
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed selective disulfide formation by reductive cross-coupling of thiosulfonates†
Tingting
Yuan
 ab,
Xiang-Yu
Chen
bc,
Tengfei
Ji
b,
Huifeng
Yue
a,
Kathiravan
Murugesan
a and
Magnus
Rueping
*a
ab,
Xiang-Yu
Chen
bc,
Tengfei
Ji
b,
Huifeng
Yue
a,
Kathiravan
Murugesan
a and
Magnus
Rueping
*a
aKAUST Catalysis Center, KCC, King Abdullah University of Science and Technology, KAUST, Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
bInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, 52074 Aachen, Germany
cSchool of Chemical Science, University of Chinese Academy of Science, Beijing 10049, China
First published on 30th August 2024
Abstract
Developing innovative methodologies for disulfide preparation is of importance in contemporary organic chemistry. Despite significant advancements in nickel-catalyzed reductive cross-coupling reactions for forming carbon–carbon and carbon–heteroatom bonds, the synthesis of S–S bonds remains a considerable challenge. In this context, we present a novel approach utilizing nickel catalysts for the reductive cross-coupling of thiosulfonates. This method operates under mild conditions, offering a convenient and efficient pathway to synthesize a wide range of both symmetrical and unsymmetrical disulfides from readily available, bench-stable thiosulfonates with exceptional selectivity. Notably, this approach is highly versatile, allowing for the late-stage modification of pharmaceuticals and the preparation of various targeted compounds. A comprehensive mechanistic investigation has been conducted to substantiate the proposed hypothesis.
Introduction
Establishing unsymmetrical reactions for S–S bond formation holds significant importance in both organic synthesis and drug discovery.1 To date, only limited methods have been developed for constructing unsymmetrical disulfides using substrates with different functional groups.2–5 In the past years researchers have focused on the reductive cross-coupling of distinct electrophiles to overcome selectivity problems. However, the exploration of electrophiles possessing identical functionalities for efficient S–S bond-forming reactions remains mainly lacking.6,7 This difficulty arises from the need to control the preference for one electrophile over the other and to circumvent the formation of by-products. To address these considerations, we have developed an approach based on nickel catalysis for accomplishing the reductive cross-coupling of thiosulfonates. This method enables the formation of symmetric and unsymmetric disulfides with a remarkable level of selectivity.Disulfides play a crucial role in life science,8 pharmaceutical science,9 and food chemistry,10 due to their distinct pharmacological and physicochemical properties (Fig. 1). Moreover, they can serve as natural connectors for creating secondary and tertiary structures in polypeptides and proteins (Fig. 1).11 Consequently, several approaches have been developed to generate structurally diverse disulfides. However, achieving their selective and efficient synthesis under mild conditions, without the need for oxidants, presents specific challenges.
 | ||
| Fig. 1 Selected the disulfide moiety in nature products and drugs. | ||
The commonly pursued method involves thiol oxidation (Scheme 1A, upper left),12 necessitates using stoichiometric oxidants and potentially hazardous and unpleasant-smelling thiols. Additionally, this methodology occasionally encounters issues such as overoxidation of the S–H bond. On the other hand, the substitution of thiolates in the presence of strong bases is incompatible with sensitive functional groups (Scheme 1A, upper right).3 An alternative approach involves the utilization of Rh catalysis for the synthesis of aryl–alkyl and alkyl–alkyl disulfides through a disulfide exchange reaction13 (Scheme 1A, below left). Furthermore, strategies for disulfide modification have been investigated4 (Scheme 1A, below right). Most of these methods are limited to aryl-substituted disulfide synthesis, the preparation of starting disulfide reagents usually requires multiple steps.
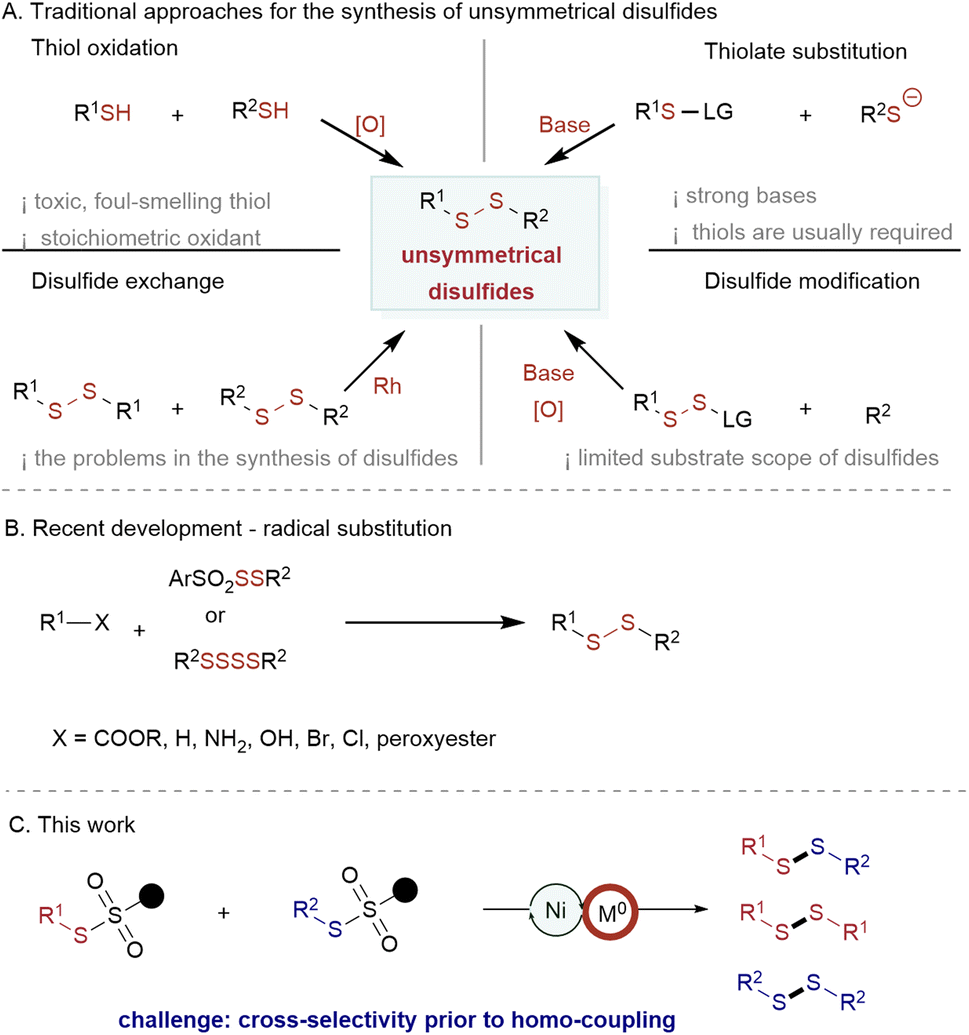 | ||
| Scheme 1 Reported methods for unsymmetric disulfides construction and this work. | ||
Recently, radical disulfuration has gained increasing attention; the Pratt,5a,b Ackermann,5c Studer,5d and Wang5e–h groups have demonstrated this radical addition to be a powerful alternative for the construction of alkyl–alkyl disulfides and/or acyl–alkyl disulfides (Scheme 1B).5
Despite this progress, a practical and efficient synthetic route allowing various substitution patterns, including aryl–aryl, aryl–alkyl, and alkyl–alkyl disulfides, is still underexplored.
Compared to conventional cross-coupling reactions, reductive cross-couplings offer the advantage of mild reaction conditions without the need to prepare unstable and expensive organometallic reagents. This feature allows for improved compatibility with various functional groups.6,7,14 Nickel-catalyzed reductive cross-coupling has emerged as a powerful and appealing method for generating multiple chemical bonds.6 As such, nickel-catalyzed reductive cross-couplings have received increasing attention for forming carbon–carbon/hetero atom bonds.15–19 Nevertheless, the exploration of other chemical bond constructions, particularly S–S bonds, remains relatively limited. This limitation may arise from the challenge of conducting reductive cross-coupling with two substrates possessing similar reactivity.6 Therefore, we developed a method to address this problem and access diverse S–S bonds in a reductive cross-coupling approach with readily available thiosulfonates (Scheme 1C). This method assembled a wide range of aryl–alkyl, aryl–aryl, and alkyl–alkyl unsymmetrical disulfides, making it one of the most versatile approaches for the preparation of disulfides. The odorless thiosulfonates are readily available, can be easily stored, and have broad substrate scopes.
We started our investigation with the reductive home-coupling of phenyl benzenesulfonothioate 1 utilizing NiCl2·glyme as the catalyst, 6,6-dimethyl-2,2-bipyridyl (6,6′-di-Me-2,2′-bpy; dmbpy) as the ligand and Mn as the reductant gave the desired product 2 in 92% yield (Table 1, entry 1). The importance of Mn, dmbpy, and Ni(II) in achieving high yields was confirmed through control experiments (Table 1, entries 2–4). Further investigations demonstrated that the reaction proceeded via Ni(0) catalysis, as evidenced by the catalytic amount of Ni(0) gave 25% yield of the desired product, and the yield was increased to 86% when 1.0 equiv. of Ni(0) was employed (entries 5 and 6).
|
|
||
|---|---|---|
| Entry | Deviation from standard condition | Yield of 2a (%) |
| a Reaction conditions: benzenesulfonothioate (0.2 mmol), NiCl2·glyme (0.01 mmol, 5 mol%), 6,6′-di-Me-2,2′-bpy (0.01 mmol, 5 mol%), Mn (0.2 mmol, 1.0 equiv.), under N2, GC yield using n-decane (0.1 mmol) as the internal standard. b Isolated yield. | ||
| 1 | None | 92 (89)b |
| 2 | Absence of Ni | 29 |
| 3 | Absence of Mn | 30 |
| 4 | Absence of 6,6′-di-Me-2,2′-bpy | 44 |
| 5 | Ni(COD)2 instead of Ni(II) + Mn | 25 |
| 6 | 1.0 equiv. of Ni(COD)2 instead of Ni(II) + Mn | 86 |
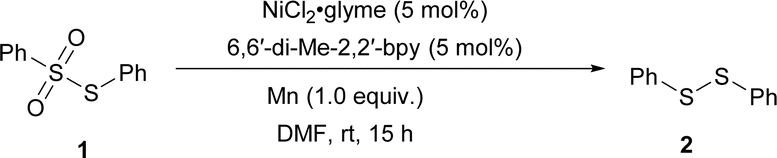
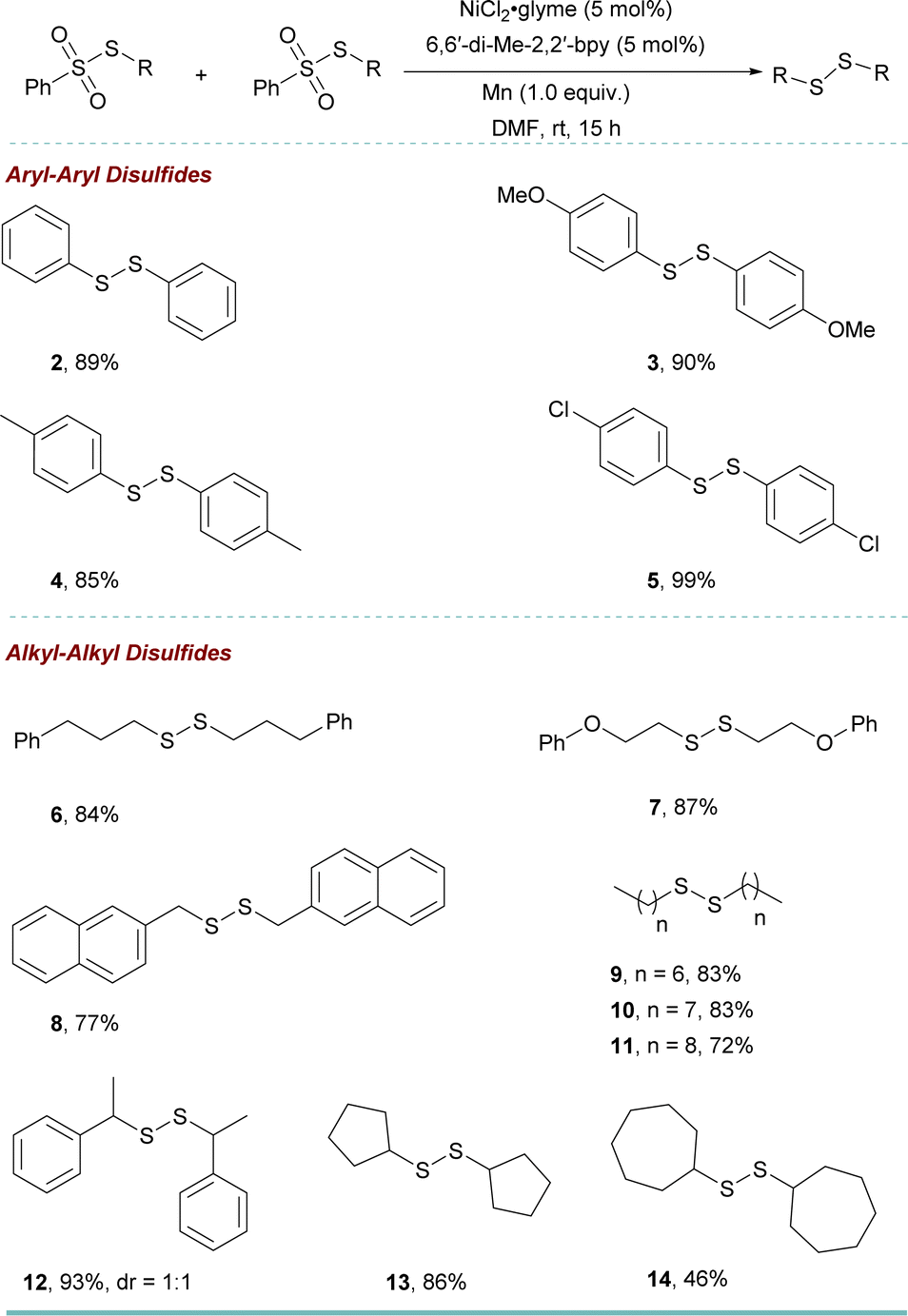
We then utilized the optimized reaction conditions to assemble a variety of disulfides (Table 2). Thiosulfonates with aryl and alkyl groups reacted smoothly to give the corresponding symmetric disulfides 2–14 in up to 99% yields. The exploration of this transformation led us to identify an efficient selective cross-coupling reaction to obtain unsymmetric disulfide 15–47 (see ESI† for the optimization details) (Table 3). Interestingly, the addition of 1 equiv. KF increased the cross-coupled product significantly over homo-coupling (refer to the ESI, see Table S8†). Benzenesulfonothioates with methoxy, methyl, and tertiary butyl substituents all underwent smoothly to afford the corresponding unsymmetric disulfides 16–18 in moderate to good yield. In addition, when a di-substituted thiosulfonate could also be employed as a coupling partner to obtain the product in good yield (19). It is noted that separating the products is not easy due to the similar polarity of the products and the side products. However, effective separation can be achieved by column chromatography using an n-hexane/ether mixture or n-hexane as the eluent.
| a Reaction conditions: benzenesulfonothioate (0.2 mmol), NiCl2·glyme (0.01 mmol, 5 mol%), 6,6′-di-Me-2,2′-bpy (0.01 mmol, 5 mol%), Mn (0.2 mmol, 1.0 equiv.), under N2; yields after purification. |
|---|
|
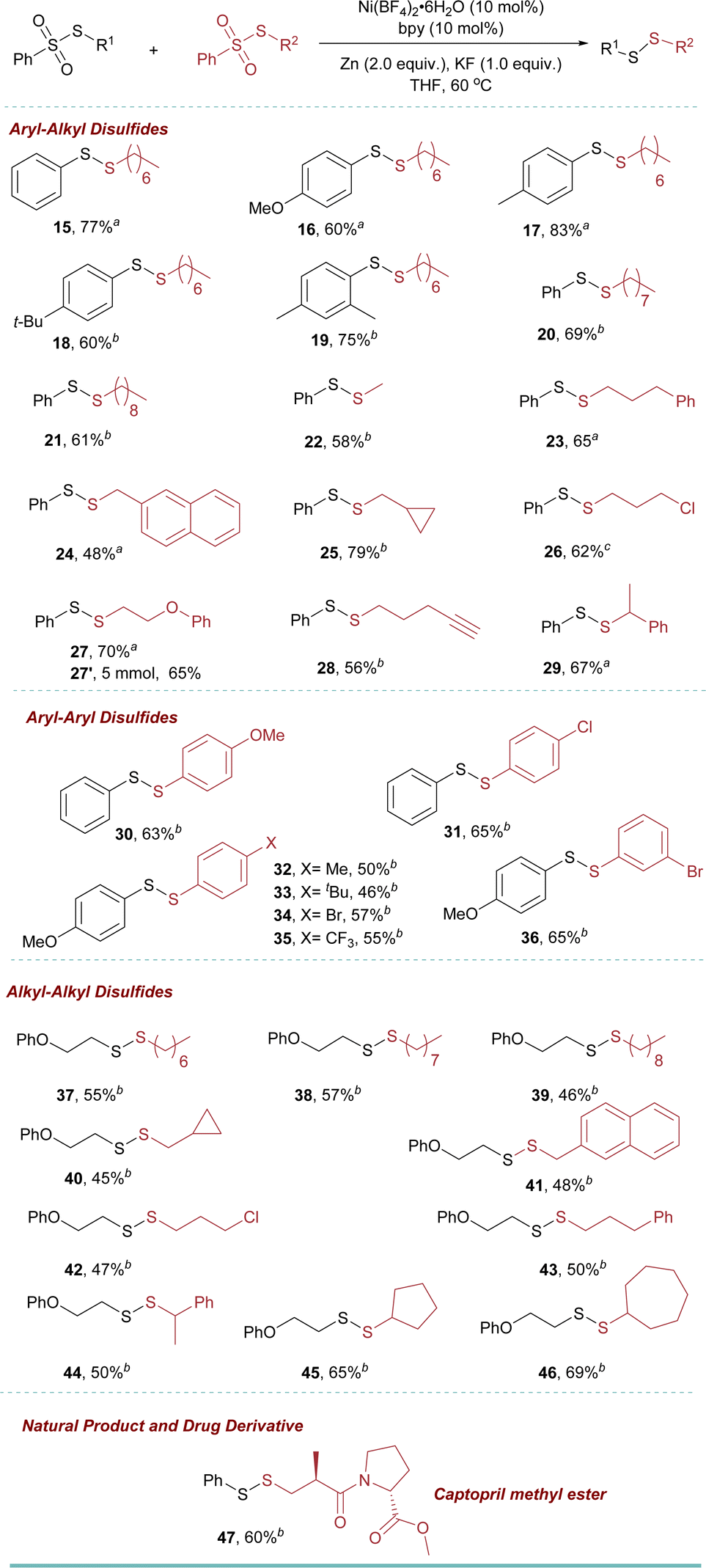
| a Reaction conditions: benzenesulfonothioate (black) (0.1 mmol), benzenesulfonothioate (red) (0.2 mmol, 2.0 equiv.), Ni(BF4)2·6H2O (0.01 mmol, 10 mol%), bpy (0.01 mmol, 10 mol%), Zn (0.2 mmol, 2.0 equiv.), KF (0.1 mmol, 1.0 equiv.), in 1 mL THF under N2; yields after purification. b Bpy (0.015 mmol, 15 mol%). c Bpy (0.005 mmol, 5 mol%) and 4′-(p-tolyl)-2,2′:6′,2′′-terpyridine (0.005 mmol, 5 mol%). |
|---|
|
The sulfonothioates bearing other alkyl groups also reacted well in the reaction (20 and 21). Approaches to biologically relevant methyl-containing disulfides “MeSS–R” moiety are still limited.9,20 However, using our new methodology, 22 was obtained in 58% yield. Furthermore, different functional groups, such as arenes, cyclopropane, alkyl chloride, ether, and alkyne (23–28) could be tolerated. This was also true for the secondary benzenesulfonothioate (29). To further showcase the utility of this approach, a 5 mmol scale-up reaction was performed. The reaction went smoothly to give the desired product 27′ in 65% yield.
Our investigation extended to the exploration of aryl–aryl disulfide synthesis. Employing the reaction conditions, a range of substituted aryl thiosulfonates readily engaged in coupling reactions, affording the respective products 30–36 with isolated yields spanning from 46% to 65%.
Subsequent to this, a diversity of alkyl–alkyl disulfides 37–46 (yielding 45–69%) were synthesized using various alkyl thiosulfonates. Notably, cyclic, acyclic, halogen-substituted alkyl, and secondary alkyl sulfonothioates were also employed, yielding the respective products (37–46) with good yields.
Late-stage modification of a drug molecule is of significant importance in drug discovery. In this context, hypertension drug captopril was successfully modified to the corresponding unsymmetrical disulfides in 60% isolated yields (47).
A comprehensive series of mechanistic studies was conducted to elucidate the reaction mechanism (Scheme 2 and refer to the ESI†). Significantly, in the presence of the radical quencher TEMPO, the yield of the desired product markedly decreased to 12% (Scheme 2a). The radical-clock experiments were performed using diene 50. The reactions of thiosulfonates with diene led to the generation of the 5-exo cyclized products 51 and 53, respectively. The 51 and 53 were confirmed by HRMS (Scheme 2b and c). The results showed that both the sulfur and sulfonyl radicals were involved (Scheme 2a–c). To validate the participation of Ni(0) in the catalytic cycle, the use of catalytic amounts of Ni(COD)2, instead of the Ni(II) precursor under standard conditions, resulted in a yield of only 10% of the desired product. However, employing 1.0 equiv. of Ni(COD)2 led to a notable increase in the product yield, reaching 78% (Scheme 2d and e). It is important to note that the utilization of a disulfide instead of a sulfonothioate led to a notable reduction in both selectivity and yield of the product (Scheme 2f and g), underscoring the pivotal role of sulfonothioates in determining product selectivity and yield. Based on the mechanistic experiments and reported literature,16d,21 the proposed reaction mechanism is illustrated in Scheme 3. The Ni(0) species is generated by in situ reduction of Ni(II) salt by Zn or Mn (A). Thiosulfonates readily undergo oxidative addition with Ni(0) to form divalent nickel B. Subsequently, the Ni(II) species B captures the radical from thiosulfonates yielding C. Intermediate C undergo reductive elimination to give the desired product (cross-coupled/homo coupled) and Ni(I) species (D). The resulting Ni(I) intermediate reacts with another thiosulfonate and furnishes the intermediate E, which is further reduced to Ni(0) species A and regenerates the catalytic cycle.
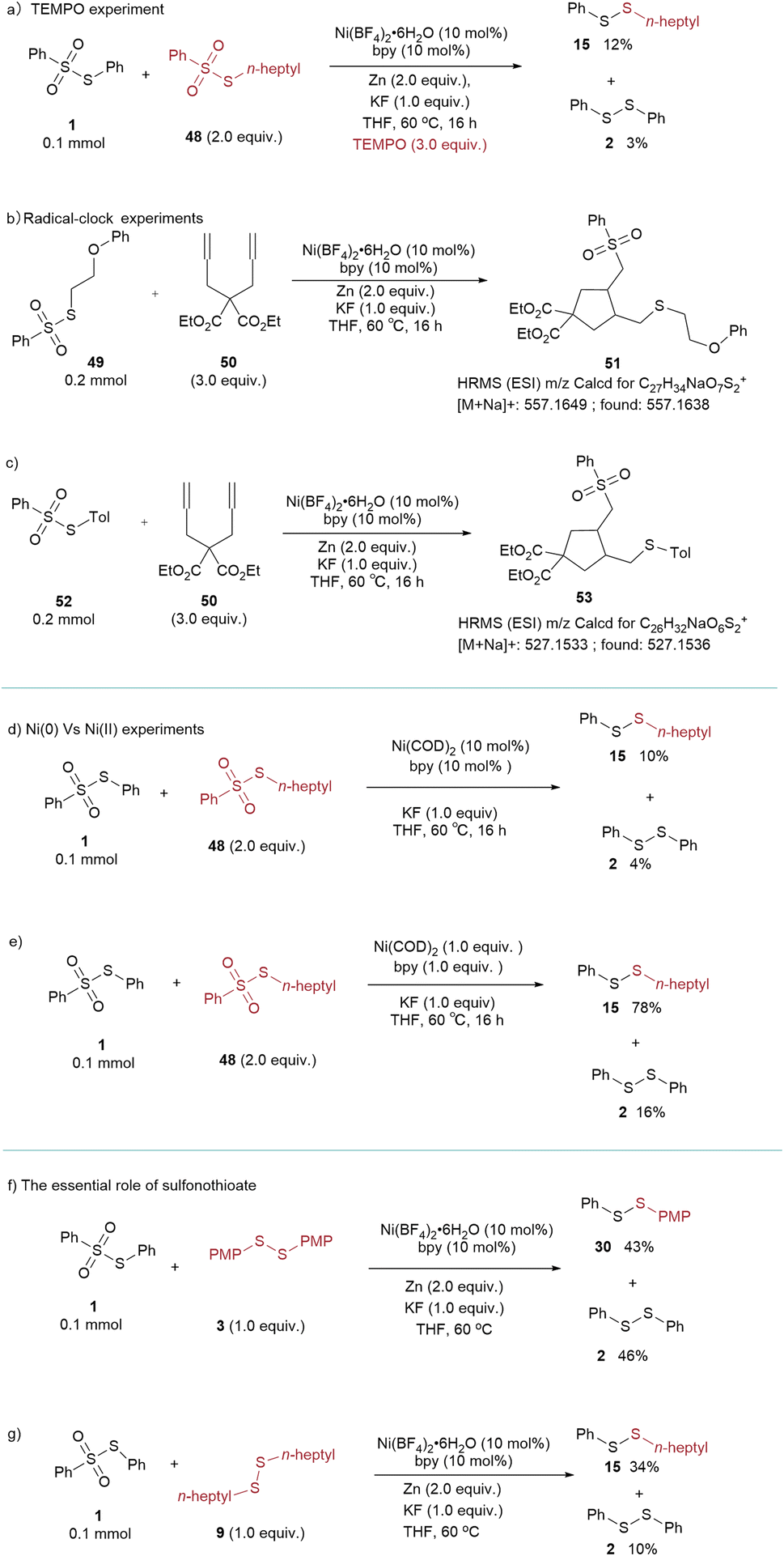 | ||
| Scheme 2 (a) TEMPO experiment (b and c) radical-clock experiments (d and e) Ni(0) vs. Ni(II) experiments (f and g) the essential role of sulfonothioate. | ||
 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In summary, we have successfully established a nickel-catalyzed reductive cross-coupling reaction of thiosulfonates, enabling the selective formation of S–S bonds and the synthesis of both symmetrical and unsymmetrical disulfides. This transformation has demonstrated exceptional selectivity under oxidant-free conditions, showcasing a broad substrate scope that encompasses aryl–aryl, aryl–alkyl, and alkyl–alkyl disulfides. The developed methodology offers practical advantages due to its versatility, generality, and reliance on readily available thiosulfonates and stable Ni(II) catalysts.Data availability
All experimental procedures, details of the optimization, and additional data can be found in the ESI.†Author contributions
T. Y., X.-Y. C. and M. R. conceived and designed the experiments. T. Y., T. J., and H. Y. performed the experiments and analyzed the data. T. Y. and K. M. wrote the manuscript and performed mechanistic experiments. M. R. directed the project. All authors discussed the experimental.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication is based upon work supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under CRG award no. URF/1/5082 and URF/1/4701. T. Y. thanks the China Scholarship Council.Notes and references
- (a) F. A. Moura, K. Q. d. Andrade, J. C. F. d. Santos and M. O. F. Goulart, Curr. Top. Med. Chem., 2015, 15, 458–483 CrossRef CAS PubMed; (b) C.-S. Jiang, W. E. G. Müller, H. C. Schrödera and Y.-W. Guo, Chem. Rev., 2012, 112, 2179–2207 CrossRef CAS PubMed; (c) B. Li, W. J. Wever, C. T. Walsh and A. A. Bowers, Nat. Prod. Rep., 2014, 31, 905–923 RSC; (d) S. Brocchini, S. Balan, A. Godwin, J.-W. Choi, M. Zloh and S. Shaunak, Nat. Protoc., 2006, 1, 2241–2252 CrossRef CAS PubMed.
- (a) C. L. Ong, S. Titinchi, J. C. Juan and N. G. Khaligh, Helv. Chim. Acta, 2021, 104, e2100053 CrossRef CAS; (b) M. Musiejuka and D. Witt, Org. Prep. Proced. Int., 2015, 47, 95–131 CrossRef; (c) G. R. Morais and R. A. Falconer, Org. Biomol. Chem., 2021, 19, 82–100 RSC; (d) M. Wang and X. Jiang, Top. Curr. Chem., 2019, 14, 285–324 Search PubMed.
- (a) M. Musiejuk, T. Klucznik, J. Rachon and D. Witt, RSC Adv., 2015, 5, 31347–31351 RSC; (b) Q. T. Doa, D. Elothmania, G. L. Guillanton and J. Simonet, Tetrahedron Lett., 1997, 38, 3383–3384 CrossRef; (c) R. D. Bach, O. Dmitrenko and C. Thorpe, J. Org. Chem., 2008, 73, 12–21 CrossRef CAS PubMed; (d) S. J. Brois, J. F. Pilot and H. W. Barnum, J. Am. Chem. Soc., 1970, 92, 7629–7631 CrossRef CAS; (e) D. Witt, M. Szymelfejnik, S. Demkowicz and J. Rachon, Synthesis, 2007, 22, 3528–3534 CrossRef.
- (a) Z. Dai, X. Xiao and X. Jiang, Tetrahedron, 2017, 73, 3702–3706 CrossRef CAS; (b) C. M. Park, B. A. Johnson, J. Duan, J. J. Park, J. J. Day, D. Gang, W. J. Qian and M. Xian, Org. Lett., 2016, 18, 904–907 CrossRef CAS PubMed; (c) W. Wang, Y. Lin, Y. Ma, C. H. Tung and Z. Xu, Org. Lett., 2018, 20, 3829–3832 CrossRef CAS PubMed; (d) X. Xiao, M. Feng and X. Jiang, Angew. Chem., Int. Ed., 2016, 55, 14121–14125 CrossRef CAS PubMed; (e) X. Xiao, J. Xue and X. Jiang, Nat. Commun., 2018, 9, 2191–2199 CrossRef PubMed; (f) Q. Zhang, Y. Li, L. Zhang and S. Luo, Angew. Chem., Int. Ed., 2021, 60, 10971–10976 CrossRef CAS PubMed.
- (a) Z. Wu and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 10284–10290 CrossRef CAS PubMed; (b) Z. Wu and D. A. Pratt, Angew. Chem., Int. Ed., 2021, 60, 15598–15605 CrossRef CAS; (c) F. Wang, Y. Chen, W. Rao, L. Ackermann and S.-Y. Wang, Nat. Commun., 2022, 13, 2588 CrossRef CAS PubMed; (d) J. Zhang and A. Studer, Nat. Commun., 2022, 13, 3886 CrossRef CAS PubMed; (e) Y. Chen, D. Sheng, F. Wang, W. Rao, S. Shen and S.-Y. Wang, Org. Chem. Front., 2022, 9, 4962–4968 RSC; (f) W. Chen, X.-Y. Liu, D. Sheng, Y.-F. Jiang, W. Rao, S.-S. Shen, Z.-Y. Yang and S.-Y. Wang, Org. Chem. Front., 2024, 11, 830–835 RSC; (g) S. Chen, S. Cao, C. Liu, B. Wang, X. Ren, H. Huang, Z. Peng and X. Wang, Org. Lett., 2021, 23, 7428–7433 CrossRef CAS PubMed; (h) C. Liu, X. Lin, D. An, X. Wang and Q. Gao, Org. Chem. Front., 2024, 11, 358–363 RSC.
- (a) D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed; (b) O. Sensuke, Nickel Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2020 Search PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed; (d) J. Liu, Y. Ye, J. L. Sessler and H. Gong, Acc. Chem. Res., 2020, 53, 1833–1845 CrossRef CAS PubMed; (e) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 2, 1411–1421 RSC; (f) X. Wang, Y. Dai and H. Gong, Top. Curr. Chem., 2016, 374, 43 CrossRef PubMed; (g) C. E. Knappke, S. Grupe, D. Gartner, M. Corpet, C. Gosmin and A. J. von Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS PubMed; (h) L. Yi, T. Ji, K.-Q. Chen, X.-Y. Chen and M. Rueping, CCS Chem., 2022, 4, 9–30 CrossRef CAS.
- (a) M.-Y. Gao and C. Gosmini, Org. Lett., 2023, 25, 7689–7693 CrossRef CAS PubMed; (b) C. Dorval, M. Tricoire, J.-M. Begouin, V. Gandon and C. Gosmini, ACS Catal., 2020, 10, 12819–12827 CrossRef CAS; (c) M. Amatore and C. Gosmini, Angew. Chem., Int. Ed., 2008, 47, 2089–2092 CrossRef CAS PubMed.
- (a) J. Alegre-Cebollada, P. Kosuri, J. A. Rivas-Pardo and J. M. Fernandez, Nat. Chem., 2011, 3, 882–887 CrossRef CAS PubMed; (b) M. Gongora-Benitez, J. Tulla-Puche and F. Albericio, Chem. Rev., 2014, 114, 901–926 CrossRef CAS PubMed; (c) S. Lu, S.-B. Fan, B. Yang, Y.-X. Li, J.-M. Meng, L. Wu, P. Li, K. Zhang, M.-J. Zhang, Y. Fu, J. Luo, R.-X. Sun, S.-M. He and M.-Q. Dong, Nat. Methods, 2015, 12, 329–331 CrossRef CAS PubMed; (d) M. Narayan, E. Welker, W. J. Wedemeyer and H. A. Scherage, Acc. Chem. Res., 2000, 33, 805–812 CrossRef CAS PubMed.
- (a) W. C. Widdison, S. D. Wilhelm, E. E. Cavanagh, K. R. Whiteman, B. A. Leece, Y. Kovtun, V. S. Goldmacher, H. Xie, R. M. Steeves, R. J. Lutz, R. Zhao, L. Wang, W. A. Blattler and R. V. J. Chari, J. Med. Chem., 2006, 49, 4392–4408 CrossRef CAS PubMed; (b) R. Y. Zhao, H. K. Erickson, B. A. Leece, E. E. Reid, V. S. Goldmacher, J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2012, 55, 766–782 CrossRef CAS PubMed.
- (a) E. Block, T. Bayer, S. Naganathan and S.-H. Zhao, J. Am. Chem. Soc., 1996, 118, 2799–2810 CrossRef CAS; (b) F. S. Hanschen, E. Lamy, M. Schreiner and S. Rohn, Angew. Chem., Int. Ed., 2014, 53, 11430–11450 CrossRef CAS PubMed.
- (a) E. Gross, C. S. Sevier, A. Vala, C. A. Kaiser and D. Fass, Nat. Struct. Biol., 2002, 9, 61–67 CrossRef CAS PubMed; (b) M. Matsumura and B. W. Matthews, Science, 1989, 243, 792–794 CrossRef CAS PubMed; (c) M. Wensien, F. R. von Pappenheim, L. M. Funk, P. Kloskowski, U. Curth, U. Diederichsen, J. Uranga, J. Ye, P. Fang, K. T. Pan, H. Urlaub, R. A. Mata, V. Sautner and K. Tittmann, Nature, 2021, 593, 460–464 CrossRef CAS PubMed.
- (a) M. Arisawa, K. Fukumoto and M. Yamaguchi, ACS Catal., 2020, 10, 15060–15064 CrossRef CAS; (b) G. A. Bagiyan, I. K. Koroleva, N. V. Soroka and A. V. Ufimtsev, Russ. Chem. Bull., 2003, 52, 1135–1141 CrossRef CAS; (c) X. Qiu, X. Yang, Y. Zhang, S. Song and N. Jiao, Org. Chem. Front., 2019, 6, 2220–2225 RSC; (d) J. M. Swan, Nature, 1957, 180, 643–645 CrossRef CAS.
- M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2003, 125, 6624–6625 CrossRef CAS PubMed.
- (a) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (c) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2023, 145, 17527–17550 CrossRef CAS PubMed; (d) B. Zhang, T.-T. Li, Z.-C. Mao, M. Jiang, Z. Zhang, K. Zhao, W.-Y. Qu, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2024, 146, 1410–1422 CrossRef CAS PubMed.
- (a) L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454–457 CrossRef CAS PubMed; (b) H. Yue, C. Zhu, L. Shen, Q. Geng, K. J. Hock, T. Yuan, L. Cavallo and M. Rueping, Chem. Sci., 2019, 10, 4430–4435 RSC; (c) D. Sun, G. Ma, X. Zhao, C. Lei and H. Gong, Chem. Sci., 2021, 12, 5253–5258 RSC; (d) J. Liu, Q. Ren, X. Zhang and H. Gong, Angew. Chem., Int. Ed., 2016, 55, 15544–15548 CrossRef CAS PubMed; (e) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., Int. Ed., 2017, 56, 11901–11905 CrossRef CAS PubMed; (f) J. Davies, D. Janssen-Muller, D. P. Zimin, C. S. Day, T. Yanagi, J. Elfert and R. Martin, J. Am. Chem. Soc., 2021, 143, 4949–4954 CrossRef CAS PubMed; (g) L.-L. Liao, G.-M. Cao, J.-H. Ye, G.-Q. Sun, W.-J. Zhou, Y.-Y. Gui, S.-S. Yan, G. Shen and D.-G. Yu, J. Am. Chem. Soc., 2018, 140, 17338–17342 CrossRef CAS PubMed; (h) H. Li, F. Wang, S. Zhu and L. Chu, Angew. Chem., Int. Ed., 2022, 61, e202116725 CrossRef CAS PubMed; (i) G. A. Molander, S. R. Wisniewski and K. M. Traister, Org. Lett., 2014, 16, 3692–3695 CrossRef CAS PubMed.
- (a) Y. Chen, F. Wang, B.-X. Liu, W.-D. Rao and S.-Y. Wang, Org. Chem. Front., 2022, 9, 731–736 RSC; (b) J. Li, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2019, 84, 16147–16156 CrossRef CAS PubMed; (c) J. Li, W. Rao, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2019, 84, 11542–11552 CrossRef CAS PubMed; (d) Y. Fang, T. Rogge, L. Ackermann, S.-Y. Wang and S.-J. Ji, Nat. Commun., 2018, 9, 2240 CrossRef PubMed.
- (a) J. Duan, K. Wang, G.-L. Xu, S. Kang, L. Qi, X.-Y. Liu and X.-Z. Shu, Angew. Chem., Int. Ed., 2020, 59, 23083–23088 CrossRef CAS PubMed; (b) J. Duan, Y. Wang, L. Qi, P. Guo, X. Pang and X.-Z. Shu, Org. Lett., 2021, 23, 7855–7859 CrossRef CAS PubMed; (c) L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 18587–18590 CrossRef CAS PubMed; (d) M. Xing, H. Cui and C. Zhang, Org. Lett., 2021, 23, 7645–7649 CrossRef CAS; (e) S. Guven, G. Kundu, A. Weßels, J. S. Ward, K. Rissanen and F. Schoenebeck, J. Am. Chem. Soc., 2021, 143, 8375–8380 CrossRef CAS PubMed.
- (a) P.-F. Su, K. Wang, X. Peng, X. Pang, P. Guo and X.-Z. Shu, Angew. Chem., Int. Ed., 2021, 60, 26571–26576 CrossRef CAS PubMed; (b) P. Guo, X. Pang, K. Wang, P.-F. Su, Q.-Q. Pan, G.-Y. Han, Q. Shen, Z.-Z. Zhao, W. Zhang and X.-Z. Shu, Org. Lett., 2022, 24, 1802–1806 CrossRef CAS PubMed.
- (a) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146–6159 CrossRef CAS PubMed; (b) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562–11565 CrossRef CAS PubMed.
- X. Xiao, M. Feng and X. Jiang, Chem. Commun., 2015, 51, 4208–4211 RSC.
- (a) A. Esparza-Ruiz, G. Gonzalez-Gomez, E. Mijangos, A. Pena-Hueso, H. Lopez-Sandoval, A. Flores-Parra, R. Contreras and N. Barba-Behrens, Dalton Trans., 2010, 39, 6302–6309 RSC; (b) J. A. Bellefeuille, C. A. Grapperhaus, R. M. Buonomo, J. H. Reibenspies and M. Y. Darensbourg, Organometallics, 1998, 17, 4813–4821 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02969k |
| This journal is © The Royal Society of Chemistry 2024 |