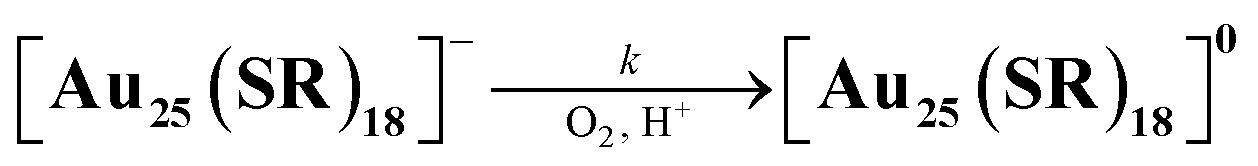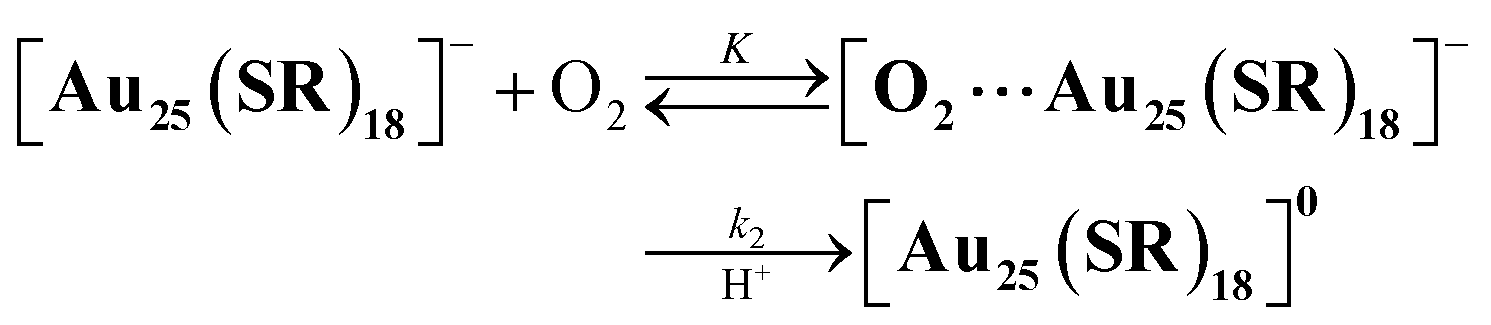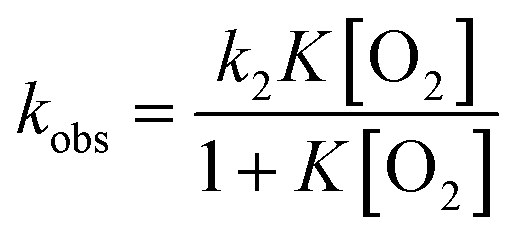DOI:
10.1039/D4SC02995J
(Edge Article)
Chem. Sci., 2024,
15, 18896-18902
Quantitative analysis of air-oxidation reactions of thiolate-protected gold nanoclusters†
Received
7th May 2024
, Accepted 5th October 2024
First published on 17th October 2024
Abstract
The interaction of dioxygen (O2) with inorganic nanomaterials is one of the most essential steps to understanding the reaction mechanism of O2-related reactions. However, quantitative analyses for O2-binding processes and subsequent oxidation reactions on the surface are still elusive, whereas the reaction of O2 with molecules such as transition metal complexes has been widely explored. Herein, we have quantitatively evaluated reaction processes of air-oxidation reactions of atomically precise thiolate-protected Au25 nanoclusters ([Au25(SR)18]−) as a model of O2 activation by inorganic nanomaterials. Kinetic analyses on the air-oxidation reaction of [Au25(SR)18]− revealed a controlling factor for O2-activation processes, which could be finely tunable by the protecting thiolate ligands.
Introduction
Dioxygen (O2) activation in O2 reduction and aerobic oxidations is one of the most fundamental and crucial reaction processes in chemistry. So far, many catalysts for O2 activation have been developed by employing molecular catalysts1–3 and inorganic nanomaterials, including metal nanoparticles (MNPs)4–6 and metal nanoclusters (MNCs).7–9 An understanding of the reaction mechanisms of O2-activation processes at the atomic level is essential to develop more efficient catalysts. Hence, mechanistic insights into O2 activation have also been investigated.10–13 For molecular catalysts such as Fe, Co and Cu complexes or organocatalysts such as porphyrinoids, O2-activation processes, including O2-binding and subsequent reduction to form reactive O2 species, were experimentally well-revealed through structural10 and kinetic11 analyses based on experimental evidence. In contrast, because reaction systems with MNPs are relatively complicated due to the distributions of size and composition of MNPs, the understanding of the reaction mechanisms for the O2-activation process at the atomic level is quite a difficult challenge. Thus, estimations of such a reaction mechanism have mainly relied on theoretical analysis until now.12
Compared with MNPs, MNCs with sizes of less than 2 nm are considered a desirable model to reveal detailed reaction mechanisms at the atomic level owing to their atomically precise structures.14 Among various MNCs, gold nanoclusters (AuNCs), such as thiolate-protected gold clusters (denoted as Aun(SR)m, SR = thiolate), are appropriate candidates to perform atomically precise analyses on O2-activation processes using inorganic nanomaterials because of their tunable reactivities, easy preparation and good stability.7,15,16 For example, the thiolate-protected Au25 cluster anion ([Au25(SR)18]−) exhibited reactivity towards O2, as seen in the catalytic oxygen reduction reaction (ORR).17 The catalytic activity of [Au25(SR)18]− in the ORR could be tuned by the type of thiolate ligand17a and hetero-atom doping.17b In addition, [Au25(SR)18]− which shows higher reactivity than other AuNCs with different sizes17c is a promising material for practical use. Together with the ORR, the reactions of [Au25(SR)18]− with O2 are also important in the reductive activation of O2 for substrate oxidation, which was seen in the aerobic oxidation of thiolates to form disulfide in Au25 polymer films.18 In both cases, the interaction of O2 on the surface of the Au25 cluster and reduction of O2, namely Au25 cluster oxidation,19,20 are proposed to be crucial processes as an initial step for O2 activation. However, whereas some reports demonstrated the interaction between O2 and [Au25(SR)18]− using mass spectrometry21 or theoretical analysis,13d,f,17d experimental evidence for the O2 binding and/or activation by AuNCs is still limited. In particular, the air-oxidation reaction of [Au25(SR)18]− was analysed just qualitatively,22 but quantitative analysis for O2 activation by AuNCs, which is important to systematically evaluate the reactivities of AuNCs, has yet to be reported.
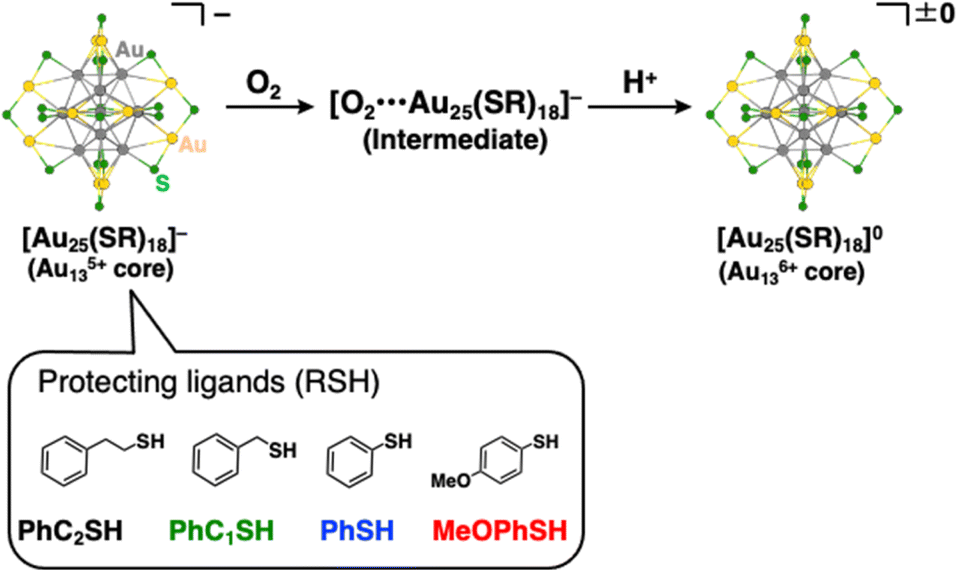
Herein, we report quantitative analyses of the air-oxidation reactions of AuNCs based on kinetic analysis for the first time. For a model of AuNCs, we chose [Au25(SR)18]− because of its reversible redox properties20 and a variety of choices for protecting ligands.14,23 The Au25 cluster anions are composed of an Au135+ core surrounded by six [Au2(SR)3]− units, and the oxidation reactions of the gold cluster occur in the Au13 core rather than in staple moieties.19b Evaluations of the reactivities of [Au25(SR)18]− with O2 were conducted in the presence of protons (Scheme 1). The addition of protons can promote the electron transfer reaction to reduce O2 through proton-coupled electron transfer (PCET)24 and this reaction system has a big advantage of facile quantitative analysis of the reactivity, as seen in the mechanistic analysis of O2 reduction by organic molecules3c,25 or transition metal complexes.26 We have controlled redox potentials and O2-affinities of [Au25(SR)18]− by changing the thiolate (SR) ligands and revealed controlling factors in air-oxidation reactions of [Au25(SR)18]− to form neutral [Au25(SR)18]0.
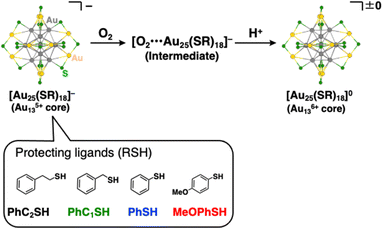 |
| | Scheme 1 Reaction scheme for air-oxidation reactions of [Au25(SR)18]− employed in this work. R groups were omitted for clarity. Atom labels: grey: gold in the Au13 core, yellow: gold on the staple, and green: sulfur. | |
Results and discussion
Synthesis and characterisation of thiolate-protected Au25 cluster anions
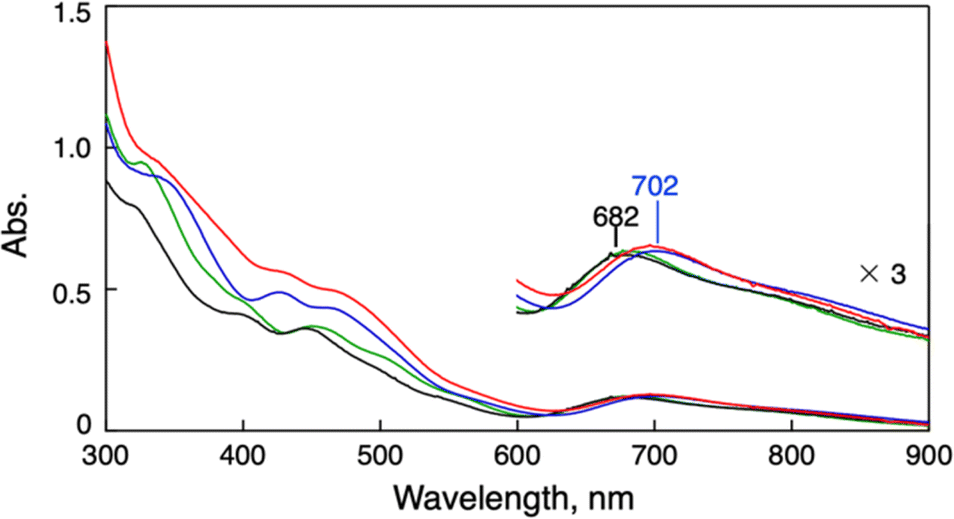
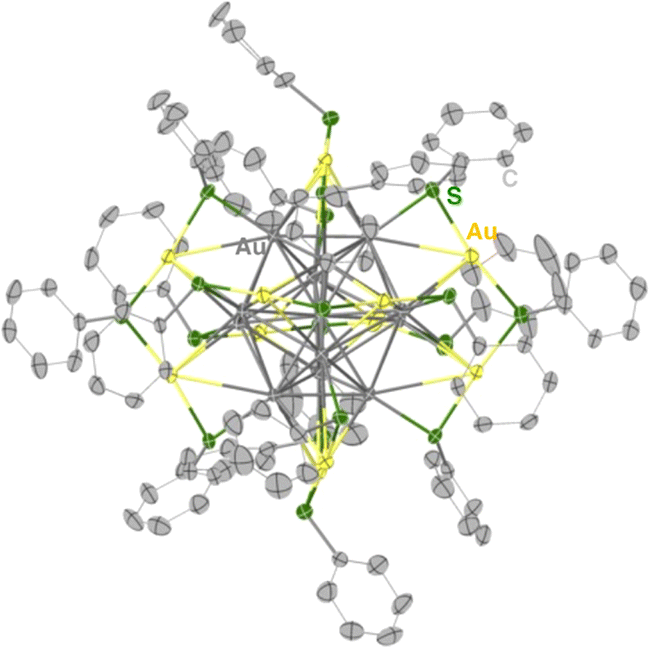
Synthesis of the phenylethanethiolate-protected Au25 cluster anion (TOA+[Au25(SC2Ph)18]−, TOA+ = tetraoctylammonium) was conducted through the reduction of Au(I)–thiolate complexes according to the previous report.27 Other Au25 clusters (TOA+[Au25(SR)18]−) with different thiolate ligands (phenylmethanethiol (PhC1SH), thiophenol (PhSH) and 4-methoxythiophenol (MeOPhSH)) were synthesized using ligand-exchange reactions. Phenylmethanethiolate-protected Au25 cluster anions (TOA+[Au25(SC1Ph)18]−) were successfully synthesized through the ligand-exchange reaction of TOA+[Au25(SC2Ph)18]− with excess amounts of PhC1SH in CH2Cl2. TOA+[Au25(SPh)18]− and TOA+[Au25(SPhOMe)18]− were prepared by the ligand-exchange reaction of the cyclohexanethiolate-protected Au23 cluster (TOA+[Au23(SCy)16]−, SCy = cyclohexanethiolate) with PhSH or MeOPhSH (see the ESI† for detailed synthetic methods).28 A series of thiolate-protected Au25 cluster anions were characterised using UV-vis absorption spectroscopy, 1H NMR, ESI-MS and X-ray absorption fine structure (XAFS) measurements (Fig. 1 and S1–S6†). In negative-mode ESI-MS spectra (Fig. S1†) of TOA+[Au25(SR)18]−, peaks from [Au25(SR)18]− were clearly observed as well as the results of 1H NMR spectroscopy (Fig. S2–S5†), supporting the high purity of the products. UV-vis absorption spectra of all products in tetrahydrofuran (THF) showed similar spectral features with a thiolate-protected Au25 cluster anion,22 that is, the absorption bands at around 400 nm and 700 nm and a shoulder peak at ca. 800 nm (Fig. 1). The absorption peak at around 700 nm, which is mainly derived from a Au13 core-to-core transition,29 was slightly red-shifted from TOA+[Au25(SC2Ph)18]− (682 nm) to TOA+[Au25(SPh)18]− (702 nm). These shifts of absorption bands were caused by the differences in the electronic effects of thiolate ligands.30 By contrast, the shape of absorption bands in the higher energy region was clearly different depending on the SR ligands because of the direct contribution of protecting ligands to the electronic transitions.29 The structural features of a series of Au25 cluster anions were compared using Au-L3 edge XAFS measurements at 10 K with transmission mode (Fig. S6 and Table S1†). Extended XAFS (EXAFS) analysis showed that structures of all Au25 cluster anions employed in this work showed similar structural parameters (Table S1†). Moreover, the solid-state structure of TOA+[Au25(SPh)18]− was clarified using single-crystal X-ray crystallographic analysis (Fig. 2 and S7†). The anionic part in TOA+[Au25(SPh)18]− has a Au13 icosahedral core (Au135+) with six dimeric staple moieties ([Au2(SR)3]−), which are typical components for the Au25 cluster anion. The average Au–Au and Au–S bond lengths, summarized in Table S2,† were comparable to those of previously reported TOA+[Au25(SC2Ph)18]−.16
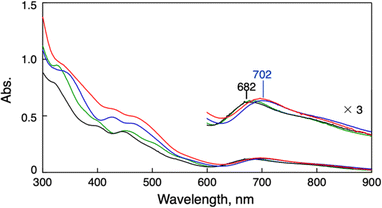 |
| | Fig. 1 UV-vis spectra of TOA+[Au25(SR)18]− (0.010 mM) in THF. SR = SC2Ph (black), SC1Ph (green), SPh (blue) and SPhOMe (red). | |
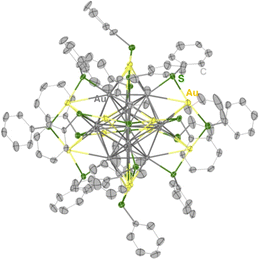 |
| | Fig. 2 A thermal ellipsoid plot (50% probability) of TOA+[Au25(SPh)18]−. The TOA cation, solvent molecules, and hydrogen atoms were omitted for clarity. Atom labels: grey: gold in the Au13 core, yellow: gold on the staple, green: sulfur, and light grey: carbon. | |
To determine the redox potentials of Au25 clusters, cyclic voltammetry (CV) and differential pulse voltammetry on a series of Au25 cluster anions were performed in THF containing TBAPF6 as an electrolyte (Fig. S8†). The cyclic voltammogram of TOA+[Au25(SC2Ph)18]− showed two reversible redox waves at E1/2 = +0.06 V and +0.41 V vs. SCE (Fig. S8a†). These two redox waves could be assigned to the redox couples of Au25−/Au250 and Au250/Au25+, respectively.20 The first E1/2 values were positively shifted with decreasing electron-donating abilities of thiolate ligands;30 for TOA+[Au25(SC1Ph)18]−, the first oxidation wave was observed at +0.16 V vs. SCE owing to the weaker electron-donating ability of SC1Ph than that of SC2Ph. The largest shift of E1/2 (=+0.26 V vs. SCE) was seen for TOA+[Au25(SPh)18]− (Fig. S8d†) with the weakest electron-donating ligands in this work. It should be noted that the E1/2 value in the redox couple of Au25−/Au250 for TOA+[Au25(SPhOMe)18]− (E1/2 = +0.17 V vs. SCE) is almost the same as that for TOA+[Au25(SC1Ph)18]− (Table 1 and Fig. S8b, c†). As well as the redox properties of [Au25(SR)18]−, the affinity with O2 should be one of the essential factors to determine reactivities with O2. The O2-affinities of inorganic nanomaterials would be affected by surface environments including structures of protecting ligands. Thus, the two kinds of [Au25(SR)18]−, with the same redox potentials but different surface ligands, could provide meaningful insights into the correlation between the O2-affinity and the reactivities of Au25 clusters.
Table 1 Summary of redox potentials of TOA+[Au25(SR)18]− in THF containing 0.1 M TBAPF6 under Ar
|
SR
|
E
1/2 (Au25−/Au250) V, vs. SCE |
E
1/2 (Au250/Au25+) V, vs. SCE |
|
SC2Ph
|
+0.06 |
+0.41 |
|
SC1Ph
|
+0.16 |
+0.50 |
|
SPhOMe
|
+0.17 |
+0.41 |
|
SPh
|
+0.26 |
+0.54 |
Air-oxidation reactions of thiolate-protected Au25 clusters
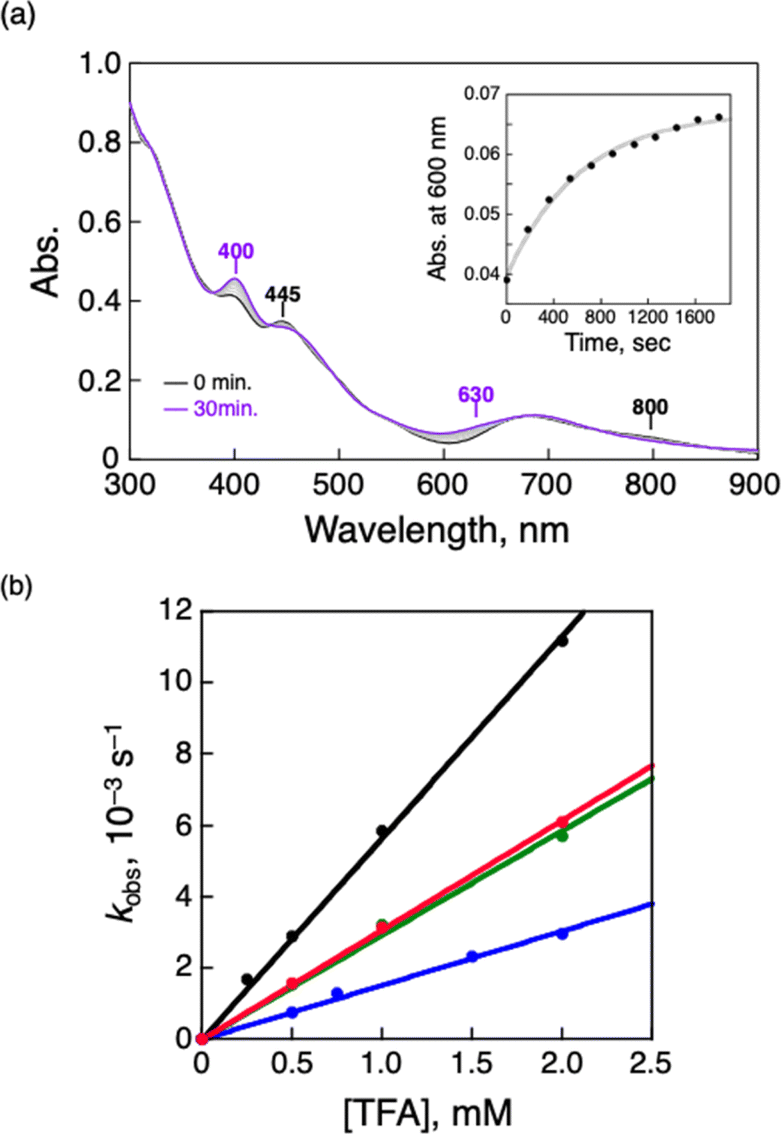
Air-oxidation reactions of TOA+[Au25(SR)18]− were monitored by UV-vis spectroscopic measurements. First, we evaluated the stability of TOA+[Au25(SR)18]− (0.010 mM) in air-saturated THF at 298 K for 24 h (Fig. S9†). The lack of spectroscopic changes indicates that all TOA+[Au25(SR)18]− are highly stable in the absence of any proton sources. Jin and co-workers reported photo-mediated oxidation reactions of [Au25(SC2Ph)18]− under ambient light exposure,13d but such oxidation reactions were not observed in our reaction system. Thus, the light irradiation from the UV-vis spectroscopic device could not induce the photo-induced oxidation reaction and we have excluded the light-mediated effect from our reaction system. By contrast, in the presence of a Brønsted acid like trifluoroacetic acid (TFA, 0.25 mM), the absorption at 445 nm and around 800 nm of TOA+[Au25(SC2Ph)18]− slightly decreased and a new absorption band at around 630 nm appeared (Fig. 3a). In addition, the absorption peak at around 400 nm became sharper. Such spectral change was also observed even in the presence of a small amount of TFA (0.050 mM, Fig. S10†). The UV-vis absorption feature of the final product indicates the formation of [Au25(SC2Ph)18]0, a one-electron oxidized product of [Au25(SC2Ph)18]− (eqn (1)).22 The formation of [Au25(SC2Ph)18]0 was also confirmed by 1H NMR (Fig. S11†) and CV measurements of [Au25(SC2Ph)18]− (Fig. S12†). In the 1H NMR spectrum, air-oxidation of TOA+[Au25(SC2Ph)18]− in THF-d8 gave a new peak at 5.17 ppm (Fig. S11a and b†), which was in good agreement with that from [Au25(SC2Ph)18]0 (Fig. S11c†), supporting the formation of an one-electron-oxidized Au25 cluster. In the electrochemical measurements on TOA+[Au25(SC2Ph)18]−, the open circuit potential (OCP) was determined to be −0.03 V vs. SCE in the absence of TFA, which was more negative than E1/2 (Au25−/Au250) of [Au25(SC2Ph)18]− (+0.06 V vs. SCE). Upon addition of TFA, OCP shifted to +0.21 V vs. SCE, which was positioned between E1/2 (Au25−/Au250) and E1/2 (Au250/Au25+). Moreover, the negative potential sweep from +0.21 V showed a clear reduction wave assigned with the reduction of [Au25(SC2Ph)18]0 to [Au25(SC2Ph)18]−. This electrochemical evidence strongly supports the formation of the neutral Au25 cluster. The resulting [Au25(SC2Ph)18]0 formed under UV-vis spectroscopic conditions could be reduced to [Au25(SC2Ph)18]− quantitatively (Fig. S13†) by reductants such as sodium borohydride (NaBH4). The reversible behaviour of Au25−/Au250 conversion was also observed even at a high TFA concentration (10 mM, Fig. S14†). This complete reversibility indicates that no side reactions such as decompositions of Au25 clusters occur, suggesting that [Au25(SC2Ph)18]− is desirable as a highly durable redox catalyst.| | 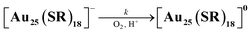 | (1) |
| | | d[Au25(SR)18]/dt = kobs[[Au25(SR)18]−] = k[TFA][[Au25(SR)18]−] | (2) |
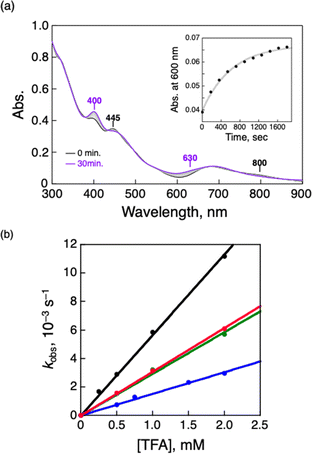 |
| | Fig. 3 (a) UV-vis spectral change of TOA+[Au25(SC2Ph)18]− (0.010 mM) in air-saturated THF containing TFA (0.25 mM) at 298 K. Inset: the time profile of absorbance at 600 nm. (b) [TFA] dependence of kobs in the air-oxidation reaction of TOA+[Au25(SR)18]− in air-saturated THF at 298 K. SR = SC2Ph (black), SC1Ph (green), SPhOMe (red) and SPh (blue). [O2] = 2.2 mM.31 | |
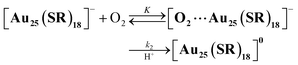
To perform the kinetic analysis in the formation of [Au25(SC2Ph)18]0, the pseudo-first-order rate constant (kobs) was determined to be (1.6 ± 0.1) × 10−3 s−1 at 298 K from the time course of the UV-vis spectral change at 600 nm (Fig. 3a, inset). A linear correlation was observed between the TFA concentration and kobs (Fig. 3b, black), indicating that only one proton was involved in the oxidation reaction. The second-order rate constant (k in eqn (1) and (2)) could be determined to be (5.65 ± 0.06) M−1 s−1 from the slope. It should be noted that no oxidation reaction of [Au25(SC2Ph)18]− occurred in deaerated THF (Fig. S15†). Therefore, O2 should be an oxidant for [Au25(SC2Ph)18]− in this reaction system. In addition, no change in optical properties of [Au25(SC2Ph)18]− upon addition of TFA under the degassed conditions suggests no interaction between the Au25 cluster and TFA (Fig. S15†). Therefore, TFA should play a role in acceleration of Au25 cluster air-oxidation, namely O2 reduction through PCET, in which an electron transfer from [Au25(SC2Ph)18]− to O2 and a proton comes from a Brønsted acid.24 Other TOA+[Au25(SR)18]− (SR = SC1Ph, SPh and SPhOMe) were also oxidized to form corresponding neutral [Au25(SR)18]0 in air-saturated THF containing TFA, as observed by the UV-vis spectral change (Fig. S16†). In addition, each [Au25(SR)18]0 could be fully recovered to the corresponding anionic species by the addition of NaBH4, indicating high durability of these Au25−/Au250 redox reaction systems (Fig. S17†). Kinetic analyses were performed to determine k values (Fig. 3b), which are summarized in Table 2. The oxidation reaction of [Au25(SC2Ph)18]− with the lowest one-electron oxidation potential (+0.06 V vs. SCE, Table 1) showed the highest k value, whereas the lowest k value was confirmed in TOA+[Au25(SPh)18]− with the highest E1/2 among TOA+[Au25(SR)18]− in this work. The reactivities of TOA+[Au25(SC1Ph)18]− and TOA+[Au25(SPhOMe)18]− were almost the same despite the different types of SR moieties (benzylic and aromatic thiolate, respectively). Considering the similar E1/2 values between TOA+[Au25(SC1Ph)18]− and TOA+[Au25(SPhOMe)18]− (Table 1), the redox potential of TOA+[Au25(SR)18]− should be one of the main controlling factors to determine the reactivities with O2.
| |  | (3) |
| | 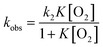 | (4) |
Table 2 Summary of rate constants (k and k2) and equilibrium constants (K) in the air oxidation of TOA+[Au25(SR)18]− in THF in the presence of TFAa
|
SR
|
k, M−1 s−1 |
k
2, s−1 |
K, mM−1 |
|
At 298 K. [Au25(SR)18]− = 0.010 mM.
|
|
SC2Ph
|
5.65 ± 0.06 |
9.9 ± 0.3 |
0.20 ± 0.01 |
|
SC1Ph
|
2.93 ± 0.08 |
4.2 ± 0.1 |
0.26 ± 0.03 |
|
SPhOMe
|
3.19 ± 0.01 |
3.67 ± 0.03 |
0.34 ± 0.01 |
|
SPh
|
1.51 ± 0.02 |
1.35 ± 0.01 |
0.55 ± 0.01 |
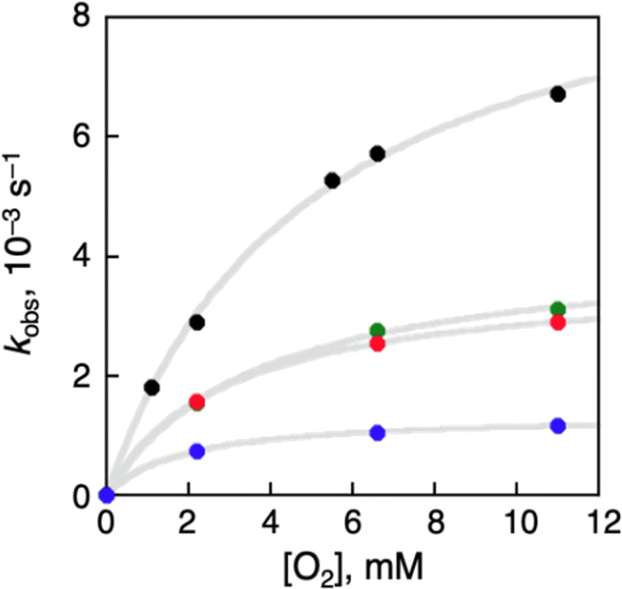
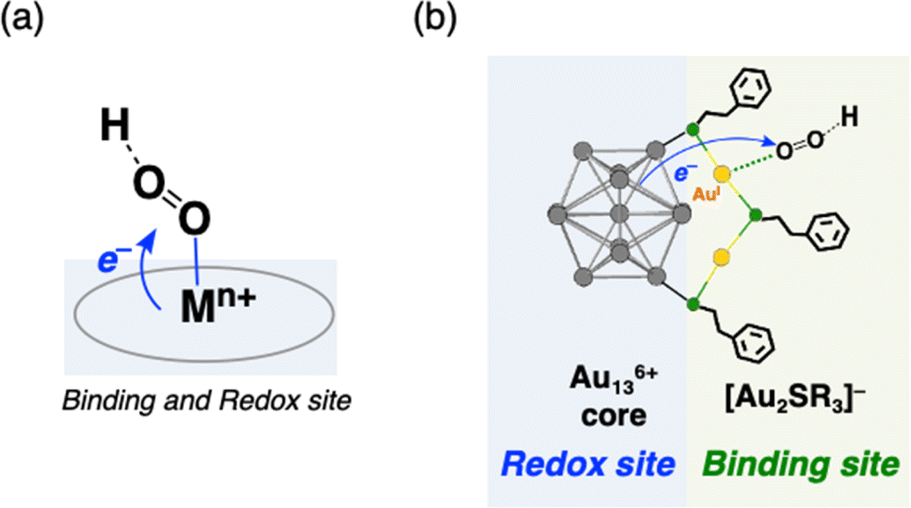
Häkkinen and co-workers demonstrated that small molecules such as O2 were able to access the surface of ligand-protected AuNCs, which would play an important role in small molecule activation.32 Then, to examine the interaction of the surface of [Au25(SR)18]− with O2 during air-oxidation reactions, kinetic analyses were performed by changing the concentration of O2 in THF (Fig. 4). The pseudo-first-order rate constants (kobs) of the air-oxidation reaction of TOA+[Au25(SR)18]− showed saturation behaviour on increasing the concentration of O2, suggesting the existence of a pre-equilibrium between TOA+[Au25(SR)18]− and O2 (eqn (3)). Based on eqn (4),33 pre-equilibrium constants (K), namely O2 binding constants with Au25 cluster anions, and second-order rate constants (k2) were determined and are summarized in Table 2. In the case of TOA+[Au25(SC2Ph)18]−, k2 and K were (9.9 ± 0.3) s−1 and (0.20 ± 0.01) mM−1 at [TFA] = 0.50 mM, respectively. When changing the concentration of TFA to 2.0 mM, the K value was retained (0.18 ± 0.04 mM−1), whereas the k2 value increased ((44 ± 4) s−1) (Fig. S18†). No influence of the acid concentration on the O2 binding constant suggests that protons are not involved in the pre-equilibrium between the Au25 cluster and O2 (eqn (3)). The highest O2 binding constant (0.55 ± 0.01 mM−1) was observed in the air-oxidation reaction of TOA+[Au25(SPh)18]−, whereas TOA+[Au25(SC2Ph)18]− showed the lowest O2-binding properties (0.20 ± 0.01 mM−1). In other words, Au25 cluster anions protected by rigid aromatic thiolate ligands showed higher O2 binding constants than those with flexible aliphatic thiolate ligands. The affinity of O2 would be regulated by the electronic effects and/or the steric effects of R moieties of thiolate ligands. In mononuclear metal complexes, more electron-donating ligands increased the O2-binding constants,34 whereas an opposite tendency was observed in our system, in that more electron-donating ligands such as SC2Ph gave reduced K values. This difference between MNCs and transition metal complexes would be caused by the binding position of O2: in mononuclear metal complexes, because both the O2-binding and the redox reactions generally take place at the same sites, namely the metal centres (Fig. 5a), the affinity of O2 should be affected by the electronic states of metal centres. Although experimental evidence for O2 binding sites is insufficient due to the weak electronic interaction between O2 and AuNCs13d and further spectral and structural analyses for O2-bound Au25 clusters are needed, some theoretical reports discussed that O2 would bind with the gold atom of staple Au2(SR)3 moieties of [Au25(SR)18]− in an end-on coordinating fashion rather than the Au13 icosahedral core.13d,17d It should be noted that the one-electron oxidation of [Au25(SR)18]− occurs at the Au13 core, not at the staple Au2(SR)3 units.19b Therefore, we could think that the O2-binding and the redox sites are different in the ligand-protected MNCs, suggesting almost no electronic effects on the O2 affinity (Fig. 5b). To evaluate the steric effects of protecting ligands, ligand flexibility such as free rotation of alkyl chains should also be considered.35 We compared the half cone angles (θ) of [Au25(SC2Ph)18]− and [Au25(SPh)18]− based on the atomic structures revealed by X-ray analysis. The θ value is defined as the averaged C–Au–S angle, where C is the carbon atom at the ortho-position of the phenyl ring of SC2Ph or SPh ligands (Fig. S19†). The θ (63.2°) for [Au25(SC2Ph)18]− is larger than that for [Au25(SPh)18]− (50.3°), indicating the sterically more crowded environment on the surface of [Au25(SC2Ph)18]−. Based on the above discussion, it would be reasonable to explain that the steric effects of thiolate moieties, their bulkiness or flexibility, are dominant factors in determining the O2-affinity with a Au25 cluster anion. This conclusion suggests that O2 would be favourably bound at a less steric gold(I) ion at the staple moiety rather than at sterically crowded metal-core gold atoms, which does not contradict with the theoretical analysis.13d,17d
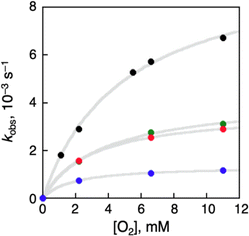 |
| | Fig. 4 [O2] dependence of kobs in oxidation reactions of TOA+[Au25(SR)18]− in THF ([TFA] = 0.50 mM) at 298 K. SR = SC2Ph (black), SC1Ph (green), SPhOMe (red) and SPh (blue). | |
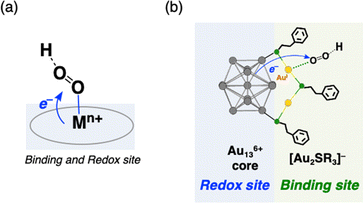 |
| | Fig. 5 Schematic illustrations of the interaction and reduction of O2 with (a) transition metal complexes and (b) [Au25(SR)18]− through PCET. In (b), only one staple moiety (Au2SR3 unit) is shown for clarity. Atom labels: grey: gold in the Au13 core, yellow: gold on the staple, and green: sulfur. | |
Finally, we considered the controlling factors in determining the reactivities of Au25 clusters. As seen in the comparison of the kinetic parameters (K and k2) of [Au25(SC2Ph)18]− with those of [Au25(SPh)18]− (Table 2), the electronic effects of Au25 clusters, evaluated using redox potentials (E1/2), are a dominant factor in regulating the reaction with O2. Indeed, a linear relationship was observed between k2 values and E1/2 (Au25−/Au250), as shown in Fig. S20.† However, interestingly, a different aspect could be seen when the influences of O2 affinity on the reactivities were extracted from the kinetic parameters of TOA+[Au25(SC1Ph)18]− and TOA+[Au25(SPhOMe)18]−: a higher K value (0.34 ± 0.01 mM−1) was observed in TOA+[Au25(SPhOMe)18]−, whereas the k2 value of TOA+[Au25(SPhOMe)18]− (3.67 ± 0.03 s−1) was slightly lower than that of TOA+[Au25(SC1Ph)18]− (4.2 ± 0.1 s−1), resulting in comparable reactivities of these two Au25 clusters. Thus, the steric effects of protecting ligands could also contribute to tuning the reactivities of the Au25 clusters against O2. For ca. 3 nm gold nanoparticles, the reactivities with O2 were simply correlated with O2-binding constants in pre-equilibrium,12c which were regulated using the accessibility to active sites. In contrast, the oxidation reactivities of [Au25(SR)18]− with O2 could be controlled over both steric and electronic effects. This dual effect on the reactivity enables us to construct finely tunable reaction systems by protecting ligand design, which should be important for precisely controlled catalytic systems based on inorganic nanomaterials.
Conclusions
We have compared the air-oxidation reactivities of a series of thiolate-protected Au25 nanoclusters bearing different ligands. Kinetic analysis for the oxidation of Au25− to Au250 has quantitatively revealed that the O2-affinity was regulated by the accessibility of surface gold atoms surrounded by protecting ligands, similar to other inorganic nanomaterials. On the other hand, the total reactivities with O2 can also be tuned by the electronic states of Au25 clusters such as transition metal complexes. This duality in controlling factors for reactivities allows us to construct precisely controlled reaction systems based on metal nanoclusters, which can be achieved by the rational design of protecting ligands. These results shed new light on ligand-protected MNCs with sizes of less than 2 nm to construct finely tunable reaction systems, in contrast to the case of reaction systems based on larger MNPs. Ligand designs, considering ligand–ligand or ligand–substrate interactions, should be effective for more precise control over the reactivities of MNCs.
Data availability
Additional experimental data supporting this article are included in the ESI.† Reasonable requests for additional information can be made to the corresponding authors.
Author contributions
W. S. designed the research, performed the synthesis and characterisation and wrote the manuscript. R. T. performed EXAFS measurements using the synchrotron. Y. M. and N. T. performed single crystal X-ray diffraction measurement and analysis. S. X. contributed to the discussion and editing the manuscript. T. T. supervised the research. All authors have approved the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
XAFS measurements were performed at the BL01B1 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). This work was financially supported by the MEXT/Japan Society for the Promotion of Science (JSPS), KAKENHI, for Scientific Research (S) (grant no. JP19H05634 and JP24H00053) (T. T.), a JSPS Research Fellowship (grant no. 20J01921), JSPS KAKENHI for Early-Career Scientists (grant no. JP23K13765), International Collaboration Research Project of the Institute for Chemical Research, Kyoto University (grant no. 2024-119), and the Tokuyama Science Foundation (W. S.). We thank Ian McNaught, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
References
-
(a) M. L. Peigs, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef PubMed;
(b) H. Sterckx, B. Morel and B. U. W. Maes, Angew. Chem., Int. Ed., 2019, 58, 7946–7970 CrossRef CAS PubMed.
-
(a) D. J. Martin, C. F. Wise, M. L. Peigs and J. M. Mayer, Acc. Chem. Res., 2020, 53, 1056–1065 CrossRef CAS PubMed;
(b) A. Rana, Y.-M. Lee, X. Li, R. Cao, S. Fukuzumi and W. Nam, ACS Catal., 2021, 11(5), 3073–3083 CrossRef CAS;
(c) S. Fukuzumi, L. Tahsini, Y.-M. Lee, K. Ohkubo, W. Nam and K. D. Karlin, J. Am. Chem. Soc., 2012, 134, 7025–7035 CrossRef CAS PubMed.
-
(a) I. Hatay, B. Su, M. A. Méndez, C. Corminboeuf, T. Khoury, C. P. Gross, M. Bourdillon, M. Meyer, J.-M. Barbe, M. Ersoz, S. Záliš, Z. Samec and H. H. Girault, J. Am. Chem. Soc., 2010, 132, 13733–13741 CrossRef CAS PubMed;
(b) K. Mase, K. Ohkubo, Z. Xue, H. Yamada and S. Fukuzumi, Chem. Sci., 2015, 6, 6496–6504 RSC;
(c) E. Aoki, W. Suzuki, H. Kotani, T. Ishizuka, H. Sakai, T. Hasobe and T. Kojima, Chem. Commun., 2019, 55, 4925–4928 RSC.
-
(a) Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed;
(b) C. Xie, Z. Niu, D. Kim, M. Li and P. Yang, Chem. Rev., 2020, 120, 1184–1249 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 405–408 CrossRef CAS.
-
(a) M. K. Carpenter, T. E. Moylan, R. S. Kukreja, M. H. Atwan and M. M. Tessema, J. Am. Chem. Soc., 2012, 134, 8535–8542 CrossRef CAS PubMed;
(b) M. Daka, M. Ferrara, M. Bevilacqua, P. Pengo, P. Rajak, R. Ciancio, T. Montini, L. Pasquato and P. Fornasiero, ACS Appl. Nano Mater., 2022, 5, 4710–4720 CrossRef CAS.
-
(a) Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed;
(b) R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
-
(a) Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915–1920 CrossRef CAS PubMed;
(b) K. Harano, S. Takano and T. Tsukuda, Chem. Commun., 2019, 55, 15033–15036 RSC;
(c) S. Tian, Y. Cao, T. Chen, S. Zang and J. Xie, Chem. Commun., 2020, 56, 1163–1174 RSC;
(d) S. Zhuang, D. Chen, W.-P. Ng, D. Liu, L.-J. Liu, M.-Y. Sun, T. Nawaz, X. Wu, Y. Zhang, Z. Li, Y.-L. Huang, J. Yang and J. He, JACS Au, 2022, 2, 2617–2626 CrossRef CAS PubMed.
-
(a) H. Tsunoyama, A. Ohnuma, K. Takahashi, A. Velloth, M. Ehara, N. Ichikuni, M. Tabuchi and A. Nakajima, Chem. Commun., 2019, 55, 12603–12606 RSC;
(b) T. Kawawaki, N. Shimizu, K. Funai, Y. Mitomi, S. Hossain, S. Kikkawa, D. J. Osborn, S. Yamazoe, G. F. Metha and Y. Negishi, Nanoscale, 2021, 13, 14679–14687 RSC.
-
(a) R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu and J. Zubieta, J. Am. Chem. Soc., 1988, 110, 3690–3692 CrossRef CAS;
(b) T. Chishiro, Y. Shimazaki, F. Tani, Y. Tachi, Y. Naruta, S. Karasawa, S. Hayami and Y. Maeda, Angew. Chem., Int. Ed., 2003, 42, 2788–2791 CrossRef CAS PubMed;
(c) J. Cho, R. Sarangi, H. Y. Kang, J. Y. Lee, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2010, 132, 16977–16986 CrossRef CAS PubMed.
-
(a) H. Kotani, T. Yagi, T. Ishizuka and T. Kojima, Chem. Commun., 2015, 51, 13385–13388 RSC;
(b) C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, J. Am. Chem. Soc., 2016, 138, 4186–4193 CrossRef CAS PubMed;
(c) W. Suzuki, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141(14), 5987–5994 CrossRef CAS PubMed;
(d) W. Suzuki, H. Kotani, T. Ishizuka and T. Kojima, Chem.–Eur. J., 2020, 26(46), 10480–10486 CrossRef CAS PubMed.
-
(a) A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed;
(b) C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2011, 133, 9938–9947 CrossRef CAS PubMed;
(c) C. G. Long, J. D. Gilbertson, G. Vijayaraghavan, K. J. Stevenson, C. J. Pursell and B. D. Chandler, J. Am. Chem. Soc., 2008, 130, 10103–10115 CrossRef CAS PubMed.
-
(a) A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9578 CrossRef;
(b) H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed;
(c) Y. Zhu, H. Qian and R. Jin, Chem.–Eur. J., 2010, 16, 11455–11462 CrossRef CAS PubMed;
(d) D. R. Kauffman, D. Alfonso, C. Matranga, G. Li and R. Jin, J. Phys. Chem. Lett., 2013, 4, 195–202 CrossRef CAS PubMed;
(e) Z. Wu, G. Hu, D. Jiang, D. R. Mullins, Q.-F. Zhang, L. F. Allard Jr, L.-S. Wang and S. H. Overbury, Nano Lett., 2016, 16, 6550–6567 Search PubMed;
(f) D. A. Pichugina, N. A. Nikitina and N. E. Kuźmenko, J. Phys. Chem. C, 2020, 124, 3080–3086 CrossRef CAS.
-
(a) Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed;
(b) S. Yamazoe, S. Takano, W. Kurashige, T. Yokoyama, K. Nitta, Y. Negishi and T. Tsukuda, Nat. Commun., 2016, 7, 10414 CrossRef CAS PubMed;
(c) I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
-
(a) G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed;
(b) R. R. Nasaruddin, Q. Yao, T. Chen, M. J. Hülsey, N. Yan and J. Xie, Nanoscale, 2018, 10, 23113–23121 RSC;
(c) Q. Yao, Z. Wu, Z. Liu, Y. Lin, X. Yuan and J. Hie, Chem. Sci., 2021, 12, 99–127 RSC;
(d) H. Shan, J. Shi, T. Chen, Y. Cao, Q. Yao, H. An, Z. Yang, Z. Wu, Z. Jiang and J. Xie, ACS Nano, 2023, 17, 2368–2377 CrossRef CAS PubMed.
-
(a) M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed;
(b) M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
-
(a) Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC;
(b) B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC;
(c) W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed;
(d) F. Sun, C. Deng, S. Tian and Q. Tang, ACS Catal., 2021, 11, 7957–7969 CrossRef CAS.
- Y. Saito, Y. Shichibu and K. Konishi, Nanoscale, 2021, 13, 9971–9977 RSC.
-
(a) T. A. Dreier, O. A. Wong and C. J. Aikens, Chem. Commun., 2015, 51, 1240–1243 RSC;
(b) M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS PubMed.
-
(a) S. Antonello, A. H. Holm, E. Instuli and F. Maran, J. Am. Chem. Soc., 2007, 129, 9836–9837 CrossRef CAS PubMed;
(b) S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585–15594 CrossRef CAS PubMed;
(c) S. Antonello, G. Arrigoni, T. Dainese, M. De Nardi, G. Parisio, L. Perotti, A. René, A. Venzo and F. Maran, ACS Nano, 2014, 8, 2788–2795 CrossRef CAS PubMed;
(d) K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- S. Bhat, R. Pradeep, N. Ananya, P. Chakraborty, G. Paramasivam, R. R. J. Methikkalam, A. Nag, G. Natarajan and T. Pradeep, J. Phys. Chem. C, 2018, 122, 19455–19462 CrossRef CAS.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CrossRef CAS.
-
(a) X. Yuan, N. Goswami, W. Chen, Q. Yao and J. Xie, Chem. Commun., 2016, 52, 5234–5237 RSC;
(b) Z. Lei, J.-J. Li, Z.-A. Nan, Z.-G. Jiang and Q.-M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14415–14419 CrossRef CAS PubMed;
(c) W. Suzuki, R. Takahata, Y. Chiga, S. Kikkawa, S. Yamazoe, Y. Mizuhata, N. Tokitoh and T. Teranishi, J. Am. Chem. Soc., 2022, 144, 12310–12320 CrossRef CAS PubMed.
-
(a) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed;
(b) S. Fukuzumi, Y.-M. Lee and W. Nam, ChemCatChem, 2018, 10, 9–28 CrossRef CAS.
- W. Suzuki, H. Kotani, T. Ishizuka and T. Kojima, Chem.–Eur. J., 2020, 26, 10480–10486 CrossRef CAS PubMed.
- K. Mase, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2800–2808 CrossRef CAS PubMed.
- J. F. Parker, J. E. F. Weaver, F. McCallum, C. A. Fields-Zinna and R. W. Murray, Langmuir, 2010, 26(16), 13650–13654 CrossRef CAS PubMed.
- M. P. Maman, A. S. Nair, A. M. A. H. Nazeeja, B. Pathak and S. Mandal, J. Phys. Chem. Lett., 2020, 11, 10052 CrossRef CAS PubMed.
- C. M. Aikens, J. Phys. Chem. Lett., 2010, 1, 2594–2599 CrossRef CAS.
-
(a) R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef CAS PubMed;
(b) Z. Liu, Y. Li, E. Kahng, S. Xue, X. Du, S. Li and R. Jin, ACS Nano, 2022, 16, 18448–18458 CrossRef CAS PubMed.
- D. Das, Y.-M. Lee, K. Ohkubo, W. Nam, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 2825–2834 CrossRef CAS PubMed.
-
(a) X. Chen and H. Häkkinen, J. Am. Chem. Soc., 2013, 135, 12944–12947 CrossRef CAS PubMed;
(b) O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329–334 CrossRef CAS PubMed.
- T. Ishizuka, S. Ohzu, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Chem. Sci., 2014, 5, 1429–1436 RSC.
- M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed.
- J. A. Bilbrey, A. H. Kazez, J. Locklin and W. D. Allen, J. Comput. Chem., 2013, 34, 1189–1197 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2299526. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02995j |
| ‡ Present Address: University of Hyogo, Himeji, 671-2280, Japan. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ryo
Takahata
a,
Ryo
Takahata