
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymeric bis(triphenylphosphine)iminium chloride as a recyclable catalyst†
Ziwei
Xu
 ab,
Meng
Wang
ab and
Michael P.
Shaver
*ab
ab,
Meng
Wang
ab and
Michael P.
Shaver
*ab
aDepartment of Materials, School of Natural Sciences, The University of Manchester, Manchester, UK. E-mail: michael.shaver@manchester.ac.uk
bSustainable Materials Innovation Hub, The University of Manchester, Royce Hub Building, Oxford Road, Manchester, UK
First published on 7th August 2024
Abstract
Metal-free catalysts have garnered considerable interest as an environmental and economical alternative to precious metal catalysts. Bis(triphenylphosphine)iminium chloride (PPNCl) has emerged as a prominent choice due to its air and thermal stability and broad reactivity, especially in applications where a bulky cation is needed. The high phosphorus content and synthetic effort required for catalyst synthesis increase environmental impact; the recyclability of PPNCl in catalytic processes remains largely unexplored. The potential development of a polymer-supported PPNCl catalysts therefore desirable to enable this recyclability. In this work, we synthesise polymeric PPNCl (poly(PPNCl)) for the first time. Poly(PPNCl) demonstrates a comparative catalytic reactivity to its small molecule variant when employed as a catalyst in halogen-exchange reactions and CO2/epoxide coupling. For the latter the effect of catalyst loading, CO2 pressure, reaction time and addition of co-catalyst on conversion and selectivity was investigated. Poly(PPNCl) was easily recovered from the crude product by simple precipitation and its catalytic reactivity was well-maintained over three reaction cycles, providing environmental and economic advantages for sustainable reaction development.
Introduction
Catalysts, often containing transition metals, are essential tools in chemical synthesis.1–3 Challenges in metal extraction, complex toxicity, and economic cost frustrate the long-term sustainability of their industrial application.4,5 Consequently, metal-free catalysts have been touted as environmentally friendly, economically viable, and less hazardous alternatives to metal-based catalysts.6–9 An emerging class of metal-free catalysts are bis(triphenylphosphine)iminium salts,10–12 thanks to their good solubility, stability at high temperatures and ease of handling.11,13Bis(triphenylphosphine)iminium chloride (PPNCl) has been utilized in carbon dioxide (CO2) valorisation14–18 to affect the coupling of CO2 and epoxides to form cyclic carbonates as key reagents for the pharmaceutical and fine chemical industry.19–21 Wing et al. demonstrated that both the PPN+ and Cl− play important roles in the reaction mechanism.22 PPNCl has also been explored as a co-catalyst in the ring-open co-polymerisation of CO2 and epoxides to form polycarbonates.23,24 Showing catalyst versatility, PPNCl can also be utilized to facilitate the hydrogenation of CO2 to yield higher alcohols,25 and in difficult halogen-exchange reactions to yield fluorinated aromatic compounds for pharmaceutical and agrochemical applications (Fig. 1a).11
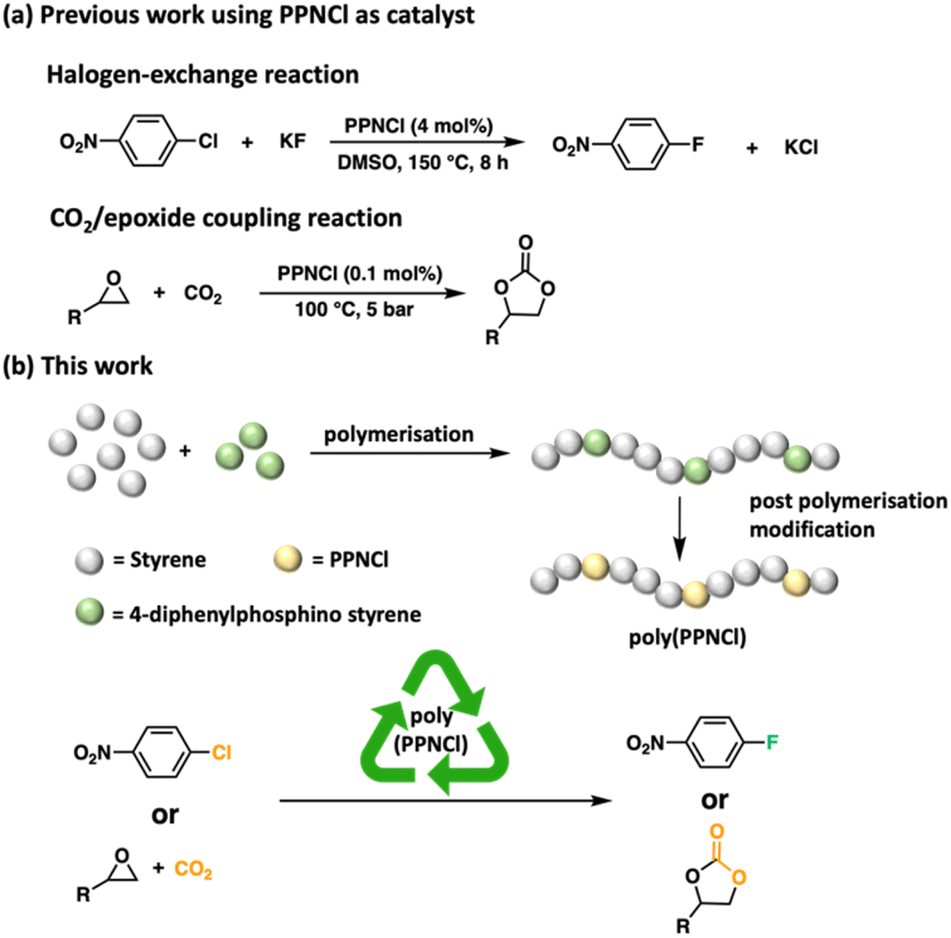 | ||
| Fig. 1 (a) Previous work using PPNCl as catalysts; (b) poly(PPNCl) as a recyclable catalyst for halogen exchange and CO2/epoxide coupling reactions. | ||
The material-intensive, multi-step synthesis of PPNCl means that catalyst recovery would be important, however, the recycling of PPNCl has thus far not been explored. In recent years, the recyclability of catalysts through incorporation into polymers has gained significant attention.26–29 Previous work in our group showed that polymer-supported catalysts featuring main-group elements including phosphorus, can be easily recovered and achieve comparable catalytic efficiencies to their small molecule counterparts.30–32
Developing a polymeric variant of PPNCl could generate a catalyst capable of maintaining high activity and new recyclability. In this work, we report for the first time, the preparation of macromolecular poly(PPNCl). We demonstrate the catalytic reactivity of poly(PPNCl) in catalyzing both halogen-exchange and CO2/epoxide coupling reactions. The catalyst is readily recovered through post-reaction precipitation and serves as an important step in non-metal catalyst recycling (Fig. 1b).
Results and discussion
Synthesis of poly(PPNCl)
Macromolecular poly(PPNCl) was prepared through post-polymerisation modification of poly(styrene-co-diphenylphosphine styrene), P1, as shown in Scheme 1. P1 was prepared by anionic co-polymerisation of styrene and p-(diphenylphosphino)styrene using n-butyllithium as the initiator in a methodology modified from our previous work on polymeric frustrated Lewis pairs.30 The styrene co-monomer serves to provide inert spacers in the polymer chains, separating the active catalyst moieties, and improving solubility. The phosphorus groups incorporated into the polymer chains served as anchors for a modified small molecule synthesis.11 As PPNCl is prepared by the reaction of a dichlorinated phosphine (Ph3PCl2), accessible in near quantitative conversion, and a lithiated amino(triphenylphosphine) (Ph3PNLi) that forms unavoidable side-products, it is essential to dichlorinate the polymer first. The reaction of P1 with C2Cl6 generates P2 with full conversion and high purity, as shown by NMR spectroscopy (Scheme 1a and Fig. S1†). A broad resonance at 63.2 ppm in the 31P NMR spectrum is comparable to the chemical shift for Ph3PCl2 (65.0 ppm).33 Separately, amination of triphenylphosphine afforded amino(triphenyl)phosphonium chloride which was subsequently lithiated to afford lithium (triphenylphosphoranylidene)azanide (Scheme 1b). Finally, the targeted poly(PPNCl) was synthezised by the reaction between P2 and lithium (triphenylphosphoranylidene)azanide (Scheme 1c). The obtained poly(PPNCl) was purified by dialysis to remove small molecule impurities. Analysis by 31P NMR spectroscopy (Fig. S1†) showed a single resonance at 20.7 ppm, confirming the full conversion of P2 to the desired product. Diffusion order spectroscopy (DOSY) showed a single diffusion coefficient, confirming that styrene and PPNCl moieties were distributed within the same polymer chains (Fig. S3†). This synthetic protocol was modified to prepare catalysts with 5 and 10 mol% PPNCl moieties (poly(PPNCl)-5 and poly(PPNCl)-10 respectively; Table S1†). For further characterisation of poly(PPNCl), thermogravimetry analysis (TGA) and differential scanning calorimetry (DSC) showed that poly(PPNCl)-5 has a lower degradation temperature and glass transition temperature compared to poly(PPNCl)-10 due its lower PPNCl loading in the polymer chains (Fig. S4†). Importantly, the evident absorptions of Fourier-transform infrared spectroscopy (FTIR) in the region between 1100 and 1480 cm−1 correspond to the vibration of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds according to the literature,34 further supporting the formation of poly(PPNCl).
N bonds according to the literature,34 further supporting the formation of poly(PPNCl).
 | ||
Scheme 1 (a) Synthetic procedures of P2 from phosphine-containing copolymer, m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n = 95:5 or 90:10; (i) n-butyllithium, toluene, −78 °C, 3 h; (ii) C2Cl6, THF, r.t., 3 h; (b) Synthesis of Ph3PNLi from triphenylphosphine; (iii) hydroxylamine-O-sulfonic acid, methanol/DCM, r.t., 1 h; (iv) n-butyllithium, THF, initiated at −50 °C, −15 °C 2 h; (c) synthesis of poly(PPNCl) (v) THF, r.t., 3 h. :n = 95:5 or 90:10; (i) n-butyllithium, toluene, −78 °C, 3 h; (ii) C2Cl6, THF, r.t., 3 h; (b) Synthesis of Ph3PNLi from triphenylphosphine; (iii) hydroxylamine-O-sulfonic acid, methanol/DCM, r.t., 1 h; (iv) n-butyllithium, THF, initiated at −50 °C, −15 °C 2 h; (c) synthesis of poly(PPNCl) (v) THF, r.t., 3 h. | ||
Halogen-exchange reaction
Halogen-exchange reactions were performed with these novel catalyst systems, poly(PPNCl)-5 and 10. The conversion of 1-chloro-4-nitrobenzene (CNB) to 1-fluoro-4-nitrobenzene (FNB) by poly(PPNCl)-10 (63%) was surprisingly lower than that of poly(PPNCl)-5 (80%) (Scheme 2a, Table S2†). While the higher loading offered a higher catalyst concentration, the increased number of PPNCl groups led to poorer solubility of the catalyst, therefore lowering conversion. Similar phenomena have been observed in the literature.35 The conversion catalysed by poly(PPNCl)-5 is comparable to that facilitated by PPNCl (81%), suggesting similar efficiency for small and macromolecular systems. With its superior solubility and performance, poly(PPNCl)-5 was chosen as the preferred catalyst for the rest of this work.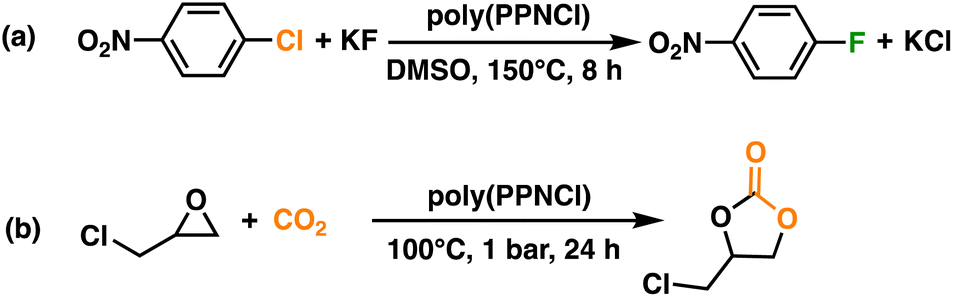 | ||
| Scheme 2 (a) Halogen-exchange reaction from CNB to FNB; (b) coupling reaction of CO2 with GC. | ||
Conversion of CNB with poly(PPNCl)-5 was systematically optimized (Table S2†). Minor increases in conversion by increasing the molar ratio of potassium fluoride (KF) added and when lower concentrations of catalyst were observed (2 mol%, 89%), further improving reaction sustainability. While the reaction worked in DMSO, MeOH and DMF, the optimal solvent was DMSO which correlated with enhanced catalyst solubility. CNB concentration is also important, with both lower (0.5, 1 M) and higher (3, 4 M) concentrations giving decreased conversions. High concentrations of CNB impact catalyst solubility in DMSO, reducing resultant reactivity.
With an optimized system in hand, we investigated whether this substrate scope could be expanded using poly(PPNCl) as the catalyst. Electrophilic aromatic substitution is favoured by the inductive and resonance effects promoted by electron-withdrawing groups (EWGs); previously this reaction was only reported for nitro-substituted aromatics. Displacing the nitro group of CNB with other para-substituted EWGs or meta-substituted electron-donating groups (EDGs) (Fig. S6†) was challenging. Of the substrates tested, only 4-chlorobenzonitrile showed conversion by 1H and 31P NMR spectroscopy (Fig. S7†).
CO2 and epoxide coupling reactions
The coupling of CO2 and epoxides is an important reaction for CO2 transformation, as the resultant cyclic carbonates are valuable intermediates for fuel additives,36 polycarbonates,37 and pharmaceuticals.38 In previous reports, the PPNCl catalyst was usually paired with a Lewis acid co-catalyst such as triphenylborane (BPh3) to enhance reactivity.39,40 The catalytic reactivity of poly(PPNCl) for the insertion of CO2 to epoxides in the absence of a solvent was investigated (Scheme 2b). Glycidyl chloride (GC) was selected as a model substrate, and the results summarized in Table S3† under standard conditions of 1 bar CO2 for 24 h with 0.1 mol% poly(PPNCl)-5.In the absence of poly(PPNCl), no conversion to chloropropene carbonate was observed. With 0.1 mol% (by catalyst moiety) poly(PPNCl) without any borane co-catalyst, 66% conversion was achieved, increasing to 98% at 0.4 mol%. Conversions did not substantially change upon alteration of CO2 pressure (1–5 bar) (Table S3,† entries 1 and 5–7), suggesting CO2 is not involved in the rate-determining step. While the rate of epoxide activation is often enhanced through the addition of BPh3, the conversion of GC surprisingly decreased to 48% when the Lewis acid is added to the system (Table S3,† entry 9).30 No reaction was observed when employing BPh3 alone. We hypothesize that the coordination of BPh3 to a key intermediate (I in Fig. S9†) would slow the rate of CO2 insertion and therefore decrease reaction rates. As the reaction works without the addition of an air-sensitive Lewis acid, we explored milder conditions under an air atmosphere (Table S3,† entry 16). However, GC conversion remained unchanged compared to inert atmosphere (Table S3,† entry 2).
The kinetics of the GC/CO2 coupling reaction catalysed by poly(PPNCl) was explored (Table S3,† entries 10–15). Conversion of GC increased from 3 h to 48 h, indicating a long-lived catalyst (Fig. S11a†) and first-order kinetics for [GC] (Fig. S11b†). A catalytic mechanism for the GC/poly(PPNCl) system is proposed (Scheme S1†). Switching the substrate to propylene oxide (PO) showed remarkably different results: PO conversion was very low (9%) when using poly(PPNCl)-5 while the introduction of a BPh3 co-catalyst increased conversion to 48% (Table S4†). This reversion of the trends observed for PO suggests a significant change in the mechanism proposed in Scheme S2.† Increased reaction time resulted in a higher conversion (61%) suggesting catalyst death is not the reason for reduced yields. The effect of CO2 pressure on PO conversion gave no significant change in conversion (1–5 bar). PO conversion was enhanced to 71% by increasing catalyst loading to 0.2 mol%. The efficacy of the borane system also allowed us to test the poly(Lewis acid) variant previously developed in our group.30 Poly(BPh3), a co-polymer of styrene and 4-styryl-diphenylborane, gave conversations comparable to poly(PPNCl)/BPh3 system, demonstrating that both PPNCl and BPh3 can be switched to recyclable polymeric systems while maintaining catalytic reactivity (Table S4,† entry 10).
Further expanding the substrate scope delineated two clear classes, evaluated by the overall catalytic reactivity of poly(PPNCl)versus poly(PPNCl)/BPh3 under conditions that avoided reaching full conversion in order to probe structure/activity relationships (Fig. 2). Only the most reactive substrate, GC, favoured poly(PPNCl) as a lone catalyst and outperformed all other tested epoxides (66%). Styrene oxide achieved the second highest conversion (34%) using only poly(PPNCl), supporting the theory that the electron-withdrawing nature of the substituents is key to ensuring reactivity. Interestingly, whilst the poly(PPNCl)/BPh3 binary system resulted in a lower conversion of GC than for poly(PPNCl) alone, this trend was reversed for all other substrates screened. As the most reactive substrate, GC can be activated by both poly(PPN+) and BPh3, the two catalytic mechanisms may compete with each other to stall catalytic turnover. The sterically hindered cyclohexene oxide had the lowest conversion, attributed to its high steric hindrance preventing associative ring-opening.
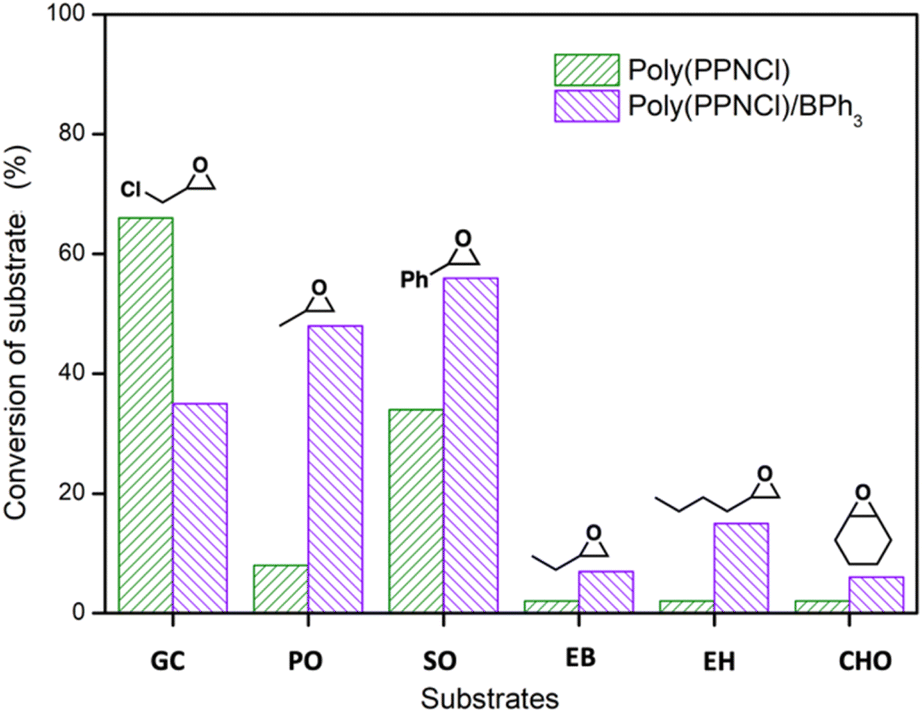 | ||
| Fig. 2 CO2/epoxides coupling reaction to yield cyclic carbonates. Conversions calculated by 1H NMR spectroscopy. Conversions for GC and PO were taken from the average value of three trials; the rest substrates were conducted once. Reactions were performed in a sealed ampoule on a 12.8 mmol scale at 0.1 mol% poly(PPNCl) with or without 0.1 mol% BPh3 (0.1 mol% as calculated from the number of expected active functional groups). | ||
Importantly, the selectivity of cyclic carbonates is 100% in nearly every case. Under no reaction conditions is polycarbonate and polyether generated in the presence of poly(PPNCl) alone. The good leaving group abilities of both poly(PPN+) and Cl− in the ring-closure step to form chloropropene carbonate likely facilitate this selectivity. For the GC/poly(PPNCl)/BPh3 system, a secondary product was observed (1H NMR: 4.7 ppm, Fig. S17 and S18†), identified as the carbonate anion adduct of BPh3 following ring opening by Cl−. This suggests that chlorine may attack the more substituted carbon to form an off-cycle product.
Finally, the catalytic selectivity and kinetic switch can be used in one-pot stepwise catalytic transformations. GC and PO could be sequentially converted into their cyclic carbonates as shown in Scheme 3. The epoxide mixture was first treated with poly(PPNCl) to yield chloropropene carbonate and propylene carbonate conversions of 97% and 2% respectively. The addition of BPh3 to the reaction led to the conversion of the unreacted PO (74%), evidencing this epoxide selectivity.
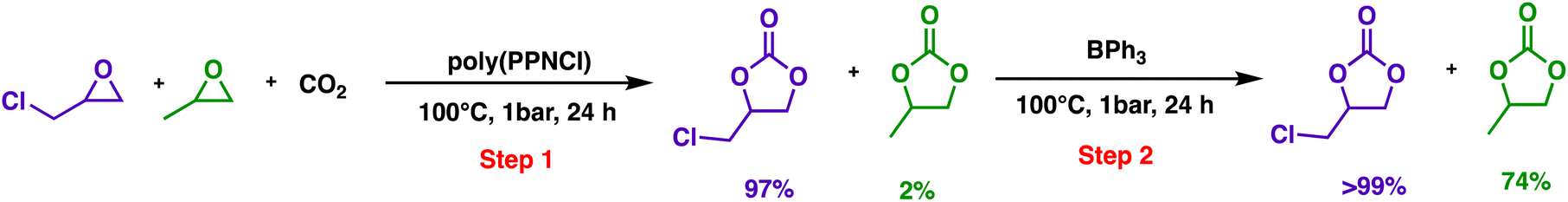 | ||
| Scheme 3 The selective one-pot, two-step reaction of epoxide conversion to cyclic carbonates. General conditions: the feed ratio of GC:PO:poly(PPNCl):BPh3 is 1000:1000:4:4. | ||
Catalyst recycling
Polymer-supported catalysts are important additions to sustainable catalyst development as their ready isolation allows for recyclability.31 We sought to explore the catalytic performance of both poly(PPNCl) and poly(PPNCl)/poly(BPh3) after repeated recovery of the polymer-supported catalysts from crude products (Fig. 3). Poly(PPNCl) can be recovered easily from the product by precipitation from n-hexane and dried before reuse in subsequent reactions. | ||
| Fig. 3 Reaction conversions over three cycles (a) halogen-exchange, (b) CO2/GC coupling, and (c) CO2/PO coupling reactions. Conversion calculated by 1H NMR spectroscopy. Conversions for recycled poly(PPNCl) were obtained from an average value of two trials. | ||
In the halogen-exchange reaction, the addition of n-hexane precipitated poly(PPNCl)-5 which was recovered from the crude product by filtration and dried under vacuum prior to weighing between each cycle. Catalytic performance was well-maintained over three reuse cycles (1st cycle: 80%; 2nd: 79%; 3rd: 77%), demonstrating excellent recyclability. For CO2/epoxide coupling, precipitation of the catalyst or catalysts from the crude chloropropene carbonate (poly(PPNCl)-5) and propylene carbonate (poly(PPNCl)-5/poly(BPh3)) reactions was conducted in the reaction vessel. The catalytic reactivity of poly(PPNCl) drops slightly over three reaction cycles of GC/CO2 coupling (1st cycle: 67%; 2nd: 61%; 3rd: 59%). A more precipitous decrease in substrate conversion is observed in the poly(PPNCl)/poly(BPh3) binary catalyst system (1st cycle: 50%; 2nd: 46%; 3rd: 30%) likely due to the mass loss and partial deactivation of air-sensitive poly(BPh3) during catalyst recovery.
Conclusions
In summary, we report the synthesis of poly(PPNCl) using post-polymerisation modification. Poly(PPNCl) can achieve high conversion at a low feeding ratio in the halogen-exchange reaction of chloroarenes to fluoroarenes, matching catalytic efficiency to the unrecoverable small molecule analogue, PPNCl. In CO2/epoxides coupling, the selective formation of cyclic carbonates is catalysed by two mechanisms. Depending on epoxide reactivity, either air-stable poly(PPNCl) can be used on its own or with the addition of BPh3 or poly(BPh3) as a co-catalyst. The robust poly(PPNCl) can be recovered by simple precipitation and reused in further reactions, with both halogen-exchange and CO2 coupling of reactive epoxides displaying excellent recyclability. The recovery and reuse of this key metal-free catalyst is an essential step to support sustainable chemical syntheses.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ziwei Xu and Meng Wang: investigation, methodology and manuscript writing. Michael P. Shaver: conceptualisation, manuscript writing – review and editing, supervision and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly thank the University of Manchester (President's Doctoral Scholar Awards, ZX) and the Leverhulme Trust (81420) for their financial support. This work was also supported by the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P. We thank Dr Christina Picken and Jordan Holland for their valuable perspectives as this manuscript developed.Notes and references
- J. Magano and J. R. Dunetz, Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- N. Saini, A. Malik and S. L. Jain, Light driven chemical fixation and conversion of CO2 into cyclic carbonates using transition metals: A review on recent advancements, Coord. Chem. Rev., 2024, 502, 215636 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Review of metal catalysts for oxygen reduction reaction: from nanoscale engineering to atomic design, Chem, 2019, 5, 1486–1511 CAS.
- H. Esteves, N. E. Morais Costa, V. K. Tomazett, J. L. S. Milani, R. P. das Chagas and Â. de Fátima, Organocatalysis for the Chemical Fixation of Carbon Dioxide to Synthesise N-Heterocycles, Curr. Green Chem., 2024, 11, 87–126 CrossRef CAS.
- D. Yang and B. C. Gates, Catalysis by metal organic frameworks: perspective and suggestions for future research, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- Y. Zhang, Q. Wang, Q. Chen, X. Li, Y. Li, M. Kang, Q. Li and J. Wang, A new boron modified carbon nitride metal-free catalyst for the cycloaddition of CO2 and bisepoxides, Appl. Catal., A, 2024, 119615 CrossRef CAS.
- R. Morodo, D. M. Dumas, J. Zhang, K. H. Lui, P. J. Hurst, R. Bosio, L. M. Campos, N. H. Park, R. M. Waymouth and J. L. Hedrick, Ring-Opening Polymerization of Cyclic Esters and Carbonates with (Thio) urea/Cyclopropenimine Organocatalytic Systems, ACS Macro Lett., 2024, 13, 181–188 CrossRef CAS.
- E. Poursaitidis, P. L. Gkizis, I. Triantafillidi and C. G. Kokotos, Organocatalytic activation of hydrogen peroxide: towards green and sustainable oxidations, Chem. Sci., 2024, 15, 1177–1203 RSC.
- K. A. Ahrendt, C. J. Borths and D. W. MacMillan, New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels−Alder reaction, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- T. W. Rave, Preparation and properties of bis(phosphoranylidene) ammonium chlorides, J. Org. Chem., 1967, 32, 3461–3466 CrossRef CAS.
- M. A. Lacour, M. Zablocka, C. Duhayon, J. P. Majoral and M. Taillefer, Efficient Phosphorus Catalysts for the Halogen-Exchange (Halex) Reaction, Adv. Synth. Catal., 2008, 350, 2677–2682 CrossRef CAS.
- A. Martinsen, J. Songstad, R. Larsson, M. Pouchard, P. Hagenmuller and A. Andresen, Preparation and properties of some bis(triphenylphosphine) iminium salts,[(Ph3P) 2N] X, Acta Chem. Scand., 1977, 31, 645–650 CrossRef CAS.
- J. Ruff and W. J. SCHLIENTZ, Inorg. Synth., 1974, 16, 84 CrossRef.
- M. Scharfenberg, J. Hilf and H. Frey, Functional polycarbonates from carbon dioxide and tailored epoxide monomers: degradable materials and their application potential, Adv. Funct. Mater., 2018, 28, 1704302 CrossRef.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Synthesis and properties of CO2-based plastics: Environmentally-friendly, energy-saving and biomedical polymeric materials, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS.
- H. Zhang and M. W. Grinstaff, Synthesis of atactic and isotactic poly(1, 2-glycerol carbonate) s: degradable polymers for biomedical and pharmaceutical applications, J. Am. Chem. Soc., 2013, 135, 6806–6809 CrossRef CAS.
- L.-Y. Wang, G.-G. Gu, T.-J. Yue, W.-M. Ren and X.-B. Lu, Semiaromatic Poly(thioester) from the Copolymerization of Phthalic Thioanhydride and Epoxide: Synthesis, Structure, and Properties, Macromolecules, 2019, 52, 2439–2445 CrossRef CAS.
- J. Huang, Y. Xu, M. Wang and Z. Duan, Copolymerization of propylene oxide and CO2 catalyzed by dinuclear salcy–CoCl complex, J. Macromol. Sci., Part A, 2020, 57, 131–138 CrossRef CAS.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Novel iron(III) catalyst for the efficient and selective coupling of carbon dioxide and epoxides to form cyclic carbonates, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- A. Sibaouih, P. Ryan, M. Leskelä, B. Rieger and T. Repo, Facile synthesis of cyclic carbonates from CO2 and epoxides with cobalt(II)/onium salt based catalysts, Appl. Catal., A, 2009, 365, 194–198 CrossRef CAS.
- F. Chen, S. Tao, N. Liu, C. Guo and B. Dai, Hemilabile N-heterocyclic carbene and nitrogen ligands on Fe(II) catalyst for utilization of CO2 into cyclic carbonate, Appl. Organomet. Chem., 2021, 35, e6099 CrossRef CAS.
- W. N. Sit, S. M. Ng, K. Y. Kwong and C. P. Lau, Coupling reactions of CO2 with neat epoxides catalyzed by PPN salts to yield cyclic carbonates, J. Org. Chem., 2005, 70, 8583–8586 CrossRef CAS PubMed.
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, A Lewis pair as organocatalyst for one-pot synthesis of block copolymers from a mixture of epoxide, anhydride, and CO2, Macromolecules, 2021, 54, 763–772 CrossRef CAS.
- C. E. Anderson, S. I. Vagin, W. Xia, H. Jin and B. Rieger, Cobaltoporphyrin-catalyzed CO2/epoxide copolymerization: selectivity control by molecular design, Macromolecules, 2012, 45, 6840–6849 CrossRef CAS.
- M. Cui, Q. Qian, Z. He, Z. Zhang, J. Ma, T. Wu, G. Yang and B. Han, Bromide promoted hydrogenation of CO2 to higher alcohols using Ru–Co homogeneous catalyst, Chem. Sci., 2016, 7, 5200–5205 RSC.
- M. Benaglia, A. Puglisi and F. Cozzi, Polymer-supported organic catalysts, Chem. Rev., 2003, 103, 3401–3430 CrossRef CAS.
- Y. Chauvin, D. Commereuc and F. Dawans, Polymer supported catalysts, Prog. Polym. Sci., 1977, 5, 95–226 CrossRef CAS.
- B. Chen and F. Jäkle, Boron-Nitrogen Lewis Pairs in the Assembly of Supramolecular Macrocycles, Molecular Cages, Polymers, and 3D Materials, Angew. Chem., 2024, 136, e202313379 CrossRef.
- U. Yolsal, T. A. Horton, M. Wang and M. P. Shaver, Cyclic ether triggers for polymeric frustrated Lewis pair gels, J. Am. Chem. Soc., 2021, 143, 12980–12984 CrossRef CAS.
- M. Wang, F. Nudelman, R. R. Matthes and M. P. Shaver, Frustrated Lewis pair polymers as responsive self-healing gels, J. Am. Chem. Soc., 2017, 139, 14232–14236 CrossRef CAS.
- T. A. Horton, M. Wang and M. P. Shaver, Polymeric frustrated Lewis pairs in CO2/cyclic ether coupling catalysis, Chem. Sci., 2022, 13, 3845–3850 RSC.
- M. Wang, M. Shanmugam, E. J. McInnes and M. P. Shaver, Light-Induced Polymeric Frustrated Radical Pairs as Building Blocks for Materials and Photocatalysts, J. Am. Chem. Soc., 2023, 145, 24294–24301 CrossRef CAS PubMed.
- Q. Yin, Y. Ye, G. Tang and Y.-F. Zhao, Studies on the structure behavior of triphenyldichlorophosphorane in different solvents, Spectrochim. Acta, Part A, 2006, 63, 192–195 CrossRef PubMed.
- C. S. Davis and R. J. Richardson, Organophosphorus compounds, Experimental and clinical neurotoxicology, 1980, vol. 1 Search PubMed.
- S. L. Regen, Triphase catalysis. Kinetics of cyanide displacement on 1-bromooctane, J. Am. Chem. Soc., 1976, 98, 6270–6274 CrossRef CAS.
- T. Chang, H. Jing, L. Jin and W. Qiu, Quaternary onium tribromide catalyzed cyclic carbonate synthesis from carbon dioxide and epoxides, J. Mol. Catal. A: Chem., 2007, 264, 241–247 CrossRef CAS.
- J. Sun, S.-i. Fujita and M. Arai, Development in the green synthesis of cyclic carbonate from carbon dioxide using ionic liquids, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- X.-B. Lu, Y.-J. Zhang, B. Liang, X. Li and H. Wang, Chemical fixation of carbon dioxide to cyclic carbonates under extremely mild conditions with highly active bifunctional catalysts, J. Mol. Catal. A: Chem., 2004, 210, 31–34 CrossRef CAS.
- K. A. Andrea and F. M. Kerton, Triarylborane-catalyzed formation of cyclic organic carbonates and polycarbonates, ACS Catal., 2019, 9, 1799–1809 CrossRef CAS.
- K. A. Andrea and F. M. Kerton, Functionalized polycarbonates via triphenylborane catalyzed polymerization-hydrosilylation, RSC Adv., 2019, 9, 26542–26546 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03119a |
| This journal is © The Royal Society of Chemistry 2024 |