
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew frontiers in alkali metal insertion into carbon electrodes for energy storage
Zachary T.
Gossage
 a,
Daisuke
Igarashi
a,
Yuki
Fujii
a,
Masayuki
Kawaguchi
b,
Ryoichi
Tatara
a,
Kosuke
Nakamoto
a and
Shinichi
Komaba
*a
a,
Daisuke
Igarashi
a,
Yuki
Fujii
a,
Masayuki
Kawaguchi
b,
Ryoichi
Tatara
a,
Kosuke
Nakamoto
a and
Shinichi
Komaba
*a
aDepartment of Applied Chemistry, Tokyo University of Science, Tokyo 162-8601, Japan. E-mail: komaba@rs.tus.ac.jp
bFundamental Electronics Research Institute, Osaka Electro-Communication University, Neyagawa, Osaka 572-8530, Japan
First published on 17th October 2024
Abstract
With rising interest in new electrodes for next-generation batteries, carbon materials remain as top competitors with their reliable performance, low-cost, low voltage reactions, and diverse tunability. Depending on carbon's structure, it can attain high cyclability as with Li+ at crystalline graphite or exceptional capacities with Na+ at amorphous, porous hard carbons. In this review, we discuss key results and research directions using carbon electrodes for alkali ion storage. We start the first section with hard carbon (HC), a leading material of interest for next-generation Na-ion batteries. Methods for tuning the HC structure towards a high capacity pore-filling mechanism are examined. The rate performance of hard carbon electrodes is further discussed. We finish this section with soft carbons that mostly remain as low performing materials compared to other carbons. In the second section, we discuss alkali ion insertion into graphite and graphite-like materials. Though graphite has a long history with Li-ion batteries, it also shows promising characteristics for K-ion batteries. We discuss the significant progress made on improving the electrolyte for high cyclability of graphite with K+. Thereafter, we evaluate B/C/N materials that have a similar structure to graphite but can attain higher capacities for both Li+ and Na+. Finally, we touch on the recent developments using alternative solvents for Na+ cointercalation at graphite and deeper knowledge on the intercalant structure. Despite steady progress, carbon electrodes continue to improve as a key group of materials for alkali energy storage.
1 Introduction
Lithium-ion batteries (LIBs) continue to have a strong hold on the battery market as the most reliable and robust energy storage technology to date. Their chemistry has seen major improvements over the years with a growing usage across the globe and in our daily lives.1 At the same time, there is significant motivation for moving “beyond” the LIB with many unique chemistries under development that posit massive capacities, improved rates, safety and even lower costs for the next-generation batteries.2–5 Following in the footsteps of the LIB, sodium ion (SIBs) and potassium ion batteries (PIBs) are steadily making progress.6 Both Na and K show significantly improved global distribution and abundance compared with Li, and their electrode materials tend to be produced from low-cost, abundant sources.7,8 At the same time, they maintain comparable high voltages and capacities to the LIB. SIBs are even becoming commercialized and have been recognized as a top emerging technology.9 Though a direct translation of our knowledge from LIBs to the other alkali ions has not proven successful,10 the effort has nevertheless stirred up significant research interests in improving the electrode materials and electrolytes for better compatibility for future alkali ion batteries.6,11,12For electrode materials in alkali ion batteries, carbon continues to show several advantages being low-cost, sustainable, and having a low redox potential for alkali ion insertion reactions. Carbon shows a wide variety of structures (e.g. 2D and 3D architectures) with unique properties (conductivity, energy storage mechanism). Hard carbons (HCs), which show great structural tunability, are proving to be the most promising negative electrode for SIBs. Their structure can incorporate pores as well as pseudo-graphitic domains (Fig. 1) that lead to fast rate performance and high reversible capacities.13 HCs are also of significant interest for PIBs and have had commercial usage in LIBs as well. Looking to the chemistry of the LIB, we find that much of its success has been reliant on the robust performance of the graphite negative electrode.1,14 With a stable layered structure, graphite shows a highly reversible intercalation process with Li+ (Fig. 1). Similarly, fast K+ (de)intercalation and low redox potentials makes graphite a promising negative electrode material for commercial PIBs.7,15,16 In the middle ground, the relatively unexplored soft carbons (SCs) can have overlapping properties with both graphite and HCs.17,18
 | ||
| Fig. 1 Illustration of the key carbon electrode materials for energy storage with alkali ions. | ||
Currently, there are many research directions to further improve carbon electrodes for higher performing SIBs and PIBs. In particular, our group has shown vast improvements in the performance and reversible capacities of HCs through various optimization strategies.19,20 The pore structure has been recognized as a key feature of HCs and accessing its full potential is drawing significant research attention. For graphite, a vast amount of literature has already been involved in improving its performance for Li+.14 More recent efforts have focused on incorporating other electrode materials with higher capacities, e.g. Si nanoparticles, directly with graphite to further boost the energy storage capabilities. Aside, there have also been significant efforts looking toward improving the cyclability of graphite in K+ electrolytes for use in PIBs.21,22 Much of this has involved engineering of the electrolyte and additive discovery. Surprisingly, graphite also shows some remarkable cycling behavior for sodium ion batteries when certain electrolytes are utilized.23 We believe carbon electrodes will continue to play a major role in energy storage for years to come, and thus we will highlight their key characteristics herein.6,24,25
This review is divided into two main sections: (1) non-graphitized carbons, and (2) graphite and graphite-like materials. In Section 1, we start by focusing on HCs due to their significant promise for next-generation alkali ion batteries, especially SIBs. The major developments by our group for improving the capacities of HCs using a template synthesis method are discussed. Other synthetic methods using modified activated carbons are also considered. We emphasize the importance of the rate performance of HCs and present effective methods for understanding their rate limitations utilized in our laboratory. We finish this section with the current knowledge and progress on soft carbons for battery applications. In Section 2, we cover the stage structures that form with graphite and graphite-like materials. We discuss recent progress toward electrolyte development for utilizing graphite as a long-life negative electrode for PIBs. Thereafter, we discuss the observed stage 1 Na+ intercalation compounds that can form with graphite-like B/C/N materials and their associated electrochemical properties. We finish this section with recent progress on understanding solvent cointercalation chemistry at graphite for application toward SIBs. There are many interesting directions that have been developing in recent years; here we provide our vantage point of the current progress and promising routes for future research focus.
2 Non-graphitized carbons
2.1 Hard carbon
Hard carbons, or non-graphitizable carbons, are some of the most promising negative electrode materials for SIBs.6,19 HCs can show very high energy densities due to their large sodium storage capacity and low working potential compared to other candidates. They are easily prepared from most organic compounds by thermal decomposition under inert atmosphere, including commonly available saccharides and biomass waste. Both the starting materials and the synthesis methods (e.g. heat-treatment) strongly impact the final HC structure and performance, and there is still significant opportunity to obtain HCs with improved structures for Na+ storage.HCs are low-crystalline carbons composed of two main types of structural domains that are involved in the energy storage process: (1) pseudo-graphitic domains and (2) (nano)pore structures between the pseudo-graphitic domains.13,26,27 Note that the pseudo-graphitic domains contains various defects such as residual heteroatoms, carbon vacancies, and Stone–Wales defects.28Fig. 2a shows the sodiation (discharge) curve of a HC electrode with the corresponding structures and mechanisms involved.29 Based on various characterizations and theoretical calculations, it is now widely accepted that the Na+ storage mechanism proceeds as follows: [1] physicochemical adsorption of Na+ on defects sites in the high potential region, [2] Na+ intercalation into pseudo-graphitic domains at lower potentials, and finally [3] pore-filling by the formation of quasi-metallic Na clusters during the low potential plateau.13,31,32 The capacity obtained during each step are well correlated with the structural characteristics of the HC, such as the amount of defect sites, the crystallinity of the pseudo-graphitic domains, and the size/volume of the nanopores. Therefore, not all HCs necessarily exhibit practical capacities for energy storage. In fact, our group has improved the specific capacity of HCs from 260 mA h g−1 to 480 mA h g−1 over the past 15 years.
 | ||
| Fig. 2 Alkali ion storage at hard carbon. (a) Sodium insertion model and typical sodiation curve of a HC electrode. Reproduced with permission from ref. 29. Copyright 2022 Elsevier. (b) Charge and discharge curves in Li, Na, or K cells, (c) SAXS patterns, and (d) calculated average pore-size of sucrose-derived HCs prepared through different HTT. (b)–(d) were reproduced with permission from ref. 30. Copyright 2020 American Chemical Society. | ||
The synthesis temperature, or heat treatment temperature (HTT), is one of factors that has a significant influence on the final HC structure and can be easily managed experimentally. Our group systematically investigated sucrose-derived HCs prepared at various HTTs as model materials in order to understand the fundamental relationship between HC structure and alkali metal storage properties.30Fig. 2b shows charge and discharge profiles of sucrose-derived hard carbons in Li, Na, or K cells. Here, the sample name denotes the HC synthesis temperature, e.g. HC1100 for HC prepared at 1100 °C. For Li cells, HC700 exhibited a sloping profile with notable potential hysteresis, but a very large reversible capacity >500 mA h g−1. HC1100 also showed a large reversible capacity and improved hysteresis. As the synthesis temperature was increased further, as in HC1500 and HC2000, the plateau region (including intercalation and pore-filling processes) diminished. This trend is fully consistent with the pioneering works by Dahn and co-workers.33 In contrast, Na cells showed an increase in the plateau region with higher synthesis temperature. Even for HC2000, a notable plateau region was observed. K cells showed a similar trend to Na, with an increasing plateau-like profile as the synthesis temperature increased. The capacities did not vary significantly from HC700 to HC2000 for the K cells, within the range of 200–250 mA h g−1. Based on these results, the impact of the HC structure on energy storage is notable, but also the structures show some preference for particular alkali ions (Na+ in this case).
Since HCs obtained at lower HTTs have many defects, it is assumed that the sloping profile as observed with HC700 is mostly derived from the chemical adsorption of the alkali ion on those defect sites. On the other hand, the plateau region has been linked to the formation of Li clusters within the nanopores by neutron/X-ray scattering and 7Li-NMR methods.34–37 Likewise, studies have reported that a major part of plateau capacity in Na cells is also derived from Na cluster formation in the nanopores.31,32,38–40 In the pore-filling process, the Na is close to the metallic state, referred to as “quasi-metallic”, which enables large-capacity Na storage without the stage-mechanism limited theoretical capacity as seen in Li–graphite system. Regulation of the nanopore structure appears to be the key to achieving higher energy densities and high performance for SIBs.
The most used method for evaluating the nanopore structures of HCs is small angle X-ray scattering (SAXS). Contrast between the electron density of the carbon matrix and nanopores is observed as a shoulder peak in the Q = 10−2–1 Å−1 range, where Q is the X-ray scattering vector.41 Our group often applies this method, where the average pore size is calculated by fitting the observed shoulder peak under the assumption that the carbon matrix can be regarded as low-density graphitic carbon and the nanopores as spherical empty space.42Fig. 2c shows the SAXS patterns for sucrose-derived HCs treated at various HTTs and Fig. 2d shows the relationship between HTT and average pore-size calculated from the SAXS patterns. A clear trend where the larger average pore size was obtained for HCs treated at higher HTT. It should be noted that HCs with larger average pore size do not necessarily have larger reversible capacity, which appears to imply that there is an optimal pore size for Na storage. Furthermore, the defect concentration and porosity also impact the coulombic efficiency.43 It has been shown that HCs prepared at high-temperatures (>2000 °C) have a shortened interlayer distance for their pseudo graphitic domains and this leads to increased resistance toward Na+ insertion and a low-potential plateau capacity.6,31,44 Moreover, Stratford et al. have suggested that the pore-filling capacity is determined by the pore size distribution rather than the average pore size, based on their total X-ray scattering measurement and corresponding pair-distribution function analysis.32 According to our experience, the observable plateau capacity of HCs may be underestimated due to increased cell resistance caused by various factors, such as cell configuration and electrode composition. It has been observed that a certain amount of Na+ insertion proceeds even at potentials below 0 V when HCs are overcharged in a two-electrode system.19,45
For the improvement of Na-storage capacity of HCs, we have recently employed ‘template’ methods for synthesis of novel HCs.19,20 Template synthesis is a well-known approach to prepare porous carbon materials, in which a composite of inorganic particles and carbon or carbon precursor is synthesized followed by the removal of the inorganic materials to produce a porous carbon.46–49 An illustration of our template method is shown in Fig. 3a. We have developed some large capacity HCs using MgO and ZnO nanoparticles as templates, which were prepared from pyrolysis of the gluconate salts of Mg and Zn (here referred to as HC-Mg and HC-Zn, respectively). In our experiments, the nano-sized MgO and ZnO particles composited with carbon is an appropriate template for formation of the nanopore structure. In contrast, we also synthesized HCs using calcium gluconate as the starting material, but large clumps of CaCO3 were observed which led to lower capacities. The SAXS patterns of HCs synthesized via template method from Mg, Zn or Ca gluconate are shown in Fig. 3b, where the intensity of the shoulder peaks indicates well developed nanopores in HC-Mg and HC-Zn. Both HC-Mg and HC-Zn exhibited large reversible capacities of around 400 mA h g−1 and a long charge/discharge plateau at low redox potentials, as shown in Fig. 3c. Furthermore, a higher capacity was observed for the material containing larger pores (HC-Zn), though an ideal or maximum size of the pores remains unknown. It is also noteworthy that these HCs demonstrated excellent cycle performance and coulombic efficiencies (CEs). Generally, plating/stripping reactions of Na metal that occur on the surface of the electrode tend to show very low CEs,50 so the pore structure appears to make such reactions more facile and block electrolyte decomposition within the pore. The relationship between pore filling with underpotential electrodeposition of sodium has recently been discussed.51
 | ||
| Fig. 3 Template synthesis of HC incorporating Zn, Ca and Mg salts. (a) Schematic of template synthesis of porous HCs. and (b) SAXS patterns of HCs synthesized from different metal gluconate salts. (c) Initial charge and discharge curves of template HCs. Reproduced with permission from ref. 20. Copyright 2023 Wiley-VCH GmbH. | ||
The distribution of inorganic templates and nanopore in the carbon matrix of template HCs can be further optimized for Na storage by adjusting the composition and processing method of starting materials. As illustrated in Fig. 4a, we found that using a freeze-drying method combined with template synthesis using MgO has produced the largest Na storage capacity among the HCs our group have explored.19 The freeze-drying method is advantageous for attaining homogeneous dispersions of MgO nanoparticles within the carbon matrix. As shown in Fig. 4b, the freeze-dried mixture of glucose and Mg gluconate appears somewhat amorphous and is different from simple powder grinding. The optimized MgO template HC exhibited a large capacity of 478 mA h g−1 as shown in Fig. 4c. This value is significantly larger than that of commercially available HCs for Li-ion batteries and other previously reported HCs obtained by heating of various organic compounds (Fig. 4d). While there are still many debates on the Na insertion mechanisms of HCs,52 the pore structure and distribution appear to be key for high capacities.
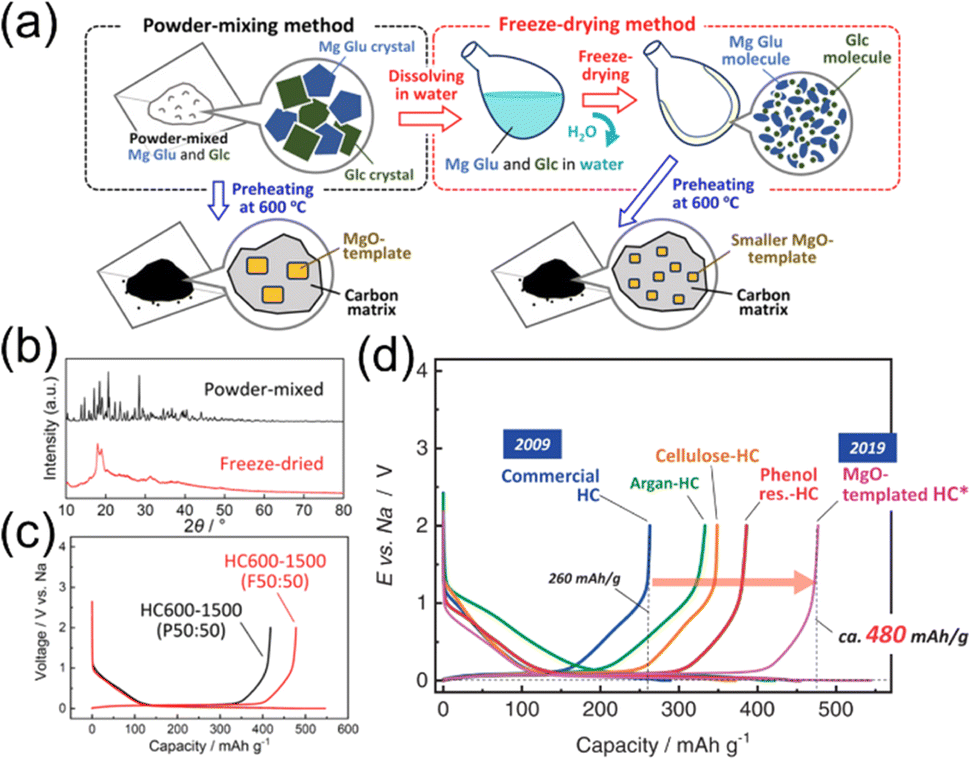 | ||
| Fig. 4 Other templated HCs. (a) Schematic illustration of mixing processes with Mg Glu and Glc for the synthesis of MgO-template HCs. (b) The XRD patterns of powder-mixed or freeze-dried Mg Glu–Glc mixtures. (c) Initial charge and discharge curves of HCs synthesized from powder-mixed or freeze-dried methods. (d) Initial charge and discharge curves of HCs which the authors' group have reported. Reproduced with permission from ref. 19. Copyright 2021 Wiley-VCH GmbH. | ||
In recent years, several groups have reported large-capacity HCs synthesized from activated carbon (AC) or other porous carbon. Micropores on activated carbon surface are generally ‘open pores’ that are exposed to the surroundings, leading to significant electrolyte decomposition instead of reversible Na+ insertion. However, these open pores can be “closed” through high-temperature treatment. Our group and others have reported that heat treatment of AC at a high temperatures can close the surface micropores and exhibit a large reversible capacity over 400 mA h g−1.53,54 However, for these materials the Na insertion and surface plating potentials can show significant overlap. We attribute this to increased Na+ diffusion resistance caused by the progressive graphitization of the pseudo graphitic domains.19 Aside, Zheng et al. reported a large-capacity HC obtained by two-step heat-treatment of a starch-based raw material.55 First the precursors were heated at 800 °C in a CO2 atmosphere to produce an AC. This was heated further at 1300 °C in an Ar atmosphere to attain the final closed-pore HC structure (Fig. 5a). In this example, a low potential plateau and large capacity of 488 mA h g−1 were observed in the potential range between 0 and 3.0 V (Fig. 5b) despite the moderate synthesis temperatures.
 | ||
| Fig. 5 Conversion of activated carbon to hard carbon. (a) Schematic illustration of the process of converting activated carbon to closed-pore-rich hard carbon through heat treatment. (b) Charge–discharge curves of the converted activated carbon. (a) and (b) were reproduced with permission from ref. 55. Copyright 2023 Wiley-VCH GmbH. (c) Illustration of converting AC using a CVD method. (d) Initial charge and discharge curves of an activated carbon and ‘filling’ carbons. (c) and (d) were reproduced with permission from ref. 56. Copyright 2023 Royal Society of Chemistry. | ||
Other novel large-capacity HCs have been prepared by tightening or closing micropores of ACs using chemical vapor deposition (CVD).56,57 These carbons have been referred to as “sieving” or “filling” carbons. Fig. 5c shows the illustration of synthesis procedure for altering the pore structure via CVD and following post-heat treatment.56 Through controlled adsorption and decomposition of common precursors, such as methane, benzene, etc., the micropore structure/opening can be modified. Due to their highly porous nature, such HCs can exhibit large capacities exceeding 400 mA h g−1 (Fig. 5d). Drawing from our wide knowledge on electric double layer capacitors, among other applications of porous carbons, further progress in these materials is anticipated.
Another major point of interest in HCs is the availability of a wide variety of raw materials for their syntheses.58,59 Significant diversity in both the micro-structure, such as crystallinity and nanopore content, as well as the macroscopic structure (i.e. the electrode geometry) can be achieved. This has led to some demonstrations of free-standing HC electrodes prepared from 2D fibrous starting materials, including tissue papers and electrospun resins.60,61 Likewise, other free-standing HC electrodes with unique morphologies have been obtained through sol–gel methods (Fig. 6a).62 As shown in Fig. 6b, these materials exhibited sufficient capacities of over 300 mA h g−1 and excellent CEs of over 90%. More recently, Katsuyama et al. developed a method to synthesize a free-standing HC electrode from a photocured resin lattice designed using a 3D printer.63 The precursor morphology, including the size of the electrode grid, can be effectively prepared through this method (Fig. 6c). So far, the charge and discharge profiles for these materials were below 250 mA h g−1 (Fig. 6d). Although the gravimetric capacities were mediocre, these materials demonstrated reversible operation with an extraordinary loading of active material at 98 mg cm−2. The idea of precisely controlling the electrode structure to improve electrolyte infiltration and transport is highly appealing.
 | ||
| Fig. 6 HC precursors with controlled structure. (a) Optical photo (inset) and SEM image of carbon monolith. (b) Charge and discharge curves of carbon monolith electrodes treated at 1200–2500 °C. (a) and (b) are reproduced with permission from ref. 62. Copyright 2015 Wiley-VCH GmbH. (c) Optical photo of 3D-printed photocured resin and corresponding carbon micro-lattice. (d) Charge and discharge curves of carbon micro-lattice electrodes (400–133) with different mass loadings. (c) and (d) are reproduced with permission from ref. 63. Copyright 2022 Wiley-VCH GmbH. | ||
Since HC is a promising negative electrode material with the various advantages and potential described above, it is utilized as the negative electrode in many cases of the research of SIB full cells. Our group recently reported a Na5/6Ni1/3Fe1/6Mn1/6Ti1/3O2//ZnO-template HC full cell with a gravimetric energy density comparable to that of LiFePO4//graphite-type LIBs (Fig. 7a).20 This result is achieved not only by the large reversible capacity, but also the high initial coulombic efficiency of 91% of the ZnO-template HC. However, the synthesis process of these novel high-capacity materials has not been optimized for mass production. Therefore, commercially available HC is often used to fabricate and evaluate large-scale prototype cells such as cylindrical or pouch-type cells. In that case, there are several challenges to be overcome including the lower capacity and initial coulombic efficiency compared to the graphite electrode of LIBs.
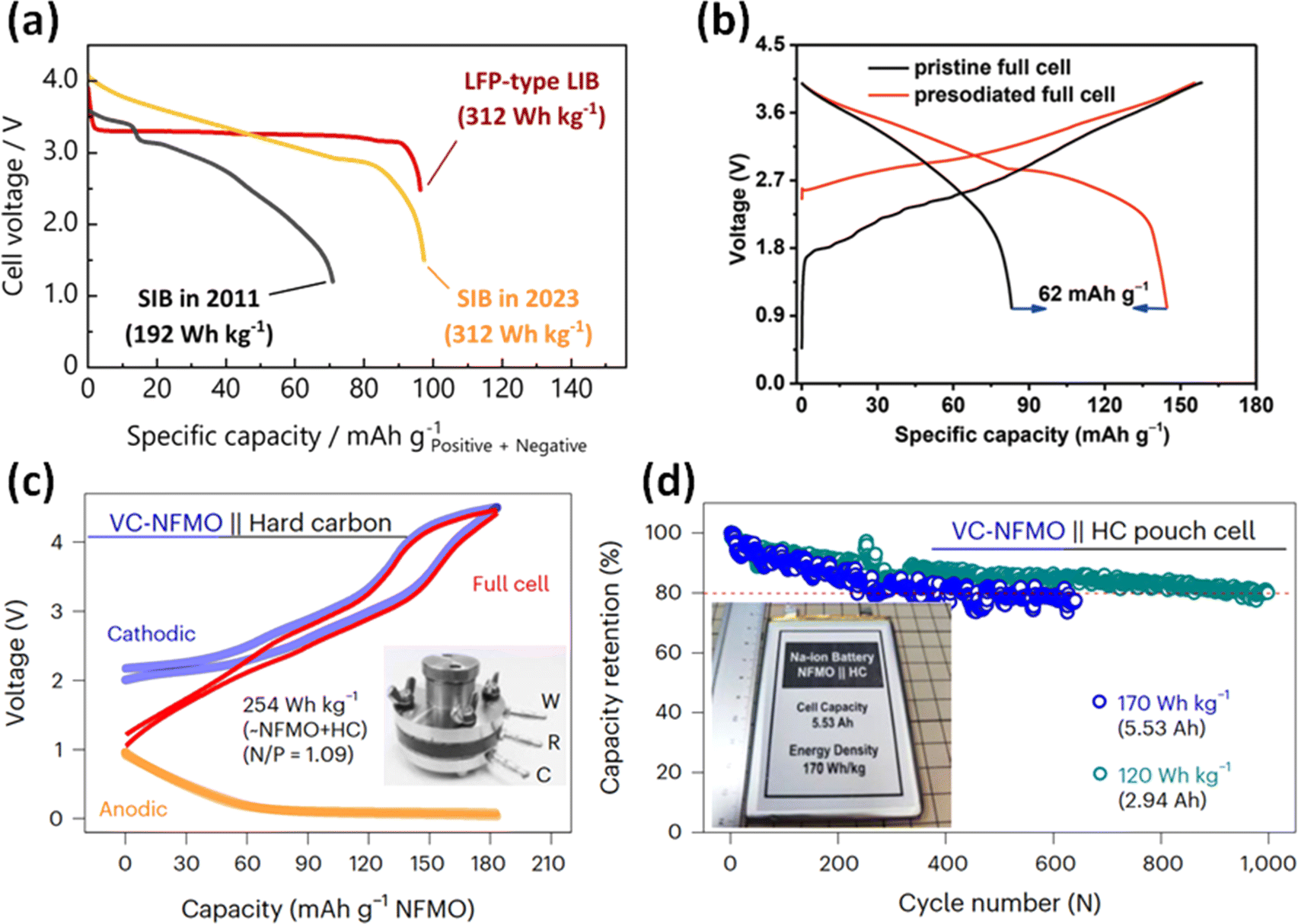 | ||
| Fig. 7 (a) Comparison of discharge curves of full cells of Na and Li-ion batteries. Reproduced with permission from ref. 20. Copyright 2023 Wiley-VCH GmbH. (b) Initial charge–discharge curves of Na0.9Cu0.22Fe0.3Mn0.48O2//HC full cells with or without presodiation of the HC electrodes. Reproduced with permission from ref. 64. Copyright 2019 Wiley-VCH GmbH. (c) Charge–discharge curves of a VC-NFMO//HC full cell in three-electrode system. (d) Cycle performance of the pouch-type VC-NFMO//HC full cells. (c) and (d) reproduced with permission from ref. 65. Copyright 2024 Springer Nature. | ||
For compensating for the initial irreversible capacity of the HC electrode in full cells, a few approaches have been proposed: chemical presodiation of the HC negative electrode,64,66 and the incorporation of a sacrificial additive into the positive electrode.67,68Fig. 7b shows the initial charge–discharge curves of SIB full cells with or without presodiation of the HC electrodes using sodium naphthalenide reported by Liu et al.64 They showed that the reversible capacity of the full cell was greatly improved by chemical reduction before cell assembly. Some important factors should be considered for practicality of the presodiation process, including capacity balance with the positive electrode and handling procedure of energetically unstable presodiated electrodes. Overall, the concept of presodiation is an interesting approach that may be feasible for improving the energy density of SIBs.
For the evaluation of a practical SIB full cell, various parameters are required, such as the mass loading of the electrode, N/P ratio, E/C ratio and the weight of entire cell. In a recent paper, Tang et al. fabricated large pouch-type full cells consisting of a Na0.7Fe0.1Mn0.75□0.15O2 (vacancy-containing (VC)-NFMO) positive electrode and a HC negative electrode (Fig. 7c) for long-term operation at 170 W h kg−1 and 120 W h kg−1 (Fig. 7d).65 Their data will be considered a benchmark for the research of SIBs that aims for mass production and practical applications in the future. Furthermore, we also expect to see interesting results applying improved hard carbons to K ion batteries, as a higher rate performance may be obtainable compared with Na.69 The weaker interactions between the solvent and anions with larger alkali ions could improve the rate of ion transport and insertion. However, tuning the HC structure may be necessary for optimal performance with other alkali ion batteries.
Another key point for practical batteries, is attaining improved charge/discharge rates. When evaluating the rate of HC electrodes, the low redox potential of alkali ion insertion at HC can be quite inhibitive due to its close proximity to Na plating potentials.51,70 As the rate is increased, the plateau potentials tend to shift to lower and higher potentials for insertion and deinsertion, respectively. This can be observed in both galvanostatic charge–discharge testing and cyclic voltammetry. Such shifting is caused by (1) polarization of the sodium metal counter electrode71 as well as (2) ion depletion and inhomogeneous state of charge (SOC) distribution across the composite electrode.72–74 These effects limit our ability to effectively evaluate the active material performance at high rates, and thus, some reported rate measurements for HC are mainly capturing limitations of the composite electrode or coin cell. Polarization of sodium (or potassium) metal can easily be overcome by using a three-electrode cell with a separate reference and counter electrode. However, limitations at the composite electrode are often overlooked, and thus alternative electrochemical measurements can provide better insight. Specifically, the application of small electrodes (e.g. micro- or nanoelectrodes) for single particle measurements75,76 or the use of thin composite electrodes77 can overcome such issues. Major analytical developments for single particle analysis have occurred in recent years,78–80 and we encourage other researchers to explore such advanced methodologies among others.81 Aside, methods that are simple and amenable with common battery analysis, e.g. coin cells and composite electrodes, are also highly desirable.
Recently, our lab has been exploring the application of the diluted electrode method, as first proposed by Ariyoshi et al.82 This method can also eliminate polarization issues in composite electrodes to provide “quasi-single particle” measurements. As seen in Fig. 8a, diluted electrodes are prepared by partially exchanging the active material, e.g. hard carbon, with inactive particles, such as aluminum oxide or nickel.72 These inert particles do not reversibly exchange host alkali ions, so the active material loading can be varied without changing the thickness or porosity of the composite electrode; hence the terminology “diluted electrode.” Typical battery electrodes (or undiluted electrodes) contain a high concentration of active material which leads to a depletion of ions and a gradient across the bulk of the electrode during insertion (Fig. 8a). This becomes especially pertinent for high capacity materials and at high charge/discharge rates. With diluted electrodes, the low active material concentration suppresses the concentration overvoltage caused by guest-ion shortage. In this way, the composite electrode can be charged more uniformly which provides improved insight when evaluating the rate performance of different electrode materials.
 | ||
| Fig. 8 Comparison of undiluted and diluted hard carbon electrodes. (a) Schematic of diluted electrodes method. (b) Cyclic voltammograms of sodium insertion/extraction of hard carbon obtained by using a three electrodes cell for diluted (10% AM) and conventional (95% AM) electrodes. | ||
The effects of diluted hard carbon electrode are shown in Fig. 8b using cyclic voltammetry and commercial hard carbon (Kureha Co., Ltd). As with our previous report,72 the diluted electrode (hard carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nickel:binder = 10:85:5 vol/vol) shows apparent “faster” kinetics, as indicated by the improved peak splitting. These measurements are in good agreement with differential capacity curves (dQ/dV) from slow rate galvanostatic charge/discharge curves.32 This is a closer reflection of HC's true insertion rate and shows the limitations of such analysis using typical composite electrodes. Overall, the diluted electrode method has indicated an excellent rate performance for HC in conventional battery electrolytes, e.g. 1 M NaPF6 EC:DEC (1:1 v/v), maintaining 80% of the full capacity when charging (electrochemical reduction) at 1000 mA g−1 (or ∼4C).72 Such quick, low-cost methods provide improved and valuable insight for evaluating the rate performance of other battery materials.
:nickel:binder = 10:85:5 vol/vol) shows apparent “faster” kinetics, as indicated by the improved peak splitting. These measurements are in good agreement with differential capacity curves (dQ/dV) from slow rate galvanostatic charge/discharge curves.32 This is a closer reflection of HC's true insertion rate and shows the limitations of such analysis using typical composite electrodes. Overall, the diluted electrode method has indicated an excellent rate performance for HC in conventional battery electrolytes, e.g. 1 M NaPF6 EC:DEC (1:1 v/v), maintaining 80% of the full capacity when charging (electrochemical reduction) at 1000 mA g−1 (or ∼4C).72 Such quick, low-cost methods provide improved and valuable insight for evaluating the rate performance of other battery materials.
2.2 Soft carbons
Soft carbon, also known as ‘graphitizable carbon’, is a carbon material that is transformed into graphite by heat treatment at high temperatures (generally around 3000 °C) and have also been studied as negative electrode materials for SIBs.17,83 To date, most SC materials reported show modest performances compared with HCs including even lower discharge capacities and CEs. The lower CE is due to the higher defect content of SC which can also include irreversible expansion of the carbon structure.83 The starting materials of SC include some low molecular weight organic compounds, such as 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA) and dibenzanthrone, polymer compounds, and fossil fuel-derived substances like pitch and coke.17,18 As shown in Fig. 9a and b, SC is a low crystalline carbon that shows broad diffraction peaks alike to HC.84 However, SC tends to show a sharper, higher intensity 002 diffraction peak and a less intensity at small scattering angles (below 2θ = 15° (CuKα)). This suggests that SC does not have well-developed nanopore structures as found in HCs.27 The diffraction peaks of SC become sharper and approach to that of graphite by further high-temperature heat treatment.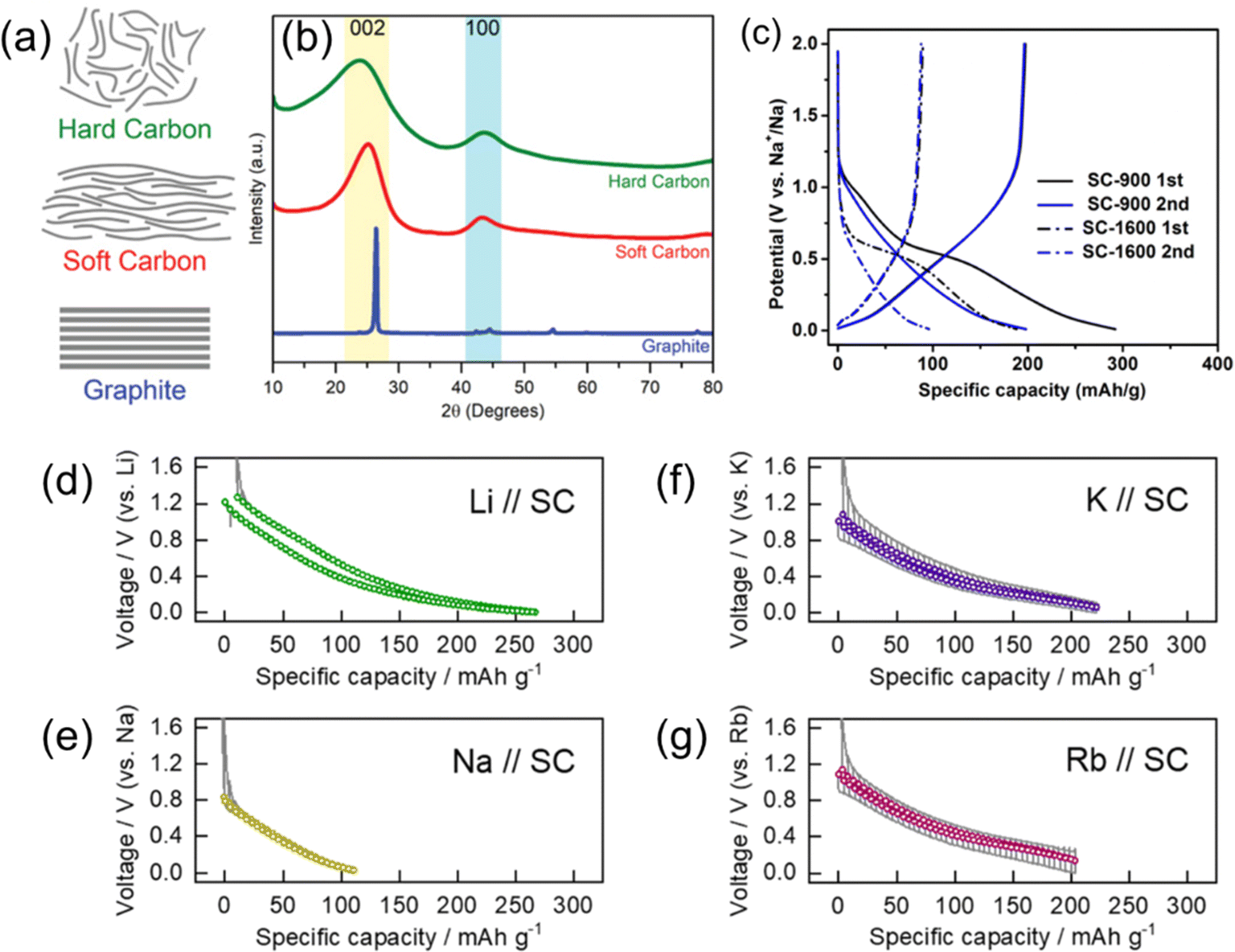 | ||
| Fig. 9 Soft carbons for alkali ion storage. (a) Structure models and (b) typical XRD patterns of hard carbon, soft carbon, and graphite. (a) and (b) are reproduced with permission from ref. 84. Copyright 2019 Wiley-VCH GmbH. (c) Charge and discharge curves of SC prepared by different HTTs. Reproduced with permission from ref. 85. Copyright 2017 American Chemical Society. Open circuit voltage curves of soft carbon electrodes in (d) Li, (e) Na, and (f) K, and (g) Rb cells. (d)–(g) are reproduced with permission from ref. 86. Copyright 2023 Royal Society of Chemistry. | ||
Fig. 9c shows charge and discharge curves of PTCDA-derived SCs treated at 900 or 1600 °C (denoted SC900 and SC1600, respectively) in Na cells.85 Both SCs exhibited sloping charge/discharge profiles, and SC1600 had less than half of the capacity of SC900. In general, the Na storage capacity of SC decreases with treatment at higher temperatures, suggesting that Na insertion into SC takes place toward non-graphitic sites such as turbostratic and expanded interlayer and defect sites. Our group compared the various alkali-ion storage properties of needle coke-derived SC.86 The charge storage properties are shown in Fig. 9d–g with open circuit voltage curves during the discharge/charge processes in non-aqueous Li, Na, K, and Rb half-cells. Only a small capacity of around 100 mA h g−1 was obtained for the Na cell, whereas moderate capacities over 200 mA h g−1 were obtained for the Li, K, and Rb cells. This trend is potentially related to structural similarities between SC and graphite, where poor performance with Na+ is observed compared to the other alkali ions.
Although the Na storage capacity in SCs has so far been insufficient for practical energy storage, several interesting studies have been made on SC–HC conversion and SC–HC composite materials. Lee and Min et al. have reported that polyethylene, a commonly used polymer material, is selectively carbonized to SC by heat treatment after thermal oxidation and to HC with heat treatment after acid treatment as illustrated in Fig. 10a.87 The obtained final products showed charge and discharge profiles typical for SC and HC, respectively (Fig. 10b). Cheng et al. also reported that carbons derived from perylene and terephthaloyl chloride can be selectively converted into SC or HC depending on the ratio of the raw materials.89 In addition, it has been reported that pitch, which is inherently carbonized to SC, was successfully converted to HC with a high carbonization yields by introducing defects to prevent graphitization.90
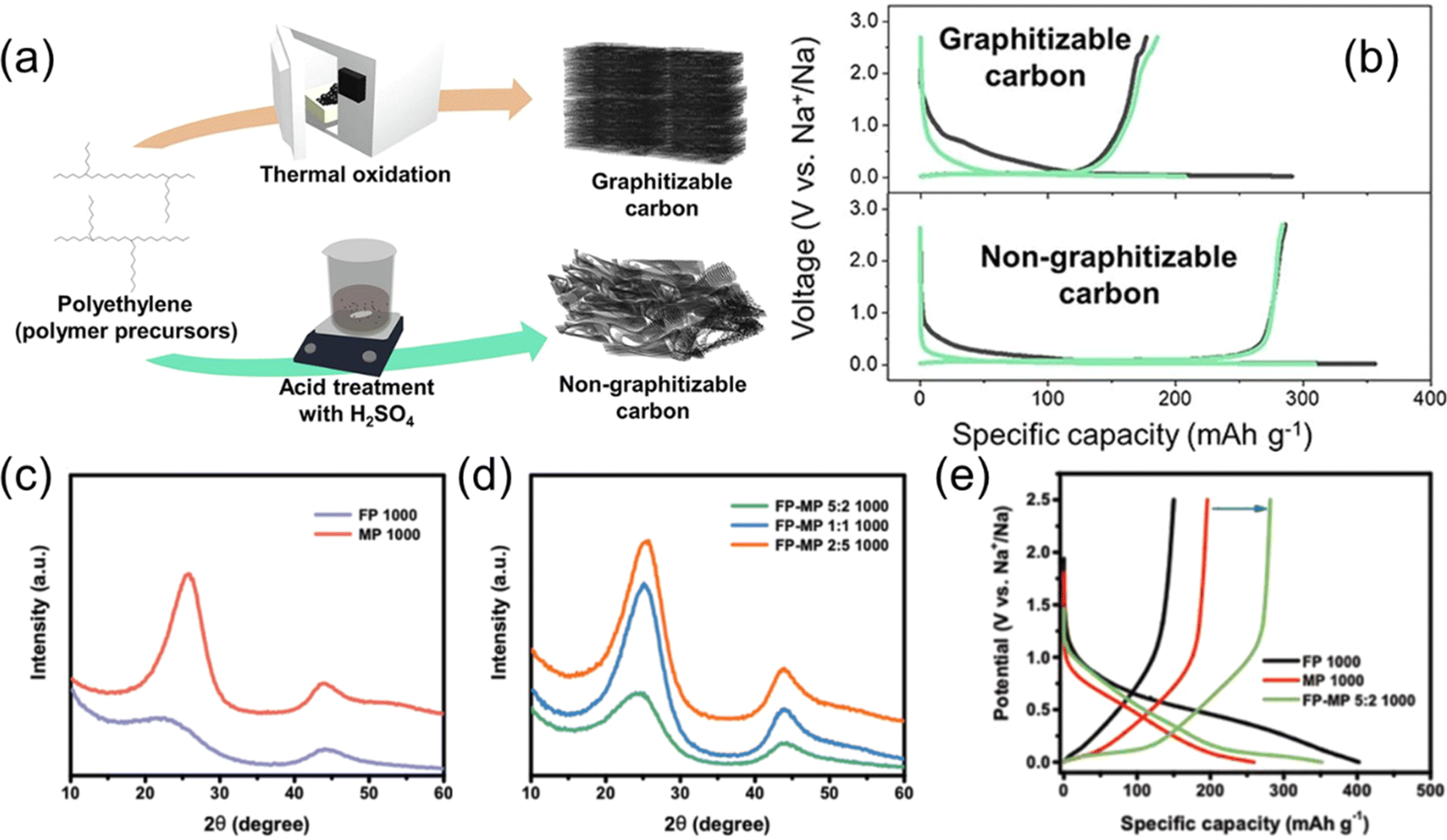 | ||
| Fig. 10 Polymer-based synthesis of SC and performance of SC–HC composites. (a) Synthesis methods and (b) typical charge and discharge curves of graphitizable or non-graphitizable carbons from polyethylene. (a) and (b) are reproduced with permission from ref. 87. Copyright 2023 the Authors. XRD patterns of (c) FP-derived HC and MP-derived SC and (d) SC–HC composite carbons derived from FP–MP mixtures with different mixing ratio. (e) Initial charge and discharge curves of FP-derived HC, MP-derived SC, and SC–HC composite carbon. (c)–(e) are reproduced with permission from ref. 88. Copyright 2019 Wiley-VCH GmbH. | ||
Xie et al. and others have reported the synthesis and battery properties of SC–HC composite materials.88,91 Their group used filter paper (FP) made of cellulose and mesophase pitch (MP) as the carbon precursors. As shown in the XRD patterns in Fig. 10c and d, FP and MP-derived carbons showed typical structural characteristics of HC and SC, respectively. The FP–MP mixture-derived carbons exhibited a continuously variable structure from SC-like to HC-like depending on the mixing ratio. The SC–HC composite showed a superior capacity compared to the FP-derived HC and a MP-derived SC (Fig. 10e). The authors conclude the addition of soft carbon decreased the surface area and number of open pores which improved the performance. Adding SC to the HC active material seems like a reasonable strategy, with some similarity to surface coatings which are often used for improving material performance in battery research.
3 Stage compounds of graphite and graphite-like materials
3.1 Stage compounds of graphite
In contrast to the high structural diversity and complex energy storage modes among non-graphitizable carbons, graphite is a highly crystalline, layered structure of sp2-hybridized carbons with one predominant storage mechanism, intercalation. For graphite, this insertion process involves alkali ions filling the interlayer space between each graphene sheet and arranging in a series of stages based on the SOC. Stage 1 refers to filled alkali ion layers separated by a single graphene sheet. In stage 2 there is an additional layer of graphene which leads to alternating alkali ion-filled and unfilled layers. With increasing stage number, the structure includes an additional graphene sheet and unfilled layer. In the Li cell, graphite is well-known to reduce to a stable, stage 1 structure of LiC6 with a reasonable capacity of 372 mA h g−1 (Fig. 11a).10 Much of the success for commercial LIBs has relied on this mechanism and its excellent cyclability.14 Unfortunately, some of graphite's exceptional qualities start to diminish when moving to other alkali ion systems.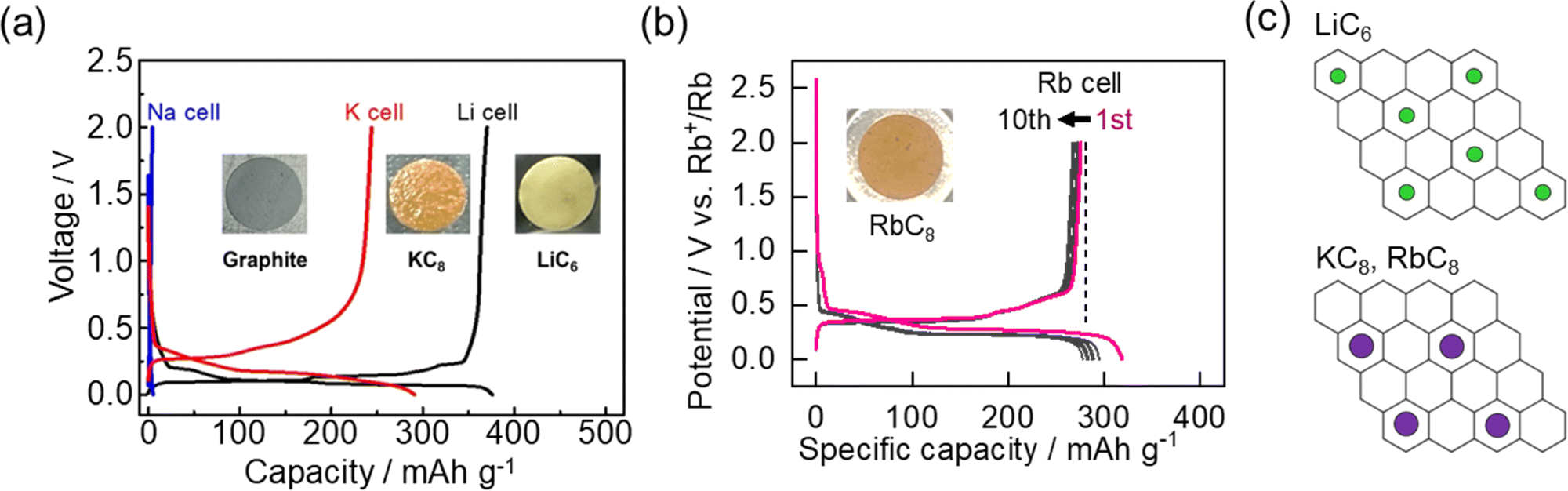 | ||
| Fig. 11 Electrochemical intercalation of alkali ions at graphite. (a) Charge/discharge curves of graphite in LiPF6, NaPF6 and KFSA electrolytes. The insets show the color change of graphite after intercalation. Reproduced with permission from ref. 10. Copyright 2015 Elsevier. (b) Charge/discharge curves of graphite in 1 mol kg−1 RbTFSA (inset: charged graphite electrode). Reproduced with permission from ref. 86. Copyright 2023 Royal Society of Chemistry. (c) Illustration of ion arrangement for LiC6 and KC8/RbC8 structures. | ||
For Na+, the formed graphite intercalation compounds (GICs) are thermodynamically unstable with positive formation energies.92–95 Aside from using a modified form of graphite,96,97 only dilute stages with low capacities are observed,98 which agrees with our experimental results (Fig. 11a). On the other hand, GICs formed with heavier alkali metals K and Rb are known to be stable and were even reported prior to Li-GICs.99 These alkali ions show an MC8 structure with a lower theoretical capacity of 279 mA h g−1 for the stage 1 compounds (Fig. 11a and b).10,86,100 In 2015, our group10 among others101,102 showed reversible electrochemical intercalation of K+ into graphite. We further confirmed reversible staging mechanism from graphite to KC8 with operando XRD.100 In addition, our group recently confirmed a reversible RbC8 structure for the stage 1 compounds of Rb with a reversible capacity close to the theoretical capacity (Fig. 11b).86 Using a rubidium bis(trifluoromethanesulfonyl) amide (RbTFSA) salt, we could observe (de)intercalation within various battery electrolytes. RbC8, RbC24, and RbC36 structures were confirmed at each stage of the electrochemical reduction process via operando X-ray diffraction and Raman spectroscopy. Few other studies have explored electrochemical insertion of Rb+,103 but this helps build a better fundamental understanding of the (de)intercalation process.
The similar behavior between Rb (ionic/atomic radius = 0.147/0.248 nm) and K (ionic/atomic radius = 0.133/0.235 nm) compared with Li (ionic/atomic radius = 0.068/0.152 nm) is an interesting point to consider, especially with the lack of stable KC6 or other higher capacity structures (Fig. 11c).104 Only under high pressures/temperatures, highly saturated structures (e.g. LiC2) have been reported.105,106 The concept of a hard-sphere model can be used to rationalize the capacity and structural limitations for GICs.107 Still, the actual intercalate size and packing limitations, as well as their relationship to bonding in the lattice are points to consider. Furthermore, DFT calculations have indicated the thermodynamic stability of K-GICs is more dependent on deformation of graphite compared with Li-GICs. According to some calculations, KC6 structures are energetically stable.94 However, no observations of this structure have been made.
From a practical perspective, it remains highly desirable to improve the capacity of graphite for not only PIBs, but even for modern LIBs.108 This has led to significant interest in incorporating higher capacity materials directly with graphite and the use graphite-like materials.109–111 Another approach is to consider plating-type reactions which can take some inspiration from HC and the major developments in harnessing the pore filling mechanism. By understanding and controlling plating reactions at graphite electrodes, significantly higher capacities can be attained.112 Lastly, graphite materials are relatively homogeneous compared with HCs and SCs, but they are polycrystalline in nature and do contain grain boundaries, pore structures, high-reactivity edge-sites and even impurities.113 These sites can be highly reactive toward ion insertion.114 Furthermore, the use of synthetic graphite opens many opportunities for tuning the structure to incorporate defects and dopants. These are all interesting approaches that may be useful for attaining higher performance with graphitic electrodes in the future.
While graphite continues to be a major material in LIBs, interest is growing for its use in PIBs where several benefits could be directly inherited from the LIB market.6,7 In addition to showing faster diffusion rates within the electrolyte compared to Li+, K+ also shows faster transport inside the graphite lattice (up to 5 orders faster), which could enable faster (de)intercalation rates.10,16 However, several early reports indicated fast capacity fading and poor first cycle CEs, especially in carbonate-based electrolytes.11,101,115 The binder was shown to strongly impact graphite's performance,10,100 linking some of the limitations to insertion of the larger K ions. During alkali ion insertion, the interlayer spacing within the graphite structure (typically 0.336 nm) expands to accommodate the ion size. This can be observed with XRD methods which show changes in the diffraction peaks during staging (Fig. 12).86 During operando XRD measurements of Li+ intercalation, we observe an apparent shift in the 002 peak to smaller angles with a final structure showing the d001 at ∼24°(2θ) for the stage 1 structure (Fig. 12a). This is indicative of a slight expansion of the graphite lattice by ∼10% (or 35 pm). In contrast, intercalation of the larger K+ tends to cause more expansion of the graphite lattice attaining a final stage 1 structure increased by ∼50% (or 0.2 nm). Likewise, a large expansion of the graphite lattice was observed with Rb as indexed with the d002 reflection in Fig. 12c. The graphite particles explored for LIBs and PIBs are often micron-sized and can handle even larger reversible expansions without compromising their crystal structure, as seen with solvent cointercalation.23,116 However, the precise structure of graphite can have some effects on performance, especially during extended cycling.
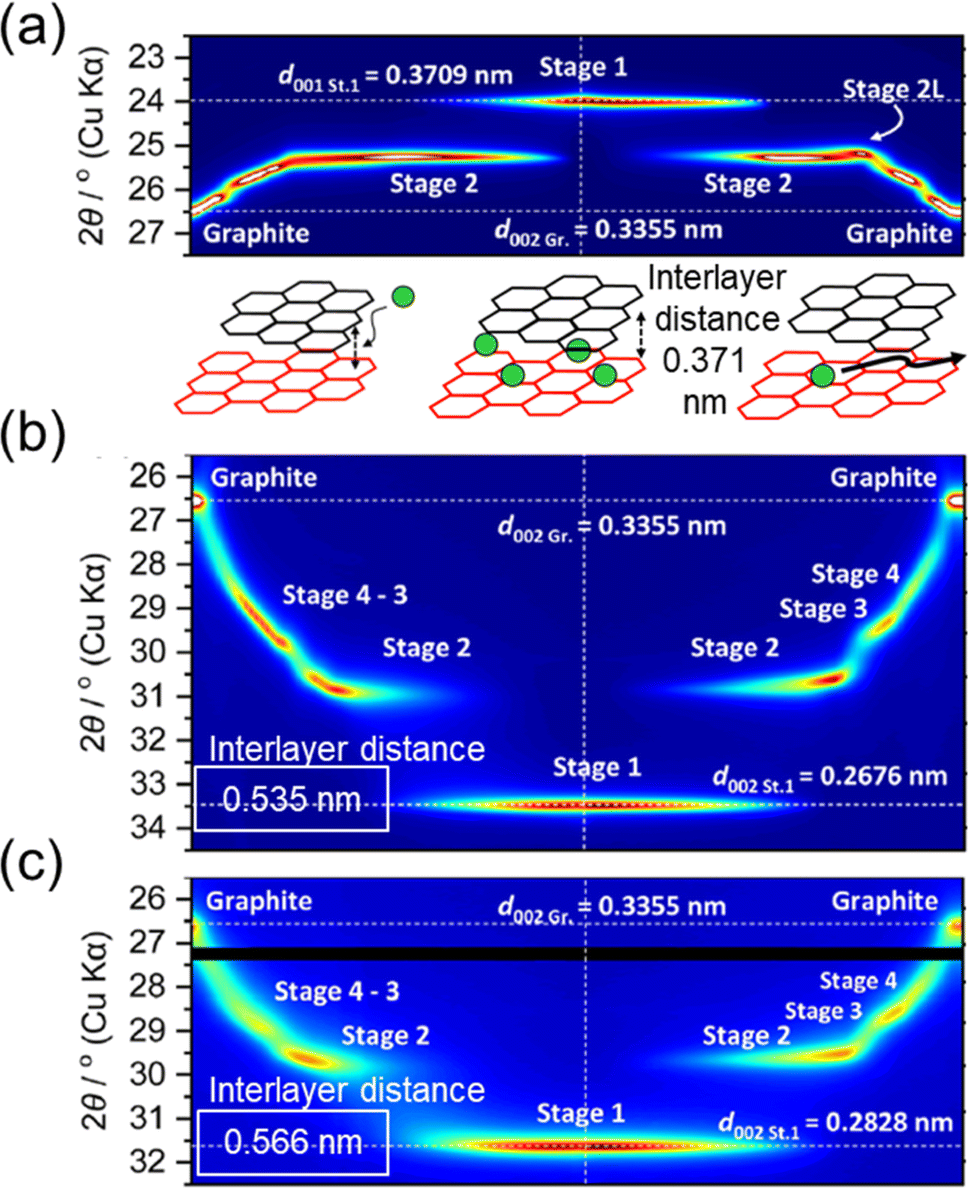 | ||
| Fig. 12 Operando XRD of alkali intercalation at graphite in (a) Li+, (b) K+ and (c) Rb+-based ionic liquids. Below (a) is an illustration of the lattice space increase with ion insertion. All XRD data are modified with permission from ref. 86. Copyright 2023 Royal Society of Chemistry. | ||
Previous works in LIBs indicated high degrees of graphitization leads to a narrower interlayer space and higher reversible capacities,117,118 as observed with highly crystalline natural graphite compared to synthetic graphite.14,119 Recently, we evaluated the K ion intercalation process at two types of synthetic graphite with low or high crystallinity.120 In this case, we did not see any difference in performance for Li intercalation with both graphite samples showing similar voltage profiles, discharge capacities, and capacity retention. For K intercalation, we observed similar discharge capacities, but only the highly crystalline graphite was stable for long term cycling. We also evaluated the impact of graphite particle size on K-ion insertion.100 In this case, the discharge capacities were not strongly impacted by particle size, but smaller particles tended to show lower initial CEs and improved capacity retention during cycling. The graphite structure can handle large distortion, but at the same time, expansion of the lattice may lead to disruptions in the SEI which would require additional interphase formation and consumption of the positive electrode capacity within full cells.
To realize improved stability and cycle life for PIBs utilizing graphite, there have been several efforts to improve the electrolyte and SEI chemistry. Our group has shown significant improvements in half and full-cells through electrolyte optimization strategies and the discovery of appropriate electrolyte additives. For example, we recently developed binary-salt electrolytes based on KPF6/KFSA in EC/DEC and EC/PC.21 By mixing of the counter anion in an optimized ratio, these electrolytes could improve the first cycle CE (Fig. 13a) and achieve a 3.6 V full cell with graphite as the negative electrode. The capacity retention remained high at 75% and 85% after 500 cycles for 1 and 5C rates, respectively.21 Much of the improvements in performance were considered to be linked to decomposition products from the N(SO2F)2− anion incorporated into the SEI structure. Indeed, we found that even an electrolyte based on 0.8 M KPF6 in EC/DEC could be improved through the addition of a sulfamoyl fluoride-type additive.22 By adding 10 wt% of dimethylsulfamoyl fluoride (DMSF) to the electrolyte, interfacial charge transfer properties were improved (Fig. 13b). When mixing DMSF with an optimized electrolyte containing KFSA, we observed further improved capacity retention of 82.4% after 500 cycles (Fig. 13c). It was assumed that the improvements are based on a thinner SEI with an altered structure that incorporates decomposition products derived from the DMSF additive.
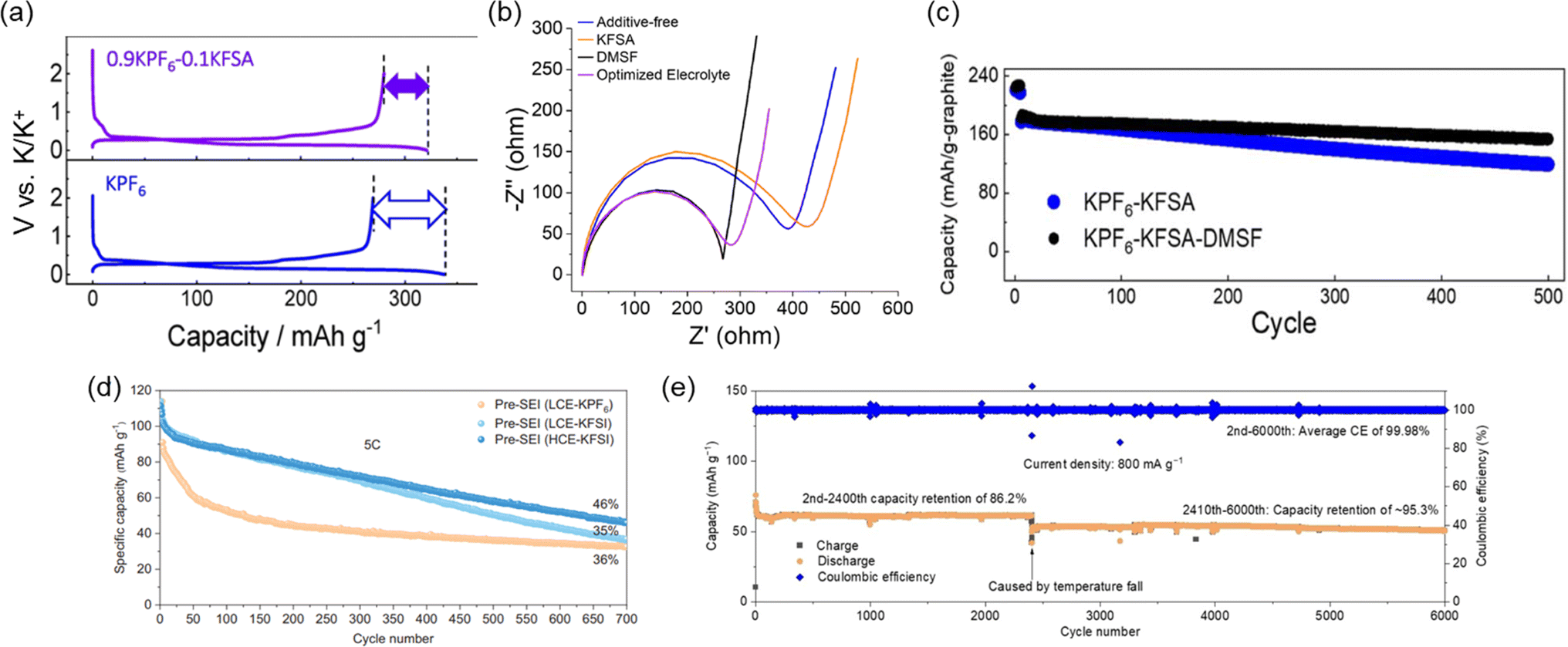 | ||
| Fig. 13 Impact of electrolyte on graphite cycling in half and full cells. (a) First cycle charge/discharge of graphite/K half-cells in KPF6 and binary 0.9 KPF6–0.1 KFSA electrolyte. Reproduced with permission from ref. 21. Copyright 2020 American Chemical Society. (b) Electrochemical impedance spectroscopy (after reducing to 0 V) in KPF6-ester carbonate-based electrolytes containing a DMSF additive. (c) Charge/discharge curves for graphite//PB full cells. (b) and (c) are reproduced with permission from ref. 22. Copyright 2023 Royal Society of Chemistry. (d) Graphite//PB full cell cycling using high and low concentrations of KFSI. Reproduced with permission from ref. 121. Copyright 2024 Wiley-VCH GmbH. (e) Long-term graphite//PB full cell cycling in a chloro-modified electrolyte. Reproduced with permission from ref. 122. Copyright 2024 Wiley-VCH GmbH. | ||
While exploring diverse additives to improve the graphite performance, we make note that not all additives show significant influence on the behavior of graphite and can even result in negative effects.115,123 For example, the well-known additive fluoroethylene carbonate for LIBs shows a negative effect incorporated into graphite/K half-cell. Key differences in the SEI structures and their properties among the alkali ions have been noted in previous works.124–126 This would suggest that the SEI, as with other aspects of batteries, requires specific tuning/optimization when exchanging the alkali ion. We also note that reactive K metal can influence the decomposition of the electrolyte by producing soluble species after reaction.123 This may extend to other reactive alkali metals which could influence the developing SEI in half cell measurements. The SEI is composed of diverse electrolyte derived components.127,128 These SEI components (e.g. fluorides, oxides, etc.) are frequently identified with various spectroscopic techniques.81 On the other hand, characterization of SEI properties (passivation, ion transfer) is rarely conducted. Recent analytical developments81 are making this process more simplified, and we expect future studies will provide valuable insight to improve the performance.114,129 While our knowledge on how to build the SEI and direct its properties remains limited, it is apparent that the SEI is a key component for attaining long cycle life, high-voltage batteries beyond the LIB.
Aside from looking for electrolyte additives to improve the performance, concentrated electrolytes and other alterations to the electrolyte seem to be other promising approaches. At high rates, the anion choice and the concentration can both impact the cycling performance. Other groups have shown the exchange of KPF6 for KFSI was quite effective for improving the cyclability at fast rates (Fig. 13d).121 They further observed that using a highly concentrated electrolyte led to a similar output but improved capacity retention. Others have observed this as well with concentrated electrolytes including ionic liquids.130 Our group recently attained excellent performance of a graphite//PB full cell using an ionic liquid. We observed a very high capacity retention of 94.7% after ∼1000 cycles at 2C.12 As with the other reports, this was strongly linked to a highly stable SEI that could protect the graphite electrode after charging the battery; i.e. in the reduced state. Aside, the recent use of functionalized solvents could also impact the SEI structure and performance. As shown in Fig. 13e, electrolytes using chloro-modified ethers could significantly improve the performance of PB//graphite full cells (Fig. 13e).122 High coulombic efficiency and over 6000 cycles was demonstrated which could be attributed to high stability at the graphite electrode and improved oxidation stability of the modified electrolyte.
3.2 Staging in B/C/N and B/C materials
As discussed in the previous section, Li+, K+, and Rb+ can easily intercalate into the graphite host, while Na+ intercalation is thermodynamically unstable at potentials above plating. Interestingly, increasing the lattice spacing by using an “expanded graphite,” as well as surface modifications have been shown to encourage Na+ insertion.96,131 By adjustment of the structure and the associated energetics, the Na+–graphite intercalate structure can be stabilized. To that end, graphite-like materials have also been explored for their intercalation of Na+. In particular, Na+ can reversibly intercalate into B/C/N and B/C materials to form stage 1 structures.97,132 Like graphite, B/C/N and B/C materials also have a layered structure with some of their carbon atoms replaced by boron and nitrogen (Fig. 14a).133 These heteroatom-substituted carbons have been prepared by the chemical vapor deposition (CVD) method and pyrolysis. Much like other carbons, they can exhibit conductive or semiconducting properties depending on their compositions and atomic arrangements within the layer structures.97,132,134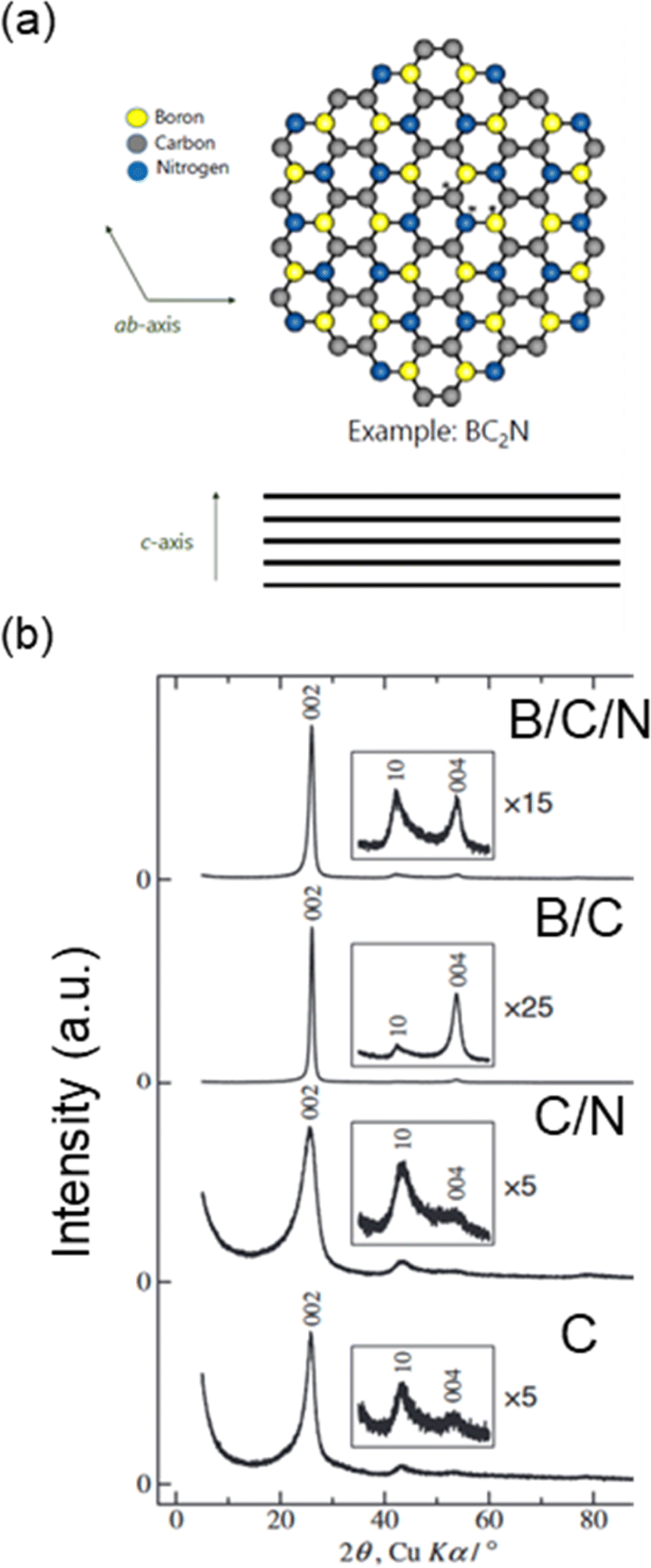 | ||
| Fig. 14 Structure of B/C/N materials. (a) Graphite-like network of B/C/N (example: BC2N). (b) XRD patterns of B/C/N, B/C, C/N and C powders prepared using chemical vapor deposition. (b) is reproduced with permission from ref. 97. Copyright 2015 Electrochemical Society of Japan. | ||
As shown in Fig. 14b, B/C and B/C/N materials show a sharp diffraction peak at a similar position (2θ = ca. 26°; interlayer spacing of ∼0.34 nm) to that of graphite 002 diffraction peak (2θ = 26.5°; interlayer spacing of 0.335 nm).97 Some small, additional peaks were observed including peaks at a similar position (2θ = ca. 54°) to that of graphite 004 diffraction peak. While this suggests improved ordering of the layer stacking, a broad 10 diffraction peak was also observed suggesting a random, turbostratic structure. For B/C prepared at higher temperatures, more regular ordering of the crystal structure was observed though the product contained a B4C impurity. The C/N material, in which some of the carbon atoms are replaced with nitrogen, showed a broader 002 diffraction peak indicative of poorer crystallinity and higher disorder. The peak was also broader than carbon powders from pyrolysis of ethylene. Together, these results suggest that B addition to the graphene structure aids in the crystallization, while N increases disorder.97
For the charge/discharge curves of B/C/N electrodes in Na half-cells (Fig. 15a), a sloping plateau is observed during reduction of the electrode (discharge) with two inflection points around 0.7 V and 0.03 V vs. Na/Na+.97 The oxidation (charge) response is also sloped achieving a reversible capacity of 190 mA h g−1, equivalent to a composition of NaC12. Similarly, reversible charge/discharge was observed for B/C materials with higher capacities (200–240 mA h g−1) and at slightly different redox potentials.135 The structure of the intercalates were further evaluated with XRD, which shows a change in the 002 diffraction peak during (de)intercalation. After intercalation of Na+ into the B/C/N material, the 002 peak shifted to lower angles (Fig. 15b) indicating an increase in the interlayer distance (d-spacing). The stage number could be estimated based on the original d-spacing of B/C/N (dhost = 0.343 nm) and the layer spacing of Na-intercalated B/C/N (di = 0.463 nm). Assuming a stage 1 structure, the interlayer spacing increased by 35%. This seems reasonable considering graphite shows an increase in the interlayer space by approximately 10% and 50% for Li+ and K+ insertion, respectively.86,100,136 Stage 2 intercalation compounds (sample held at 0.7 V vs. Na/Na+) further confirmed these results indicating an increase in the interlayer distance dependent on the amount of intercalated Na.135 Similar XRD results and stage 1 structures were observed for the intercalated B/C materials.
 | ||
| Fig. 15 Electrochemical insertion of Na+ into B/C/N (Na-B/C/N). (a) Galvanostatic charge–discharge curves of B/C/N materials in 1 M NaPF6 EC:DEC (1:1 v/v). (b) Ex situ XRD patterns of Na-B/C/N at 0.003 V and 0.70 V vs. Na+/Na, respectively. Modified with permission from ref. 97. Copyright 2015 Electrochemical Society of Japan. | ||
When a heteroatom such as boron is substituted in a graphene layer, the orbitals of carbon and the heteroatom (boron) become hybridized. This leads to a lowering of the LUMO and conduction band, consistent with the differences for the XANES spectra of B/C/N and HOPG (highly oriented pyrolytic graphite).132 In other words, boron doping improves electron donation from Na to the host B/C/N material compared with Na+ inserted into graphite. Interestingly, when B/C materials are applied in Li+ cells, capacities of 580 mA h g−1 can be attained.134 This is significantly larger than Li+ insertion into graphite (LiC6) and corresponds to an equivalent composition of LiC4. The interlayer expansion of the B/C layers was a bit higher than Li+ intercalation into graphite, but overall, boron-doped carbons seem to improve packing of the intercalation compounds inside the layered host. The upper limit of such packing is still questionable, but a highly relevant topic for future research to explore. Despite the good performances observed when using B/C/N and B/C as anode materials for Li-ion and Na-ion secondary batteries, there are some current drawbacks. For instance, the current preparation method using CVD requires expensive and highly reactive BCl3 gas as the starting material. The discovery of low-cost precursors or alternative synthetic methods may provide a more cost-effective route for growing B/C and B/C/N materials for battery applications.
3.3 Solvent cointercalation
Aside from using modified graphite or graphite-like materials for SIBs, a reversible ion-solvent cointercalation mechanism was identified by Adelhelm's and Kang's groups.23,137 In this case, solvent molecules continue to solvate the alkali ions during the (de)intercalation process, forming ternary graphite intercalation compounds (T-GICs). Most works have focused on diethylene glycol dimethyl ether (a.k.a. diglyme or G2) as the cointercalating solvent, but a variety of other glymes are also being explored including dimethyl ether (monoglyme), tetraethylene glycol dimethyl ether (tetraglyme) and others. This has opened up opportunities for using the graphite negative electrode even within SIBs. In contrast to the poor Na+ storage capacity of graphite in carbonate electrolytes,10,124 Na+ dissolved in glyme solvents can attain reversible capacities up to ∼120 mA h g−1 with excellent cyclability (>1000 cycles), good CEs and full cells already demonstrated. Furthermore, using cointercalation at graphite can enable rapid kinetics for high power applications.138 Previous reports have indicated higher rate capabilities for cointercalation compared to ion intercalation at graphite as well as when using such solvents during insertion at HC.116,139,140 The high rate capabilities are thought to originate from a lack of an SEI and faster desolvation kinetics with fewer desolvating solvent molecules.As seen in Fig. 16a, the (dis)charge profile shows some similarity to that observed during Li+ insertion at graphite, though the reactions occur at more positive potentials (0.5–0.8 V vs. Na/Na+).138 As with alkali ion intercalation, the solvated Na+ ions fill the various layers of graphite via a staging mechanism, and a stage 1 structure of the approximate formula, Na(glyme)xC20, has been be confirmed.23,142 The difference in potential compared with alkali ion insertion suggests improved stability for the formation of T-GICs. This has been attributed to shielding of the ion by the solvent as it may impact interlayer repulsion and interactions of Na+ with the graphite host. Interestingly, other interactions have also been noted to impact the redox potential of cointercalation such as: length of the glyme and activity of free glyme in the solvent structure.143
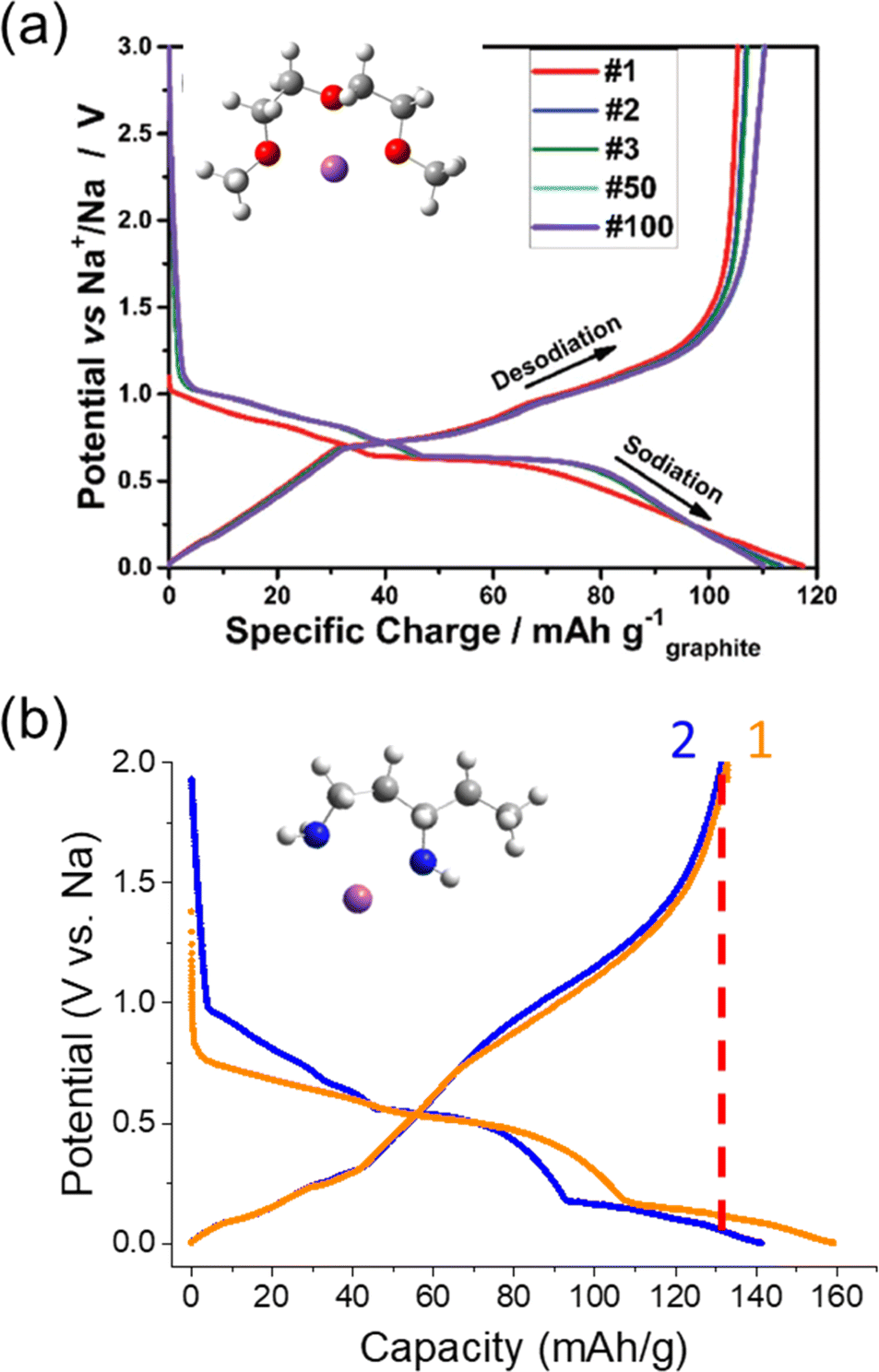 | ||
| Fig. 16 Electrochemical cointercalation of Na+ in graphite. Charge/discharge curves of graphite in half-cells containing (a) NaOTf in diglyme and (b) NaPF6 in 1,3-diaminopentane. (a) was reproduced with permission from ref. 138. Copyright 2018 Wiley-VCH GmbH. (b) was reproduced with permission from ref. 141. Copyright 2024 The Authors. | ||
Despite gradual progress and interesting chemistry involved, there are notable issues for using cointercalation for battery applications. At the forefront, the cointercalation mechanism leads to large changes in the graphite interplanar distance (a 200% increase) during insertion and deinsertion.138 This is quite significant compared with bare ion insertion (e.g. ∼10% for Li+ and ∼50% for K+).100,136 Expansion of the actual composite electrode tends to be a bit smaller,144 but this factor is expected to become problematic at the battery level.145 Furthermore, such expansion leads to a decrease in the volumetric capacity/energy density of the system. A few recent works by Lerner and Adelhelm's groups explored diamines for alleviating the graphite expansion.144,146 Using in situ electrochemical dilatometry, Escher et al. showed that the addition of 10 vol% ethylenediamine (EN) to the diglyme electrolyte reduced the initial electrode expansion from 275% to 200% and further reduced expansion/contraction during charge/discharge.144 The EN also clearly impacted the charge/discharge profile with additional plateaus compared with diglyme suggesting its involvement during the de/intercalation process. Unfortunately, EN tended to decrease the reversible capacity and showed poor ICEs of 38%, which may be due in part to the usage of half-cell measurements (containing Na metal). The authors noted poor stability for EN and Na metal, and as such, they only explored EN as a co-solvent but not as an individual solvent to prepare the electrolyte. Despite the promise of using solvent mixtures and additives as commonly applied in LIBs,128 few other works have explored means for improving the reversible capacities while minimizing electrode expansion.
Building on the interesting results with EN, our group started exploring other stable cointercalates based on diamines. Several chemically prepared T-GICs using diamines were reported by Gotoh et al., but not explored for electrochemical applications.147,148 We found the use of branched diamines, such as 1,3-diaminopentane (13DAP), showed improved stability compared to EN when in contact with Na metal and slightly higher reversible capacities up to 130 mA h g−1.141 As seen in the charge/discharge curve (Fig. 16b), the profile is different from diglyme and shows redox reactions occurring at lower redox potentials (<0.5 V vs. Na/Na+). Though the capacities were limited in this region, these potentials are quite similar to insertion/intercalation reactions of bare alkali ions. For another branched diamine, 2-methyl-1,5-diaminopentane (2M15DAP), we observed a lower reversible capacity and a completely different charge/discharge profile. This was more in agreement with the charge/discharge profile observed for the EN mixture with diglyme, but again showed a redox plateau at low potentials during oxidation.144 From these initial results, we see that changing the amine structure can strongly impact the cointercalation electrochemistry.
Much like EN, we found that expansion of the graphite lattice was drastically reduced during intercalation to only ∼135% for both 13DAP and 2M15DAP based on XRD analysis.141 Comparing with the reported expansion of the graphite lattice in diglyme, we can estimate almost double the volumetric capacity when using 13DAP. Likewise, 2M15DAP showed an improved volumetric capacity over DG based on this metric. Though these diamine-based electrolytes demonstrated notable opportunities, we found their cycling performance and oxidation stability still needs to be improved for practical use as a battery solvent. Only by incorporating 5 vol% DG cosolvent into the diamine electrolyte enabled 82% capacity retention after 200 cycles. Considering the vast variety of amines, as well as mixed structures, there remains a vast landscape of unexplored cointercalation systems that may help improve the performance for SIBs.
Overall, the coordination structure appears to be key for improving the packing density for higher capacities while at the same time minimizing electrode expansion. There has been some debate on the number of solvent molecules that remain coordinated to Na+ during intercalation. For 2G, measurements of the electrode mass as well as theoretical investigations suggested only one diglyme molecule per Na+.142,149 Other larger glymes, 3G, 4G, etc., are also expected to only require coordination of a single glyme per Na+.150 On the other hand, Na+ prefers a coordination number of 4–7 which would suggest two 2G molecules per Na+.150,151 More recently, the use of various in situ NMR methods has proven more complex dynamics involved during cointercalation.152–154 The results suggest cointercalated Na+ can be coordinated with either one or two 2G molecules at room temperature depending on the SOC.154 As seen in Fig. 17, the NMR data also indicate uncoordinated 2G molecules within the graphite host. While Na+ does not appear to hop in between these 2G molecules, it does provide some sense that a pillaring-type effect could occur and prevent continuous expansion/contraction of the electrode.146 Indeed, 2G can also induce cointercalation of other solvent structures, such as cyclic ethers, which do not typically cointercalate on their own at room temperature.153 Exciting new directions in plausible additives or co-solvents are starting to emerge.
 | ||
| Fig. 17 Solvent dynamics during solvent cointercalation. (a) Illustration of Na+ and diglyme interactions with graphite during cointercalation. (b) NMR response indicating the ratio of each solvent species. Reproduced with permission from ref. 154. Copyright 2022 Wiley-VCH GmbH. | ||
To date, there have been several reports incorporating cointercalation at graphite for Na full cells.143,155 Output voltages ≥3 V and good capacity retentions over 1000 cycles have already been demonstrated. As seen in the past work by Kang's group, excellent cyclability was obtained for a cell constructed from Na1.5VPO4.8F0.7 and graphite in an NaPF6 electrolyte.143 Likewise, other works have shown promise for assembling full cells with other positive electrodes.155,156 Cointercalation can provide very long cycle lifetimes and at very fast charge/discharge rates, but there are some notable drawbacks. Aside from the relatively high redox potential, low capacities and electrode expansion, there is another issue with solvent uptake. Due to this process, it is necessary to incorporate additional electrolyte into the cell, which impacts energy density as well as battery weight.
Aside from its use for Na+ cointercalation, other alkali ions can likewise cointercalate with strongly bound solvent species. The case of dimethyl sulfoxide (DMSO) and Li+ is well-known,157,158 while propylene carbonate–Li+ cointercalation is not reversible due to cracking of graphite.159 For glyme based electrolytes, similar charge/discharge profiles and capacities near 100–110 mA h g−1 are observed regardless of using Li+, K+, or Rb+.86,150,160 However, the cyclability and rate performance are strongly impacted by the alkali ion with Li+ showing poor performance compared with Na+ and K+.160 Previous reports have indicated higher rate capabilities for larger ions, when considering Li, Na and K. Larger K ions can move faster during cointercalation, because the graphite interlayer space is more expanded.160 This has also been attributed to the favorable interactions of Li+ with the graphite host, as well as differences in the solvation structures which impacts diffusion.161 Interestingly, at high electrolyte concentrations, the insertion mechanism can be changed from favoring cointercalation toward ion intercalation for both Li+ and K+ electrolytes.11,162 This may be related to formation of SEI structures which block the solvent from insertion, as well as the decrease in available solvent molecules which may impact desolvation processes at the interface. Still, cells containing NaPF6 electrolytes in diglyme up to 3 M show a stable and reversible cointercalation mechanism.143 The ICE of these electrolytes decreased with increased salt concentration for numerous electrolytes, but it is peculiar that the solvated ions seem to traverse the SEI while solvated Li+ and K+ seem to be blocked. This is in good alignment with other mysterious aspects of potential SEI structures in cointercalating solvents.138,163
So far, there has been much speculation on the nature of SEI structures when cointercalating solvents are applied. Considering the high CEs observed, the concepts of very thin SEI143 or even SEI-free systems seem plausible.138,163 A lack of SEI seems more convincing when considering the large changes in volume for cointercalation and the difficulty of solvated ions transporting across an SEI. At the same time, the electrolyte anion has been shown to strongly impact the CEs and cyclability,163 and in all cases there are still first cycle losses observed. Glymes may show higher cathodic stability than carbonate esters (which reduce near 1.0 V vs. Li/Li+),164,165 but this would also mean no decomposition of the anion species. This becomes especially questionable for highly concentrated electrolytes where reduction of the anion tends to become more facile. Reactions at highly reactive edge sites or impurities may also be involved, but the point at which this is recognized as SEI chemistry remains ambiguous. Nevertheless, the prospects of an SEI-free electrode while maintaining prevention of self-discharge with the electrolyte is highly appealing for long-life batteries.
Clearly layered graphite and graphite-like materials are quite versatile negative electrodes for batteries. When looking at the various chemistries of graphite, as summarized in Table 1, we see distinct differences based on the electrolyte system. The highest capacities are currently observed for modified graphite (B/C/N) for both Li+ and Na+ insertion. However, Na+ insertion in B/C/N is not so far advanced beyond the solvent cointercalation mechanism, especially considering cyclability. As new cointercalating electrolytes are discovered, it may become possible to further improve the capacities and energy densities for more practical usage. We hope to see further advancements in modified graphite materials and the solvent cointercalation mechanism as future research progresses.
| Li | Na | K | Rb | |
|---|---|---|---|---|
| Graphite | LiC6 >350 mA h g−1 | Dilute stage only 10–30 mA h g−1 | KC8 ∼240 mA h g−1 | RbC8 276 mA h g−1 |
| B/C/N and B/C | LiC4 580 mA h g−1 | NaC12 190 mA h g−1 | — | — |
| Co-intercalation (graphite) | Li(glyme)xC20 110 mA h g−1 | Na(13DAP)xC17 130 mA h g−1 | K(glyme)xC20 105 mA h g−1 | Rb(glyme)xC20 100 mA h g−1 |
| Na(glyme)xC20 110 mA h g−1 |
4 Concluding remarks
Overall, the opportunity for applying and improving carbon electrodes for alkali energy storage remains rich. In Table 2, we have compiled some of the most promising results for the various carbons beings explored for energy storage. While the highest capacities are mainly being observed for HC electrodes, the cyclability and capacity retention of intercalation and cointercalation mechanisms at graphitic carbons should not be dismissed. The usage of carbon electrodes within Li-ion batteries remains at the top of energy storage technologies, but many of these emerging chemistries may take hold of the market due to their lower price and greener components.| Electrode | Capacity (mA h g−1); # of cycles; retention (%); rate (mA g−1); half or full cell | |
|---|---|---|
| Na | K | |
| Hard carbon | ||
| 295, 6000, 83, 2000, half140 | ∼160, 1000, 73, 4900, half167 | |
| 283, 3000, 72, 1000, half166 | ||
| Template method | 464; 200; 93; 25, half20 | 381; 50; 25; half20 |
| Mod. activated carbon | 350; 1000; 80, 500, half56 | — |
|
||
| Graphitic carbons | ||
| Intercalation | — | 230, 1000, 95, 558, half12 |
| 180, 500, 85, 279, full22 | ||
| ––, 6000, 66, 800, full122 | ||
| Cointercalation | 119, 10000, 87, 1000, half168 |
90, 100, 89, 2000, half116 |
| ∼100, 8000, 94, 10000, half168 |
∼100, 60, 91, –––, half160 | |
| With co-solvent | 134, 1800, 94, 500, half155 | — |
|
||
| B/C/N and B/C carbons | ∼200, 100, –––, half135 | — |
In this review, we have made an effort to provide the reader with sufficient fundamental information as well as practical insight on the various carbon-based energy storage technologies under development. Starting with hard carbon, we dove into the main points of interest: the pore structure. This topic will remain a valuable point of interest for years ahead as we approach high cyclability for long life batteries. Maximizing the pore system is a major goal that could further improve the capacities to as high as 500–600 mA h g−1.169 At the same time, reversible and facile transport to these pores is very important for battery development. From there, we discussed the relatively limited progress on soft carbons. Currently, these materials need to see some further improvements for practical usage in batteries, and their prospects are somewhat unknown. Finally, we discussed the place that graphite holds in next generation batteries. While its promising characteristics shine brightest when used in LIBs, it is also a notable electrode for PIBs. Many interesting concepts still remain relatively unexplored including further modified graphite materials and cointercalation chemistry, as well as controlling plating reactions which could also boost the performance of graphite. Some mysteries of graphite may still remain hidden and only future research will show if this is the case. In all, with the vast structures and properties of carbon electrodes, the opportunities for energy storage continue to grow.
Data availability
Data are partly available upon request from the authors because this is a review paper.Author contributions
Z. T. G, D. I., Y. F., and M. K. wrote and edited the manuscript. S. K. guided the writing, organization and planning of this work. R. T. and K. N. helped with editing the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was partly funded by JST-GteX (JPMJGX23S4), the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Program: Data Creation and Utilization Type Materials Research and Development Project (JPMXP1122712807), JST-CREST (JPMJCR21O6), and JST-ASPIRE (JPMJAP2313).References
- M. Winter, B. Barnett and K. Xu, Before Li ion batteries, Chem. Rev., 2018, 118, 11433–11456 CrossRef PubMed.
- S. Kalnaus, N. J. Dudney, A. S. Westover, E. Herbert and S. Hackney, Solid-state batteries: The critical role of mechanics, Science, 2023, 381, eabg5998 CrossRef PubMed.
- J. Qin, R. Wang, P. Xiao and D. Wang, Engineering cooperative catalysis in Li–S batteries, Adv. Energy Mater., 2023, 13, 2300611 CrossRef.
- K. Chen, D.-Y. Yang, G. Huang and X.-B. Zhang, Lithium–air batteries: Air-electrochemistry and anode stabilization, Acc. Chem. Res., 2021, 54, 632–641 CrossRef PubMed.
- D.-H. Liu, Z. Bai, M. Li, A. Yu, D. Luo, W. Liu, L. Yang, J. Lu, K. Amine and Z. Chen, Developing high safety Li-metal anodes for future high-energy Li-metal batteries: strategies and perspectives, Chem. Soc. Rev., 2020, 49, 5407–5445 RSC.
- E. J. Kim, P. R. Kumar, Z. T. Gossage, K. Kubota, T. Hosaka, R. Tatara and S. Komaba, Active material and interphase structures governing performance in sodium and potassium ion batteries, Chem. Sci., 2022, 13, 6121–6158 RSC.
- T. Hosaka, K. Kubota, A. S. Hameed and S. Komaba, Research development on K-ion batteries, Chem. Rev., 2020, 120, 6358–6466 CrossRef PubMed.
- D. Larcher and J.-M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7, 19–29 CrossRef PubMed.
- https://iupac.org/iupac-2022-top-ten/ .
- S. Komaba, T. Hasegawa, M. Dahbi and K. Kubota, Potassium intercalation into graphite to realize high-voltage/high-power potassium-ion batteries and potassium-ion capacitors, Electrochem. Commun., 2015, 60, 172–175 CrossRef.
- T. Hosaka, K. Kubota, H. Kojima and S. Komaba, Highly concentrated electrolyte solutions for 4 V class potassium-ion batteries, Chem. Commun., 2018, 54, 8387–8390 RSC.
- H. Onuma, K. Kubota, S. Muratsubaki, T. Hosaka, R. Tatara, T. Yamamoto, K. Matsumoto, T. Nohira, R. Hagiwara and H. Oji, Application of ionic liquid as K-ion electrolyte of graphite//K2Mn[Fe(CN)6] cell, ACS Energy Lett., 2020, 5, 2849–2857 CrossRef.
- Y. Youn, B. Gao, A. Kamiyama, K. Kubota, S. Komaba and Y. Tateyama, Nanometer-size Na cluster formation in micropore of hard carbon as origin of higher-capacity Na-ion battery, npj Comput. Mater., 2021, 7, 48 CrossRef.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, The success story of graphite as a lithium-ion anode material–fundamentals, remaining challenges, and recent developments including silicon (oxide) composites, Sustain. Energy Fuels, 2020, 4, 5387–5416 RSC.
- S. Dhir, S. Wheeler, I. Capone and M. Pasta, Outlook on K-ion batteries, Chem, 2020, 6, 2442–2460 Search PubMed.
- Z. Wang, A. P. Ratvik, T. Grande and S. M. Selbach, Diffusion of alkali metals in the first stage graphite intercalation compounds by vdW-DFT calculations, RSC Adv., 2015, 5, 15985–15992 RSC.
- D. Saurel, B. Orayech, B. Xiao, D. Carriazo, X. Li and T. Rojo, From charge storage mechanism to performance: a roadmap toward high specific energy sodium-ion batteries through carbon anode optimization, Adv. Energy Mater., 2018, 8, 1703268 Search PubMed.
- Y. Fujii, K. Sugata, Y. Omura, N. Kubota, K. Kisa, H. Sofuji and J. Suzuki, Optimization of Soft Carbon Negative Electrode in Sodium-Ion Batteries Using Surface-Modified Mesophase-Pitch Carbon Fibers, Electrochem, 2023, 91, 077008 CrossRef.
- A. Kamiyama, K. Kubota, D. Igarashi, Y. Youn, Y. Tateyama, H. Ando, K. Gotoh and S. Komaba, MgO-template synthesis of extremely high capacity hard carbon for Na-ion battery, Angew. Chem., Int. Ed., 2021, 60, 5114–5120 CrossRef.
- D. Igarashi, Y. Tanaka, K. Kubota, R. Tatara, H. Maejima, T. Hosaka and S. Komaba, New Template Synthesis of Anomalously Large Capacity Hard Carbon for Na- and K-Ion Batteries, Adv. Energy Mater., 2023, 13, 2302647 Search PubMed.
- T. Hosaka, T. Matsuyama, K. Kubota, S. Yasuno and S. Komaba, Development of KPF6/KFSA binary-salt solutions for long-life and high-voltage K-ion batteries, ACS Appl. Mater. Interfaces, 2020, 12, 34873–34881 CrossRef PubMed.
- Z. T. Gossage, T. Hosaka, T. Matsuyama, R. Tatara and S. Komaba, Fluorosulfonamide-type electrolyte additives for long-life K-ion batteries, J. Mater. Chem. A, 2023, 11, 914–925 RSC.
- B. Jache and P. Adelhelm, Use of graphite as a highly reversible electrode with superior cycle life for sodium-ion batteries by making use of co-intercalation phenomena, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 Search PubMed.
- Y. Xu, M. Titirici, J. Chen, F. Cora, P. L. Cullen, J. S. Edge, K. Fan, L. Fan, J. Feng and T. Hosaka, 2023 roadmap for potassium-ion batteries, JPhys Energy, 2023, 5, 021502 CrossRef.
- N. Tapia-Ruiz, A. R. Armstrong, H. Alptekin, M. A. Amores, H. Au, J. Barker, R. Boston, W. R. Brant, J. M. Brittain and Y. Chen, 2021 roadmap for sodium-ion batteries, JPhys Energy, 2021, 3, 031503 CrossRef.
- R. E. Franklin, The structure of graphitic carbons, Acta Crystallogr., 1951, 4, 253–261 CrossRef.
- R. E. Franklin, Crystallite growth in graphitizing and non-graphitizing carbons, Proc. R. Soc. A, 1951, 209, 196–218 Search PubMed.
- P.-c. Tsai, S.-C. Chung, S.-k. Lin and A. Yamada, Ab initio study of sodium intercalation into disordered carbon, J. Mater. Chem. A, 2015, 3, 9763–9768 RSC.
- M. Song, Z. Yi, R. Xu, J. Chen, J. Cheng, Z. Wang, Q. Liu, Q. Guo, L. Xie and C. Chen, Towards enhanced sodium storage of hard carbon anodes: Regulating the oxygen content in precursor by low-temperature hydrogen reduction, Energy Storage Mater., 2022, 51, 620–629 CrossRef.
- K. Kubota, S. Shimadzu, N. Yabuuchi, S. Tominaka, S. Shiraishi, M. Abreu-Sepulveda, A. Manivannan, K. Gotoh, M. Fukunishi and M. Dahbi, Structural analysis of sucrose-derived hard carbon and correlation with the electrochemical properties for lithium, sodium, and potassium insertion, Chem. Mater., 2020, 32, 2961–2977 CrossRef.
- Y. Morikawa, S. i. Nishimura, R. i. Hashimoto, M. Ohnuma and A. Yamada, Mechanism of sodium storage in hard carbon: an X-ray scattering analysis, Adv. Energy Mater., 2020, 10, 1903176 CrossRef.
- J. M. Stratford, A. K. Kleppe, D. S. Keeble, P. A. Chater, S. S. Meysami, C. J. Wright, J. Barker, M.-M. Titirici, P. K. Allan and C. P. Grey, Correlating local structure and sodium storage in hard carbon anodes: insights from pair distribution function analysis and solid-state NMR, J. Am. Chem. Soc., 2021, 143, 14274–14286 CrossRef PubMed.
- J. R. Dahn, T. Zheng, Y. Liu and J. S. Xue, Mechanisms for lithium insertion in carbonaceous materials, Science, 1995, 270, 590–593 CrossRef.
- K. Tatsumi, J. Conard, M. Nakahara, S. Menu, P. Lauginie, Y. Sawada and Z. Ogumi, Low temperature 7Li-NMR investigations on lithium inserted into carbon anodes for rechargeable lithium-ion cells, J. Power Sources, 1999, 81, 397–400 CrossRef.
- D. A. Stevens and J. R. Dahn, The mechanisms of lithium and sodium insertion in carbon materials, J. Electrochem. Soc., 2001, 148, A803 CrossRef.
- S. E. Hayes, R. A. Guidotti, W. R. Even Jr, P. J. Hughes and H. Eckert, 7Li solid-state nuclear magnetic resonance as a probe of lithium species in microporous carbon anodes, J. Phys. Chem. A, 2003, 107, 3866–3876 CrossRef.
- M. Nagao, C. Pitteloud, T. Kamiyama, T. Otomo, K. Itoh, T. Fukunaga, K. Tatsumi and R. Kanno, Structure characterization and lithiation mechanism of nongraphitized carbon for lithium secondary batteries, J. Electrochem. Soc., 2006, 153, A914 CrossRef.
- J. S. Weaving, A. Lim, J. Millichamp, T. P. Neville, D. Ledwoch, E. Kendrick, P. F. McMillan, P. R. Shearing, C. A. Howard and D. J. L. Brett, Elucidating the sodiation mechanism in hard carbon by operando raman spectroscopy, ACS Appl. Energy Mater., 2020, 3, 7474–7484 CrossRef.
- L. Kitsu Iglesias, E. N. Antonio, T. D. Martinez, L. Zhang, Z. Zhuo, S. J. Weigand, J. Guo and M. F. Toney, Revealing the Sodium Storage Mechanisms in Hard Carbon Pores, Adv. Energy Mater., 2023, 13, 2302171 CrossRef.
- K. Gotoh, T. Ishikawa, S. Shimadzu, N. Yabuuchi, S. Komaba, K. Takeda, A. Goto, K. Deguchi, S. Ohki and K. Hashi, NMR study for electrochemically inserted Na in hard carbon electrode of sodium ion battery, J. Power Sources, 2013, 225, 137–140 CrossRef.
- D. Saurel, J. Segalini, M. Jauregui, A. Pendashteh, B. Daffos, P. Simon and M. Casas-Cabanas, A SAXS outlook on disordered carbonaceous materials for electrochemical energy storage, Energy Storage Mater., 2019, 21, 162–173 CrossRef.
- K. Omote, Y. Ito and S. Kawamura, Small angle x-ray scattering for measuring pore-size distributions in porous low-κ films, Appl. Phys. Lett., 2003, 82, 544–546 CrossRef.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Low-defect and low-porosity hard carbon with high coulombic efficiency and high capacity for practical sodium ion battery anode, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- H. Au, H. Alptekin, A. C. S. Jensen, E. Olsson, C. A. O'Keefe, T. Smith, M. Crespo-Ribadeneyra, T. F. Headen, C. P. Grey and Q. Cai, A revised mechanistic model for sodium insertion in hard carbons, Energy Environ. Sci., 2020, 13, 3469–3479 RSC.
- K. Gotoh, T. Yamakami, I. Nishimura, H. Kometani, H. Ando, K. Hashi, T. Shimizu and H. Ishida, Mechanisms for overcharging of carbon electrodes in lithium-ion/sodium-ion batteries analysed by operando solid-state NMR, J. Mater. Chem. A, 2020, 8, 14472–14481 RSC.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. Cui, I. Khayrullin, S. O. Dantas, J. Marti and V. G. Ralchenko, Carbon structures with three-dimensional periodicity at optical wavelengths, Science, 1998, 282, 897–901 CrossRef PubMed.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Ordered nanoporous arrays of carbon supporting high dispersions of platinum nanoparticles, Nature, 2001, 412, 169–172 CrossRef PubMed.
- T. Morishita, T. Tsumura, M. Toyoda, J. Przepiórski, A. W. Morawski, H. Konno and M. Inagaki, A review of the control of pore structure in MgO-templated nanoporous carbons, Carbon, 2010, 48, 2690–2707 CrossRef.
- H. Nishihara and T. Kyotani, Zeolite-templated carbons–three-dimensional microporous graphene frameworks, Chem. Commun., 2018, 54, 5648–5673 RSC.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, On the comparative stability of Li and Na metal anode interfaces in conventional alkyl carbonate electrolytes, J. Electrochem. Soc., 2015, 162, A7060 CrossRef CAS.
- Y. Li, A. Vasileiadis, Q. Zhou, Y. Lu, Q. Meng, Y. Li, P. Ombrini, J. Zhao, Z. Chen and Y. Niu, Origin of fast charging in hard carbon anodes, Nat. Energy, 2024, 1–9 Search PubMed.
- X. Chen, C. Liu, Y. Fang, X. Ai, F. Zhong, H. Yang and Y. Cao, Understanding of the sodium storage mechanism in hard carbon anodes, Carbon Energy, 2022, 4, 1133–1150 CrossRef.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Towards K-ion and Na-ion batteries as “beyond Li-ion”, Chem. Rec., 2018, 18, 459–479 CrossRef PubMed.
- C. Zhao, Q. Wang, Y. Lu, B. Li, L. Chen and Y.-S. Hu, High-temperature treatment induced carbon anode with ultrahigh Na storage capacity at low-voltage plateau, Sci. Bull., 2018, 63, 1125–1129 CrossRef.
- Z. Zheng, S. Hu, W. Yin, J. Peng, R. Wang, J. Jin, B. He, Y. Gong, H. Wang and H. J. Fan, CO2-Etching Creates Abundant Closed Pores in Hard Carbon for High-Plateau-Capacity Sodium Storage, Adv. Energy Mater., 2024, 14, 2303064 CrossRef.
- X. Chen, N. Sawut, K. Chen, H. Li, J. Zhang, Z. Wang, M. Yang, G. Tang, X. Ai and H. Yang, Filling carbon: a microstructure-engineered hard carbon for efficient alkali metal ion storage, Energy Environ. Sci., 2023, 16, 4041–4053 RSC.
- Q. Li, X. Liu, Y. Tao, J. Huang, J. Zhang, C. Yang, Y. Zhang, S. Zhang, Y. Jia and Q. Lin, Sieving carbons promise practical anodes with extensible low-potential plateaus for sodium batteries, Natl. Sci. Rev., 2022, 9, nwac084 CrossRef PubMed.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, High-density sodium and lithium ion battery anodes from banana peels, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- X. Dou, I. Hasa, M. Hekmatfar, T. Diemant, R. J. Behm, D. Buchholz and S. Passerini, Pectin, hemicellulose, or lignin? Impact of the biowaste source on the performance of hard carbons for sodium-ion batteries, ChemSusChem, 2017, 10, 2668–2676 CrossRef PubMed.
- W. Li, L. Zeng, Z. Yang, L. Gu, J. Wang, X. Liu, J. Cheng and Y. Yu, Free-standing and binder-free sodium-ion electrodes with ultralong cycle life and high rate performance based on porous carbon nanofibers, Nanoscale, 2014, 6, 693–698 RSC.
- B. H. Hou, Y. Y. Wang, Q. L. Ning, W. H. Li, X. T. Xi, X. Yang, H. J. Liang, X. Feng and X. L. Wu, Self-supporting, flexible, additive-free, and scalable hard carbon paper self-interwoven by 1D microbelts: superb room/low-temperature sodium storage and working mechanism, Adv. Mater., 2019, 31, 1903125 CrossRef PubMed.
- G. Hasegawa, K. Kanamori, N. Kannari, J. i. Ozaki, K. Nakanishi and T. Abe, Hard carbon anodes for Na-ion batteries: toward a practical use, Chemelectrochem, 2015, 2, 1917–1920 Search PubMed.
- Y. Katsuyama, A. Kudo, H. Kobayashi, J. Han, M. Chen, I. Honma and R. B. Kaner, A 3D-Printed, Freestanding Carbon Lattice for Sodium Ion Batteries, Small, 2022, 18, 2202277 CrossRef PubMed.
- X. Liu, Y. Tan, T. Liu, W. Wang, C. Li, J. Lu and Y. Sun, A simple electrode-level chemical presodiation route by solution spraying to improve the energy density of sodium-ion batteries, Adv. Funct. Mater., 2019, 29, 1903795 CrossRef.
- Y. Tang, Q. Zhang, W. Zuo, S. Zhou, G. Zeng, B. Zhang, H. Zhang, Z. Huang, L. Zheng and J. Xu, Sustainable layered cathode with suppressed phase transition for long-life sodium-ion batteries, Nat. Sustain., 2024, 7, 348–359 Search PubMed.
- H. Fang, S. Gao, M. Ren, Y. Huang, F. Cheng, J. Chen and F. Li, Dual-function presodiation with sodium diphenyl ketone towards ultra-stable hard carbon anodes for sodium-ion batteries, Angew. Chem., Int. Ed., 2023, 62, e202214717 CrossRef PubMed.
- M. Sathiya, J. Thomas, D. Batuk, V. Pimenta, R. Gopalan and J.-M. Tarascon, Dual stabilization and sacrificial effect of Na2CO3 for increasing capacities of Na-ion cells based on P2-NaxMO2 electrodes, Chem. Mater., 2017, 29, 5948–5956 CrossRef.
- M. Matsuzaki, R. Tatara, K. Kubota, K. Kuroki, T. Hosaka, K. Umetsu, N. Okada and S. Komaba, Application of Na2CO3 as a Sacrificial Electrode Additive in Na-ion Batteries to Compensate for the Sodium Deficiency in Na2/3[Fe1/2Mn1/2]O2, Batteries Supercaps, 2024, 7, e202400009 CrossRef.
- Z. Jian, Z. Xing, C. Bommier, Z. Li and X. Ji, Hard carbon microspheres: potassium-ion anode versus sodium-ion anode, Adv. Energy Mater., 2016, 6, 1501874 Search PubMed.
- H. Hijazi, Z. Ye, L. Zhang, J. Deshmukh, M. B. Johnson, J. R. Dahn and M. Metzger, Impact of sodium metal plating on cycling performance of layered oxide/hard carbon sodium-ion pouch cells with different voltage cut-offs, J. Electrochem. Soc., 2023, 170, 070512 Search PubMed.
- Z. Li, Z. Jian, X. Wang, I. A. Rodríguez-Pérez, C. Bommier and X. Ji, Hard carbon anodes of sodium-ion batteries: undervalued rate capability, Chem. Commun., 2017, 53, 2610–2613 RSC.
- Y. Fujii, R. Tatara, D. Igarashi, T. Hosaka, R. Takaishi, E. Shiiyama, T. Matsuyama and S. Komaba, Application of Diluted Electrode Method to Sodium-ion Insertion into Hard Carbon Electrode, Electrochem, 2023, 91, 077002 CrossRef.
- Y. Orikasa, Y. Gogyo, H. Yamashige, M. Katayama, K. Chen, T. Mori, K. Yamamoto, T. Masese, Y. Inada and T. Ohta, Ionic conduction in lithium ion battery composite electrode governs cross-sectional reaction distribution, Sci. Rep., 2016, 6, 26382 CrossRef PubMed.
- S. Tardif, N. Dufour, J.-F. Colin, G. Gébel, M. Burghammer, A. Johannes, S. Lyonnard and M. Chandesris, Combining operando X-ray experiments and modelling to understand the heterogeneous lithiation of graphite electrodes, J. Mater. Chem. A, 2021, 9, 4281–4290 RSC.
- M. Umeda, K. Dokko, Y. Fujita, M. Mohamedi, I. Uchida and J. R. Selman, Electrochemical impedance study of Li-ion insertion into mesocarbon microbead single particle electrode: Part I. Graphitized carbon, Electrochim. Acta, 2001, 47, 885–890 CrossRef.
- K. Dokko, N. Nakata, Y. Suzuki and K. Kanamura, High-rate lithium deintercalation from lithiated graphite single-particle electrode, J. Phys. Chem. C, 2010, 114, 8646–8650 Search PubMed.
- N. Takami, K. Hoshina and H. Inagaki, Lithium diffusion in Li4/3Ti5/3O4 particles during insertion and extraction, J. Electrochem. Soc., 2011, 158, A725 CrossRef.
- Z. T. Gossage, N. B. Schorr, K. Hernández-Burgos, J. Hui, B. H. Simpson, E. C. Montoto and J. Rodríguez-López, Interrogating charge storage on redox active colloids via combined Raman spectroscopy and scanning electrochemical microscopy, Langmuir, 2017, 33, 9455–9463 CrossRef CAS PubMed.
- Z. T. Gossage, K. Hernández-Burgos, J. S. Moore and J. Rodríguez-López, Impact of charge transport dynamics and conditioning on cycling efficiency within single redox active colloids, Chemelectrochem, 2018, 5, 3006–3013 CrossRef CAS.
- B. Tao, L. C. Yule, E. Daviddi, C. L. Bentley and P. R. Unwin, Correlative Electrochemical Microscopy of Li-Ion (De) intercalation at a Series of Individual LiMn2O4 Particles, Angew. Chem., Int. Ed., 2019, 58, 4606–4611 CrossRef CAS PubMed.
- J. Hui, Z. T. Gossage, D. Sarbapalli, K. Hernández-Burgos and J. Rodríguez-López, Advanced electrochemical analysis for energy storage interfaces, Anal. Chem., 2018, 91, 60–83 CrossRef PubMed.
- K. Ariyoshi, S. Mizutani, T. Makino and Y. Yamada, A clue to high rate capability of lithium-ion batteries obtained by an electrochemical approach using “diluted” electrode, J. Electrochem. Soc., 2018, 165, A3965 CrossRef CAS.
- W. Luo, Z. Jian, Z. Xing, W. Wang, C. Bommier, M. M. Lerner and X. Ji, Electrochemically expandable soft carbon as anodes for Na-ion batteries, ACS Cent. Sci., 2015, 1, 516–522 CrossRef CAS PubMed.
- R. A. Adams, A. Varma and V. G. Pol, Carbon anodes for nonaqueous alkali metal-ion batteries and their thermal safety aspects, Adv. Energy Mater., 2019, 9, 1900550 CrossRef.
- Z. Jian, C. Bommier, L. Luo, Z. Li, W. Wang, C. Wang, P. A. Greaney and X. Ji, Insights on the mechanism of Na-ion storage in soft carbon anode, Chem. Mater., 2017, 29, 2314–2320 CrossRef.
- D. Igarashi, R. Tatara, R. Fujimoto, T. Hosaka and S. Komaba, Electrochemical intercalation of rubidium into graphite, hard carbon, and soft carbon, Chem. Sci., 2023, 14, 11056–11066 RSC.
- G. Lee, K. B. Min, M. E. Lee, Y.-K. Lee, H. R. Lee, S.-S. Kim, S. Y. Cho, H.-I. Joh, Y.-K. Kim and S. Lee, Selective synthesis of soft and hard carbons from a single precursor through tailor-made stabilization for anode in sodium-ion batteries, Chem. Eng. J., 2024, 479, 147766 CrossRef.
- F. Xie, Z. Xu, A. C. S. Jensen, H. Au, Y. Lu, V. Araullo-Peters, A. J. Drew, Y. S. Hu and M. M. Titirici, Hard-soft carbon composite anodes with synergistic sodium storage performance, Adv. Funct. Mater., 2019, 29, 1901072 CrossRef.
- D. Cheng, X. Zhou, H. Hu, Z. Li, J. Chen, L. Miao, X. Ye and H. Zhang, Electrochemical storage mechanism of sodium in carbon materials: A study from soft carbon to hard carbon, Carbon, 2021, 182, 758–769 CrossRef.
- J. Wang, L. Yan, B. Liu, Q. Ren, L. Fan, Z. Shi and Q. Zhang, A solvothermal pre-oxidation strategy converting pitch from soft carbon to hard carbon for enhanced sodium storage, Chin. Chem. Lett., 2023, 34, 107526 Search PubMed.
- Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, A superior low-cost amorphous carbon anode made from pitch and lignin for sodium-ion batteries, J. Mater. Chem. A, 2016, 4, 96–104 Search PubMed.
- H. Moriwake, A. Kuwabara, C. A. J. Fisher and Y. Ikuhara, Why is sodium-intercalated graphite unstable?, RSC Adv., 2017, 7, 36550–36554 RSC.
- Y. Liu, B. V. Merinov and W. A. Goddard, Origin of low sodium capacity in graphite and generally weak substrate binding of Na and Mg among alkali and alkaline earth metals, Proc. Natl. Acad. Sci. U.S.A., 2016, 113, 3735–3739 CrossRef CAS PubMed.
- O. Lenchuk, P. Adelhelm and D. Mollenhauer, New insights into the origin of unstable sodium graphite intercalation compounds, Phys. Chem. Chem. Phys., 2019, 21, 19378–19390 RSC.
- K. Nobuhara, H. Nakayama, M. Nose, S. Nakanishi and H. Iba, First-principles study of alkali metal-graphite intercalation compounds, J. Power Sources, 2013, 243, 585–587 Search PubMed.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Expanded graphite as superior anode for sodium-ion batteries, Nat. Commun., 2014, 5, 4033 CrossRef CAS.
- K. Yamada, H. Ishikawa, C. Kamiwaki and M. Kawaguchi, The role of boron in B/C/N and B/C materials as an anode of sodium ion batteries, Electrochem, 2015, 83, 452–458 CrossRef CAS.
- Y. Kondo, T. Fukutsuka, K. Miyazaki, Y. Miyahara and T. Abe, Investigation of electrochemical sodium-ion intercalation behavior into graphite-based electrodes, J. Electrochem. Soc., 2019, 166, A5323 CrossRef CAS.
- K. Fredenhagen and G. Cadenbach, Die bindung von kalium durch kohlenstoff, Z. Anorg. Allg. Chem., 1926, 158, 249–263 CrossRef CAS.
- H. Onuma, K. Kubota, S. Muratsubaki, W. Ota, M. Shishkin, H. Sato, K. Yamashita, S. Yasuno and S. Komaba, Phase evolution of electrochemically potassium intercalated graphite, J. Mater. Chem. A, 2021, 9, 11187–11200 Search PubMed.
- Z. Jian, W. Luo and X. Ji, Carbon electrodes for K-ion batteries, J. Am. Chem. Soc., 2015, 137, 11566–11569 CrossRef CAS PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu and V. Barone, Potassium ion batteries with graphitic materials, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- A. Yadav, H. Kobayashi, T. Yamamoto and T. Nohira, Electrochemical Rubidium Storage Behavior of Graphite in Ionic Liquid Electrolyte, Electrochem., 2023, 91, 017002 CrossRef CAS.
- Y. Okamoto, Density functional theory calculations of alkali metal (Li, Na, and K) graphite intercalation compounds, J. Phys. Chem. C, 2014, 118, 16–19 CrossRef CAS.
- V. A. Nalimova, D. Guerard, M. Lelaurain and O. V. Fateev, X-ray investigation of highly saturated Li-graphite intercalation compound, Carbon, 1995, 33, 177–181 CrossRef CAS.
- J. Sangster, C-Na (carbon-sodium) system, J. Phase Equilibria Diffus., 2007, 28, 571–579 CrossRef CAS.
- M. S. Dresselhaus and G. Dresselhaus, Intercalation compounds of graphite, Adv. Phys., 1981, 30, 139–326 CrossRef CAS.
- F. Wu, J. Maier and Y. Yu, Guidelines and trends for next-generation rechargeable lithium and lithium-ion batteries, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Silicon-based anodes for lithium-ion batteries: from fundamentals to practical applications, Small, 2018, 14, 1702737 CrossRef.
- S. Yamazaki, R. Tatara, H. Mizuta, K. Kawano, S. Yasuno and S. Komaba, High-performance SiO electrodes for lithium-ion batteries: merged effects of a new polyacrylate binder and an electrode-maturation process, Mater. Adv., 2023, 4, 1637–1647 RSC.
- Q. Cheng, Y. Okamoto, N. Tamura, M. Tsuji, S. Maruyama and Y. Matsuo, Graphene-like-graphite as fast-chargeable and high-capacity anode materials for lithium ion batteries, Sci. Rep., 2017, 7, 14782 CrossRef.
- X. Yue, J. Zhang, Y. Dong, Y. Chen, Z. Shi, X. Xu, X. Li and Z. Liang, Reversible Li Plating on Graphite Anodes through Electrolyte Engineering for Fast-Charging Batteries, Angew. Chem., Int. Ed., 2023, 62, e202302285 CrossRef CAS.
- N. A. Kaskhedikar and J. Maier, Lithium storage in carbon nanostructures, Adv. Mater., 2009, 21, 2664–2680 CrossRef CAS PubMed.
- Z. T. Gossage, J. Hui, Y. Zeng, H. Flores-Zuleta and J. Rodríguez-López, Probing the reversibility and kinetics of Li+ during SEI formation and (de)intercalation on edge plane graphite using ion-sensitive scanning electrochemical microscopy, Chem. Sci., 2019, 10, 10749–10754 Search PubMed.
- X. Bie, K. Kubota, T. Hosaka, K. Chihara and S. Komaba, A novel K-ion battery: hexacyanoferrate(II)/graphite cell, J. Mater. Chem. A, 2017, 5, 4325–4330 RSC.
- L. Li, L. Liu, Z. Hu, Y. Lu, Q. Liu, S. Jin, Q. Zhang, S. Zhao and S. L. Chou, Understanding high-rate K+-solvent co-intercalation in natural graphite for potassium-ion batteries, Angew. Chem., Int. Ed., 2020, 59, 12917–12924 CrossRef PubMed.
- A. Satoh, N. Takami and T. Ohsaki, Electrochemical intercalation of lithium into graphitized carbons, Solid State Ionics, 1995, 80, 291–298 CrossRef.
- M. Endo, Y. Nishimura, T. Takahashi, K. Takeuchi and M. S. Dresselhaus, Lithium storage behavior for various kinds of carbon anodes in Li ion secondary battery, J. Phys. Chem. Solids, 1996, 57, 725–728 CrossRef.
- F. Cao, I. V. Barsukov, H. J. Bang, P. Zaleski and J. Prakash, Evaluation of Graphite Materials as Anodes for Lithium-Ion Batteries, J. Electrochem. Soc., 2000, 147, 3579 CrossRef.
- D. Igarashi, K. Kubota, T. Hosaka, R. Tatara, T. Inose, Y. Ito, H. Inoue, M. Takeuchi and S. Komaba, Effect of Crystallinity of Synthetic Graphite on Electrochemical Potassium Intercalation into Graphite, Electrochem, 2021, 21–00062 Search PubMed.
- W. Zhou, Y. Mo, P. Gao, K. Wang, J. Ke, Z. Liu, S. Chen and J. Liu, Decoupling Interfacial Kinetics Realizes 5C Fast Charging of Potassium-Ion Batteries Using Graphite Anode, Adv. Funct. Mater., 2024, 34, 2312994 CrossRef.
- Y. Hu, H. Fu, Y. Geng, X. Yang, L. Fan, J. Zhou and B. Lu, Chloro-Functionalized Ether-Based Electrolyte for High-Voltage and Stable Potassium-Ion Batteries, Angew. Chem., Int. Ed., 2024, 63, e202403269 Search PubMed.
- T. Hosaka, T. Matsuyama, R. Tatara, Z. T. Gossage and S. Komaba, Impact of electrolyte decomposition products on the electrochemical performance of 4 V class K-ion batteries, Chem. Sci., 2023, 14, 8860–8868 RSC.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Electrochemical Na insertion and solid electrolyte interphase for hard-carbon electrodes and application to Na-Ion batteries, Adv. Funct. Mater., 2011, 21, 3859–3867 Search PubMed.
- Y. Zeng, Z. T. Gossage, D. Sarbapalli, J. Hui and J. Rodríguez-López, Tracking Passivation and Cation Flux at Incipient Solid-Electrolyte Interphases on Multi-Layer Graphene using High Resolution Scanning Electrochemical Microscopy, Chemelectrochem, 2022, 9, e202101445 CrossRef CAS.
- J. Hui, N. B. Schorr, S. Pakhira, Z. Qu, J. L. Mendoza-Cortes and J. Rodríguez-López, Achieving fast and efficient K+ intercalation on ultrathin graphene electrodes modified by a Li+ based solid-electrolyte interphase, J. Am. Chem. Soc., 2018, 140, 13599–13603 CrossRef.
- E. Peled and S. Menkin, SEI: past, present and future, J. Electrochem. Soc., 2017, 164, A1703 CrossRef.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood Iii, The state of understanding of the lithium-ion-battery graphite solid electrolyte interphase (SEI) and its relationship to formation cycling, Carbon, 2016, 105, 52–76 CrossRef.
- Z. T. Gossage, N. Ito, T. Hosaka, R. Tatara and S. Komaba, In situ Observation of Evolving H2 and Solid Electrolyte Interphase Development at Potassium Insertion Materials within Highly Concentrated Aqueous Electrolytes, Angew. Chem., Int. Ed., 2023, 62, e202307446 CrossRef PubMed.
- M. Fiore, S. Wheeler, K. Hurlbutt, I. Capone, J. Fawdon, R. Ruffo and M. Pasta, Paving the way toward highly efficient, high-energy potassium-ion batteries with ionic liquid electrolytes, Chem. Mater., 2020, 32, 7653–7661 CrossRef.
- D. Sarbapalli, Y.-H. Lin, S. Stafford, J. Son, A. Mishra, J. Hui, A. Nijamudheen, A. Romo, Z. T. Gossage and A. M. van der Zande, A Surface Modification Strategy Towards Reversible Na-ion Intercalation on Graphitic Carbon Using Fluorinated Few-Layer Graphene, J. Electrochem. Soc., 2022, 169(10), 106522 CrossRef.
- M. Kawaguchi, K. Ohnishi, K. Yamada and Y. Muramatsu, Intercalation chemistry and electronic structure of graphite-like layered material BC2N, J. Electrochem. Soc., 2010, 157, P13–P17 CrossRef.
- A. Y. Liu, R. M. Wentzcovitch and M. L. Cohen, Atomic arrangement and electronic structure of BC2N, Phys. Rev. B, 1989, 39, 1760 CrossRef PubMed.
- S. Nagakura, Y. Kouge, M. Yoshida, K. Gotoh, I. Nishimura and M. Kawaguchi, Electrochemical lithium intercalation into and de-intercalation out of B/C materials used as Li-ion battery anodes, Carbon Rep., 2023, 2, 123–129 CrossRef.
- K. Yamada, Electrochemical intercalation of heteroatom-substituted carbon materials and application to secondary batteries, Doctoral dissertation, Osaka Electro-Communication University, 2017, http://id.nii.ac.jp/1148/00000185/ Search PubMed.
- D. Aurbach and Y. Ein-Eli, The study of Li-graphite intercalation processes in several electrolyte systems using in situ X-ray diffraction, J. Electrochem. Soc., 1995, 142, 1746 CrossRef.
- H. Kim, J. Hong, Y. U. Park, J. Kim, I. Hwang and K. Kang, Sodium storage behavior in natural graphite using ether-based electrolyte systems, Adv. Funct. Mater., 2015, 25, 534–541 CrossRef.
- M. Goktas, C. Bolli, E. J. Berg, P. Novak, K. Pollok, F. Langenhorst, M. V. Roeder, O. Lenchuk, D. Mollenhauer and P. Adelhelm, Graphite as cointercalation electrode for sodium-ion batteries: electrode dynamics and the missing solid electrolyte interphase (SEI), Adv. Energy Mater., 2018, 8, 1702724 CrossRef.
- H. Kim, K. Lim, G. Yoon, J. H. Park, K. Ku, H. D. Lim, Y. E. Sung and K. Kang, Exploiting lithium-ether co-intercalation in graphite for high-power lithium-ion batteries, Adv. Energy Mater., 2017, 7, 1700418 CrossRef.
- N. Jiang, L. Chen, H. Jiang, Y. Hu and C. Li, Introducing the Solvent Co-Intercalation Mechanism for Hard Carbon with Ultrafast Sodium Storage, Small, 2022, 18, 2108092 CrossRef.
- Z. T. Gossage, T. Hosaka, R. Tatara and S. Komaba, Branched Diamine Electrolytes for Na+-Solvent Cointercalation into Graphite, ACS Appl. Energy Mater., 2024, 7(3), 845–849 CrossRef.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Sodium intercalation chemistry in graphite, Energy Environ. Sci., 2015, 8, 2963–2969 RSC.
- Z.-L. Xu, G. Yoon, K.-Y. Park, H. Park, O. Tamwattana, S. Joo Kim, W. M. Seong and K. Kang, Tailoring sodium intercalation in graphite for high energy and power sodium ion batteries, Nat. Commun., 2019, 10, 2598 CrossRef.
- I. Escher, Y. Kravets, G. A. Ferrero, M. Goktas and P. Adelhelm, Strategies for alleviating electrode expansion of graphite electrodes in sodium-ion batteries followed by in situ electrochemical dilatometry, Energy Technol., 2021, 9, 2000880 CrossRef CAS PubMed.
- K. Subramanyan and V. Aravindan, Towards Commercialization of Graphite as an Anode for Na-ion Batteries: Evolution, Virtues, and Snags of Solvent Cointercalation, ACS Energy Lett., 2022, 8, 436–446 CrossRef.
- H. Zhang, Z. Li, W. Xu, Y. Chen, X. Ji and M. M. Lerner, Pillared graphite anodes for reversible sodiation, Nanotechnol, 2018, 29, 325402 CrossRef PubMed.
- T. Maluangnont, M. M. Lerner and K. Gotoh, Synthesis of ternary and quaternary graphite intercalation compounds containing alkali metal cations and diamines, Inorg. Chem., 2011, 50, 11676–11682 CrossRef CAS PubMed.
- K. Gotoh, C. Sugimoto, R. Morita, T. Miyatou, M. Mizuno, W. Sirisaksoontorn, M. M. Lerner and H. Ishida, Arrangement and dynamics of diamine, etheric, and tetraalkylammonium intercalates within graphene or graphite oxide galleries by 2H NMR, J. Phys. Chem. C, 2015, 119, 11763–11770 CrossRef CAS.
- G. Yoon, H. Kim, I. Park and K. Kang, Conditions for reversible Na intercalation in graphite: theoretical studies on the interplay among guest ions, solvent, and graphite host, Adv. Energy Mater., 2017, 7, 1601519 CrossRef.
- B. Jache, J. O. Binder, T. Abe and P. Adelhelm, A comparative study on the impact of different glymes and their derivatives as electrolyte solvents for graphite co-intercalation electrodes in lithium-ion and sodium-ion batteries, Phys. Chem. Chem. Phys., 2016, 18, 14299–14316 RSC.
- L. Seidl, N. Bucher, E. Chu, S. Hartung, S. Martens, O. Schneider and U. Stimming, Intercalation of solvated Na-ions into graphite, Energy Environ. Sci., 2017, 10, 1631–1642 RSC.
- G. Åvall, G. A. Ferrero, K. A. Janßen, M. Exner, Y. Son and P. Adelhelm, In Situ Pore Formation in Graphite Through Solvent Co-Intercalation: A New Model for The Formation of Ternary Graphite Intercalation Compounds Bridging Batteries and Supercapacitors, Adv. Energy Mater., 2023, 13, 2301944 CrossRef.
- Y. Son, G. Åvall, G. A. Ferrero, A. I. Freytag, I. Escher, K. A. Janßen, M. Jauregui, D. Saurel, M. Galceran and P. Adelhelm, Diglyme as a promoter for the electrochemical formation of quaternary graphite intercalation compounds containing two different types of solvents, Batteries Supercaps, 2024, e202300506 CrossRef CAS.
- I. Escher, A. I. Freytag, J. M. López del Amo and P. Adelhelm, Solid-State NMR Study on the Structure and Dynamics of Graphite Electrodes in Sodium-Ion Batteries with Solvent Co-Intercalation, Batteries Supercaps, 2023, 6, e202200421 CrossRef CAS.
- H. J. Liang, Z. Y. Gu, X. X. Zhao, J. Z. Guo, J. L. Yang, W. H. Li, B. Li, Z. M. Liu, W. L. Li and X. L. Wu, Ether-Based Electrolyte Chemistry Towards High-Voltage and Long-Life Na-Ion Full Batteries, Angew. Chem., Int. Ed., 2021, 60, 26837–26846 CrossRef CAS PubMed.
- S. Gourang Patnaik, I. Escher, G. A. Ferrero and P. Adelhelm, Electrochemical Study of Prussian White Cathodes with Glymes-Pathway to Graphite-Based Sodium-Ion Battery Full Cells, Batteries Supercaps, 2022, 5, e202200043 CrossRef CAS.
- T. Abe, N. Kawabata, Y. Mizutani, M. Inaba and Z. Ogumi, Correlation between cointercalation of solvents and electrochemical intercalation of lithium into graphite in propylene carbonate solution, J. Electrochem. Soc., 2003, 150, A257 CrossRef CAS.
- Y. Yamada, Y. Takazawa, K. Miyazaki and T. Abe, Electrochemical lithium intercalation into graphite in dimethyl sulfoxide-based electrolytes: effect of solvation structure of lithium ion, J. Phys. Chem. C, 2010, 114, 11680–11685 CrossRef CAS.
- D. Aurbach, M. Koltypin and H. Teller, In situ AFM imaging of surface phenomena on composite graphite electrodes during lithium insertion, Langmuir, 2002, 18, 9000–9009 CrossRef CAS.
- H. Kim, G. Yoon, K. Lim and K. Kang, A comparative study of graphite electrodes using the co-intercalation phenomenon for rechargeable Li, Na and K batteries, Chem. Commun., 2016, 52, 12618–12621 RSC.
- S. C. Jung, Y.-J. Kang and Y.-K. Han, Origin of excellent rate and cycle performance of Na+-solvent cointercalated graphite vs. poor performance of Li+-solvent case, Nano Energy, 2017, 34, 456–462 CrossRef CAS.
- Y. Yamada, M. Yaegashi, T. Abe and A. Yamada, A superconcentrated ether electrolyte for fast-charging Li-ion batteries, Chem. Commun., 2013, 49, 11194–11196 RSC.
- M. Goktas, C. Bolli, J. Buchheim, E. J. Berg, P. Novák, F. Bonilla, T. f. Rojo, S. Komaba, K. Kubota and P. Adelhelm, Stable and unstable diglyme-based electrolytes for batteries with sodium or graphite as electrode, ACS Appl. Mater. Interfaces, 2019, 11, 32844–32855 CrossRef CAS PubMed.
- X. Zhang, R. Kostecki, T. J. Richardson, J. K. Pugh and P. N. Ross, Electrochemical and infrared studies of the reduction of organic carbonates, J. Electrochem. Soc., 2001, 148, A1341 CrossRef CAS.
- K. Westman, R. Dugas, P. Jankowski, W. Wieczorek, G. Gachot, M. Morcrette, E. Irisarri, A. Ponrouch, M. R. Palacín and J. M. Tarascon, Diglyme based electrolytes for sodium-ion batteries, ACS Appl. Energy Mater., 2018, 1, 2671–2680 CrossRef CAS.
- R. Dong, L. Zheng, Y. Bai, Q. Ni, Y. Li, F. Wu, H. Ren and C. Wu, Elucidating the mechanism of fast Na storage kinetics in ether electrolytes for hard carbon anodes, Adv. Mater., 2021, 33, 2008810 CrossRef CAS.
- H. Dai, Z. Zeng, X. Yang, M. Jiang, Y. Wang, Q. Huang, L. Liu, L. Fu, P. Zhang and Y. Wu, Superior potassium storage behavior of hard carbon facilitated by ether-based electrolyte, Carbon, 2021, 179, 60–67 CrossRef CAS.
- J. Wang, H. Wang, R. Zhao, Y. Wei, F. Kang and D. Zhai, Mechanistic insight into ultrafast kinetics of sodium cointercalation in few-layer graphitic carbon, Nano Lett., 2022, 22, 6359–6365 CrossRef CAS PubMed.
- S. Komaba, Sodium-driven rechargeable batteries: an effort towards future energy storage, Chem. Lett., 2020, 49, 1507–1516 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |