
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated “all-in-one” strategy to construct highly efficient Pd catalyst for CO2 transformation†
Lingfang
Kong‡
a,
Zekun
Tao‡
a,
Yunjia
Li
a,
Huiwen
Gong
a,
Yun
Bai
a,
Longbin
Li
a,
Xianjin
Zhang
b,
Zhonggao
Zhou
 *a and
Yiwang
Chen
*a
*a and
Yiwang
Chen
*a
aCollege of Chemistry and Chemical Engineering/Analysis and Testing Center/Key Laboratory of Jiangxi University for Functional Materials Chemistry, Gannan Normal University, Ganzhou 341000, P. R. China. E-mail: zhgzhou@foxmail.com; ywchen@ncu.edu.cn
bInstitute of Chemistry Education, Fujian Institute of Education, Fuzhou 350025, P. R. China
First published on 27th August 2024
Abstract
The synthesis of high-value chemicals featuring C–C and/or C–heteroatom bonds via CO2 is critically important, yet efficiently converting thermodynamically stable and kinetically inert linear CO2 and propargylic amine to the heterocyclic compound 2-oxazolidinone with an integrated catalytic system continues to pose a considerable challenge. Herein, we have designed an “all-in-one” (AIO) palladium (Pd) catalyst (Cat1), distinguished by its co-coordination with acetylglucose (AcGlu) and bis(benzimidazolium) units at the Pd center, which promotes the cyclization of CO2 and propargylic amine achieving a highest turnover frequency (TOF) of up to 3456 h−1. Moreover, Cat1 demonstrates excellent stability across various temperatures, with its catalytic activity remaining unchanged even after 10 cycles. The catalyst Cat1 simultaneously activates propargylic amine and CO2, facilitating the formation of N-heterocyclic carbene (NHC)-CO2 adducts and AcGlu-CO2 philes from CO2 in simulated flue gas, a key factor in reaching unprecedented TOF values. The catalytic mechanism was elucidated through quasi-in-situ NMR and 13C-isotope labeling experiments. Notably, this is the first instance of an AIO Pd catalyst that enables the simultaneous capture, activation, and catalytic conversion of in-situ activated CO2 along with propargylic amine. The design strategy of this AIO catalyst introduces a novel approach to overcoming the challenges in the efficient conversion of inert CO2.
Introduction
The substantial consumption of fossil fuels accelerates carbon dioxide (CO2) emissions and causes a severe environmental crisis, prompting significant efforts to mitigate, capture, and utilize excess CO2.1–7 Among various conversion approaches, the preparation of 2-oxazolidinone through CO2 conversion plays a critical role as a heterocyclic compound in the realms of chemistry, medicine, and polymer science.8–10 Recent advances in ionic liquids (ILs), N-heterocyclic carbenes (NHCs), N-heterocyclic olefins (NHOs), metal–organic frameworks (MOFs), organic acids and bases, and metal catalysts have facilitated the synthesis of 2-oxazolidinones (Fig. 1a, such as PNU-85055, Spirooxazolidinone C, etc.) from propargylic amines and CO2.11–13 As present in Fig. 1b, for example, Inagaki et al. reported that the gold catalyst [Au(dpbF)]X, featuring a Z-type ligand, exhibited a turnover frequency (TOF) value of 5.3 h−1, possibly attributed to the σ-acceptance of the borane atom.14 Zhao and co-authors discovered that the noble-metal-free lantern-like, eco-friendly [Zn116] nanocages catalyst achieved a TOF value of 31 h−1,15 and a mixed valence copper-based cationic MOFs catalyst achieved an impressive TOF value of 230 h−1.16 Our group also demonstrated that Pd catalyst AcGlu-MeIm-PdCl4 can achieve a TOF value of 1440 h−1.17 Zhu et al. highlighted the exceptional catalytic activity of a low-nuclear customized Ag4 NC, with turnover number (TON) and TOF values reaching 5746.2 and 2873.1 h−1 respectively.11 This catalytic system demonstrated concurrent capture, activation, and catalytic conversion of in-situ activated CO2 alongside propargylic amine, indicating its efficacy in facilitating these reactions. However, due to the kinetic inertness and thermodynamic stability of linear CO2, significant challenges persist in its efficient activation and conversion. Clearly, the activation of inert CO2 is a crucial step in reducing the energy required for subsequent reactions involving CO2 as a reactant. From a practical perspective, the direct transformation of low-concentration CO2 (such as simulated flue gas, CO2 concentration 15–30%) into desirable products should be an ideal alternative as it can avoid the high energy consumption from the separation and concentration of anthropogenic CO2.18,19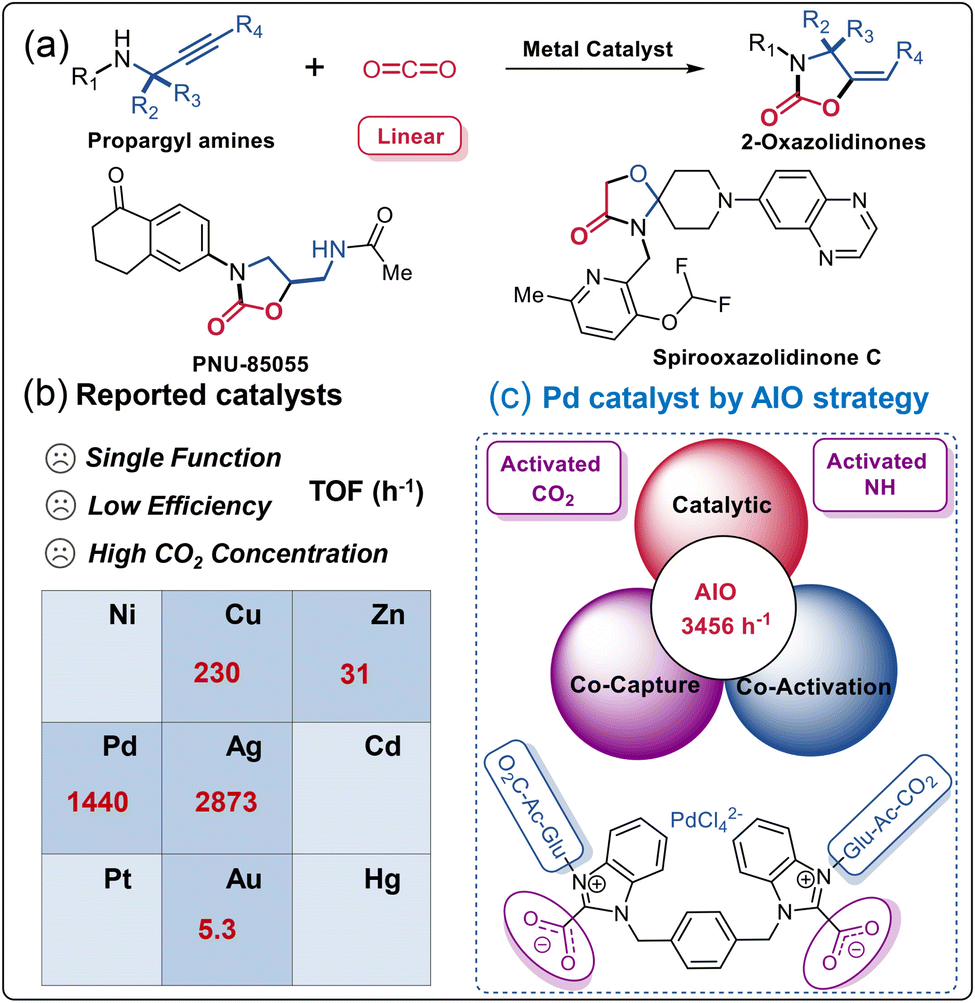 | ||
| Fig. 1 (a) The metal-catalyzed cyclization of propargylic amines and CO2. (b) The reported metal catalysts with different TOFs, see attached Table S1† for more detailed parameters. (c) Pd catalyst construct by integrated AIO strategy herein. | ||
Numerous Lewis base (LB)-CO2 adducts have been employed to activate CO2 and developed as organocatalysts, including organic phosphine, organic amine, alkoxide-functionalized imidazolium betaines, free NHOs, and phosphorus-ylide systems.20–23 The strong σ-donor character of free NHC has also been utilized for CO2 capture, and the resulting NHC-carboxylates or NHC-CO2 adducts have been applied as effective organocatalysts and activated CO2 sources.24–27 However, the in-situ capture of CO2 as LB-CO2 adducts and its subsequent transformation via metal catalysis remain challenging, with limited studies.17,28 Consequently, the pursuit of developing highly efficient integrated metal catalysts that can transform CO2, specifically from in-situ formed LB-CO2, into value-added chemicals without requiring extra additives, stands as a paramount goal for the scientific community. The “all-in-one” (AIO) strategy for constructing efficient catalysts is an emerging approach that combines the unique properties of traditional metallic, organic, and inorganic analogs due to its hybrid nature.29–31 ILs possess many excellent characteristics, such as the ability to dissolve acidic gases, high thermal stability, and low vapor pressure, which meet the requirements in CO2 capture.32–36 Metal-containing imidazolium ILs have been recognized as potential NHC ligand, NHC-metal catalyst precursor, and reaction reagents for various transformations.37–39 However, the in-situ formation of NHC-CO2 adducts from metal-containing imidazolium ILs for catalytic CO2 conversion shows great promise, yet has not been realized.
Sugar acetates (SAs) featuring acetyl (Ac) units present a unique strategy for the capturing and activating of CO2 through CO2-philes.40–44 Tsivintzelis et al. found that composite membranes made from cellulose acetate, which exhibited high CO2 affinity, are utilized for separating CO2 from natural gas or in CO2/N2 mixed gas.45 The interactions between CO2 and a combination of α-D-glucose pentaacetate (GPA) and [Bmim][BF4] ILs (−25.3 kJ mol−1) are notably stronger than those in GPA⋯CO2 (−10.7 kJ mol−1) and [Bmim][BF4]⋯CO2 (−17.8 kJ mol−1) when examined independently. These interactions offer additional benefits including non-toxicity and cost-effectiveness.40 In addition, integrating SAs into ILs significantly enhances the interactions between metals and SAs, thereby greatly improving their effectiveness in metal-catalyzed reactions with increased catalytic activity.17,46–50 It is thus plausible to propose that SAs and ILs can jointly activate CO2 and stabilize metal catalytic species during the catalysis process. The utilization of Pd catalysts for synthesizing important, biologically active heterocyclic compounds has attracted considerable research attention.51–53 Despite substantial efforts to develop various Pd catalysts for CO2 transformation,17 challenges such as low turnover frequency (TOF), catalytic efficiency, high CO2 concentration, and high catalyst loading persist. Therefore, developing a robust, high-performance AIO Pd catalyst for the capture, activation, and catalytic transformation (CACT) of CO2 remains a formidable and intriguing research area.
Herein, we designed a series of metal-containing bis(benzimidazolium) ILs (Cat1-5), specifically engineered for the CACT of CO2 and propargylic amines (Fig. 1a, R4 = H). These chloropalladate-containing ILs were facilitated synthesized via a biphasic ion exchange reaction between the respective bis(benzimidazolium) bromide salts (Im-Br1-4) and Na2PdCl4. We further evaluated the catalytic performance of Cat1-5 in the CACT of CO2 and propargylic amines. The integrated Cat1 catalytic system, containing AcGlu and bis(benzimidazolium) units, demonstrated a synergistic effect with Pd in the CACT of in situ activated-CO2, forming NHC-CO2 adducts and AcGlu-CO2-philes. This stimulation activation of CO2 and propargylic amine ensures reached a highest reported TOF value of 3456 h−1 (Fig. 1c). In addition, Cat1 exhibits good stability across a range of temperatures, effectively integrating the advantages of homogeneous catalysis and catalyst recycling, and can be reused for at least 10 cycles without significant loss in catalytic performance. 1H NMR experimentals confirmed that AcGlu and bis(benzimidazolium) co-coordinated Cat1, providing multiple LA-LB sites for the activation of CO2, propargylic amine, and stabilization of Pd catalyst species. The catalytic mechanism was elucidated through quasi-in-situ NMR and 13C-isotope-labeling experiments.
Results and discussion
Reasonable design, preparation, and characterization of Pd catalysts
At the outset of our study, acetylglucoside bis(benzimidazolium) tetrachloropalladate salts (Cat1-3 and 5) were synthesized in four steps from natural D-glucose. The final step involved a straightforward ion exchange of the corresponding bromide salts (Im-Br1-3 and 5) with Na2PdCl4 (Fig. 2a). For comparative purposes, a bis(benzimidazolium) tetrachloropalladate salt (Cat4) featuring an n-octyl (–nC8H15) side chain was synthesized using a similar method. Cat5 and Im-Br5 were also obtained by deprotecting the Ac groups from the corresponding Cat1 and Im-Br1 (Fig. 2b), primarily to investigate their roles in the catalytic process. Cat1-5 can be synthesized with high purity and are readily available in large quantities with high yields. It is worth noting that the use of 2-bromoethyl glucoside is intended to alleviate steric hindrance between the anomeric equatorial carbon of the sugar and the axial C2–H of the imidazolium ring by introducing an ethyl group to separate them.49 This design also helps to minimize potential interactions between the imidazolium ring and sugar units in order to evaluate their role during the catalytic process.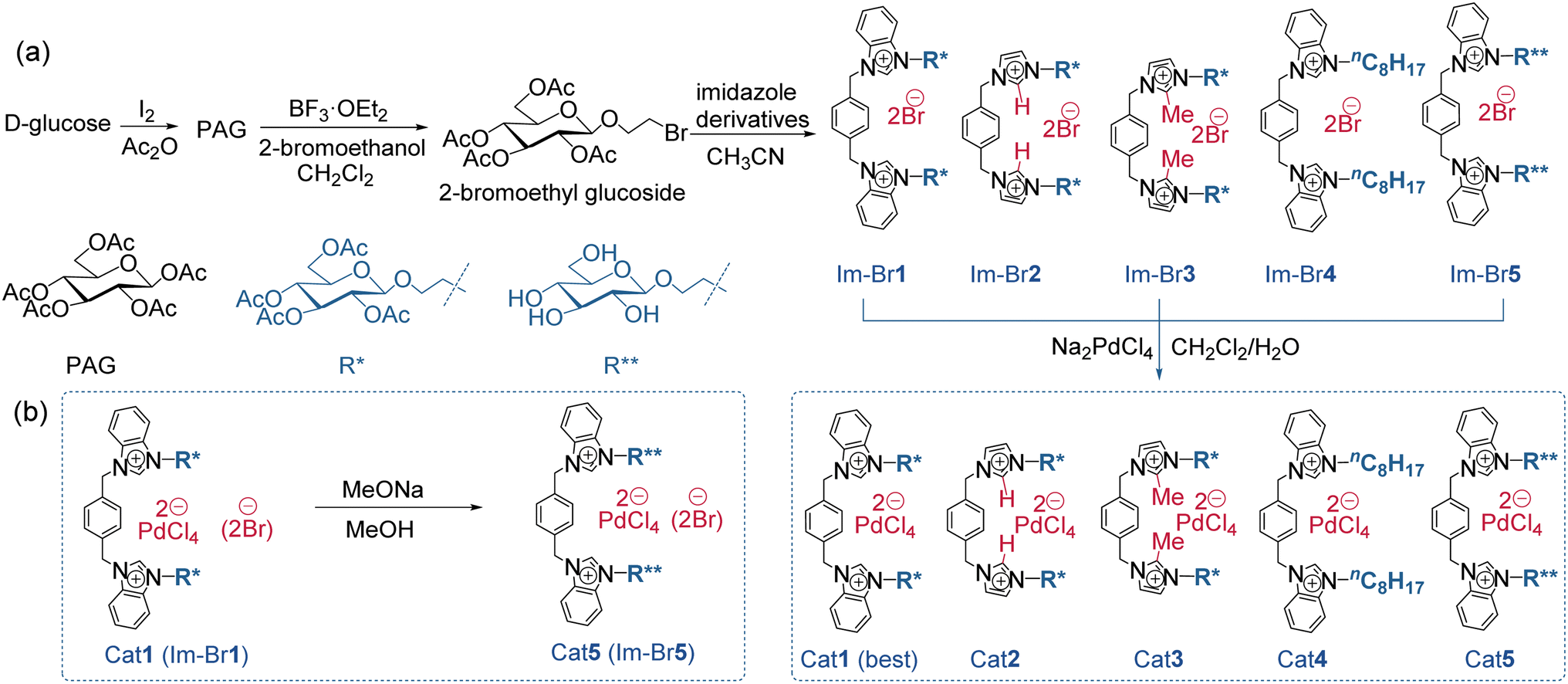 | ||
| Fig. 2 (a) The synthetic routes of Pd catalysts Cat1-5. (b) The synthesis route of bromide salt Im-Brc1 and Pd catalyst Cat5. | ||
All these compounds were thoroughly characterized using 1H and 13C nuclear magnetic resonance spectra (NMR, Fig. S1–S10†), Fourier-transform infrared spectra (FT-IR, Fig. S11–S15†), and liquid chromatography-high-resolution mass spectrometry (LC-HR/MS, Fig. S16–S25†). Furthermore, X-ray photoelectron spectroscopy (XPS, Fig. S26†) was employed to discern the distinct chemical coordination environments of Cat1 and Cat5, with and without Ac protecting groups. Meanwhile, thermal stability of Cat1-5 was also studied using thermogravimetric analysis (TGA, Fig. S27†). The NMR signals of Cat1-5 closely resembled those of their corresponding bromide salts (Im-Br1-5), with the most notable peaks appearing in similar regions across spectra (Fig. S1–S10†). Specifically, the characteristic imidazolium C2–H singlet peak in the bromide salts Im-Br1-2 and 4-5 was observed at δ 9.80∼10.40 ppm, shifting to δ 9.3∼10.10 ppm in Cat1-2 and 4-5 after ion exchange. The multiplets for C1–6–H in glucoside of Im-Br1-3 and 5, ranging from δ 3.20–5.90 ppm, were comparable to those in Cat1-3 and 5, also shifting to δ 9.3∼10.10 ppm. 13C NMR spectra revealed a significant peak for C2–C in Im-Br1-5 at δ 137.0∼145.4 ppm, slightly altered to δ 136.4∼145.4 ppm in Cat1-5. NMR results showed that the introduction of PdCl42− anion not only maintained the original structure of the parent compound, but also enhanced its interaction with AcGlu groups and benzimidazolium cation. FT-IR spectra indicated absorption peaks at approximately 1563 and 1630 cm−1, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretch vibrations of the imidazolium unit in Cat1-5. Additional signals at about 1172, 1126, and 1035 cm−1 were consistent with C–H bending vibrations in the imidazolium rings of Cat1-5 (Fig. S11–S15†). LC-HR/MS analysis in positive mode confirmed the presence of only imidazolium cations, with indistinguishable test signals for Cat1-5 and Im-Br1-5 (Fig. S16–S25†), corroborating our previously reported results.17 The chemical environment of the key elements in the near-surface region of Cat1 and Cat5 was further characterized using XPS. Carbon was the predominant element, with the XPS spectrum of Cat5 resolving into component parts corresponding to C–C (∼284.8 eV), C–N (∼286.7 eV), and CO/O–CO (∼289.2 eV).31 Pd 3d XPS spectra for Cat5 displayed peaks at 337.7 (Pd 3d5/2) and 343.2 eV (Pd 3d3/2), indicative of Pd in the +2 oxidation state and substantial electron donation from AcGlu and bis(benzimidazolium) units to the PdCl42− anions (Fig. S26†).54 Notably, the Pd 3d XPS spectra of Cat5 without Ac protecting groups also showed significant shifts (0.6–0.7 eV) in the binding energies of benzimidazolium heterocycles, suggesting increased electron delocalization after ion exchange. These experimental results confirmed the successful synthesis of the target products Im-Br1-5 and Cat1-5. The TGA curves show that the decomposition temperatures of catalysts Cat1-5 range from 186 °C to 225 °C (Fig. S27†), indicating their good thermal stability. Furthermore, Cat1 was dissolved in dimethyl sulfoxide (DMSO-d6) and maintained at various temperatures (−22 °C, 25∼30 °C, and 70 °C) for 10 h to assess its stability in solution. Remarkably, no significant changes were observed in its 1H, 13C NMR spectrum, and color across these different environments (Fig. S28†). These results suggest that Cat1 is stable and does not undergo decomposition during the catalytic reaction.
N and CC stretch vibrations of the imidazolium unit in Cat1-5. Additional signals at about 1172, 1126, and 1035 cm−1 were consistent with C–H bending vibrations in the imidazolium rings of Cat1-5 (Fig. S11–S15†). LC-HR/MS analysis in positive mode confirmed the presence of only imidazolium cations, with indistinguishable test signals for Cat1-5 and Im-Br1-5 (Fig. S16–S25†), corroborating our previously reported results.17 The chemical environment of the key elements in the near-surface region of Cat1 and Cat5 was further characterized using XPS. Carbon was the predominant element, with the XPS spectrum of Cat5 resolving into component parts corresponding to C–C (∼284.8 eV), C–N (∼286.7 eV), and CO/O–CO (∼289.2 eV).31 Pd 3d XPS spectra for Cat5 displayed peaks at 337.7 (Pd 3d5/2) and 343.2 eV (Pd 3d3/2), indicative of Pd in the +2 oxidation state and substantial electron donation from AcGlu and bis(benzimidazolium) units to the PdCl42− anions (Fig. S26†).54 Notably, the Pd 3d XPS spectra of Cat5 without Ac protecting groups also showed significant shifts (0.6–0.7 eV) in the binding energies of benzimidazolium heterocycles, suggesting increased electron delocalization after ion exchange. These experimental results confirmed the successful synthesis of the target products Im-Br1-5 and Cat1-5. The TGA curves show that the decomposition temperatures of catalysts Cat1-5 range from 186 °C to 225 °C (Fig. S27†), indicating their good thermal stability. Furthermore, Cat1 was dissolved in dimethyl sulfoxide (DMSO-d6) and maintained at various temperatures (−22 °C, 25∼30 °C, and 70 °C) for 10 h to assess its stability in solution. Remarkably, no significant changes were observed in its 1H, 13C NMR spectrum, and color across these different environments (Fig. S28†). These results suggest that Cat1 is stable and does not undergo decomposition during the catalytic reaction.
Catalytic activity evaluation
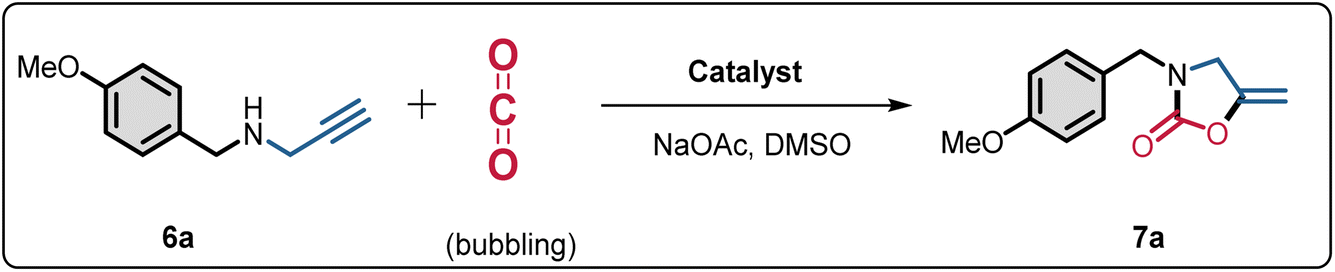
Initially, the catalytic performance of Cat1 was evaluated in the model reaction of N-(4-methoxybenzyl)propargylic amine (6a) and CO2 [simulated flue gas, V(CO2)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V(N2) = 15:85] at 70 °C with NaOtBu in different solvents (Fig. 3a). No reaction was observed in tetrahydrofuran (THF), and a 10% yield of the product 3-(4-methoxybenzyl)-5-methyleneoxazolidin-2-one (7a) was observed in acetonitrile (CH3CN). However, good to excellent yields of 7a (80–92%) were obtained in 1,4-dioxane (Diox), ethanol (EtOH), and methanol (MeOH). Remarkably, nearly 100% yields were achieved in N,N-dimethylformamide (DMF) and DMSO within 2.0 h. Further optimization of the reaction time revealed that DMSO was the superior solvent, yielding a higher TOF (89.6 h−1) compared to DMF (25.6 h−1). These results align with those previously reported by C. Nevado using PdCl2(dppf),55 and by D. Bourissou using a Pd SCS complex.56 Additionally, the interaction energies of the DMSO-CO2 complexes at the MP2 level indicate effective CO2 activation,44 facilitating faster reaction kinetics in DMSO. Subsequently, we optimized various bases in DMSO (Fig. 3b). In the presence of inorganic bases such as NaOH, NaOAc, Na3PO4·12H2O, and Na2CO3, substrate 6a was completely consumed, yielding product 7a in approximately 100% yields. KOH, K2CO3, and NaHCO3 produced yields of 96%, 95%, and 10%, respectively. The weak organic base triethylamine (TEA) afforded only a 29% yield. In contrast, the strong organic bases 1,8-diazabicycloundec-7-ene (DBU) and NaOtBu favorably promoted the reaction, yielding 88% and approximately 100% yields, respectively.
:V(N2) = 15:85] at 70 °C with NaOtBu in different solvents (Fig. 3a). No reaction was observed in tetrahydrofuran (THF), and a 10% yield of the product 3-(4-methoxybenzyl)-5-methyleneoxazolidin-2-one (7a) was observed in acetonitrile (CH3CN). However, good to excellent yields of 7a (80–92%) were obtained in 1,4-dioxane (Diox), ethanol (EtOH), and methanol (MeOH). Remarkably, nearly 100% yields were achieved in N,N-dimethylformamide (DMF) and DMSO within 2.0 h. Further optimization of the reaction time revealed that DMSO was the superior solvent, yielding a higher TOF (89.6 h−1) compared to DMF (25.6 h−1). These results align with those previously reported by C. Nevado using PdCl2(dppf),55 and by D. Bourissou using a Pd SCS complex.56 Additionally, the interaction energies of the DMSO-CO2 complexes at the MP2 level indicate effective CO2 activation,44 facilitating faster reaction kinetics in DMSO. Subsequently, we optimized various bases in DMSO (Fig. 3b). In the presence of inorganic bases such as NaOH, NaOAc, Na3PO4·12H2O, and Na2CO3, substrate 6a was completely consumed, yielding product 7a in approximately 100% yields. KOH, K2CO3, and NaHCO3 produced yields of 96%, 95%, and 10%, respectively. The weak organic base triethylamine (TEA) afforded only a 29% yield. In contrast, the strong organic bases 1,8-diazabicycloundec-7-ene (DBU) and NaOtBu favorably promoted the reaction, yielding 88% and approximately 100% yields, respectively.
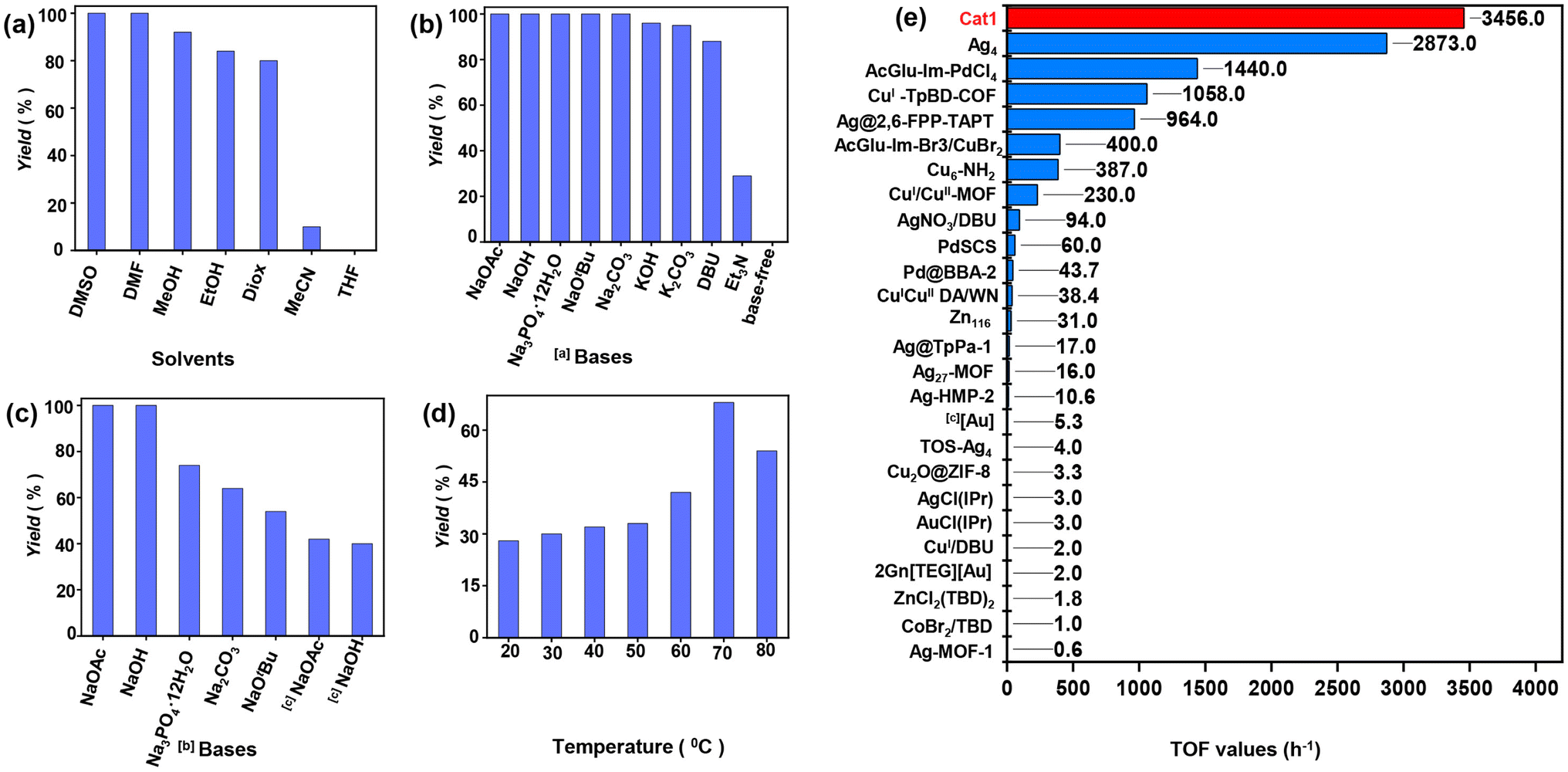 | ||
| Fig. 3 Screening the reaction of 6a with CO2 (simulated flue gas, bubbling) under various conditions, Pd catalyst (0.5 mol%), base (0.15 mmol), solvent (2.0 mL), and stirred at 70 °C for 1.0 h, unless noted otherwise, yields were determined by 1H NMR. The reactions were carried out with different solvents (a), bases (b and c), and (d) temperatures. [a] DMSO (2.0 mL). [b] 0.25 h. (e) Comparison with other reported catalysts. TOF was calculated using optimal conditions reported in the literature. Detailed reaction conditions are provided in Table S1.† | ||
As expected, no product was detected in the absence of a base. By optimizing the reaction time and catalyst dosage, we determined that the weak inorganic base NaOAc, which contains acetate anions (OAc−, Fig. 3c), was the optimal choice, achieving a TOF of 100 h−1 with 100% yield, which could further increase to 672 h−1. Rogers et al. observed that the introducing CO2 into 1,3-dialkylimidazolium acetate ILs via bubbling facilitated the formation of corresponding NHC-CO2 adducts.57 Consequently, it is plausible that the activity of CO2 manifested in the form of NHC-CO2 adducts, arising from free NHC generated in situ following the reaction between benzimidazolium cations and NaOAc. Further assessment of various temperatures revealed a notable impact on the reaction process (Fig. 3d). At room temperature (RT, approximately 20–25 °C), only 28% yield of 7a was obtained, with yields increasing to 30–68% as the temperature rose from RT to 70 °C. However, a significant reduction in yield to 54% occurred when the temperature was increased to 80 °C. These results indicate that high temperatures are not conducive to the reaction, primarily due to the decomposition of the in-situ formed NHC-CO2 adducts and DMSO-CO2 complexes at elevated temperatures.17,27,28,58
By comparing the catalytic activities of AIO Pd catalyst Cat1-5 (Table 1, entries 1–5), we observed that Cat1, containing AcGlu and bis(benzimidazolium) units, exhibited superior performance with a TOF of 2400 h−1, 100% yield, and TON of 400 (entry 1). Other catalysts, such as Cat2, Cat3, and Cat4, were less efficient, yielding 94%, 91%, and 52% respectively (entries 2–4). Cat5 without Ac protecting groups, when used as a catalyst, yielded only 36% with a TOF of 864 h−1. This comparison highlights the critical role of Ac units in enhancing catalytic performance. To elucidate the role of functional units in Cat1, various Pd catalyst systems were evaluated (entries 6–11). When commercial catalysts like K2PdCl4, Pd(OAc)2, and PdCl2 were used directly, they achieved only 28%, 44%, and 37%, respectively (entries 6–8). When combined with Im-Br1, these catalysts' activities significantly improved, resulting in 70%, 64%, and 71% yields (entries 9–11). Furthermore, the yield rose significantly to 56% (entry 12) when glucose pentaacetate (PAG) was used in conjunction with the pure K2PdCl4 catalyst, which had a yield of only 28% (entry 6). This suggests that both PAG and Im-Br1 enhance the activity of palladium species. In the absence of any Pd catalyst (entry 13), and with only Im-Br1 used, no product was detected. In addition, it was also observed that increasing the amount of NaOAc from 1.5 to 4.0 equivalents significantly boosted the yield to 74% and achieved the highest reported TOF (3456 h−1) and TON (288, entry 1), underscoring the pivotal role of OAc− herein. The TOF value of Cat1 surpasses that of all other previously reported homogeneous and heterogeneous catalytic systems for the reaction of propargylic amines and dilute CO2, as detailed in Fig. 3e and Table S1.†
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Yieldsb [%] | TOF [h−1] | TON |
| a The reactions were carried out with 6a (0.1 mmol), Pd catalyst (0.5 mol%), CO2 (simulated flue gas, bubbling), NaOAc (0.15 mmol), DMSO (1.0 mL) and stirred at 70 °C for 1.0 h, unless noted otherwise. b Determined by 1H NMR. c Catalyst (0.25 mol%), CO2 (1.0 atm, bubbling), 10 min. d Catalyst (0.25 mol%), CO2 (simulated flue gas, bubbling), NaOAc (0.15 mmol), 5 min. e 7a was not detected (n.d.). | ||||
| 1 | Cat1 | 100c, 74d | 2400c, 3456d | 400c, 288d |
| 2 | Cat2 | 94c | 2256 | 376 |
| 3 | Cat3 | 91c | 2184 | 364 |
| 4 | Cat4 | 52c | 1242 | 207 |
| 5 | Cat5 | 36c | 864 | 144 |
| 6 | K2PdCl4 | 28 | 56 | 56 |
| 7 | Pd(OAc)2 | 44 | 88 | 88 |
| 8 | PdCl2 | 37 | 74 | 74 |
| 9 | Im-Br1/K2PdCl4 | 70 | 140 | 140 |
| 10 | Im-Br1/Pd(OAc)2 | 64 | 128 | 128 |
| 11 | Im-Br1/PdCl2 | 71 | 142 | 142 |
| 12 | PAG/K2PdCl4 | 56 | 112 | 112 |
| 13 | Blank, Im-Br1 | n.d.e | — | — |
During the optimization process, we observed significantly variation in the conversion of 6a as reaction time and temperature increased. Consequently, a series of detailed kinetic experiments were thus performed to validate the impact of various on the conversion of 6a. Derived simplified kinetic equations based on existing literature reports:59,60
| ln(1 − x) = −kobst + C | (1) |
With the optimal reaction conditions established, we further evaluated substrates with various substituents on the benzyl ring (Fig. 4). Both electron-withdrawing and electron-donating groups, regardless of their positions, were well tolerated, delivering the desired products 7a-h yields ranging from 90% to 100%. This protocol proved efficient for 3-methoxy (7b), 2,6-difluoro (7c), 4-fluoro (7d), 4-bromo (7e), 4-chloro (7f), and 4-trifluoromethyl (7g) substituents. Propargylic amines with 3-butyl (6i) and 3-cyclohexylmethyl (6j) substituents were also efficiently converted into the corresponding 2-oxazolidinones (7i and 7j) in excellent yields (up to approximately 100%). Notably, the reaction proceeded smoothly with propargylic amines bearing both electron-withdrawing and electron-donating groups.
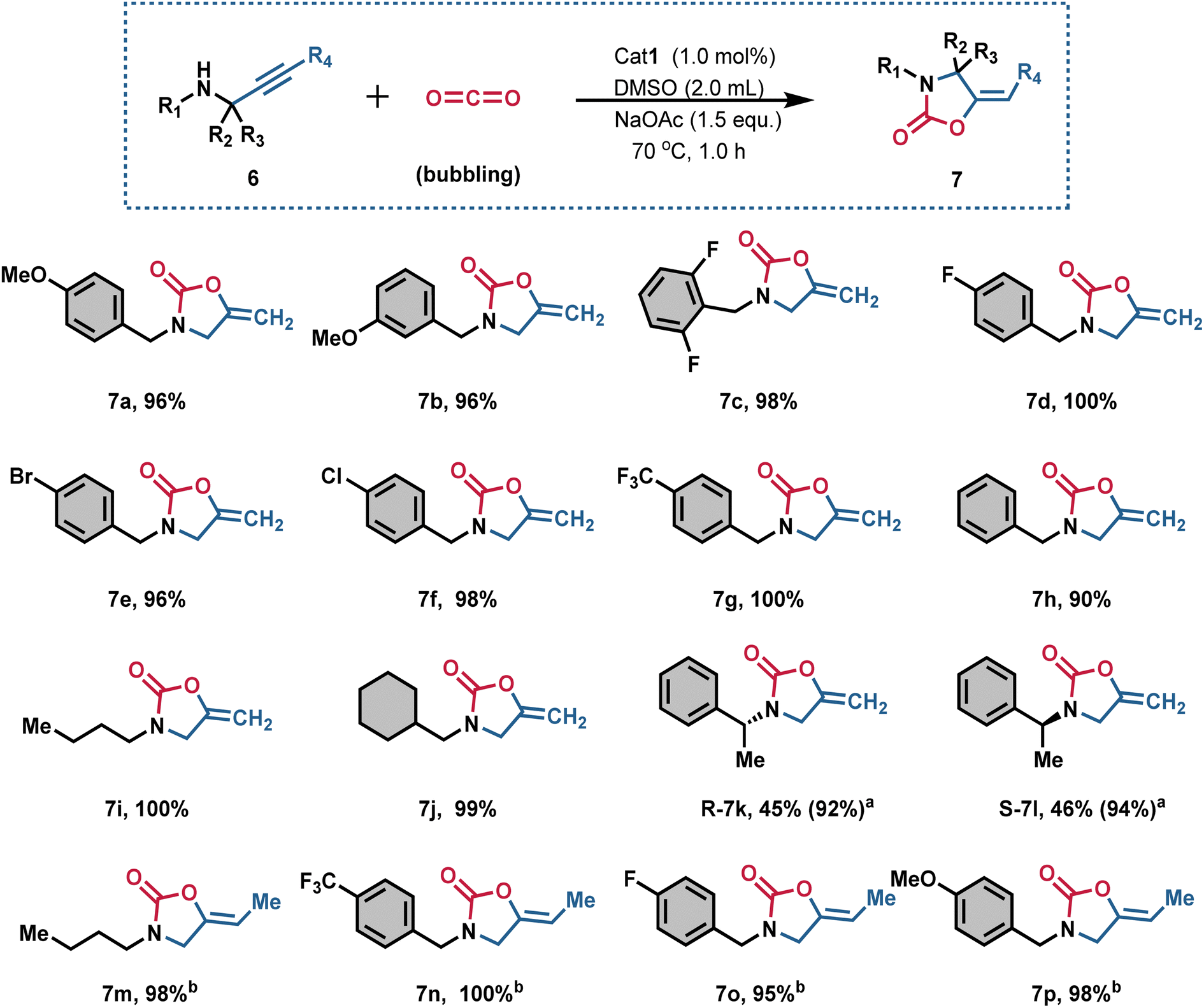 | ||
| Fig. 4 Scope of the carboxylative cyclization of propargylic amines and CO2 [simulated flue gas, V(CO2):V(N2) = 15:85]. Reaction conditions: Cat1 (1.0 mol%), substrate 6 (0.05 mmol), and NaOAc (1.5 equiv., 0.075 mmol) were added into DMSO (2.0 mL) in the reaction tube, and then stirred at 70 °C for 1.0 h. aReact for 2.0 h. bCat1 (1.5 mol%), react for 3.0 h. | ||
The introduction of a methyl substituent at the benzylic position, increasing steric hindrance, notably affected the yields, producing desired products (7k and 7l) in moderate yields. Extending the reaction time to 2.0 h also facilitated the efficient conversion of propargylic amines 6k and 6l into the corresponding products 7k and 7l in excellent yields (92% and 94%). Unfortunately, when R4 was a Me- group, the substrates 6n–6p exhibited relatively low reactivity, illustrating the negative impact of a Me- group at the terminal alkyne on the reactivity of propargyl amines. Therefore, extending the reaction time to 3.0 h and increasing the amount of catalyst to 1.5 mol% resulted in the formation of target products 7n–7p with yields ranging from 95% to 100%. Consequently, Cat1, containing AcGlu and bis(benzimidazolium) units, can be considered a novel AIO functionalized, efficient Pd catalyst for the carboxylative cyclization of propargylic amines and dilute CO2. The synthetic utility was further demonstrated by reaction, product conversion, and the synthesis of compound on a gram-scale. Additionally, the gram-scale synthesis was also achieved with good yield. It is worth mentioning that Cat1-catalyzed conversion of 6a and CO2 into gram-scale 7a with around 100% yield (Fig. S30†).
Catalyst cycle evaluation
Cat1 was selected for the recycling reaction to evaluate its stability in the synthesis of 2-oxazolidinone 7a. Vacuum distillation was utilized to separate the products and Cat1 after each cycle, while fresh 6a was introduced into the catalytic system, allowing the solution containing Cat1 to be recycled for subsequent runs. Then catalyst mixture was recycled for the synthetic reaction and the catalytic activity only decreased by only approximately 10% after 10 cycles by 1H NMR (Fig. S31a and c†). Moreover, the color of the catalytic system remained virtually unchanged throughout runs 1 to 10 (Fig. S31b†), indicating Cat1 was stabilized during recycling. These results demonstrating that catalyst Cat1 could be recycled. In contrast, the recyclability of Cat5 without Ac- units is much lower, whose yield dropped below 85% just after 4 runs (Fig. S32†). These results indicate that the Ac- groups enhance the hydrogen bonding and Lewis acid-Lewis base interactions with PdCl42−, thereby increasing the stability of the catalytic species during the catalytic process.The FT-IR and 1H NMR spectra of the Cat1 were measured before and after recycle (Fig. S33†). The 1H NMR results indicated that the disappearance of bis(benzimidazolium) units after the reaction, while AcGlu units maintained. Furthermore, changes in the signal peaks corresponding to CN and CC in the FT-IR spectrum were observed, while the peaks for CO, C–O, and C–O–C remained unchanged, further validating our judgments. This would explain the changes in cations and anions would lead to decreased catalytic activity, which further indicated that cations and anions would have a synergistic effect on the catalytic reaction. This would explain the disappearance of the bis(benzimidazolium) units would lead to decreased catalytic activity, which further indicated that among the bis(benzimidazolium), AcGlu, and palladium catalyst species would have a synergistic effect on the catalytic reaction.
Possible mechanism of AIO Cat1-catalyzed carboxylative cyclization
To deepen our understanding of the catalytic mechanism of the AIO Cat1-catalyzed carboxylative cyclization reaction, we conducted numerous quasi-in-situ NMR and 13C-isotope-labeling experiments. We conducted 13C-isotopic-labeling NMR and FT-IR experiments to verify that the CO group in product 7a is derived from CO2. The results clearly indicate that the peak intensity of the CO unit in the 13C-labeled 7a at 155.7 ppm is considerably higher than that of the unlabeled 7a (Fig. S34a†). Furthermore, from the FT-IR spectra of 7a, the signal of the CO unit shifted from 1758 cm−1 to 1783 cm−1 (Fig. S34b†). These findings provide strong support for our proposed pathway for CO2 conversion, which entails the initial insertion of CO2 into the carbonate group of intermediates, leading to the formation of the final 2-oxazolidinones. A broad peak at 2.41 ppm was identified as the NH of substrate 6a by hydrogen–deuterium exchange 1H NMR experiment in DMSO-d6 with two drops (70 μL) of D2O (Fig. S35†).
It is necessary to elucidate the roles of Cat1 and NaOAc in conjunction with substrate 6a within the reaction system. Upon the independent addition of Cat1 and NaOAc, a broad signal at 3.35 ppm appeared while the peak at 2.41 ppm disappeared, suggesting that both components can activate the NH group (Fig. S36†). When Cat1 and NaOAc were added simultaneously, the shift of the NH peak was very similar to that observed with the addition of only Cat1 to substrate 6a, indicating synergistic activation of the NH group by Cat1 and NaOAc. As illustrated in Fig. S37,† with the progression of the reaction time, the characteristic proton peaks of substrate 6a (3.08, 3.24, 3.66, and 3.72 ppm) gradually diminished and ultimately vanished, while new characteristic signals for 2-oxazolidinone 7a (3.74, 4.08, 4.33, and 4.65 ppm) emerged and intensified. These peak changes signify the gradual conversion of substrate 6a into product 7a.13 The dynamics and interactions among Cat1, NaOAc, CO2, and substrate 6a were further explored using quasi-in-situ1H and 13C NMR in DMSO-d6 (Fig. 5 and S38†). Notably, the proton signals of C2–H and Ac in Cat1 shifted upfield significantly by approximately 20 Hz and 5.0 Hz, respectively, after CO2 was bubbled into DMSO-d6 (Fig. S38†). This observation underscores the pronounced interactions between Cat1 and CO2.
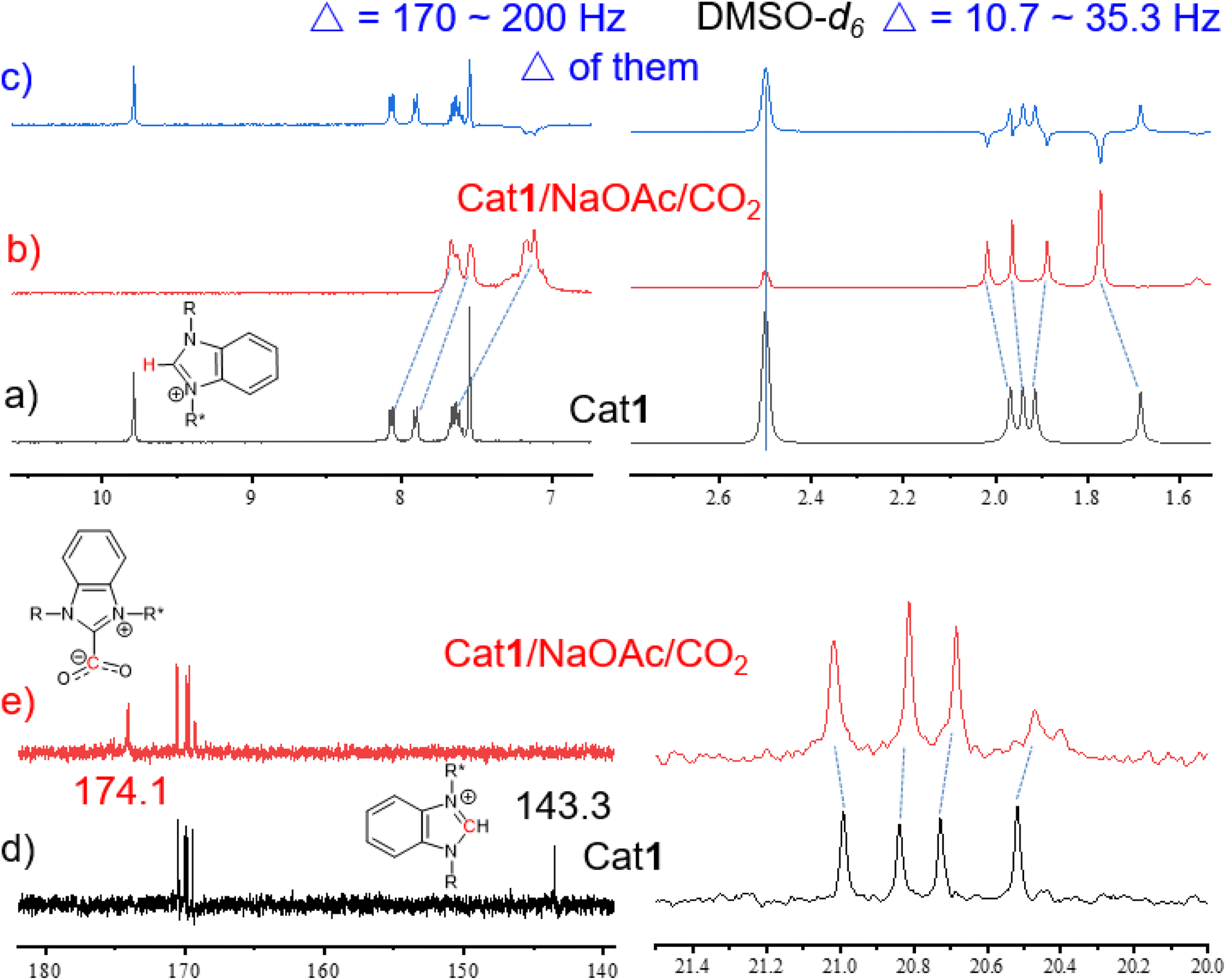 | ||
| Fig. 5 (a–c) Partially 1H NMR spectra of Cat1 and Cat1/NaOAc/CO2. (d and e) Partially 13C NMR spectra of Cat1 and Cat1/NaOAc/CO2. | ||
The pKa values of benzimidazolium salts rang between 21 and 24, positioning them between the ethyl acetate and neutral carbonyl carbon acids such as acetone. This indicates that the least basic NHC is more basic than the most basic phosphine [P(tBu)3, pKa∼10], suggesting a high proton (C2–H) affinity.61–63 Remarkably, the signal at 9.79 ppm assigned to the C2–H of Cat1 disappeared upon the introduction of NaOAc, indicating the formation of free NHC even at RT (Fig. 5b). Similarly, the C2–H peak of imidazolium salt Cat2 split into a doublet and subsequently disappeared (Fig. S39†), while the proton signal at C2–Me of Cat3 remained unchanged (Fig. S40†). The proton signals of Ac groups shifted by 10.7 to 35.3 Hz, and the signals in the benzimidazolium heterocycle broadened and shifted upfield significantly by approximately 170 to 200 Hz (Fig. 5a–c). These results indicate that the weak base NaOAc not only interacts with Cat1 but also generates free NHC in-situ. When CO2 was bubbled into the mixture of Cat1/NaOAc, the proton signals of Ac groups exhibited shifts both upfield and downfield by approximately 10.7 to 35.3 Hz (Fig. 5a–c), indicating the formation of AcGlu-CO2-philes between CO2 and Ac groups. Meanwhile, the C2–C signal at 143.3 ppm disappeared, and a new peak at 174.1 ppm appeared, assigned to the CO of the in-situ formed NHC-CO2 adducts (Fig. 5d and e).28 Hence, CO2 is co-activated in the form of AcGlu-CO2-philes and NHC-CO2 adducts, thereby promoting the catalytic process.
As a comparison, when NaOAc was added into the DMSO-d6 solution of Cat3 (may be considered as NHO precursor), the 1H NMR signals maintained unchanged (Fig. S40†). This indicates that the alkalinity of NaOAc is insufficient to generate free NHO from Cat3, preventing further reaction with CO2 to form NHO-CO2 adducts, resulting in lower reaction activity compared to Cat1. Herein, the acidity of C2–H in benzimidazolium salt was found to be superior to that in corresponding Cat2 and Cat3, primarily due to the absence of interfering acidic protons on the ring. It is reasonable to suggest that the formation of NHC-CO2 adducts from Cat1 is easier than from Cat2, and NHO-CO2 adducts from Cat3.57 Furthermore, the interactions between Cat5, NaOAc, and CO2 were further investigated via1H NMR. When NaOAc was added to the DMSO-d6 solution of Cat5, the singlet C2–H peak of the imidazolium ring split into doublet and then disappeared. However, when CO2 was bubbled into the mixture of Cat5 and NaOAc, all signals remained unchanged, indicating no interaction between them (Fig. S41†).
Based on these quasi-in-situ NMR, 13C-isotope-labeling experiments, and previous studies,2,11,17 a plausible mechanism has been proposed in Fig. 6. Initially, with the assistance of the base NaOAc, Cat1 is deprotonated in-situ into free NHC (intermediate A). Subsequently, the intermediate A activates CO2 in the form of NHC-CO2 adducts (intermediate B). Simultaneously, the AcGlu groups in Cat1 capture and activate CO2 in the form of AcGlu-CO2-philes (intermediate C). In this study, the proton of NH in the propargylic amine is co-deprotonated by NaOAc and Cat1, facilitating co-activated CO2 from intermediates B and C electrophilic attack the amide anion, forming a carbamate intermediate D. The homogeneous PdCl42− anion activates the triple bond (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) of terminal alkyne with the assistance of NaOAc, yielding a Pd acetylide intermediate E. Subsequently, the negatively charged oxygen from carbamate intermediate E attacks the Pd acetylide, resulting in intramolecular cyclization (intermediate F). The proton transfer from NaOAc-H+ to the CC bond leads to the formation of the product 2-oxazolidinone G and regenerates Cat1. The initial sequestration and activation of CO2 in the form of NHC-CO2 adducts and AcGlu-CO2-philes significantly facilitated the catalytic process. This integrated catalyst demonstrated an interesting multi-dimensional synergistic effect of CO2 co-sequestration, co-activation, and catalytic transformation in an AIO catalytic system, achieving the highest reported TOF value.
C) of terminal alkyne with the assistance of NaOAc, yielding a Pd acetylide intermediate E. Subsequently, the negatively charged oxygen from carbamate intermediate E attacks the Pd acetylide, resulting in intramolecular cyclization (intermediate F). The proton transfer from NaOAc-H+ to the CC bond leads to the formation of the product 2-oxazolidinone G and regenerates Cat1. The initial sequestration and activation of CO2 in the form of NHC-CO2 adducts and AcGlu-CO2-philes significantly facilitated the catalytic process. This integrated catalyst demonstrated an interesting multi-dimensional synergistic effect of CO2 co-sequestration, co-activation, and catalytic transformation in an AIO catalytic system, achieving the highest reported TOF value.
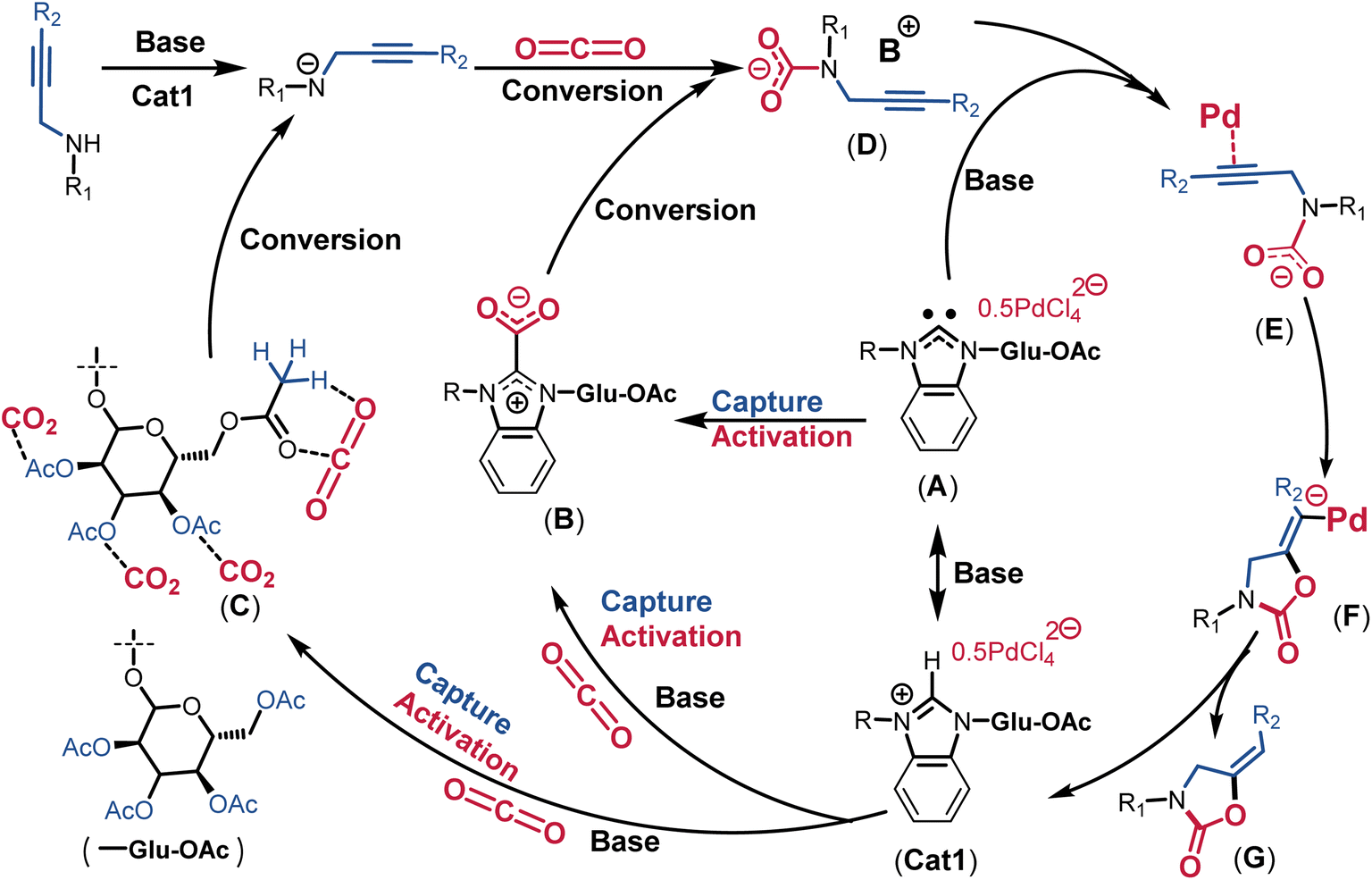 | ||
| Fig. 6 Proposed mechanism for Cat1-catalyzed carboxylative cyclization. | ||
Conclusions
In summary, we have designed a highly efficient Pd catalyst (Cat1) through an AIO strategy, functionalized with sugar acetates (AcGlu) and bis(benzimidazolium) units. Cat1-catalyzed the cyclization of simulated flue gas and propargylic amine to synthesize 2-oxazolidinone with the highest TOF of 3456 h−1, surpassing all previously reported catalysts. Through a series of quasi-in situ NMR and 13C-isotope-labeling experiments, it was observed that the C2–H in the benzimidazolium heterocycle forms free NHC more readily with the assistance of NaOAc compared to the imidazolium and 2-methyl-imidazolium heterocycles. In comparison to Cat5, which lacks Ac protective groups, the catalytic activity of Cat1 was significantly enhanced in the cyclization reaction of CO2 and propargylic amines, demonstrating that the Ac groups enhance CO2 transformation. The resulting NHC-CO2 adducts and AcGlu-CO2-philes served as effective activated CO2 sources for further conversion. The metal Pd center activated the CC bond to generate the Pd acetylide intermediate, leading to the production of a series of 2-oxazolidinones with yields between 90% and 100%. Meanwhile, Cat1 demonstrated good stability across various temperatures, with catalytic activity decreasing by approximately 10% after 10 cycles. A gram-scale experiment was also conducted to demonstrate the practical scalability of this reaction. This strategy of designing high-performance catalysts with capabilities for activating reaction substrates and stabilizing metal catalytic centers shows great promise for advancing CO2 conversion technologies. Furthermore, sugar acetates, being low-cost, nontoxic, nonvolatile, biocompatible, and renewable compounds, contribute to making the corresponding CO2 catalytic reaction process environmentally friendly.
Data availability
The data that support the findings of this study are available in the main text and the ESI.†Author contributions
Lingfang Kong: investigation, formal analysis, visualization, writing – original draft, writing – review & editing; Zekun Tao: investigation, methodology, formal analysis, writing – original draft; Yunjia Li: investigation, formal analysis; Huiwen Gong: methodology, formal analysis; Yun Bai: methodology, formal analysis, resources, writing – review & editing; Longbin Li: formal analysis, writing – review & editing; Xianjin Zhang: formal analysis, writing – review & editing; Zhonggao Zhou: resources, project administration, supervision, writing – review & editing; Yiwang Chen: resources, project administration, supervision, project administration, writing – review & editing. All the authors discussed the experimental results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China Grant Numbers 22261002 (Z. Z.), 22301044 (Y. B.), the Key Project of Jiangxi Provincial Natural Science Foundation 20232ACB203016 (Z. Z.), the Key Laboratory of Jiangxi University for Functional Materials Chemistry Grant Number FMC21702 (Z. Z.), the Innovative Special Fund Project for Graduate Students of Jiangxi Province Grant Number YC2023–S845 (L. K.).Notes and references
- F. D. Bobbink, S. Das and P. J. Dyson, N-formylation and N-methylation of amines using metal-free N-heterocyclic carbene catalysts and CO2 as carbon source, Nat. Protoc., 2017, 12, 417–428 CrossRef CAS PubMed.
- S.-L. Hou, J. Dong, X.-Y. Zhao, X.-S. Li, F.-Y. Ren, J. Zhao and B. Zhao, Thermocatalytic conversion of CO2 to valuable products activated by noble-metal-free metal-organic frameworks, Angew. Chem., Int. Ed., 2023, 62, e202305213 CrossRef CAS PubMed.
- L. Wang, C. Qi, W. Xiong and H. Jiang, Recent advances in fixation of CO2 into organic carbamates through multicomponent reaction strategies, Chin. J. Catal., 2022, 43, 1598–1617 CrossRef CAS.
- J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Radical carboxylative cyclizations and carboxylations with CO2, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed.
- R. Cauwenbergh, V. Goyal, R. Maiti, K. Natte and S. Das, Challenges and recent advancements in the transformation of CO2 into carboxylic acids: straightforward assembly with homogeneous 3d metals, Chem. Soc. Rev., 2022, 51, 9371–9423 RSC.
- X. Shang, X. Yang, G. Liu, T. Zhang and X. Su, A molecular view of single-atom catalysis toward carbon dioxide conversion, Chem. Sci., 2024, 15, 4631–4708 RSC.
- G. Ciamician, The Photochemistry of the Future, Science, 1912, 36, 385–394 CrossRef CAS PubMed.
- T. A. Mukhtar and G. D. Wright, Streptogramins, Oxazolidinones, and Other Inhibitors of Bacterial Protein Synthesis, Chem. Rev., 2005, 105, 529–542 CrossRef CAS PubMed.
- X. Yang, L. Xu, Y. Zhu, S. Zhang, G. Jia and J. Du, Efficient fabrication of oxazolidinones for the carboxylative cyclization with carbon dioxide, J. CO2 Util., 2023, 74, 102531 CrossRef CAS.
- S. Wang and C. Xi, Recent advances in nucleophile-triggered CO2-incorporated cyclization leading to heterocycles, Chem. Soc. Rev., 2019, 48, 382–404 RSC.
- L. Li, Y. Lv, H. Sheng, Y. Du, H. Li, Y. Yun, Z. Zhang, H. Yu and M. Zhu, A low-nuclear Ag4 nanocluster as a customized catalyst for the cyclization of propargylamine with CO2, Nat. Commun., 2023, 14, 6989 CrossRef CAS PubMed.
- X. L. Jiang, Y. E. Jiao, S. L. Hou, L. C. Geng, H. Z. Wang and B. Zhao, Green conversion of CO2 and propargylamines triggered by triply synergistic catalytic effects in metal-organic frameworks, Angew. Chem., Int. Ed., 2021, 60, 20417–20423 CrossRef CAS PubMed.
- A.-L. Gu, Y.-X. Zhang, Z.-L. Wu, H.-Y. Cui, T.-D. Hu and B. Zhao, Highly efficient conversion of propargylic alcohols and propargylic amines with CO2 activated by noble-metal-free catalyst Cu2O@ZIF-8, Angew. Chem., Int. Ed., 2022, 61, e202114817 CrossRef CAS PubMed.
- F. Inagaki, K. Maeda, K. Nakazawa and C. Mukai, Construction of the Oxazolidinone Framework from Propargylamine and CO2 in Air at Ambient Temperature: Catalytic Effect of a Gold Complex Featuring an L2/Z-Type Ligand, Eur. J. Org Chem., 2018, 2018, 2972–2976 CrossRef CAS.
- C.-S. Cao, S.-M. Xia, Z.-J. Song, H. Xu, Y. Shi, L.-N. He, P. Cheng and B. Zhao, Highly Efficient Conversion of Propargylic Amines and CO2 Catalyzed by Noble-Metal-Free [Zn116] Nanocages, Angew. Chem., Int. Ed., 2020, 59, 8586–8593 CrossRef CAS PubMed.
- X. Xu, Z. Li, H. Huang, X. Jing and C. Duan, A novel copper metal–organic framework catalyst for the highly efficient conversion of CO2 with propargylic amines, Inorg. Chem. Front., 2022, 9, 3839–3844 RSC.
- Z.-G. Zhou, P. He, J. Li, J. Zhang, G. Xu, S.-Y. Zhang, X.-X. Deng, Z.-Y. Du, G.-T. Luo, H.-Y. Zhen, Y. Chen and C.-T. He, Integration of CO2 Capture, Activation, and Conversion with Ternary Acetylglucosyl 2-Methyl-imidazolium Modified Pd Catalyst, Org. Chem. Front., 2023, 10, 2045–2053 RSC.
- W. Lyu, Y. Liu, J. Zhou, D. Chen, X. Zhao, R. Fang, F. Wang and Y. Li, Modulating the Reaction Configuration by Breaking the Structural Symmetry of Active Sites for Efficient Photocatalytic Reduction of Low-concentration CO2, Angew. Chem., Int. Ed., 2023, 62, e202310733 CrossRef CAS PubMed.
- M. Wang, B. Wang, J. Zhang, S. Xi, N. Ling, Z. Mi, Q. Yang, M. Zhang, W. R. Leow, J. Zhang and Y. Lum, Acidic media enables oxygen-tolerant electrosynthesis of multicarbon products from simulated flue gas, Nat. Commun., 2024, 15, 1218 CrossRef CAS PubMed.
- H. Zhou and X. Lu, Lewis base-CO2 adducts as organocatalysts for CO2 transformation, Sci. China: Chem., 2017, 60, 904–911 CrossRef CAS.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Coupling electrochemical CO2 conversion with CO2 capture, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- J. Septavaux, C. Tosi, P. Jame, C. Nervi, R. Gobetto and J. Leclaire, Simultaneous CO2 capture and metal purification from waste streams using triple-level dynamic combinatorial chemistry, Nat. Chem., 2020, 12, 202–212 CrossRef CAS PubMed.
- Y. B. Wang, Y. M. Wang, W. Z. Zhang and X. B. Lu, Fast CO2 sequestration, activation, and catalytic transformation using N-heterocyclic olefins, J. Am. Chem. Soc., 2013, 135, 11996–12003 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- K. Miharu, N. Bunta, F. Hayato, Y. Kosuke, K. Takuya and T. Mamoru, Single–carbon atom transfer to α,β-unsaturated amides from N-heterocyclic carbenes, Science, 2023, 379, 484–488 CrossRef PubMed.
- C.-Y. Wu, W. J. Wolf, Y. Levartovsky, H. A. Bechtel, M. C. Martin, F. D. Toste and E. Gross, High-spatial-resolution mapping of catalytic reactions on single particles, Nature, 2017, 541, 511–515 CrossRef CAS PubMed.
- C. He, D.-H. Si, Y.-B. Huang and R. Cao, A CO2-masked carbene functionalized covalent organic framework for highly efficient carbon dioxide conversion, Angew. Chem., Int. Ed., 2022, 61, e202207478 CrossRef CAS PubMed.
- X. Guo, F. Zhang, Z. Yang, L. Gao, R. Wei and G. Xiao, Construction of covalent organic framework functionalized with carbene dual active sites for enhancing CO2 carboxylation conversion, Chem. Eng. J., 2023, 475, 146453 CrossRef CAS.
- L. Wan, M. Pang, J. Le, Z. Xu, H. Zhou, Q. Xu and B. Wang, Oriented intergrowth of the catalyst layer in membrane electrode assembly for alkaline water electrolysis, Nat. Commun., 2022, 13, 7956 CrossRef CAS PubMed.
- T. Jia, Y.-X. Li, X.-H. Ma, M.-M. Zhang, X.-Y. Dong, J. Ai and S.-Q. Zang, Atomically precise ultrasmall copper cluster for room-temperature highly regioselective dehydrogenative coupling, Nat. Commun., 2023, 14, 6877 CrossRef CAS PubMed.
- A. Brzeczek-Szafran, K. Erfurt, A. Blacha-Grzechnik, M. Krzywiecki, S. Boncel and A. Chrobok, Carbohydrate Ionic Liquids and Salts as All-in-One Precursors for N-Doped Carbon, ACS Sustain. Chem. Eng., 2019, 7, 19880–19888 CrossRef CAS.
- X. Q. Li, K. Chen, R. L. Guo and Z. Wei, Ionic Liquids Functionalized MOFs for Adsorption, Chem. Rev., 2023, 123, 10432–10467 CrossRef CAS PubMed.
- L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Green processing using ionic liquids and CO2, Nature, 1999, 399, 28–29 CrossRef.
- B. Yoon and G. A. Voth, Elucidating the Molecular Mechanism of CO2 Capture by Amino Acid Ionic Liquids, J. Am. Chem. Soc., 2023, 145, 15663–15667 CrossRef CAS PubMed.
- A. Weilhard, S. P. Argent and V. Sans, Efficient carbon dioxide hydrogenation to formic acid with buffering ionic liquids, Nat. Commun., 2021, 12, 231 CrossRef CAS PubMed.
- Q. Li, F. Yan and J. Texter, Polymerized and Colloidal Ionic Liquids – Syntheses and Applications, Chem. Rev., 2024, 124, 3813–3931 CrossRef CAS PubMed.
- D. Prodius and A.-V. Mudring, Rare earth metal-containing ionic liquids, Coord. Chem. Rev., 2018, 363, 1–16 CrossRef CAS.
- P. Migowski, P. Lozano and J. Dupont, Imidazolium based ionic liquid-phase green catalytic reactions, Green Chem., 2023, 25, 1237–1260 RSC.
- T. Zhou, C. Gui, L. Sun, Y. Hu, H. Lyu, Z. Wang, Z. Song and G. Yu, Energy Applications of Ionic Liquids: Recent Developments and Future Prospects, Chem. Rev., 2023, 123, 12170–12253 CrossRef CAS PubMed.
- X.-B. Hu, Y.-X. Li, K. Huang, S.-L. Ma, H. Yu, Y.-T. Wu and Z.-B. Zhang, Impact of α-D-glucose pentaacetate on the selective separation of CO2 and SO2 in supported ionic liquid membranes, Green Chem., 2012, 14, 1440–1446 RSC.
- P. Raveendran and S. L. Wallen, Sugar acetates as novel, renewable CO2-philes, J. Am. Chem. Soc., 2002, 124, 7274–7275 CrossRef CAS PubMed.
- S.-L. Ma, Y.-T. Wu, M. L. Hurrey, S. L. Wallen and C. S. Grant, Sugar Acetates as CO2-philes: Molecular Interactions and Structure Aspects from Absorption Measurement Using Quartz Crystal Microbalance, J. Phys. Chem. B, 2010, 114, 3809–3817 CrossRef CAS PubMed.
- M. A. Blatchford, P. Raveendran and S. L. Wallen, Spectroscopic Studies of Model Carbonyl Compounds in CO2: Evidence for Cooperative C–H⋯O Interactions, J. Phys. Chem. A, 2003, 107, 10311–10323 CrossRef CAS.
- P. Raveendran and S. L. Wallen, Cooperative C–H⋯O Hydrogen Bonding in CO2-Lewis Base Complexes: Implications for Solvation in Supercritical CO2, J. Am. Chem. Soc., 2002, 124, 12590–12599 CrossRef PubMed.
- G. Kontos, C. Tsioptsias and I. Tsivintzelis, Cellulose Acetate-Ionic Liquid Blends as Potential Polymers for Efficient CO2 Separation Membranes, Polymers, 2024, 16, 554 CrossRef CAS PubMed.
- A. Annunziata, A. Amoresano, M. E. Cucciolito, R. Esposito, G. Ferraro, I. Iacobucci, P. Imbimbo, R. Lucignano, M. Melchiorre, M. Monti, C. Scognamiglio, A. Tuzi, D. M. Monti, A. Merlino and F. Ruffo, Pt(II) versus Pt(IV) in Carbene Glycoconjugate Antitumor Agents: Minimal Structural Variations and Great Performance Changes, Inorg. Chem., 2020, 59, 4002–4014 CrossRef CAS PubMed.
- A. Annunziata, M. E. Cucciolito, M. Di Ronza, G. Ferraro, M. Hadiji, A. Merlino, D. Ortiz, R. Scopelliti, F. Fadaei Tirani, P. J. Dyson and F. Ruffo, Ruthenium(II)–Arene Complexes with Glycosylated NHC-Carbene Co-Ligands: Synthesis, Hydrolytic Behavior, and Binding to Biological Molecules, Organometallics, 2023, 42, 952–964 CrossRef CAS.
- J. Wolfs, R. Nickisch, L. Wanner and M. A. R. Meier, Sustainable One-Pot Cellulose Dissolution and Derivatization via a Tandem Reaction in the DMSO/DBU/CO2 Switchable Solvent System, J. Am. Chem. Soc., 2021, 143, 18693–18702 CrossRef CAS PubMed.
- T. Hussain Ali, A. Washeel Salman, R. Sabah Abdulhussein, L. Persoons, D. Daelemans, W. Dehaen and L. Van Meervelt, New sugar-incorporated N-heterocyclic carbene precursors and their Ag(I) and Pd(II) complexes: Synthesis, characterization, and cytotoxicity studies, J. Mol. Liq., 2024, 393, 123618 CrossRef CAS.
- J. P. Byrne, L. Delgado, F. Paradisi and M. Albrecht, Carbohydrate-Functionalized Triazolylidene Iridium Complexes: Hydrogenation Catalysis in Water with Asymmetric Induction, ChemCatChem, 2022, 14, e202200086 CrossRef CAS PubMed.
- M. Ghosh and S. Khan, N-Heterocyclic Carbenes Capped Metal Nanoparticles: An Overview of Their Catalytic Scope, Chem. Rev., 2023, 13, 9313–9325 CAS.
- C. He, J. Liang, Y.-H. Zou, J.-D. Yi, Y.-B. Huang and R. Cao, Metal-organic frameworks bonded with metal N-heterocyclic carbenes for efficient catalysis, Natl. Sci. Rev., 2022, 9, nwab157 CrossRef CAS PubMed.
- P. Onnuch, K. Ramagonolla and R. Y. Liu, Aminative Suzuki–Miyaura coupling, Science, 2024, 383, 1019–1024 CrossRef CAS PubMed.
- C.-T. Yue, Q. Xing, P. Sun, Z.-L. Zhao, H. Lv and F.-W. Li, Enhancing stability by trapping palladium inside N-heterocyclic carbene-functionalized hypercrosslinked polymers for heterogeneous C-C bond formations, Nat. Commun., 2021, 12, 1875 CrossRef CAS PubMed.
- P. García-Domínguez, L. Fehr, G. Rusconi and C. Nevado, Palladium-catalyzed incorporation of atmospheric CO2: efficient synthesis of functionalized oxazolidinones, Chem. Sci., 2016, 7, 3914–3918 RSC.
- P. Brunel, J. Monot, C. E. Kefalidis, L. Maron, B. Martin-Vaca and D. Bourissou, Valorization of CO2: preparation of 2-oxazolidinones by metal–ligand cooperative catalysis with SCS indenediide Pd complexes, ACS Catal., 2017, 7, 2652–2660 CrossRef CAS.
- G. Gurau, H. Rodríguez, S. P. Kelley, P. Janiczek, R. S. Kalb and R. D. Rogers, Demonstration of Chemisorption of Carbon Dioxide in 1,3-Dialkylimidazolium Acetate Ionic Liquids, Angew. Chem., Int. Ed., 2011, 50, 12024–12026 CrossRef CAS PubMed.
- D. Yu and Y. Zhang, Copper- and copper-N-heterocyclic carbene-catalyzed C–H activating carboxylation of terminal alkynes with CO2 at ambient conditions, Proc. Natl. Acad. Sci. U.S.A., 2010, 107, 20184–20189 CrossRef CAS PubMed.
- P. Liu, K. Cai, D.-J. Tao and T. Zhao, The mega-merger strategy: M@COF core-shell hybrid materials for facilitating CO2 capture and conversion to monocyclic and polycyclic carbonates, Appl. Catal., B, 2024, 341, 123317 CrossRef CAS.
- K. Cai, P. Liu, Z. Chen, P. Chen, F. Liu, T. Zhao and D.-J. Tao, Construction of bifunctional triazine-based imidazolium porous ionomer polymers by a post-crosslinking tactic for efficient CO2 capture and conversion, Chem. Eng. J., 2023, 451, 138946 CrossRef CAS.
- C. Wang, X. Luo, H. Luo, D.-e. Jiang, H. Li and S. Dai, Tuning the Basicity of Ionic Liquids for Equimolar CO2 Capture, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS PubMed.
- S. Gaillard, C. S. J. Cazin and S. P. Nolan, N-Heterocyclic Carbene Gold(I) and Copper(I) Complexes in C–H Bond Activation, Acc. Chem. Res., 2011, 45, 778–787 CrossRef PubMed.
- X. li, S. Zeng, A. Wang, G. Li, L. Yuan, K. Peng, C. Jiang, H. Zhang, X. Zhang and S. Zhang, Modulating CO2 electroreduction to syngas by protic-nonprotic ionic liquid composite electrolytes, Chem. Eng. Sci., 2024, 292, 119961 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03106g |
| ‡ L. Kong and Z. Tao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |