while negative electron density peaks (red) are found at the original position of the metal ions before light exposure. Both photodifference maps evidence the displacement of both Fe ions in the approximately [1 1 1] direction, indicating that there is no difference in the direction of atomic shifts at 200 ps and 500 ps. This suggests that during the photoinduced step, there is a singular trend towards the structural change. This opposes what has been reported in FE3 and FE4Me, where the strongly connected metal centres show a continuous reorganisation during the first 1 ns after light irradiation. Nevertheless, this agrees with what has been reported in mononuclear SCO complexes.24 Since FE2 can be considered a mononuclear complex with two crystallographically independent metal centres whose ligands strands are arranged in a grid-like shape, but without strongly connected metal centres, it is expected that the material behaves like the previously reported mononuclear materials and not like the FE3 and FE4Me metallogrids that have strongly elastically linked metal ions. | ||
| Fig. 8 Photodifference maps of FE2 during the photoinduced step: (a) 200 ps and (b) 500 ps delay times. Isosurfaces (green positive, red negative) and delay times are shown in each figure. Arrows give an approximate direction of the electron density shift. Hydrogen atoms, counter ions and solvent molecules are omitted for clarity. | ||
Several attempts to obtain an accurate model of FE2 upon excitation were made without success. The best model incorporates the information from both the 200 ps and 500 ps datasets, as detailed in the methods section. Before combining the datasets, the consistence of the data was checked by plotting the experimental ION/IOFF ratios of the two datasets against each other in “correlation plots”,62 obtained with the LaueUtil tool kit.49,50 The correlation between the ratios is quite reasonable as shown in Fig. S11.† Structural changes were explored using various I/σ(I) and resolution cut-offs. The findings are reported in Table S4.† These results suggest a bond-length expansion of ∼0.002 (8)Å in both crystallographic-symmetry-independent metal ions during the first 500 ps. This expansion corresponds to roughly 1% of photoconversion in each metal centre, as calculated using the equation from our previous work:31–33
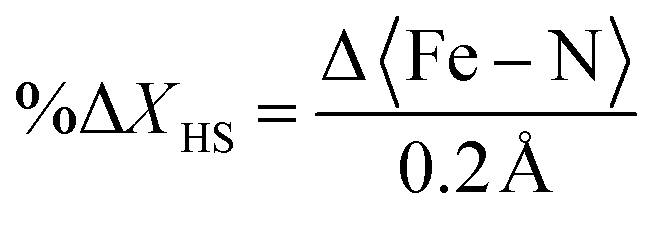
Although the bond-length expansion is comparable with those reported in literature,27 this value is below the standard deviation. Therefore, we take these results only as a suggestion of the possible bond-length expansion and photoconversion.
An estimate of the temperature difference between the laser-ON and the laser-OFF datasets was obtained using the Photo-Wilson plots.63 The slope of the plots corresponds to twice the difference of the Debye–Waller factors (ΔBON-OFF) between laser-ON and laser-OFF datasets. This factor is associated with the change in the isotropic atomic motion. Therefore, a significant increase in temperature, or a thermal motion, due to laser exposure, would increase the value of ΔB, while a non-detectable heating would lead to values close to zero.
The Photo-Wilson plot for the laser-ON dataset is presented in Fig. S12,† showing a value of ΔBON-OFF close to 0. This value indicates that no significant heating is detectable and consequently, no thermal effects are operative during the reported time range. This ensures that the reported structural response at both delay times is caused solely by the LS to HS photoswitching.
Discussion
TPE occurs when a molecule absorbs two long-wavelength photons simultaneously, promoting the molecule into an electronically excited state.64–67 Unlike OPE techniques, TPE enables deeper penetration and minimises photodamage.68,69 Despite these benefits, researchers primarily employ OPE for studying the photoinduced SCO phenomenon, while the application of TPE remains limited.Investigation of the photoinduced dynamics using TPE has been performed in [Ru(bpy)3]2+ and [Ru(bpy)3-dbc]2+ complexes (bpy = 2,2′-bipyridine, dcb = 2,2c-bipyridine-4,4c-dicarboxylic acid), revealing no discernible difference compared to results using OPE.52 However, these complexes do not show a photoinduced spin transition, and there is insufficient direct evidence to ascertain whether the LS to HS transition or the relaxation to the LS state occurs similarly under OPE or TPE. Our present results confirm that identical dynamics are observed in SCO complexes using either excitation method. In both cases, the fast formation of the HS state occurs in less than 2 ps, with a lifetime of the photoinduced HS state of 147 ns.
While these results seem attractive for applications in near-infrared optoelectronics, there is an important disadvantage. The pump fluences required to achieve excitation of a substantial proportion of molecules into the HS state, and a distinct GSB signal, are significantly higher for the TPE process (>60 times by comparing the GSB signal at 0.25 mJ cm−2 in OPE with that at 17.3 mJ cm−2 in TPE). This presents an important disadvantage as large pump fluencies can induce substantial photodamage upon surpassing a certain threshold.70 Employing the same fluences for OPE and TPE can result in a significantly lower response, as observed in TRXRD experiments. In our previous work, we observed that applying a pump fluency of 2.0 (mJ mm−2)/pulse with OPE on the solid FE4Me grid achieved a photoconversion of ∼12%,32 whereas in this work, employing the same experimental procedure with TPE on the FE2 grid, resulted in only about 1% conversion to the HS state. While this decrease in photoconversion may pose challenges for the application of TPE in optoelectronics devices utilising SCO complexes, it proves valuable for gaining insights into the out-of-equilibrium dynamics in solids.
Out-of-equilibrium studies in solid mononuclear and oligonuclear SCO FeII complexes have demonstrated a multistep response, where each step, namely photoinduced, elastic and thermal switching, takes place at particular time scales, occurring in the ps, ns and μs regimes, respectively.22–30 The major difference between both types of complexes has been observed at the photoinduced step.31–33 When a mononuclear SCO is impacted by a laser pulse, it leads to a LS → HS transition in less than one picosecond. Then, the new molecules in the HS state reorganise and preserve such geometry, at least during the whole photoinduced step.24 In contrast, oligonuclear SCO complexes with strongly linked metal centres show a continuous reorganisation during the photoinduced step. When the metal ions are tightly linked, the photoswitching of one metal ion in an oligonuclear molecule triggers a short-range elastic distortion that propagates throughout the molecule. The propagation of this distortion through the molecule causes a series of molecular rearrangements that last until the elastic step, where the long-range elastic distortion of the lattice supersedes the initial short-range distortion of the molecule. The latter behaviour is observed in the FE3 and FE4Me grids, whose elastic communication, provided by strong linking of metal centres, allows the propagation of the elastic distortion through the molecule.31–33 In the FE2 grid, this elastic communication is interrupted by the metal-devoid vertexes at the opposite corners. Hence, each of the crystallographic-symmetry-independent metal centres in FE2 behaves as a mononuclear entity, undergoing reorganisation only once during the photoinduced step as observed in the photodifference maps. This observation conclusively highlights that the continuous reorganisation during the photoinduced step is primarily driven by the strong-linking of metal ions in the oligonuclear grid platform, rather than the specific geometric arrangement of metal centres or the intermolecular interactions.
These findings prove the importance of the metal-to-metal communication for the photoinduced phenomena at out-of-equilibrium conditions. While intermolecular interactions play a significant role for the long-range cooperativity observed during the elastic step,23 the strong elastic linking of metal centres provides a metal-to-metal communication that may lead to intramolecular cooperativity during the photoinduced step. Therefore, the design of new materials with enhanced SCO properties must consider not only the strength of the intermolecular interactions and the geometrical arrangement of metal centres but also the efficiency of the elastic communication between metal ions.
It is important to highlight that the present study has been confined solely to the photoinduced transition in a SCO dimer, wherein the thermal spin transition has been supressed by the solvent included in the crystal lattice. To our knowledge, there is no clear understanding of the effects of the solvent on the out-of-equilibrium dynamics and the photoconversion, whether employing OPE or TPE. Additionally, there is a lack of information concerning the impact of the TPE in the elastic and thermal steps. The deeper penetration and larger pump fluences used in the TPE may lead to interesting effects in these particular steps. Therefore, further investigation into the out-of-equilibrium dynamics of SCO complexes is essential for a deeper understanding of this phenomenon and to identify key factors essential for designing new SCO materials with tailored properties.
Conclusions
The results of this work advance our understanding of the dynamics of light-responsive materials, particularly in the context of the photoinduced SCO phenomenon under TPE, and the importance of the metal-to-metal communication in the structural response upon excitation. We have demonstrated that both OPE and TPE induce comparable dynamics in the photoinduced SCO phenomenon. This involves the rapid formation of the High Spin (HS) state within 2 ps, with a subsequent lifetime of 147 ns. However, the practical application of TPE may encounter challenges due to the significant pump fluences required to stimulate a sufficient number of metal centres into the HS state. Nevertheless, TPE can be useful in the exploration of the structural changes upon excitation using TRXRD.The TRXRD data suggest a single reorganisation upon TPE, similar to that found in other mononuclear SCO complexes. Since there is no strong connection between the two metal centres in the FE2 grid, there is no continuous reorganisation during the photoinduced step of the out-of-equilibrium dynamics of the solid sample, contrary to that observed in the previously reported FE4Me and FE3 grids. This demonstrates that the elastic metal-to-metal communication in SCO complexes plays a key role in designing materials with enhanced SCO properties.
This knowledge is essential for developing more efficient optoelectronic devices, data storage technologies and sensors, which may operate in the near-infrared region. Additionally, our findings shed light on the advantages and challenges associated with employing TPE compared to the traditional OPE, providing valuable guidance for researchers in selecting experimental methods. By addressing the practical challenges of TPE and uncovering the importance of the metal-to-metal communication in the photoinduced SCO phenomenon, our study contributes to the ongoing efforts to optimise the design and application of new SCO materials for a wide range of technological applications.
Data availability
The authors declare that data supporting the findings of this study are available within the paper and its ESI.† Crystallographic data for FE2 at different temperatures have been deposited at the CCDC: 2332817–2332822, 2332894–2332898, 2332925–2332929, 2333003–2333006, 2333030 and 2333040–2333043. Photodifference maps were added to data with CCDC number 2332819.Author contributions
JJVG drafted the manuscript. JJVG and KB performed the time-resolved crystallographic experiment with assistance from RH and IK in beamline operation at APS. ST and FM conceived the study. FM and SD designed the molecule. SD made the magnetic susceptibility measurements. YS and HL performed the transient optical spectroscopy experiment. JW synthesised the material. All authors provided input and agreed on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Use of BioCARS was also supported by the National Institute of General Medical Sciences of the National Institutes of Health under grant number P41 GM118217. Time-resolved set-up at Sector 14 was funded in part through a collaboration with Philip Anfinrud (NIH/NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Portions of this research were carried out at the light source PETRA-III at DESY, a member of the Helmholtz Association (HGF). We would like to thank P11 staff for assistance in using beamline P11. The current work has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 217133147/SFB 1073, projects B06, C02. HG-recruitment, HG-Innovation “ECRAPS”, HG-Innovation DSF/DASHH and CMWS.References
-
P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I, Springer, 2004, vol. 233, pp. 1–47 Search PubMed
.
- J. R. Thompson, R. J. Archer, C. S. Hawes, A. Ferguson, A. Wattiaux, C. Mathonière, R. Clérac and P. E. Kruger, Dalton Trans., 2012, 41, 12720 RSC
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed., 1994, 33, 2024–2054 CrossRef
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC
- E. A. Bousseksou, C. R. Chim., 2018, 21, 1055–1300 CrossRef
- A. B. Gaspar, M. Seredyuk and P. Gütlich, J. Mol. Struct., 2009, 924–926, 9–19 CrossRef CAS
- P. Gütlich and A. Hauser, Coord. Chem. Rev., 1990, 97, 1–22 CrossRef
- P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342–391 CrossRef PubMed
- O. A. Qamar, F. Jamil, M. Hussain, M. Mustafa, R. U. Rehman, A. Inayat, M. S. Habib and M. Sajid, Chem. Pap., 2023, 77, 7331–7359 CrossRef CAS
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS
- G. Ke, C. Duan, F. Huang and X. Guo, InfoMat, 2020, 2, 92–112 CrossRef CAS
- A. Cannizzo, C. J. Milne, C. Consani, W. Gawelda, Ch. Bressler, F. Van Mourik and M. Chergui, Coord. Chem. Rev., 2010, 254, 2677–2686 CrossRef CAS
- J. J. McGarvey, I. Lawthers, K. Heremans and H. Toftlund, J. Chem. Soc., Chem. Commun., 1984, 1575–1576 RSC
- M. Khalil, M. A. Marcus, A. L. Smeigh, J. K. McCusker, H. H. W. Chong and R. W. Schoenlein, J. Phys. Chem. A, 2006, 110(1), 38–44 CrossRef CAS PubMed
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS PubMed
- W. Gawelda, A. Cannizzo, V.-T. Pham, F. van Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed
- M. M. N. Wolf, Phys. Chem. Chem. Phys., 2008, 10, 4264–4273 RSC
-
C. Brady, J. J. McGarvey, J. K. McCusker, H. Toftlund and D. N. Hendrickson, in Spin Crossover in Transition Metal Compounds III, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, pp. 1–22 Search PubMed
- C. Consani, M. Prémont-Schwarz, A. ElNahhas, C. Bressler, F. van Mourik, A. Cannizzo and M. Chergui, Angew. Chem., Int. Ed., 2009, 48, 7184–7187 CrossRef CAS PubMed
- J. K. McCusker, K. N. Walda, R. C. Dunn, J. D. Simon, D. Magde and D. N. Hendrickson, J. Am. Chem. Soc., 1992, 114, 6919–6920 CrossRef CAS
- I. Lawthers and J. J. McGarvey, J. Am. Chem. Soc., 1984, 106, 4280–4282 CrossRef CAS
- R. Bertoni, M. Lorenc, A. Tissot, M.-L. Boillot and E. Collet, Coord. Chem. Rev., 2015, 282–283, 66–76 CrossRef CAS
- R. Bertoni, M. Lorenc, H. Cailleau, A. Tissot, J. Laisney, M.-L. Boillot, L. Stoleriu, A. Stancu, C. Enachescu and E. Collet, Nat. Mater., 2016, 15, 606–610 CrossRef CAS PubMed
- E. Collet, N. Moisan, C. Baldé, R. Bertoni, E. Trzop, C. Laulhé, M. Lorenc, M. Servol, H. Cailleau, A. Tissot, M.-L. Boillot, T. Graber, R. Henning, P. Coppens and M. B.-L. Cointe, Phys. Chem. Chem. Phys., 2012, 14, 6192 RSC
- M. Lorenc, J. Hébert, N. Moisan, E. Trzop, M. Servol, M. Buron-Le Cointe, H. Cailleau, M. L. Boillot, E. Pontecorvo, M. Wulff, S. Koshihara and E. Collet, Phys. Rev. Lett., 2009, 103, 028301 CrossRef CAS PubMed
- M. Lorenc, C. Balde, W. Kaszub, A. Tissot, N. Moisan, M. Servol, M. B.-L. Cointe, H. Cailleau, P. Chasle, P. Czarnecki, M. L. Boillot and E. Collet, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054302 CrossRef
- H. Cailleau, M. Lorenc, L. Guérin, M. Servol, E. Collet and M. Buron-Le Cointe, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 189–197 CrossRef CAS PubMed
- R. Bertoni, E. Collet, H. Cailleau, M.-L. Boillot, A. Tissot, J. Laisney, C. Enachescu and M. Lorenc, Phys. Chem. Chem. Phys., 2019, 21, 6606–6612 RSC
- R. Bertoni, M. Lorenc, T. Graber, R. Henning, K. Moffat, J.-F. Létard and E. Collet, CrystEngComm, 2016, 18, 7269–7275 RSC
- E. Collet, M. Lorenc, M. Cammarata, L. Guérin, M. Servol, A. Tissot, M.-L. Boillot, H. Cailleau and M. B.-L. Cointe, Chem.–Eur. J., 2012, 18, 2051–2055 CrossRef CAS PubMed
- J. de J. Velazquez-Garcia, K. Basuroy, D. Storozhuk, J. Wong, S. Demeshko, F. Meyer, R. Henning and S. Techert, Dalton Trans., 2022, 51, 6036–6045 RSC
- J. de J. Velazquez-Garcia, K. Basuroy, D. Storozhuk, J. Wong, S. Demeshko, F. Meyer, R. Henning and S. Techert, Dalton Trans., 2022, 51, 17513–17922 RSC
- J. de J. Velazquez-Garcia, K. Basuroy, D. Storozhuk, J. Wong, S. Demeshko, F. Meyer, R. Henning and S. Techert, Dalton Trans., 2023, 52, 12224–12234 RSC
- S. Pillet, V. Legrand, H.-P. Weber, M. Souhassou, J.-F. Létard, P. Guionneau and C. Lecomte, Z. Kristallogr.–Cryst. Mater., 2008, 223, 235–249 CrossRef CAS
- A. S. Rury, L. E. Goodrich, M. G. I. Galinato, N. Lehnert and R. J. Sension, J. Phys. Chem. A, 2012, 116, 8321–8333 CrossRef CAS PubMed
- B. Freyer, F. Zamponi, V. Juvé, J. Stingl, M. Woerner, T. Elsaesser and M. Chergui, J. Chem. Phys., 2013, 138, 144504 CrossRef PubMed
- M. D. Zidan, M. M. Al-Ktaifani and A. Allahham, Opt. Laser Technol., 2017, 90, 174–178 CrossRef CAS
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 4912–4914 CrossRef CAS PubMed
- M. A. Naumova, A. Kalinko, J. W. L. Wong, M. Abdellah, H. Geng, E. Domenichini, J. Meng, S. A. Gutierrez, P.-A. Mante, W. Lin, P. Zalden, A. Galler, F. Lima, K. Kubicek, M. Biednov, A. Britz, S. Checchia, V. Kabanova, M. Wulff, J. Zimara, D. Schwarzer, S. Demeshko, V. Murzin, D. Gosztola, M. Jarenmark, J. Zhang, M. Bauer, M. L. Lawson Daku, W. Gawelda, D. Khakhulin, C. Bressler, F. Meyer, K. Zheng and S. E. Canton, J. Phys. Chem. Lett., 2020, 11, 2133–2141 CrossRef CAS PubMed
- M. A. Naumova, A. Kalinko, J. W. L. Wong, S. Alvarez Gutierrez, J. Meng, M. Liang, M. Abdellah, H. Geng, W. Lin, K. Kubicek, M. Biednov, F. Lima, A. Galler, P. Zalden, S. Checchia, P.-A. Mante, J. Zimara, D. Schwarzer, S. Demeshko, V. Murzin, D. Gosztola, M. Jarenmark, J. Zhang, M. Bauer, M. L. Lawson Daku, D. Khakhulin, W. Gawelda, C. Bressler, F. Meyer, K. Zheng and S. E. Canton, J. Chem. Phys., 2020, 152, 214301 CrossRef CAS PubMed
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2010, 49, 9274–9277 CrossRef CAS PubMed
- B. Schneider, S. Demeshko, S. Neudeck, S. Dechert and F. Meyer, Inorg. Chem., 2013, 52, 13230–13237 CrossRef CAS PubMed
- M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2014, 53, 6135–6139 CrossRef CAS PubMed
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
-
E. Bill, julX, Max Planck Institute for Bioinorganic Chemistry, Mulheim an der Ruhr, 2008, https://ewww.mpi-muelheim.mpg.de/bac/logins/bill/julX_en.php Search PubMed
- J. A. Kalinowski, A. Makal and P. Coppens, J. Appl. Crystallogr., 2011, 44, 1182–1189 CrossRef CAS PubMed
- J. A. Kalinowski, B. Fournier, A. Makal and P. Coppens, J. Synchrotron Radiat., 2012, 19, 637–646 CrossRef CAS PubMed
-
J. Z. Ren, Precognition User Guide with Reference and Tutorials, Renz Research, Inc., Westmont, IL, U.S.A., 2006 Search PubMed
- F. N. Castellano, H. Malak, I. Gryczynski and J. R. Lakowicz, Inorg. Chem., 1997, 36, 5548–5551 CrossRef CAS PubMed
- V. Šebelík, V. Kuznetsova, H. Lokstein and T. Polívka, J. Phys. Chem. Lett., 2021, 12, 3176–3181 CrossRef PubMed
- X. Zhang, Y. Xia, R. H. Friend and C. Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 245201 CrossRef
- J. Chen, K. Žídek, P. Chábera, D. Liu, P. Cheng, L. Nuuttila, M. J. Al-Marri, H. Lehtivuori, M. E. Messing, K. Han, K. Zheng and T. Pullerits, J. Phys. Chem. Lett., 2017, 8, 2316–2321 CrossRef CAS PubMed
- T. Zhang, G. Lu, J. Liu, H. Shen, P. Perriat, M. Martini, O. Tillement and Q. Gong, Appl. Phys. Lett., 2012, 101, 051109 CrossRef
- D.-F. Zhang, S. Li, Q.-H. Xu and Y. Cao, Langmuir, 2020, 36, 4721–4727 CrossRef CAS PubMed
- M. D. Carducci, M. R. Pressprich and P. Coppens, J. Am. Chem. Soc., 1997, 119, 2669–2678 CrossRef CAS
- S. Techert, F. Schotte and M. Wulff, Phys. Rev. Lett., 2001, 86, 2030–2033 CrossRef CAS PubMed
- J. Hallmann, W. Morgenroth, C. Paulmann, J. Davaasambuu, Q. Kong, M. Wulff and S. Techert, J. Am. Chem. Soc., 2009, 131, 15018–15025 CrossRef CAS PubMed
- C. D. Kim, S. Pillet, G. Wu, W. K. Fullagar and P. Coppens, Acta Crystallogr., Sect. A: Found. Crystallogr., 2002, 58, 133–137 CrossRef PubMed
- P. Coppens, A. Makal, B. Fournier, K. N. Jarzembska, R. Kamiński, K. Basuroy and E. Trzop, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 23–26 CrossRef CAS
- M. S. Schmøkel, R. Kamiński, J. B. Benedict and P. Coppens, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 632–636 CrossRef PubMed
- M. J. Wirth and H. O. Fatunmbi, Anal. Chem., 1990, 62, 973–976 CrossRef CAS
- R. G. Freeman, D. L. Gilliland and F. E. Lytle, Anal. Chem., 1990, 62, 2216–2219 CrossRef CAS
- R. R. Birge, Acc. Chem. Res., 1986, 19, 138–146 CrossRef CAS
- A. A. Rehms and P. R. Callis, Chem. Phys. Lett., 1987, 140, 83–89 CrossRef CAS
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef CAS
- A. Hopt and E. Neher, Biophys. J., 2001, 80, 2029–2036 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2332817–2332822, 2332894–2332898, 2332925–2332929, 2333003–2333006, 2333030 and 2333040–2333043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02933j |
| This journal is © The Royal Society of Chemistry 2024 |