
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phosphasilene mediated CO activation and deoxygenative homo coupling of CO molecules in reactions with metal carbonyls†
Zohreh
Hendi
a,
Renuka
Pradhan
b,
Katharina
Rachuy
a,
Soheil
Mahmoudi
 cd,
Madhusudan K.
Pandey
a,
Saroj Kumar
Kushvaha
a,
Regine
Herbst-Irmer
a,
Upakarasamy
Lourderaj
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
cd,
Madhusudan K.
Pandey
a,
Saroj Kumar
Kushvaha
a,
Regine
Herbst-Irmer
a,
Upakarasamy
Lourderaj
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Göttingen, 37077, Germany. E-mail: hroesky@gwdg.de; dstalke@chemie.uni-goettingen.de
bSchool of Chemical Sciences, National Institute of Science Education and Research (NISER) Bhubaneswar, Bhubaneswar, India. E-mail: u.lourderaj@niser.ac.in
cUniversity of Vienna, Faculty of Chemistry, Institute of Inorganic Chemistry, Waehringer Str. 42, Vienna 1090, Austria
dUniversity of Vienna, Vienna Doctoral School in Chemistry (DoSChem), Waehringer Str. 42, 1090 Vienna, Austria
First published on 10th October 2024
Abstract
Herein, we report the synthesis of a new sterically demanding hyper-coordinate phosphasilene (Mes*PSi(SiMe3)(PhC(NtBu)2) (1) and its unprecedented reactivity with metal carbonyls (M = Fe, Mo, W). The reaction of 1 with Fe(CO)5 involves the deoxygenative homocoupling of two CO molecules, forming a rare ketene (μ-CCO) inserted Fe complex 2. Contrastingly, reactions with M(CO)6 (M = Mo, W) entail the deoxygenated activation of one CO molecule, with the second CO molecule being trapped between Si and P atoms. All the compounds including their crystal structures, are thoroughly characterized and potential energy profiles for the reaction mechanisms are also explored.
Introduction
Phosphasilenes (RP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiR2), heavier analogs of imines, are highly reactive compounds that show unique reactivity with the main-group elements and transition metals.1,2 Following the first spectroscopic analysis by Bickelhaupt in 1984 (ref. 3) and the subsequent structural determination by Niecke in 1993,4 several efforts have been undertaken to isolate stable phosphasilenes and investigate their reactivity.2,5–11 These investigations mainly concentrate on two types: (i) with the main-group and transition metals such as Au, Zn, Cr, Mo, W, and Pb, phosphasilenes often exhibit κ1-P-coordination to the metal center12–14 whereas in the case of M(0) salts (M = Ni, Pd, Pt), rearrangement occurs through cleavage of the phosphorus-silicon double bond (Scheme 1a), and (ii) with small organic molecules such as ketones, aldehydes, P4, acetylenes, nitriles, and azides which resulted in the insertion of these molecules into the SiP double bond.2,15–18
SiR2), heavier analogs of imines, are highly reactive compounds that show unique reactivity with the main-group elements and transition metals.1,2 Following the first spectroscopic analysis by Bickelhaupt in 1984 (ref. 3) and the subsequent structural determination by Niecke in 1993,4 several efforts have been undertaken to isolate stable phosphasilenes and investigate their reactivity.2,5–11 These investigations mainly concentrate on two types: (i) with the main-group and transition metals such as Au, Zn, Cr, Mo, W, and Pb, phosphasilenes often exhibit κ1-P-coordination to the metal center12–14 whereas in the case of M(0) salts (M = Ni, Pd, Pt), rearrangement occurs through cleavage of the phosphorus-silicon double bond (Scheme 1a), and (ii) with small organic molecules such as ketones, aldehydes, P4, acetylenes, nitriles, and azides which resulted in the insertion of these molecules into the SiP double bond.2,15–18
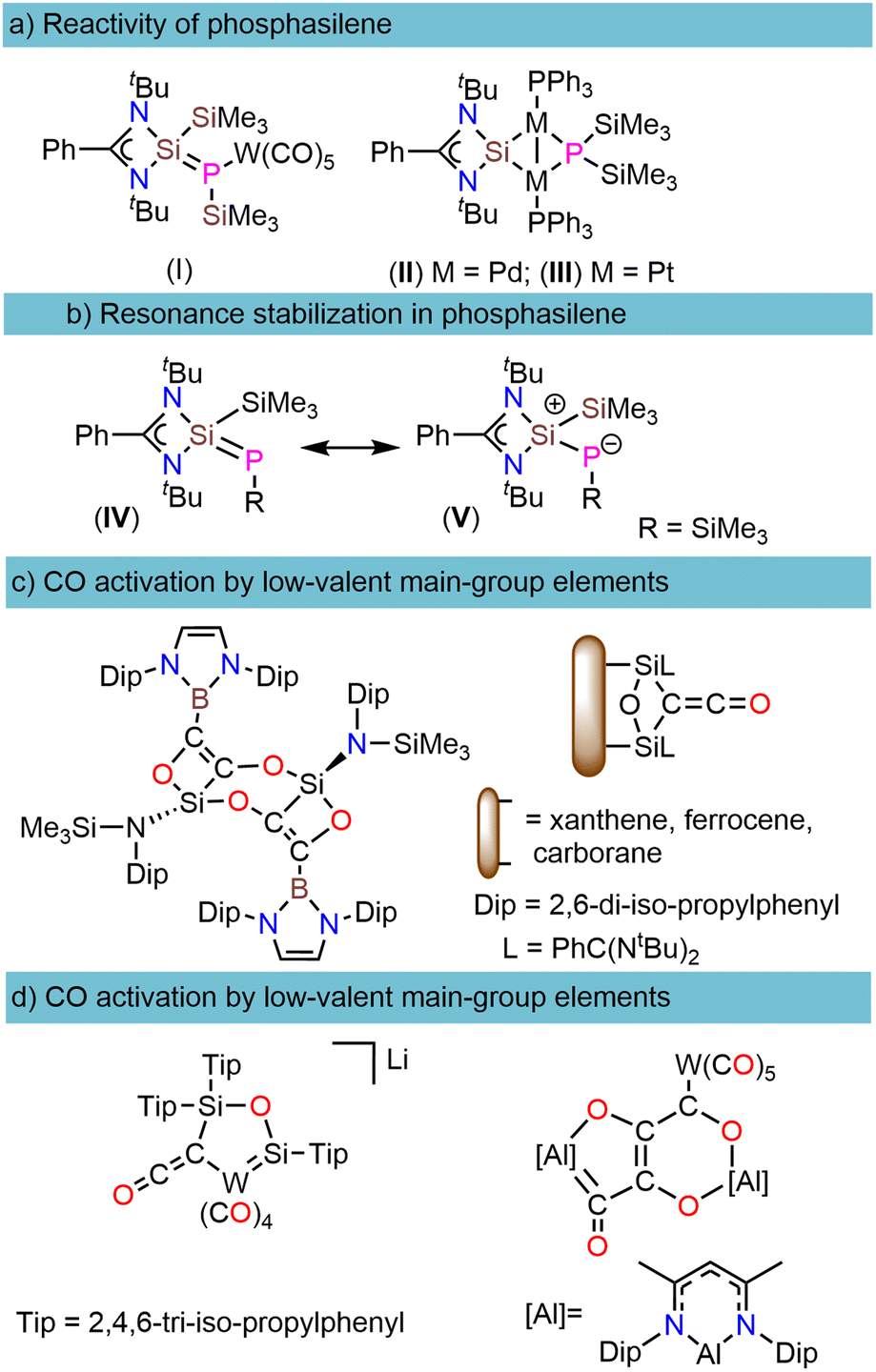 | ||
| Scheme 1 (a) Reactivity of phosphasilenes with early and late transition metals. (b) Resonance stabilization in phosphasilene (c) CO activation by low-valent main group B and Si elements. (d) CO activation by low-valent main group Al and Si elements assisting metal carbonyls and a reductant. | ||
The unusual reactivities of phosphasilenes can be attributed to the polarization of the (–P(δ−)Si(δ+)<) double bond (Scheme 1b)16 and small HOMO–LUMO energy gap, which is achieved through modifications of the substituents at the P and Si atoms, and by the coordination of a P lone pair to transition metals or main-group elements.2 However, to the best of our knowledge, reactivity studies of phosphasilenes with small molecules such as CO, CO2, N2, and H2 are currently lacking. Among these small molecules, CO activation/functionalization represents one of the most important strategies for utilizing CO molecule as a C1 source in producing bulk and fine chemicals.19
It is common knowledge that transition metal complexes play a major role in the activation of the CO bond due to its high C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond dissociation energy (BDE = 1077 kJ mol−1).20,21 In recent years, low-valent main-group elements have shown tremendous development in CO activation chemistry.20 Among different types of main group compounds that demonstrate the capability of CO activation, such as B, Si, and Al, silicon in the form of silylenes have exhibited exceptional efficiency that can be attributed to the small HOMO–LUMO energy gap, a lone pair, and a free p-orbital at the silicon center.22–26 As depicted in (Scheme 1c), in such reactions, carbon monoxide (CO) reduction is succeeded by CO homologation, leading to the formation of (CnOx) chains, which play a pivotal role in generating carbon building blocks.24–26 It is worth noting that CO homologation can be achieved not only through the use of transition metal complexes or low-valent main group elements but also in conjunction with auxiliary metal carbonyls and a reductant (Scheme 1d).27,28
O bond dissociation energy (BDE = 1077 kJ mol−1).20,21 In recent years, low-valent main-group elements have shown tremendous development in CO activation chemistry.20 Among different types of main group compounds that demonstrate the capability of CO activation, such as B, Si, and Al, silicon in the form of silylenes have exhibited exceptional efficiency that can be attributed to the small HOMO–LUMO energy gap, a lone pair, and a free p-orbital at the silicon center.22–26 As depicted in (Scheme 1c), in such reactions, carbon monoxide (CO) reduction is succeeded by CO homologation, leading to the formation of (CnOx) chains, which play a pivotal role in generating carbon building blocks.24–26 It is worth noting that CO homologation can be achieved not only through the use of transition metal complexes or low-valent main group elements but also in conjunction with auxiliary metal carbonyls and a reductant (Scheme 1d).27,28
These reductants encompass a spectrum ranging from metallocenes (M = Zr, Hf, Sm) to KC8.27,29 For these types of reactions, there are reports documenting the use of M(I) (M = Al, Mg) complexes and compounds with SiSi bond, which pose considerable synthetic challenges.28,30,31 To the best of our knowledge, there have been no reports of using phosphasilenes in this type of reaction, either directly or in conjunction with another transition metal.
Given the fact that the polarization of the double bond in phosphasilenes can be adjusted and since they have important characteristics for CO activation, such as a low HOMO–LUMO energy gap, an electrophilic silicon center, and the ability to interact with transition metals, we synthesized a sterically demanding base-stabilized phosphasilene (Mes*PSi(SiMe3)(PhC(NtBu)2) (1) and studied its reactivity with CO (gas) and metal carbonyls (M = Fe, Mo and W). Although base-stabilized phosphasilene 1 does not show any reaction with CO (gas), it interestingly facilitates CO activation and deoxygenative homo coupling of CO molecules when treated with metal carbonyls, leading to the formation of the rare complexes 2–4. The details are described.
Results and discussion
The heterolyptic chlorosilylene [(PhC(NtBu)2SiCl]32 and Mes*PLi(SiMe3) (Mes* = 2,4,6-tBu3C6H2) were first chosen as the starting precursors. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt metathesis reaction of [(PhC(NtBu)2SiCl] with an in situ generated Mes*PLi(SiMe3) salt in THF unexpectedly resulted in the formation of phosphasilene (Mes*PSi(SiMe3)(PhC(NtBu)2) (1) as orange-yellow solid in good yield, in place of phosphinosilylene (Mes*P(SiMe3)Si(PhC(NtBu)2) (Scheme 2a). This contrasts with the previous report, where phosphasilene formation could only be achieved by heating the phosphinosilylene over 100 °C for approximately four days, facilitating the transfer of the SiMe3 group from phosphorus to the Si atom.33 This interesting difference in reactivity might be due to the presence of the bulky Mes* group at the phosphorus center. Interestingly, phosphasilene 1 can be readily synthesized in a single step by heating a toluene solution of (Mes*)P(SiMe3)2 and [(PhC(NtBu)2SiCl] in a 1:1 molar ratio for 8 hours (see ESI† for details). This straightforward synthesis of 1 contrasts prior studies, where phosphasilene formation was typically achieved under harsh reaction conditions, using harsh reducing agents or with the help of catalysts and bases during the reaction.10,34–36 It should be noted that 1 is a highly air and moisture-sensitive compound. However, it is stable in solid and solution states under an inert atmosphere at room temperature for months, and no decomposition was observed. Furthermore, the phosphasilene 1 exhibits very high thermal stability and shows no signs of decomposition when heated up to 120 °C. Considering the mentioned properties, we decided to treat compound 1 with transition metal carbonyl complexes (Scheme 2b) and compare its reactivity with previously reported investigations. Surprisingly, unlike most reported reactions involving phosphasilenes that primarily exhibit simple κ1-P-coordination, this compound undergoes a unique reaction with CO groups released during the reaction with metal carbonyls, resulting in the formation of compounds 2–4 and a side product, which according to NMR and IR spectroscopies and mass spectrometry data, is considered to be an oxygenated silylene compound which will be discussed later. Although phosphasilene 1 does not react with CO (gas), interestingly, when treated with Fe(CO)5, it afforded a rare Fe complex 2. In this reaction, two CO molecules released during the reaction couple together to form a ketene μ-(CCO). This ketene is trapped between Fe and Si atoms in 2. Additionally, silanone derivative either in the form of monomer or dimer/trimer {PhC(NtBu)2Si(SiMe3)O–}n (n = 1–3) is eliminated as a side product during the reaction.
:1 salt metathesis reaction of [(PhC(NtBu)2SiCl] with an in situ generated Mes*PLi(SiMe3) salt in THF unexpectedly resulted in the formation of phosphasilene (Mes*PSi(SiMe3)(PhC(NtBu)2) (1) as orange-yellow solid in good yield, in place of phosphinosilylene (Mes*P(SiMe3)Si(PhC(NtBu)2) (Scheme 2a). This contrasts with the previous report, where phosphasilene formation could only be achieved by heating the phosphinosilylene over 100 °C for approximately four days, facilitating the transfer of the SiMe3 group from phosphorus to the Si atom.33 This interesting difference in reactivity might be due to the presence of the bulky Mes* group at the phosphorus center. Interestingly, phosphasilene 1 can be readily synthesized in a single step by heating a toluene solution of (Mes*)P(SiMe3)2 and [(PhC(NtBu)2SiCl] in a 1:1 molar ratio for 8 hours (see ESI† for details). This straightforward synthesis of 1 contrasts prior studies, where phosphasilene formation was typically achieved under harsh reaction conditions, using harsh reducing agents or with the help of catalysts and bases during the reaction.10,34–36 It should be noted that 1 is a highly air and moisture-sensitive compound. However, it is stable in solid and solution states under an inert atmosphere at room temperature for months, and no decomposition was observed. Furthermore, the phosphasilene 1 exhibits very high thermal stability and shows no signs of decomposition when heated up to 120 °C. Considering the mentioned properties, we decided to treat compound 1 with transition metal carbonyl complexes (Scheme 2b) and compare its reactivity with previously reported investigations. Surprisingly, unlike most reported reactions involving phosphasilenes that primarily exhibit simple κ1-P-coordination, this compound undergoes a unique reaction with CO groups released during the reaction with metal carbonyls, resulting in the formation of compounds 2–4 and a side product, which according to NMR and IR spectroscopies and mass spectrometry data, is considered to be an oxygenated silylene compound which will be discussed later. Although phosphasilene 1 does not react with CO (gas), interestingly, when treated with Fe(CO)5, it afforded a rare Fe complex 2. In this reaction, two CO molecules released during the reaction couple together to form a ketene μ-(CCO). This ketene is trapped between Fe and Si atoms in 2. Additionally, silanone derivative either in the form of monomer or dimer/trimer {PhC(NtBu)2Si(SiMe3)O–}n (n = 1–3) is eliminated as a side product during the reaction.
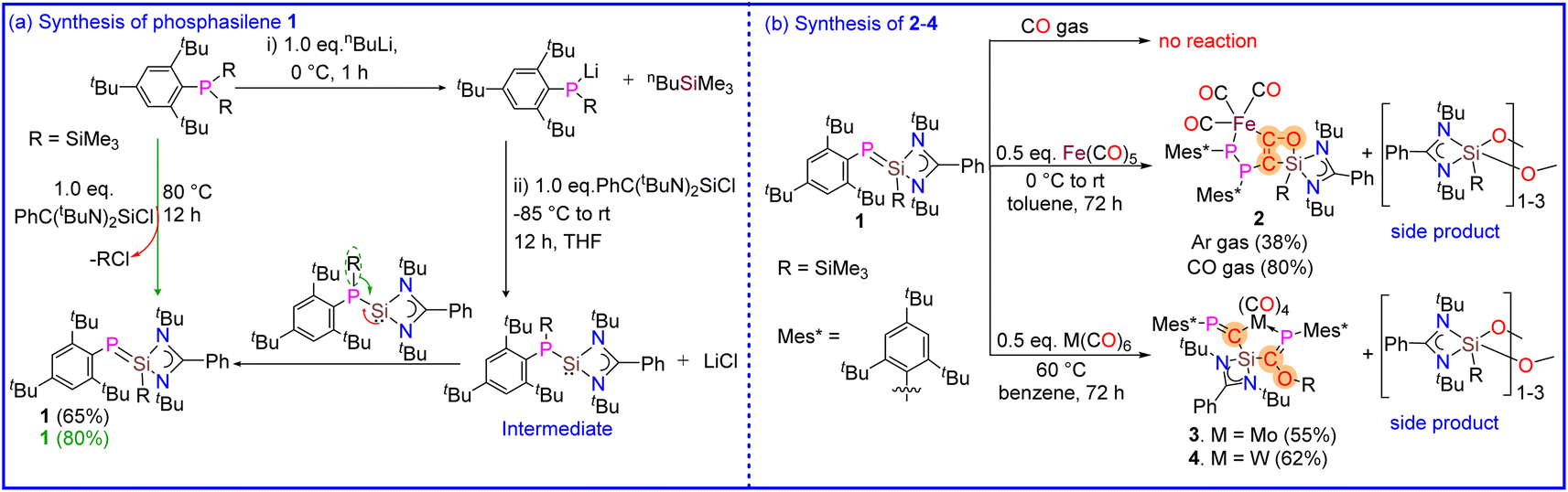 | ||
| Scheme 2 (a) Synthesis of 1. (b) Synthesis of 2–4. | ||
In the case of -treatments with M(CO)6 (M = Mo and W), the reactions were carried out at 60 °C, resulted in the formation of two interesting complexes (3 and 4) where the deoxygenation of one CO molecule without CO–CO homocoupling is observed, which is in contrast with the previous reports that the complete deoxygenation has been afforded with the help of homologation with another CO molecule.
To further investigate the effect of CO on the reaction yield, the reaction of 1 with metal carbonyls were conducted in the presence of CO gas, which resulted in the enhancement of the product yields. Compounds (1–4) were thoroughly characterized using NMR spectroscopies, mass spectrometry, elemental analysis, and X-ray diffraction analysis37–41(Fig. 1–4).
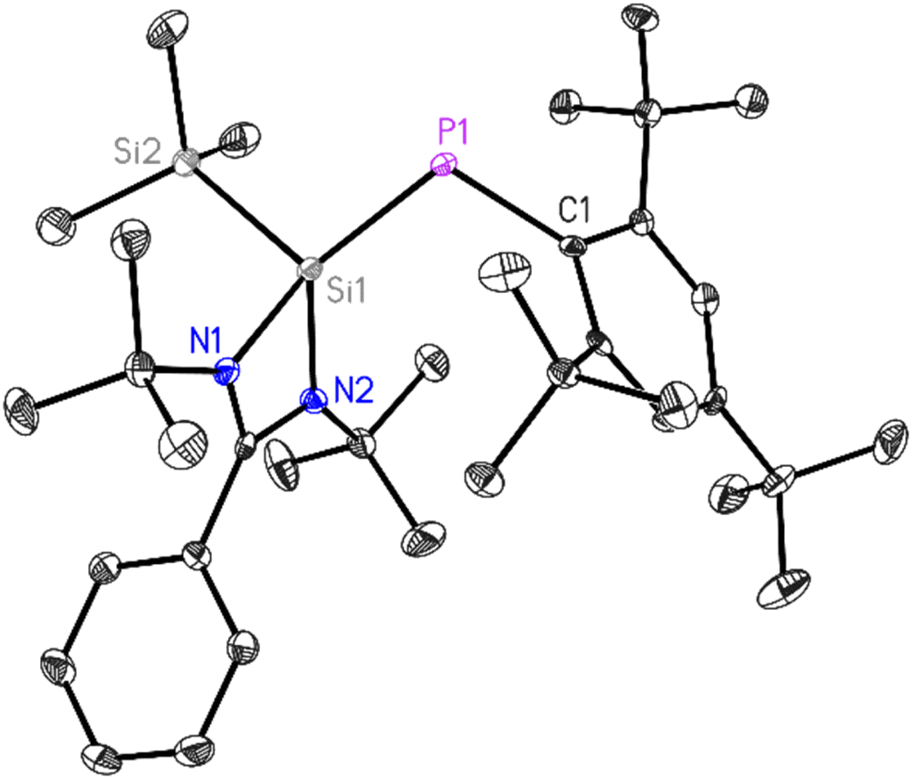 | ||
| Fig. 1 Molecular structure of 1. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°). P1–C1 1.874(2), P1–Si1 2.1250(10), N1–Si1 1.888(2), N2–Si1 1.833(2), Si1–Si2 2.3501(12), N1–Si1–N2 70.39(9), N1–Si1–Si2 106.67(7), N2– Si1–Si2 110.16(8), P1–Si1–Si2 104.80(4), C1–P1–Si1 109.30(8). | ||
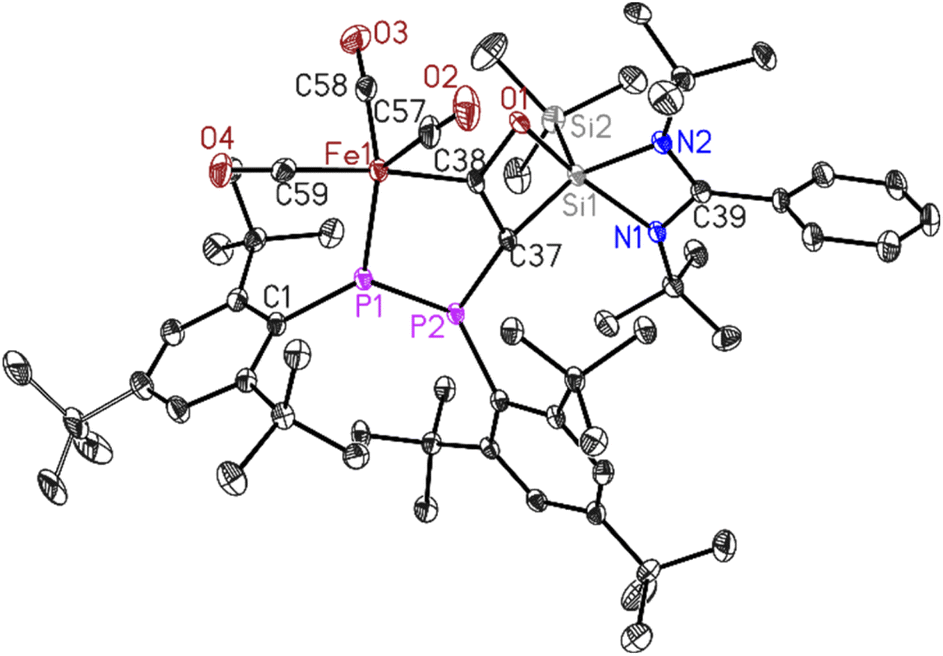 | ||
| Fig. 2 Molecular structure of 2. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°). Fe1–P1 2.1133(7), Fe1–C38 1.982(3), Fe1–C57 1.803(3), C57–O2 1.134(3), O1–Si1 1.8093(19), Si1–Si2 2.3453(10), C37–C38 1.371(4), C38–O1 1.366(3)Å, P1–P2 2.2028(9), C1–P1–P2 116.60(8). | ||
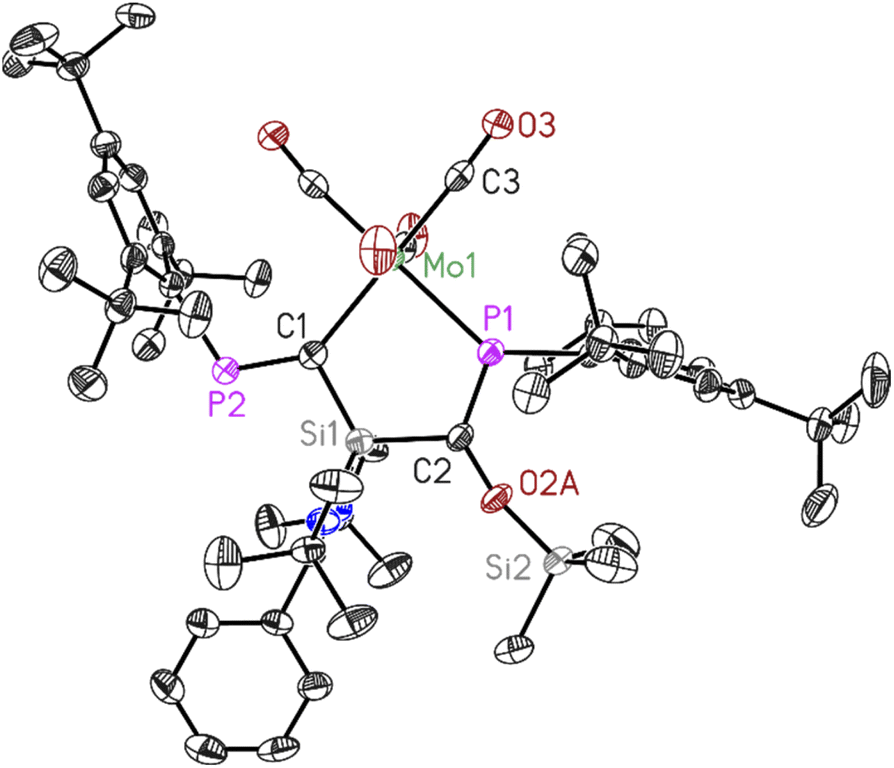 | ||
| Fig. 3 Molecular structure of 3. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°). Mo1–P1 2.5051(11), Mo1–C1 2.323(4), C1–P2 1.679(4), C2–P1 1.678(4), C1–Si1 1.817(4), C2–Si1 1.868(4), C2–O2A 1.401(10), O2A–Si2 1.653(9), P1–Mo1–C1 86.24(10), C1–Si1–C2 115.66(18), P1–C2–Si1 112.7(2), Mo1–C1–Si1 111.93(19), Mo1–P1–C2 113.43(14). | ||
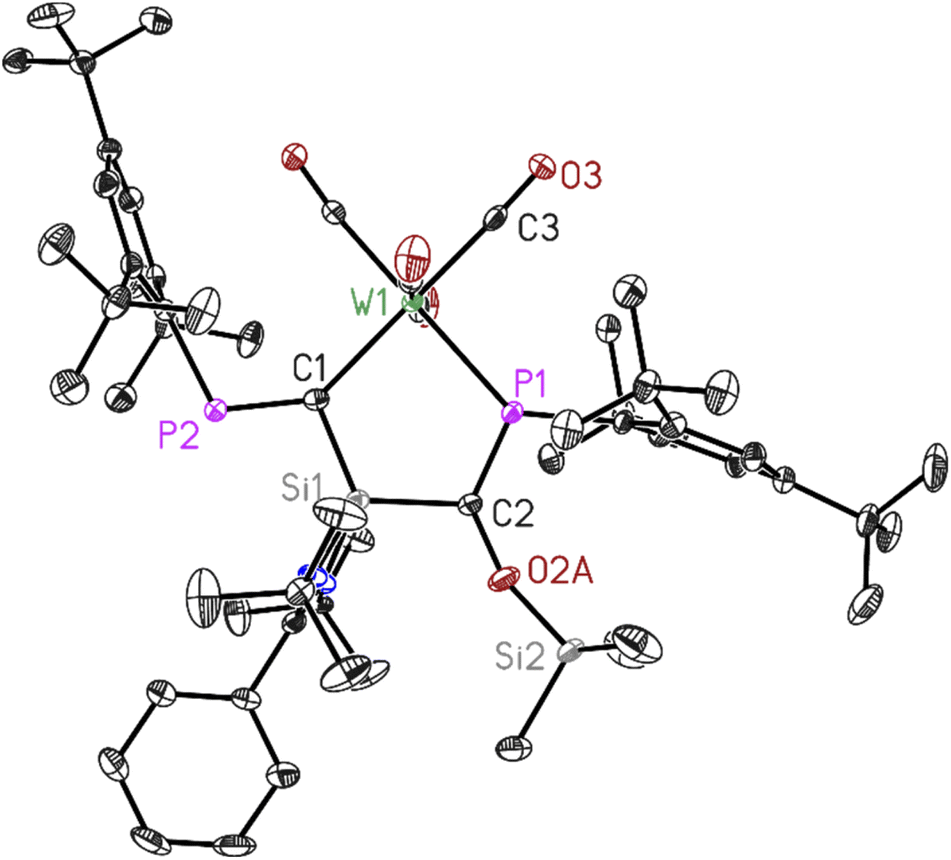 | ||
| Fig. 4 Molecular structure of 4. The anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°). W1–P1 2.4933(7), W1–C1 2.294(2), C1–P2 1.690(3), C2–P11.686(3), C1–Si1 1.821(2), C2–Si1 1.861(3), C2–O2A 1.389(9), O2A–Si2 1.657(8), P1–W1–C1 86.12(6), C1–Si1–C2 115.01(11), P1–C2–Si1 112.28(13), W1–C1–Si1 112.86(12), W1–P1–C2 113.65(9). | ||
The 31P NMR spectrum of 1 (Fig. S3†) displayed a singlet resonance at δ −92 ppm with two types of characteristic 29Si satellites attributed to the coupling with Si1 [PhC(tBuN)2Si], (1JPSi; 241.37 Hz) and Si2 [SiMe3], (3JPSi; 54.26 Hz) atoms. Whereas, the 29Si NMR spectrum of 1 (Fig. S4†) revealed two doublets centered at δ 16.59 ppm (Si1 [PhC(tBuN)2Si], 1JSiP; 241.37 Hz) and δ −16.04 ppm (Si2, [SiMe3], 3JSiP; 54.26 Hz)) resulting from the coupling with the phosphorus atom. The LIFDI mass spectrum (Fig. S7†), showed molecular ion for [M]+ at m/z = 608.5, which is well-matched with its simulated isotopic pattern confirming phosphasilene 1 formation. The UV-vis spectrum of 1 in pentane demonstrated a broad absorption band with a maximum at λ = 353 nm, a value that is similar to those observed for polarized phosphasilenes.42
The block-shaped orange-yellow color crystals of 1 suitable for X-ray diffraction analysis were grown from a saturated diethyl ether solution of 1 at room temperature over 24 h. Compound 1 crystallizes in the monoclinic P21/n space group (see Table S1†), with one molecule in the asymmetric unit. The P1–Si1 bond length in 1 is 2.1250(10) Å, slightly longer compared to the same bond (2.095(3) Å) in ((SiMe3)PSi(SiMe3)(PhC(NtBu)2) and shorter than the P–Si bond lengths (2.2264(13) and 2.2321(12) Å) in ((SiMe3)2P–Si(PhC(NtBu)2).33 This provides additional evidence for double bond formation in phosphasilene 1. The P1–Si1 bond length of 2.1250(10) Å is consistent with values reported for phosphasilenes with three-coordinate silicon atoms.5–7
Dark green crystals of 2 suitable for X-ray diffraction analysis were grown from saturated n-hexane solution at room temperature over three weeks. Compound 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (see ESI Table S1†) with one molecule in the asymmetric unit. The molecular structure of 2 confirms the deoxygenated homo-coupling of two CO molecules resulting in the formation of unique five- and four-membered rings containing P–P–Fe–C–C and C–C–O–Si atoms, respectively. The bond length of C37–C38 is 1.371(4) Å, falling within the range typical for CC double bonds, while the C38–O1 bond length of 1.366(3) Å corresponds to C–O single bonds. The 31P NMR spectrum of 2 (Fig. S11†) revealed two doublets centered at δ 54.5 and 387.6 ppm with 1JPP coupling of 460 Hz for (Mes*P–PMes*) group. This significant coupling aligns with reported compounds containing phosphorus–phosphorus bonds.43 Whereas, the 29Si NMR spectrum of 2 (Fig. S12†) showed two shielded singlets around δ −14.33 and −86.43 ppm, assigned to the (SiMe3) group and [PhC(NtBu)2Si] group respectively.
(see ESI Table S1†) with one molecule in the asymmetric unit. The molecular structure of 2 confirms the deoxygenated homo-coupling of two CO molecules resulting in the formation of unique five- and four-membered rings containing P–P–Fe–C–C and C–C–O–Si atoms, respectively. The bond length of C37–C38 is 1.371(4) Å, falling within the range typical for CC double bonds, while the C38–O1 bond length of 1.366(3) Å corresponds to C–O single bonds. The 31P NMR spectrum of 2 (Fig. S11†) revealed two doublets centered at δ 54.5 and 387.6 ppm with 1JPP coupling of 460 Hz for (Mes*P–PMes*) group. This significant coupling aligns with reported compounds containing phosphorus–phosphorus bonds.43 Whereas, the 29Si NMR spectrum of 2 (Fig. S12†) showed two shielded singlets around δ −14.33 and −86.43 ppm, assigned to the (SiMe3) group and [PhC(NtBu)2Si] group respectively.
The up-field shift is attributed to the change in the oxidation state of Si1 from (II) to (IV) and the presence of electron-withdrawing atoms, such as oxygen and carbon, around this five-coordinate silicon center. The LIFDI mass spectrum of 2 (Fig. S16†) showed molecular ion peak at m/z 1064.3 confirming the formation of 2.
Dark red crystals of 3 and 4 suitable for X-ray diffraction analysis were grown from saturated benzene-d6 solution at room temperature over one day. Both compounds 3 and 4 crystallize isomorphously in the orthorhombic space group P212121 (see ESI Table S1†) with one molecule and five benzene molecules as lattice solvent in the asymmetric unit. The molecular structures confirm the deoxygenation of one CO molecule followed by trapping another CO molecule in between the Si–P bond resulting in the formation of unique five-membered ring containing M–C–Si–C–P atoms (M = Mo, W). The important bond lengths for 3 and 4 are summarised in (Table 1). According to the data presented there, the P–C bond lengths align with the typical range observed for PC double bonds, whereas the C–O bond lengths are consistent with C–O single bonds.
| Bond length (Å) | P1–C2 | P2–C1 | C2–O2A | M–P1 |
|---|---|---|---|---|
| 3 | 1.678(4) | 1.679(4) | 1.401(10) | 2.5051(11) |
| 4 | 1.686(3) | 1.690(3) | 1.389(9) | 2.4933(7) |
Both compounds 3 and 4 are stable in an inert atmosphere for a prolonged period. However, they undergo rapid decomposition in polar solvents such as tetrahydrofuran (THF) and toluene, within 4–5 hours. Due to their limited solubility in deuterated benzene (C6D6), nuclear magnetic resonance (NMR) spectroscopy was conducted using toluene (d8) and THF (d8) (Fig. S18–S21 and S24–S30†).
Despite the suboptimal solubility of compound 3 in toluene-d8, it was selected as the solvent due to the rapid decomposition observed in other deuterated solvents such as THF-d8 and dichloromethane (d2) (CD2Cl2). The 31P NMR spectrum of 3 (Fig. S20†) revealed two peaks centered at δ 481.41 and 249.7 ppm, and 29Si-NMR spectrum displayed two peaks at δ 14.30 (attributed to Si2, OSiMe3) and −16.43 (attributed to Si1 [(Si(PhC(NtBu)2])). The phosphorous–phosphorous and silicon-phosphorous couplings were not clearly observed in the 31P and 29Si NMR spectra due to poor solubility of 3.
With compound 4, the 31P-NMR spectrum showed two doublets about δ 466.9 and 216.8 ppm with a 3JPP coupling of 7.8 Hz. The peak at δ 216.8 ppm exhibited two characteristic satellites due to coupling with the silicon atoms (2JPSi = 247.53 Hz and 3JPSi = 112.23 Hz) (Fig. S29†).
The 29Si NMR spectrum of 4 (Fig. S30†) showed two peaks, one singlet at δ 16.28 ppm and a doublet of doublets centred at δ −11.48 ppm, assigned to the (SiMe3) group and [PhC(NtBu)2Si] group, respectively.
The LIFDI mass spectra of compounds 3 and 4 (Fig. S22 and S31†) revealed molecular ion peaks at m/z 1106.3 and 1192.3, respectively, indicating the loss of one CO molecule during ionization for both compounds. As it is obvious from the characteristic results, this reactivity pattern diverges from the reaction of 1 with Fe(CO)5 and the reactions of the same category, where CO deoxygenation typically proceeds via CO–CO homogenization.17,23 Importantly, our investigations with M(CO)6 (M = Mo, W) did not manifest homocoupling of CO molecules.
Given the initial compounds and the reaction products, it is evident that one oxygen atom from the metal carbonyls and the [(PhC(NtBu)2Si(TMS)] fragment of the phosphasilene compound are missing. To characterize the side product, we analyzed the residual reaction mixture using 29Si-NMR spectroscopy. Compression of the29 Si-NMR (Fig. S33†) of all three residual mixtures revealed two signals in the same region in all the cases suggesting the similar side product in all of them. Fortunately, we could purify the side product from the residual reaction mixture of the iron complex and characterize it using NMR, IR spectroscopies and mass spectrometry (Fig. S34–S37†). The 29Si NMR data of the residual mixture suggest that the side product is a silanone derivative, which was further confirmed by 1H NMR spectrum and mass spectrometry data.44–46 However, all attempts to grow a single crystal for X-ray diffraction analysis were unsuccessful; we consistently obtained a white solid that was amorphous and, therefore, non-diffracting. Although we are not very sure about it, we rationalize that this most probably is due to the polymerization of the silanone side product during the reaction. Silanones lacking bulky substituents are challenging to stabilize and tend to polymerize readily to form polysiloxane. However in the following reaction, the bulky substituents on the silicon in PhC(NtBu)2Si(SiMe3)O likely hinder extensive polymerization, favoring the formation of dimers or trimers of the silanone {PhC(NtBu)2Si(SiMe3)O–}n (n = 1–3), rather than a polymer with n = ∞.47–49
To understand the detailed atomic-level mechanisms for the formation of products 2, 3, and 4 from the reaction of 1 with Fe(CO)5, Mo(CO)6, and W(CO)6, respectively, free energy profiles were mapped at the B3LYP-D3/6-31G* Mo, W (LanL2DZ) level of theory (refer to the ESI† for computational details).50,51 The free energies for the reaction of 1 with Fe(CO)5 were calculated at room temperature, whereas for the reaction of 1 with W(CO)6 and Mo(CO)6, they were calculated at 333.15 K. For simplicity of the calculations, the tert-butyl substituents of the aryl groups attached to the phosphorus atoms of 1 and nitrogen atoms of the benzamidinate ligand were replaced by methyl groups, and the resulting structure was labelled as 1′. The final products thus formed are denoted as 2′, 3′, and 4′, respectively. The free energy profile for the reaction of 1′ with Fe(CO)5 is shown in (Fig. S45†). The HOMO (−4.11 eV) molecular orbital of 1 corresponds to a SiP π-type orbital, while the LUMO (−0.83 eV) is a π*-type orbital of the phenyl ring of the benzamidinate ligand (Fig. S45(b)).† NBO analysis of 1 reveals that the HOMO orbital has a predominant electron density at the phosphorus atom, which is available for bond formation with incoming Fe(CO)5. Initially, 1′ and Fe(CO)5 combine to form the phosphasilene–iron complex, Fe-int1, releasing one CO molecule.
The CO molecule released from the previous step can be inserted into the P–Si bond of Fe-int1 through the transition state Fe-ts1′, producing Fe-int2′. However, this process requires a high energy barrier of 66.88 kcal mol−1, and hence is unfeasible under the experimental condition. An alternative scenario, in which Fe-int1 reacts with a second 1′ molecule to generate Fe-int2 while releasing another CO molecule, has been studied. It is worth noting that the in situ monitoring of the reaction by 31P NMR spectroscopy showed the formation of Fe complex 2 together with a new peak at δ 200 ppm, which can be attributed to Fe-int2 (Fig. S38†). The large downfield shift in the 31P NMR resonance signal compared to phosphasilene 1 might be due to the coordination of P lone pair to the Fe(CO)3 moiety which results in reduced electron density on the phosphorus atom. A similar trend is observed in the 31P NMR spectrum for the M(0) complexes of phosphasilenes, further suggesting Fe-int2 formation.5,52 Although monitoring of 31P-NMR suggests the formation of Fe-int1 and Fe-int2, the high energy barrier between Fe-int1 and Fe-ts1 (+43.68 kcal mol−1) led us to include the mechanism diagram and its discussion in the ESI (see Fig. S45†).
In the reaction of 1′ with Mo(CO)6 and W(CO)6, deoxygenative CO activation of one carbonyl group and the trapping of another CO molecules in between the Si and P atoms was observed which results in the products 3 and 4, respectively. In order to understand the atomic-level mechanisms for the formation of the products 3 and 4, the free energy profiles for the CO insertion between the P–Si bonds were mapped (Fig. 5 and 6) for different pathways. As can be seen from Fig. 5, the approach of 1′ and Mo(CO)6 or W(CO)6 leads to the formation of the phosphasilene-X complex, X-int1, with the release of one CO molecule, where X = Mo and W. The CO molecule released from the previous step may be inserted into the P–Si bond of X-int1 through the transition state X-ts1′ to form X-int2′. The CO insertion energy barrier for Mo and W is 70.51 and 69.97 kcal mol−1, respectively. The energy barrier for the insertion of CO into the P–Si bond is very high, and this reaction is not viable under the experimental conditions. An alternative multi-step mechanism involving a homo-coupling of CO moieties followed by CO insertion was also mapped (Fig. 6) This mechanism involves the initial formation of an activated complex X-int3, similar to that obtained for the reaction of 1′ with Fe(CO)5, by the reaction of X-int1 with a second molecule of 1′. It is important to note that the X-int3 structure formed here is quite different from that of Fe-int3. In X-int3, the P–X–P angle is about 77°, and the Si groups are well-separated due to crowding (see ESI Table S6†). However, in Fe-int3, the P–Fe–P angle is 170°, and a CO group attached to the Si(7) atom has weak interactions with the Fe atom. These structural differences in X-int3 result in different pathways being followed in the reactions involving Fe and Mo/W. From X-int3, due to the close proximity of the P(6) to the Si(3) group, a six-membered intermediate X-int4 is formed by the homo-coupling of CO moieties viaX-ts1. From X-int4, CO insertion into the P(4)–Si(3) happens viaX-ts2 to result in the bicylic X-int5. Then the migration of the –Si(Me)3 group from the Si(3) moiety to the carbonyl O(1) results in the formation of X-int6. This is followed by the rearrangement of X-int6via the breaking of the W(5)–P(6) and C(8)–O(9) bonds and the formation of W(5)–C(8) and O(9)–Si(3) bonds to result in X-int7. In X-int7, the P(6)–Si(7) bond is stretched (2.37 Å), leading to the elimination of the silanone (S) in the following step. The elimination of silanone (S) from X-int7 gives the product 4′. The rate-determining transition state is X-ts2, which corresponds to the insertion of CO into the P(4)–Si(3) bond, which has a barrier of ∼28 kcal mol−1 with respect to the activated complex X-int3. The reaction of 1′ with Mo(CO)6 follows a route similar to that of the reaction of 1′ with W(CO)6 to form the product 3′ (Fig. S46†).
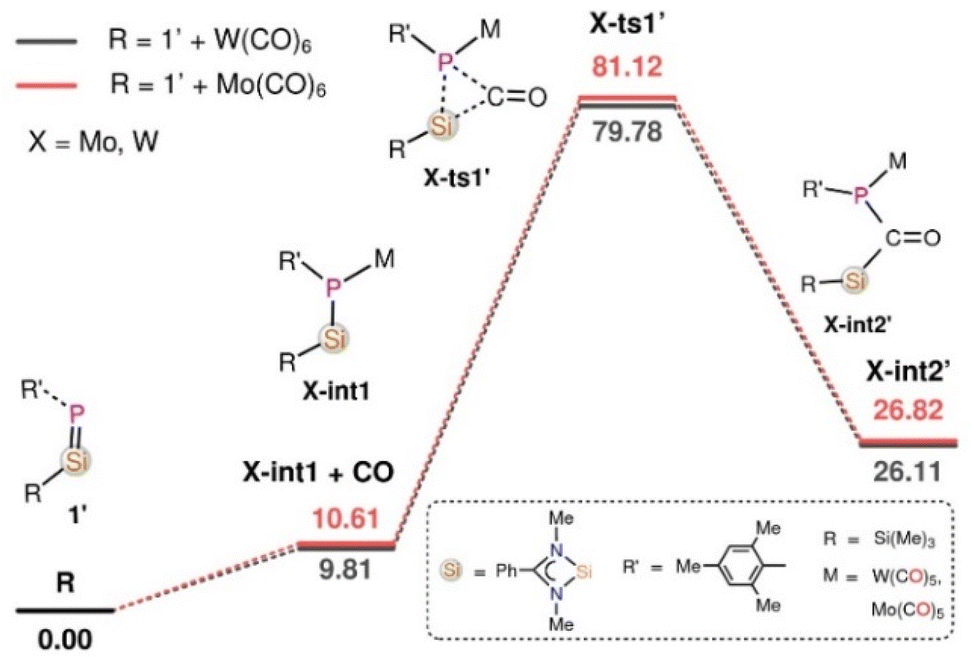 | ||
| Fig. 5 The Gibbs free energy profile for the reaction of 1′ with Mo(CO)6 and W(CO)6 obtained at the B3LYP-D3/6-31G* Mo, W (LanL2DZ) level of theory for the direct insertion of CO molecules into the Si–P bonds. The energies are given in kcal mol−1. | ||
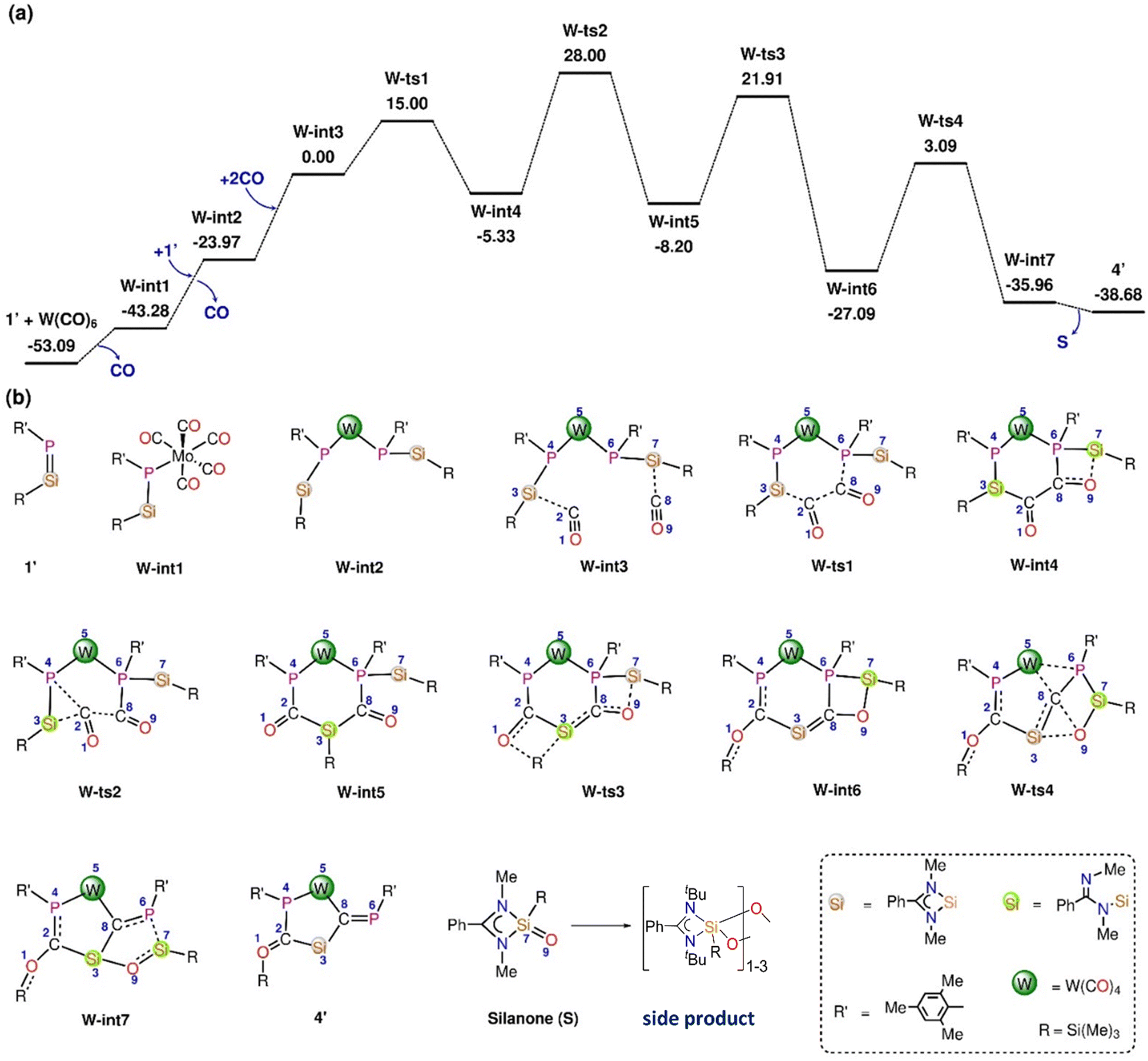 | ||
| Fig. 6 (a) The Gibbs free energy profile for the reaction of 1′ with W(CO)6 was obtained at the B3LYP-D3/6-31G* level of theory. (b) Different stationary point structures existing along the reaction pathway. The substituents connected to Si, W, and P are omitted in the images for clarity. The energies are given in kcal mol−1. | ||
Bubbling CO gas into the THF solution of compound 1 was undertaken to assess its capability to activate CO molecules. The 31P-NMR monitoring revealed no discernible changes in the spectrum. This underscores that the activation of CO occurs exclusively in the presence of two adjacent silicone atoms, consistent with observations in the reaction mechanism. The formation of int2 is imperative for this reaction, providing additional evidence supporting the validity of the proposed mechanism.
Conclusions
In summary, this work presents a new sterically demanding phosphasilene (Mes*PSi(SiMe3)(PhC(NtBu)2)) and its unexpected reaction of SiP double bond with metal carbonyls (M = Fe, Mo and W). The coordination of P lone pair to M center resulted in polarization of SiP double bond which in turn increased the electrophilicity of the Si center, therefore in the case of treatment with Fe(CO)5, it mediates the deoxygenative homocoupling of two CO molecules, yielding the ketene-inserted rare Fe complex 2. Contrastingly, reaction with Mo(CO)6 and W(CO)6 result in the deoxygenation of one carbonyl group while the second carbonyl group placed in between silicon and phosphorous atoms without any homocoupling with the first CO molecule, resulting in the formation of complexes 3 and 4, respectively. Density Functional Theory (DFT) calculations support a mechanism involving the formation of bis-phosphasilene metal complexes, M-int2 (M = Fe, Mo, W). The variation in geometrical arrangement of the two adjacent phosphasilene molecules within M-int2 leads to the formation of distinct products, with the iron complex (2) differing from the molybdenum and tungsten complexes (3 and 4).
Data availability
The data that support the finding of this article has been included as part of the ESI.† Crystallographic data for compounds 1–4 has been deposited as the CCDC 2311389, 2279235, 2370052 and 2370053.Author contributions
Z. H.: conceptualization, methodology, formal analysis, investigation, writing – original draft, review & editing, visualization, R. P.: DFT calculation, writing – review & editing, K. R.: single-crystal measurement, S. M.: single-crystal measurement, M. K. P.: writing – review & editing, S. K. K.: writing – review & editing, R. H.-I.: supervision, U. L.: supervision, writing – review & editing, D. S.: supervision, writing – review & editing, H. W. R.: writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge analytical facilities of the Georg-August-Universität Göttingen for help in recording the data related to this work.Notes and references
- M. Driess, Coord. Chem. Rev., 1995, 145, 1–25 CrossRef CAS.
- V. Nesterov, N. C. Breit and S. Inoue, Chem.–Eur. J., 2017, 23, 12014–12039 CrossRef CAS.
- C. N. Smit, F. M. Lock and F. Bickelhaupt, Tetrahedron Lett., 1984, 25, 3011–3014 CrossRef CAS.
- H. R. G. Bender, E. Niecke and M. Nieger, J. Am. Chem. Soc., 1993, 115, 3314–3315 CrossRef CAS.
- P. Willmes, L. Junk, V. Huch, C. B. Yildiz and D. Scheschkewitz, Angew. Chem., Int. Ed., 2016, 55, 10913–10917 CrossRef CAS.
- Y. Heider, P. Willmes, D. Mühlhausen, L. Klemmer, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019, 58, 1939–1944 CrossRef CAS.
- P. Willmes, M. J. Cowley, M. Hartmann, M. Zimmer, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2014, 53, 2216–2220 CrossRef CAS.
- S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786–11789 CrossRef CAS.
- S. S. Sen, S. Khan, H. W. Roesky, D. Kratzert, K. Meindl, J. Henn, D. Stalke, J.-P. Demers and A. Lange, Angew. Chem., Int. Ed., 2011, 50, 2322–2325 CrossRef CAS.
- S. Kundu, C. Mohapatra, P. P. Samuel, J. Kretsch, M. G. Walawalkar, R. Herbst-Irmer, D. Stalke, S. De, D. Koley and H. W. Roesky, Chem. Commun., 2017, 53, 192–195 RSC.
- V. Y. Lee, M. Kawai, A. Sekiguchi, H. Ranaivonjatovo and J. Escudié, Organometallics, 2009, 28, 4262–4265 CrossRef CAS.
- M. Driess, S. Block, M. Brym and M. T. Gamer, Angew. Chem., Int. Ed., 2006, 45, 2293–2296 CrossRef CAS.
- B. Li, T. Matsuo, T. Fukunaga, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, Organometallics, 2011, 30, 3453–3456 CrossRef CAS.
- N. C. Breit, T. Szilvási and S. Inoue, Chem. Commun., 2015, 51, 11272–11275 RSC.
- M. Driess, H. Pritzkow, S. Rell and U. Winkler, Organometallics, 1996, 15, 1845–1855 CrossRef CAS.
- N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958–17968 CrossRef CAS.
- Y. Xiong, S. Dong, S. Yao, C. Dai, J. Zhu, S. Kemper and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202209250 CrossRef CAS.
- Y. Xiong, S. Dong, S. Yao, C. Dai, J. Zhu and M. Driess, Angew. Chem., Int. Ed., 2022, 61, e202205358 CrossRef CAS.
- J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CAS.
- S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981–8995 CrossRef CAS.
- Y. Xiong, S. Yao, T. Szilvási, A. Ruzicka and M. Driess, Chem. Commun., 2020, 56, 747–750 RSC.
- A. Heilmann, M. M. D. Roy, A. E. Crumpton, L. P. Griffin, J. Hicks, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 12942–12953 Search PubMed.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS.
- M.-P. Luecke, A. Kostenko, Y. Wang, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 12940–12944 CrossRef CAS.
- Y. Wang, A. Kostenko, T. J. Hadlington, M.-P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 Search PubMed.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC.
- R. Y. Kong, M. Batuecas and M. R. Crimmin, Chem. Sci., 2021, 12, 14845–14854 RSC.
- D. Singh, B. J. Knight, V. J. Catalano, R. García-Serres, V. Maurel, J. M. Mouesca and L. J. Murray, Angew. Chem., Int. Ed., 2023, 62, e202308813 CrossRef CAS.
- K. Yuvaraj, J. C. Mullins, T. Rajeshkumar, I. Douair, L. Maron and C. Jones, Chem. Sci., 2023, 14, 5188–5195 Search PubMed.
- M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 8746–8750 CrossRef CAS.
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS.
- S. Inoue, W. Wang, C. Präsang, M. Asay, E. Irran and M. Driess, J. Am. Chem. Soc., 2011, 133, 2868–2871 Search PubMed.
- Y. van den Winkel, H. M. M. Bastiaans and F. Bickelhaupt, J. Organomet. Chem., 1991, 405, 183–194 CrossRef CAS.
- K. Hansen, T. Szilvási, B. Blom, S. Inoue, J. Epping and M. Driess, J. Am. Chem. Soc., 2013, 135, 11795–11798 CrossRef CAS.
- B. Li, T. Matsuo, D. Hashizume, H. Fueno, K. Tanaka and K. Tamao, J. Am. Chem. Soc., 2009, 131, 13222–13223 CrossRef CAS.
- SAINT v8.37 A, Bruker AXS Inc., Madison, 2016 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., 2015, A71, 3–8 Search PubMed.
- G. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 Search PubMed.
- N. C. Breit, T. Szilvási and S. Inoue, Chem.–Eur. J., 2014, 20, 9312–9318 Search PubMed.
- M. A. M. Forgeron, M. Gee and R. E. Wasylishen, J. Phys. Chem. A, 2004, 108, 4895–4908 Search PubMed.
- M. M. Linden, H. P. Reisenauer, D. Gerbig, M. Karni, A. Schäfer, T. Müller, Y. Apeloig and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12404–12409 Search PubMed.
- S. Ishida, T. Abe, F. Hirakawa, T. Kosai, K. Sato, M. Kira and T. Iwamoto, Chem.–Eur. J., 2015, 21, 15100–15103 Search PubMed.
- D. Wendel, D. Reiter, A. Porzelt, P. J. Altmann, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 17193–17198 Search PubMed.
- Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302–4311 CrossRef CAS.
- T. Iwamoto, H. Masuda, S. Ishida, C. Kabuto and M. Kira, J. Am. Chem. Soc., 2003, 125, 9300–9301 Search PubMed.
- W. Li, N. J. Hill, A. C. Tomasik, G. Bikzhanova and R. West, Organometallics, 2006, 25, 3802–3805 CrossRef CAS.
- E. Solel, N. Tarannam and S. Kozuch, Chem. Commun., 2019, 55, 5306–5322 Search PubMed.
- S. Kozuch, Understanding Organometallic Reaction Mechanisms and Catalysis: Computational and Experimental Tools, ed. V. P. Ananikov, Wiley-VCH: Weinheim, Germany, 2014, pp. 217–248 Search PubMed.
- K. Hansen, T. Szilvási, B. Blom and M. Driess, Organometallics, 2015, 34, 5703–5708 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2311389, 2279235, 2370052 and 2370053. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05491a |
| This journal is © The Royal Society of Chemistry 2024 |