DOI:
10.1039/D4SC03207A
(Edge Article)
Chem. Sci., 2024,
15, 11847-11855
A facile strategy for the construction of a phage display cyclic peptide library for the selection of functional macrocycles†
Received
16th May 2024
, Accepted 16th June 2024
First published on 1st July 2024
Abstract
Cyclic peptides represent invaluable scaffolds in biological affinity, providing diverse collections for discovering functional molecules targeting challenging biological entities and protein–protein interactions. The field increasingly focuses on developing cyclization strategies and chemically modified combinatorial libraries in conjunction with M13 phage display, to identify macrocyclic peptide inhibitors for traditionally challenging targets. Here, we introduce a cyclization strategy utilizing ortho-phthalaldehyde (OPA) for the discovery of active macrocycles characterized by asymmetric scaffolds with side-chain cyclization. Through this approach, aldehyde groups attached to free molecules sequentially attack the ε-amine of lysine and the thiol of cysteine, facilitating the rapid cyclization of genetically encoded linear precursor libraries displayed on phage particles. The construction of a 109-member library and subsequent screening successfully identified cyclic peptide binders targeting three therapeutically relevant proteins: PTP1B, NEK7, and hKeap1. The results confirm the efficacy in rapidly obtaining active ligands with micromolar potency. This work provides a fast and efficient operable high-throughput platform for screening functional peptide macrocycles, which hold promise for broad application in therapeutics, chemically biological probes, and disease diagnosis.
Introduction
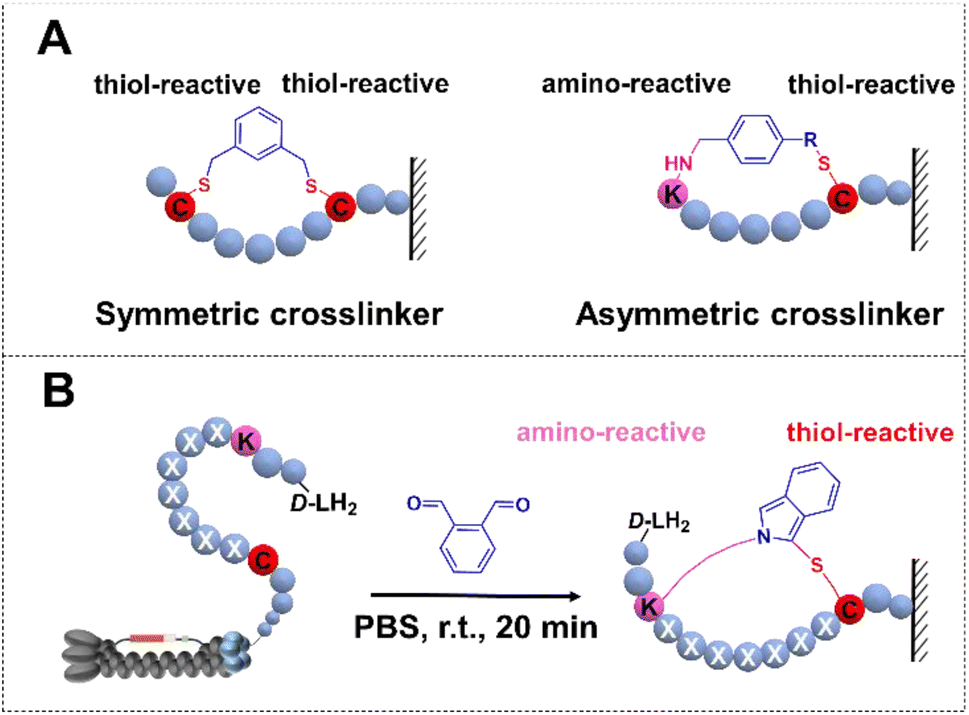
Cyclic peptides have assumed pronounced significance in therapeutic applications and as biological mediators, thereby enabling the generation of diverse libraries for the exploration of functional molecules targeted towards challenging biological entities and protein–protein interactions.1–3 Growing interest is focused on developing cyclization strategies4–6 and constructing chemically modified combinatorial libraries combined with M13 phage display to discover macrocyclic peptide inhibitors of undruggable targets.7–9 In recent years, efforts towards optimizing strategies have led to several cyclization methods involving disulfide bridges,10 non-canonical amino acid encoded linkage,11,12 and chemical cross-linkers.13,14 Notably, one chemical approach is two or three Cys residues with symmetric cross-linkers to form monocycles or bicycle phage display,15,16 in contrast, asymmetric molecular scaffolds were successfully developed for the cyclization including NCys–Cys17 and N-terminal amine–Cys18 (Fig. 1A). Nevertheless, the exploration of cyclization methods remains in its nascent stages.
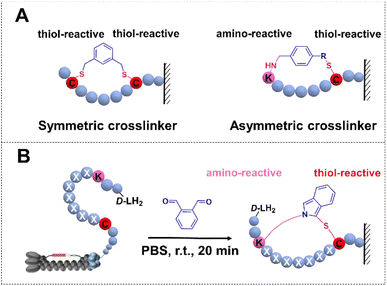 |
| | Fig. 1 Phage libraries with the cyclization strategy. (A) Over-view of cyclic peptide libraries. Cyclization by a symmetric crosslinker. (B) Lys–Cys crosslinking through OPA-cyclization. | |
Ortho-phthalaldehyde (OPA) is a versatile bio-orthogonal proximal cross-linker.19,20 Portoghese demonstrated the selective application of OPA and its derivatives in cross-linking proximal cysteines and lysines, resulting in a fluorescent probe for opioid receptors.21 Subsequently, the OPA–amine–thiol reaction was harnessed for selective bioconjugation, facilitating the identification of kinase–substrate pairs.22 More recently, the proximal cross-linking reaction of OPA has been refined for the chemoselective conjugation of native peptides and proteins, as well as the synthesis of cyclic peptides, all performed under mild, biocompatible aqueous conditions.23–25
Based on these observations, we rationalized the utilization of OPA as a bio-orthogonal cross-linker for late-stage functionalization of phage particles under mild, aqueous, and physiological conditions. Herein, we established a robust high-throughput integration platform by introducing OPA—a symmetric linker facilitating rapid cyclization of linear peptides through a cyclization reaction in physiological settings (PBS 7.4, room temperature, 20 minutes)—onto phage particles. This strategy enabled the construction of a huge cyclic peptide library featuring a rigid asymmetric scaffold (Fig. 1B). Subsequent validation of this methodology was achieved through the identification of macrocyclic peptide ligands targeting Protein Tyrosine Phosphatase 1B (PTP1B), NIMA-related kinase 7 (NEK7), and Homo Kelch-like ECH-associated protein 1 (hKeap1). These findings indicate that the cross-linker encoded phage display has tremendous value in the exploration and evolution of cyclic peptides with the ability to selectively modulate proteins and serve as promising lead compounds in early drug discovery.
Verification of peptide cyclization on phages using the OPA cross-linker
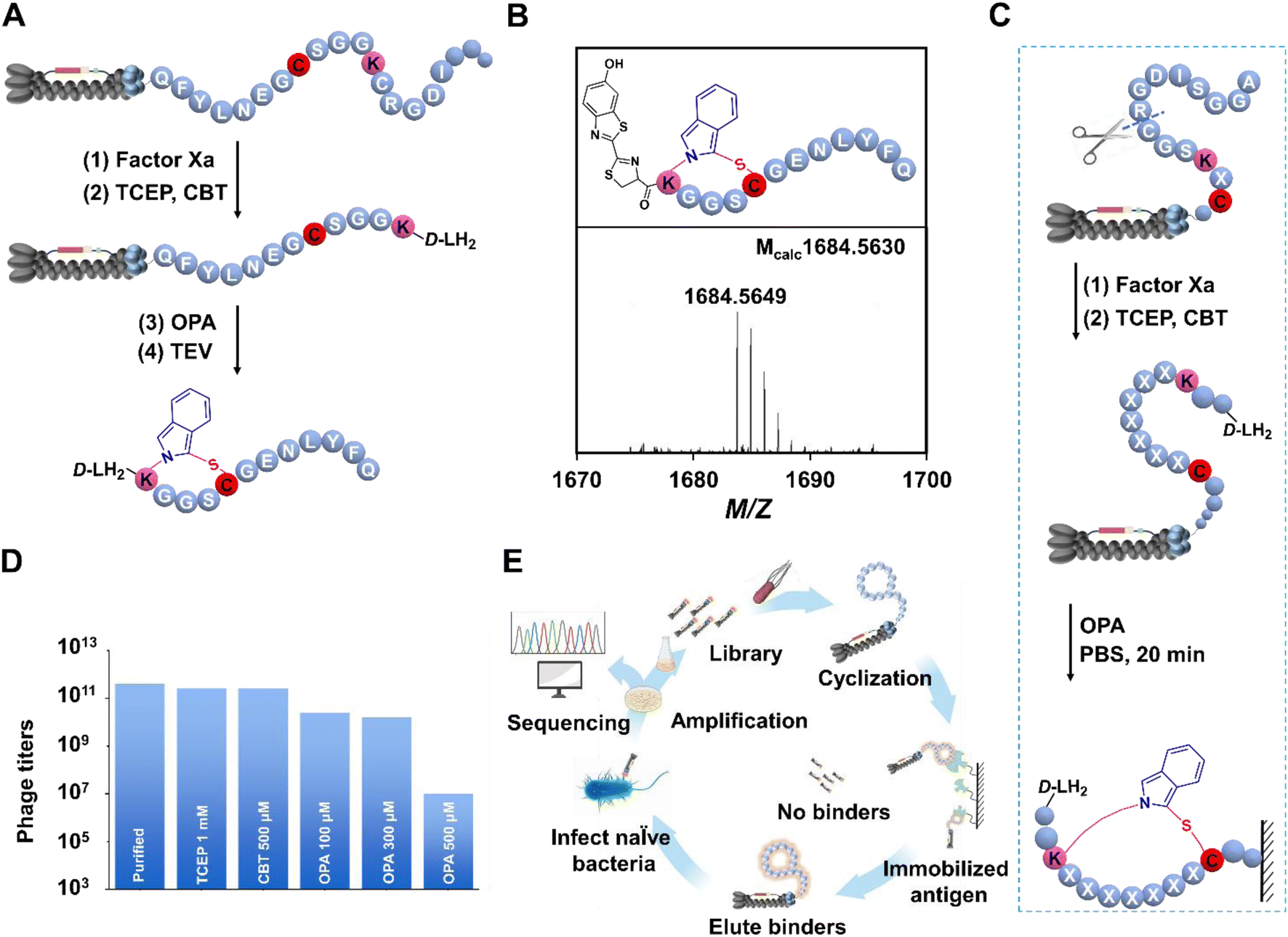
The OPA has been a subject of investigation for almost half a century in the realm of biological detection.26 Recent in vitro cyclization studies have illuminated that OPA enables facilitation of the swift generation of cyclic peptides under mild conditions, primarily through reacting with amino or sulfhydryl groups.23 However, the reliable application of this highly efficient cyclization strategy in phage selection for discovering bioactive cyclic peptides has not been thoroughly reported. To address this gap, we developed a phage display platform compatible with OPA cyclization, constructing a library with randomized sequences flanked by a lysine residue and a cysteine residue on M13 bacteriophages for selecting peptide ligands targeting several proteases (Fig. 2A). The utility of OPA in modifying linear peptides on the phage surface was meticulously examined. A defined peptide sequence (KGGSC) was encoded in the N-terminal of the phage coat protein III (pIII), integrating a factor Xa recognition site to circumvent non-selective recognition of the N-terminal amino group. Following expression and purification of bacteriophages by Escherichia coli TG1, we employed a blocking step with 6-hydroxy-1,3-benzothiazole-2-carbonitrile27 (CBT, 0.5 mM) after enzyme digestion and TCEP reduction. CBT is known for stable reactivity with NCys under physiological conditions. Rapid reaction of CBT with NCys leads to the production of D-luciferin (D-LH2). Here, CBT was applied to block N-terminal cysteine produced by enzyme digestion.
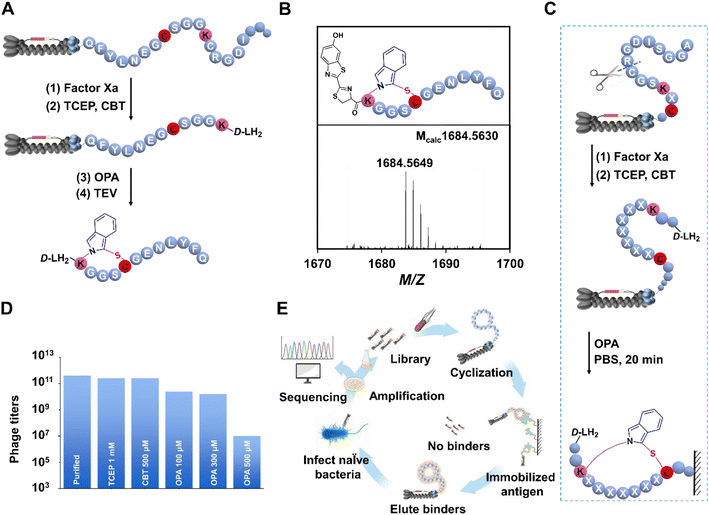 |
| | Fig. 2 Construction of the OPA cyclized phage library. (A) Multi-enzyme digestion strategy for verifying the conversion of linear peptides on the phage surface. (B) Representative raw mass spectra of crude reaction mixtures obtained for peptide KGGSC (M + Na)+ following modification and cyclization. (C) The construction phage library of cyclic peptides by the OPA crosslinker. Factor Xa cleavage digestion, TCEP reduction, CBT blocking, and OPA-cyclization lead to the formation of cyclic peptides. (D) Measured cfus by infecting E. coli TG1 cells with modified phages by the OPA-cyclization strategy. The items measured were, in order, phages with PEG purification, phages with TCEP reduction, phages after digestion and CBT blocking and phages with OPA treatment under different concentrations. Cyclization units (up to 300 μM) result in a comparable effect with PEG purification and TCEP reduction, which is insignificant for the life cycle of phages. (E) Graphical representation of the panning cycle of the OPA cyclized phage library against several proteins. | |
The subsequent removal of CBT, along with buffer exchange, facilitated the introduction of OPA (0.3 mM) into the phage solution at pH 7.4. This resulted in the efficient conversion of linear peptides into cyclic counterparts within 20 min at room temperature. To ensure accurate assessment, a TEV cleavage site was strategically inserted between the peptide sequence and pIII (Fig. 2A). In addition, TEV digestion and subsequent analysis via ESI-MS confirmed the successful and selective cyclization of the target peptide (M + Na)+ on the phage surface (Fig. 2B). In previous reports, the fluorescein, isoindole, could be generated by OPA, as expected, the fluorescence of cyclic reaction production was observed with the naked eye (Fig. S22a†). To further verify the generation of cyclo-peptide on phages, the fluorescence detection experiment was carried out at 405 nm using laser scanning confocal microscopy. The modified phage aggregates could be observed with relatively clear blue fluorescence in high resolution fluorescence images. And the results show that the phages with “KGGSC” modified by OPA activated obvious fluorescence under confocal fluorescence microscopy, while the fluorescence produced by wild phages treated with OPA in the control group was very weak (Fig. S22b and c†), which revealed the contribution of the “KGGSC” sequence to the OPA-cyclization and provided evidence in the other direction for the feasibility of this strategy on phages. An essential aspect of constructing the OPA-cyclized library was the incorporation of a factor Xa cleavage site in proximity to the randomized sequence. This strategy allowed the exposure of free NCys on the displayed peptides post factor Xa cleavage and TCEP reduction. Following the blocking of NCys, OPA-induced peptide cyclization on the phage surface was executed in PBS 7.4 over 20 min at room temperature (Fig. 2C), with the cross-linker dosage determined in subsequent tests.
Phage compatibility of OPA-cyclization
In the exploration of OPA-cyclization compatibility with the reproductive cycle of the phage display platform, a series of staged phage titer tests were conducted. The methodology involved the incorporation of pIII, laden with a random peptide sequence, into the M13 phage genome. This was followed by the cultivation of phage particles within TG1 cells, adhering to standard protocols (see the ESI†). Typically, the observable phage particles expressed per round ranged from 1011 to 1012 (Fig. 2D). The treated phage solution underwent successive infections with E. coli to quantify phage titers in colony forming units (cfus) and assess alterations in phage infectivity before and after OPA-based modification (Fig. 2E). Successive additions of OPA at concentrations of 100 μM, 300 μM, and 500 μM to the phage supernatant triggered reactions in PBS at pH 7.4 over 20 min at room temperature. The TCEP-reduced and CBT-blocked samples maintained their initial infective capacity. Notably, titer results indicated no significant impact on M13 phages in cfus when the OPA concentration was below 300 μM, demonstrating the robustness of the phage display platform under these conditions. It was only upon surpassing the 500 μM threshold that a notable more than 20-fold reduction in infectivity was observed compared to the untreated phage supernatant (Fig. 2D). The collective findings emphasize that an OPA concentration of 300 μM stands as the optimal dose for modifying the peptide library while preserving phage activity.
Discovering cyclic peptide ligands for protein tyrosine phosphatase
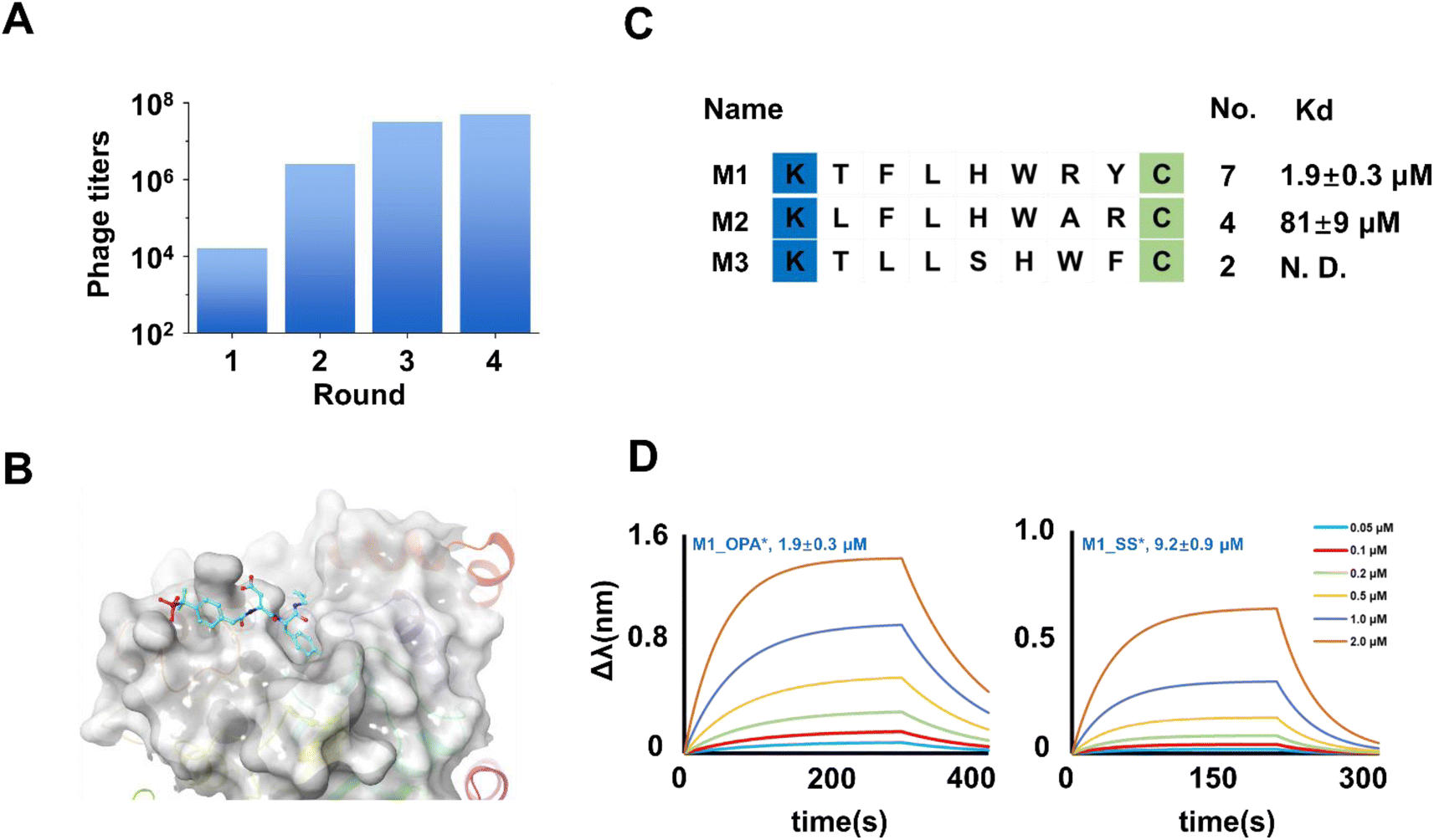
The successful compatibility of the OPA-cyclization strategy with phage life cycles encourages us to explore the phage display against proteins (Fig. 2D). A designed 7-mer phage library, KX7C, was engineered to the pIII protein (Fig. 2C). Subsequent breeding, modification, and panning (Fig. 2E) of the library are carried out according to the fixed procedures (see ESI†). PTP1B was chosen as the prime target due to its pivotal role in various cellular functions,28,29 including the regulation of the endoplasmic reticulum unfolded protein response by dephosphorylating EIF2AK3/PERK,30 thereby inhibiting its kinase activity. It exerts influence over CKII- and p60c-src-induced signal cascades.31 Notably, PTP1B negatively modulates the insulin metabolizing pathway, and its overexpression is implicated in the reduction of phosphorylation of the insulin receptor.32,33 Previous studies have associated mutations in the PTP1B gene with the development of diabetes.34–36 These multifaceted roles position PTP1B inhibition as an attractive strategy for developing therapeutics targeting type 2 diabetes and multiple cancers.37,38 Recombinantly expressed PTP1B, featuring an N-terminal His tag (Fig. S1†), was immobilized on an Elisa plate for panning the cyclized KX7C library. Real-time monitoring of phage titers revealed a significant increase after each round of panning during continuous biological screening and reinfection (Fig. 3A). In order to improve the specificity of the peptide, it is essential to reduce the amount of protein to improve the rigor of the screening. After four rounds of screening, 20 selected samples were sequenced to characterize potential PTP1B binders (Fig. S2†). Sequencing results identified several enriched peptides, including a highly enriched sequence denoted as M1. To assess the affinity of the selected peptides, two representative sequences (M1 and M2) with a single LHW motif (Fig. 3B) were chosen for linear precursor synthesis. Cyclization reactions were then performed, and their ability to bind PTP1B was characterized using Bio-Layer Interferometry (BLI): a detector immobilized with biotinylated PTP1B was sequentially added into the polypeptide solution at increased concentrations eliciting a variation of wavelength shift and kinetic parameters of the interaction. The experiments demonstrated that the tested cyclic M1 (M1_OPA) interacted with PTP1B with a Kd of 1.9 ± 0.3 μM (Fig. 3D). Assessing the role of cyclization for binding, the linear precursor (M1_SS, Kd = 9.2 ± 0.9 μM) showed a four-fold reduction in affinity compared to its cyclic counterpart (Fig. 3D and S4†). Despite sequence similarity, M2_OPA (Kd = 81 ± 9 μM) displayed a significant reduction in PTP1B binding affinity (Fig. S5†), while its linear precursor, M2_SS, was unable to bind to target proteins. Then, an in vitro enzyme inhibition experiment was performed to determine the inhibition activity of M1_OPA to PTP1B. The activity of M1_OPA was detected by measuring the changes in the yield of p-nitrophenyl generated by hydrolyzing pNPP using PTP1B. The results revealed that cyclic peptides could inhibit the dephosphoric function of PTP1B to some extent (IC50 = 10.43 ± 1.06 μM) in vitro, while its linear precursor, M1_SS, lacks this ability with an IC50 of 54.76 ± 4.21 μM (Fig. S6†).
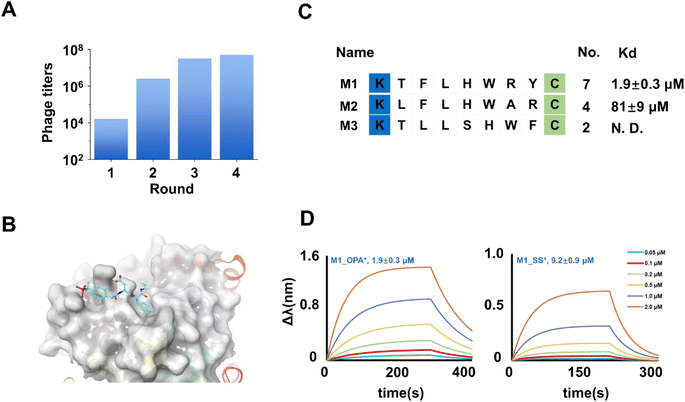 |
| | Fig. 3 Identification of cyclic peptide binders of PTP1B. (A) The phage titers were measured from the phage that remained after each round of screening target PTP1B. The increasing phage titers revealed the continuous enrichment of peptides after each round of panning. (B) Enriched peptide sequences were obtained by the final round of screening against PTP1B. The Kd value of the binder with PTP1B via curve fitting is shown on the right side of the diagram. (C) Crystal structure of the non-peptide inhibitor binding to PTP1B (pdb 1PXH). (D) Bio-layer interferometry assay results (fit) of M1_OPA and M1_SS binding PTP1B. | |
The exploration of interaction pattern of cyclic peptide with PTP1B
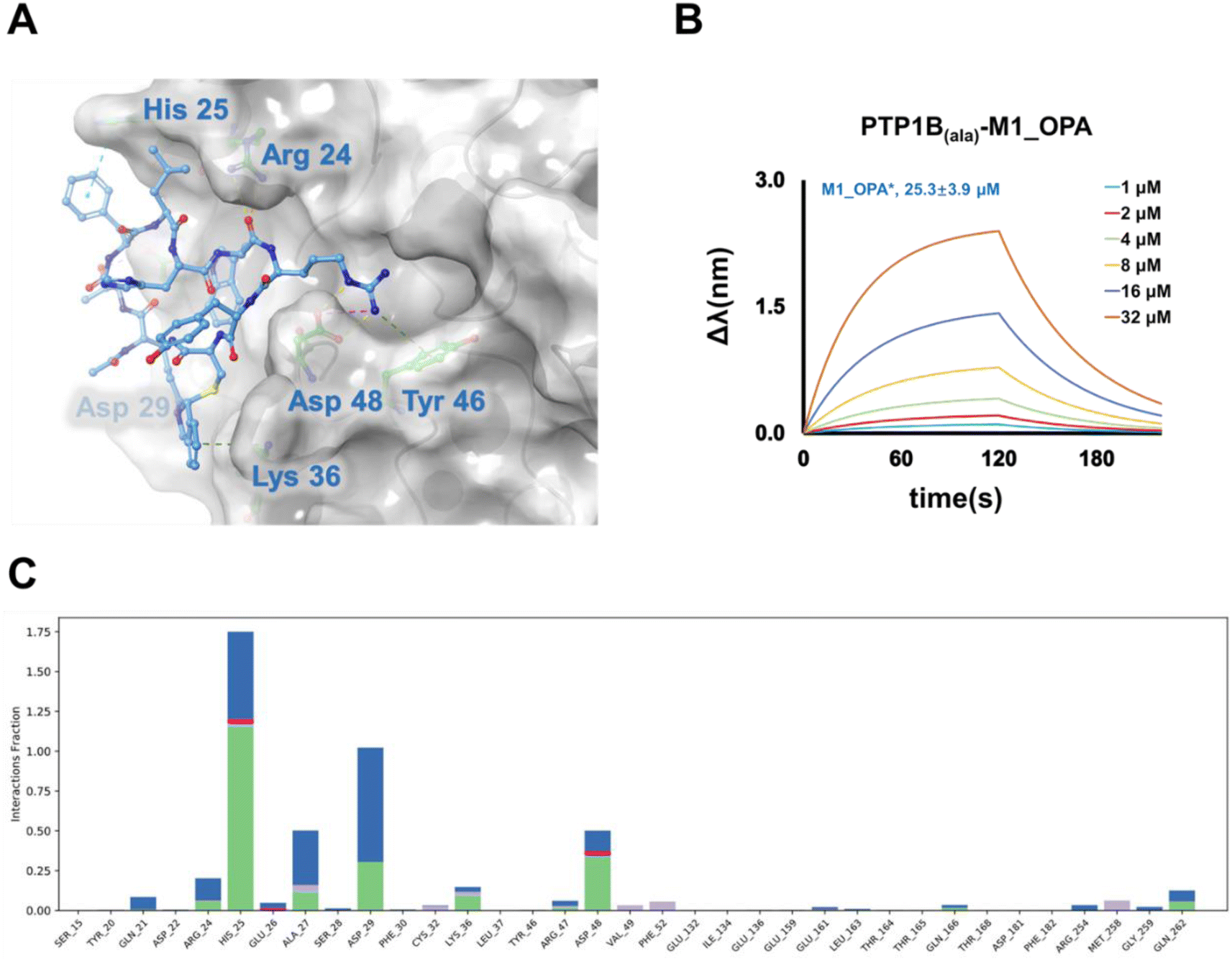
PTP1B, a critical regulatory unit in multiple signaling pathways, exerts both positive and negative regulatory effects on related proteases through its various catalytic domains. Four active sites, namely the catalytic (A) site, and B, C, and D sites, have been identified as promising targets for the development of highly selective and effective PTP1B inhibitors.39 The A site represents the primary function of binding the dianionic pTyr residue, while the B site is a noncatalytic-binding pocket, and the C site features a wide, flat area that is fully exposed to the surface of the protein and is suitable for binding inhibitors with large molecular size.40 To elucidate the active pocket holding the selected cyclic binder, M1_OPA, we conducted molecular docking studies and molecular dynamics simulation with PTP1B using the known crystal structure (Fig. 3C), focusing on the A, B, C, and D sites. Anticipatedly, the high-scoring patterns predominantly featured ligands occupying the B site and extending into the A site. Published complex structures of the PTP1B-substrate obtained by X-ray diffraction revealed that ABC-type small molecule inhibitors could establish salt bridges and hydrogen bonds with specific residues (Arg 24, Asp 29, and Asp 48) at the B site and the C site, simultaneously binding to the groove of the A site. Similarly, the chimerism of M1_OPA to these two grooves prevented the tyrosine-phosphorylated substrate (insulin receptor kinase, IRK) from contacting PTP1B (Fig. 4A). The cyclic peptide extended toward the edge of the active pocket, with the indole benzene ring forming hydrogen bonds and conjugation interactions with residues Asp 29, Arg 24, His 25, and Asp 48. In particular, the cyclization group, isoindole, contributed to affinity by providing conjugated interactions (Fig. 4A). MD simulation study also showed a similar binding mechanism of the ligand–receptor complex with docking (Fig. 4C and S8†). To further confirm this hypothesis, three key amino acids (His25, Asp 29 and Asp 48) of PTP1B that appeared in the simulation result were mutated to H25A, D29A and D48A using site-directed mutagenesis. Subsequent affinity characterization showed that the binding ability of the mutant protein to the M1_OPA (25.3 ± 3.9 μM) was significantly reduced (Fig. 4B). This preliminary analysis sheds light on the inhibitory mechanism of the cyclic peptide binding to PTP1B. Considering the unique mechanisms by which PTP1B induces IRK dephosphorylation via the B site and C site, targeting this catalytic reaction becomes crucial for discovering highly specific cyclic peptide drugs for the treatment of type 2 diabetes. The insights gained from this study lay the groundwork for the development of tight-binding inhibitors in the subsequent exploration.
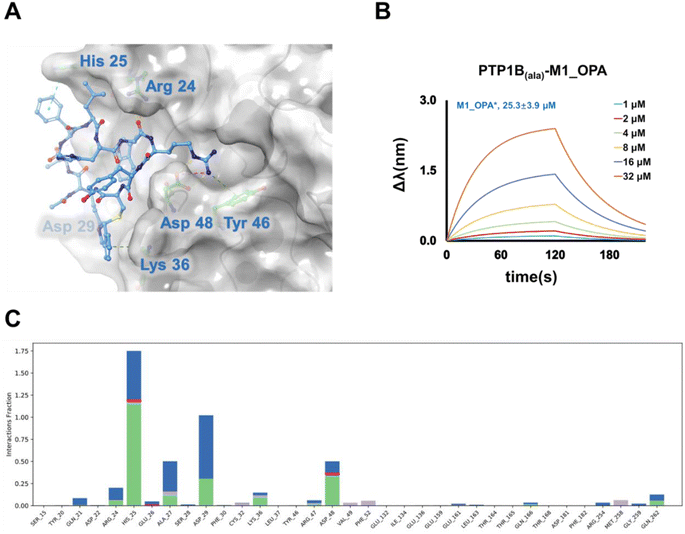 |
| | Fig. 4 Confirmation of interaction pattern of M1_OPA binding to the PTP1B. (A) Binding interface in dimensional space. M1_OPA is adapted to the B and C sites of PTP1B. (B) Bio-layer interferometry assay results (fit) of M1_OPA binding to PTP1Bala mutant (H25A, D29A and D48A). (C) PTP1B-M1_OPA contact histogram (salt bridges, water bridges, hydrophobic interaction, and hydrogen bonds) from 120 ns MD trajectories. The ordinate represents the percentage of time that the protein interacts with the ligand during the simulation time. | |
Selection of cyclic peptides binding to NEK7
Our attention turned to NEK7 as we sought to uncover the potential of the cyclic peptide library. NEK7 plays a pivotal role in regulating the NLRP3 inflammasome within macrophages,41,42 and its dysregulation has been implicated in inflammatory responses through the activation of inflammasomes in various pathways, including NF-κB signaling, ROS signaling, and lysosomal destabilization.43–45 Perturbation of these signaling pathways accelerates cancers.46 Recognizing NEK7 as a key factor, efforts are underway to develop potent anti-inflammatory and anticancer drugs, with a current emphasis on Computer-Aided Drug Design (CADD) for developing non-peptide inhibitors (Fig. 5A).47 In our approach, purified NEK7 (Fig. S9†) was immobilized on microplates, and the cyclized KX7C library underwent screening over four rounds. The identification of 20 randomized clones (Fig. S10†) revealed multiple repeated sequences, indicating their enrichment after panning (Fig. 5B and S11†).
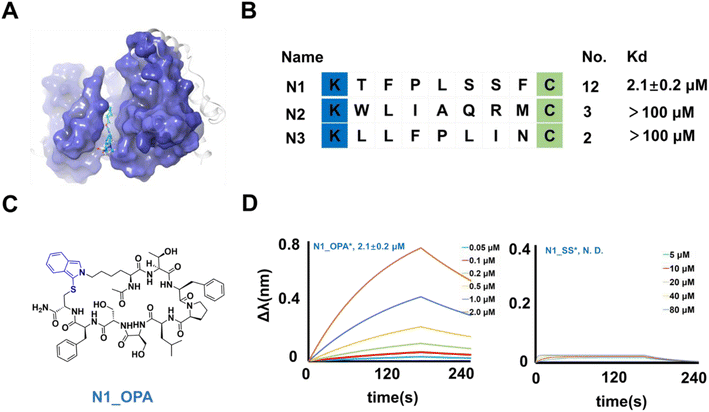 |
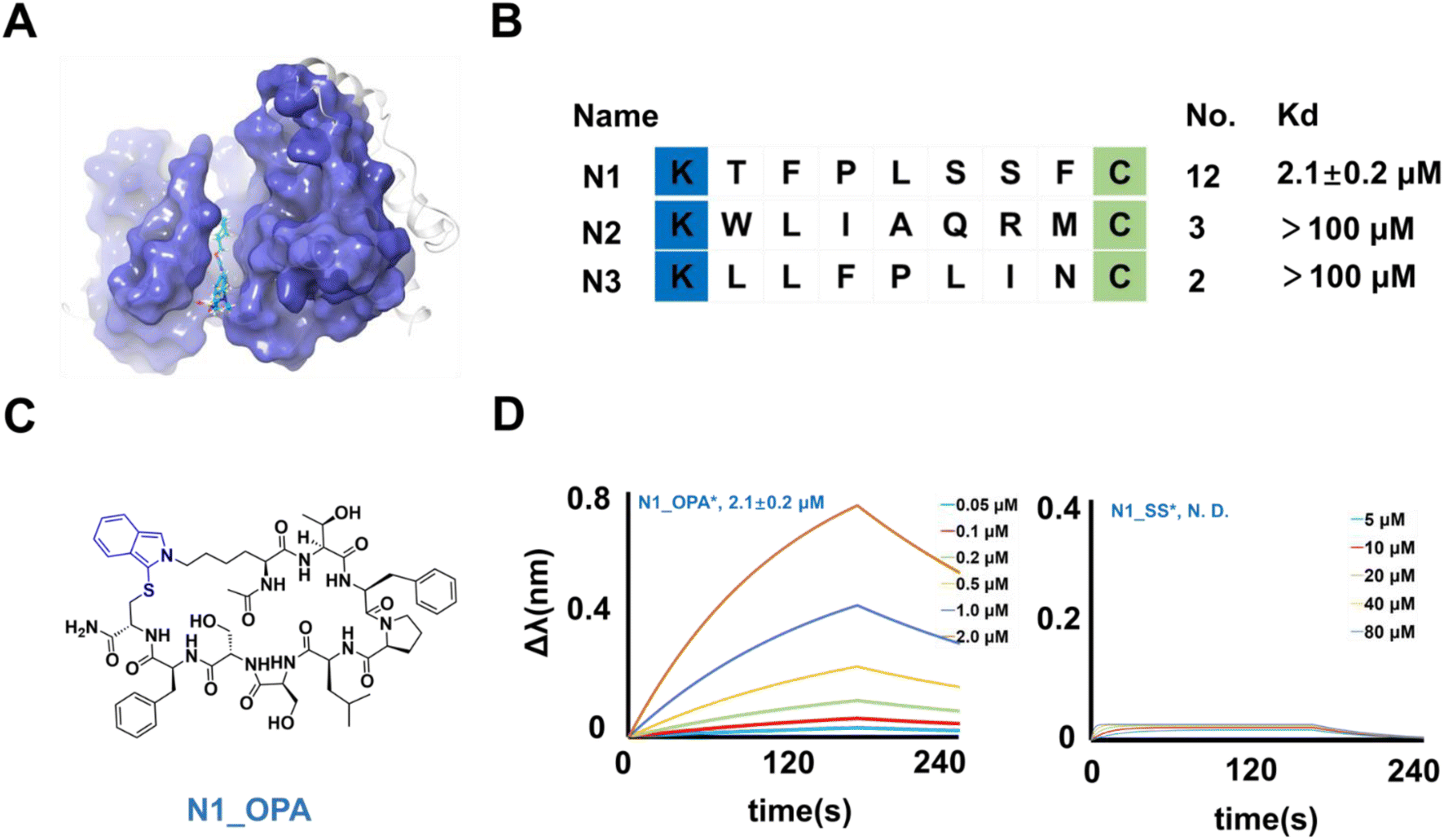
| | Fig. 5 Binders of NEK7 from selection. (A) Crystal structure of the NEK7 binding non-peptide inhibitor (pdb 6S75). (B) Enriched peptide sequences from the cyclic peptide library. The Kd value of the binder with NEK7 via curve fitting is listed at the right of the diagram. (C) Chemical structure of N1_OPA. (D) The fitted curves from bio-layer interferometry assay results of N1_OPA and N1_SS binding NEK7. | |
To characterize their activity, the linear structures of these sequences were synthesized, and the corresponding cyclic peptides were prepared. The peptides were then assessed for NEK7 binding using BLI (Fig. S11†). The results demonstrated that N1_OPA (Fig. 5C) exhibited binding to NEK7 with an affinity of 2.1 ± 0.2 μM (Fig. 5D). Subsequent glide docking also revealed the potential pockets of N1_OPA binding to NEK7, which was formed by the Pro 92, Lys 96, ASP 175, Thr 172 and Lys 176 (Fig. S12†). Surprisingly, the lack of interaction observed with N1_SS underscored the necessity of OPA-cyclization for effective binding (Fig. 5D). The remaining peptide sequences, N2 (Fig. S13†) and N3 (Fig. S14†) were considered false positives due to their low binding capacity.
Screening for cyclic peptides of the hKeap1
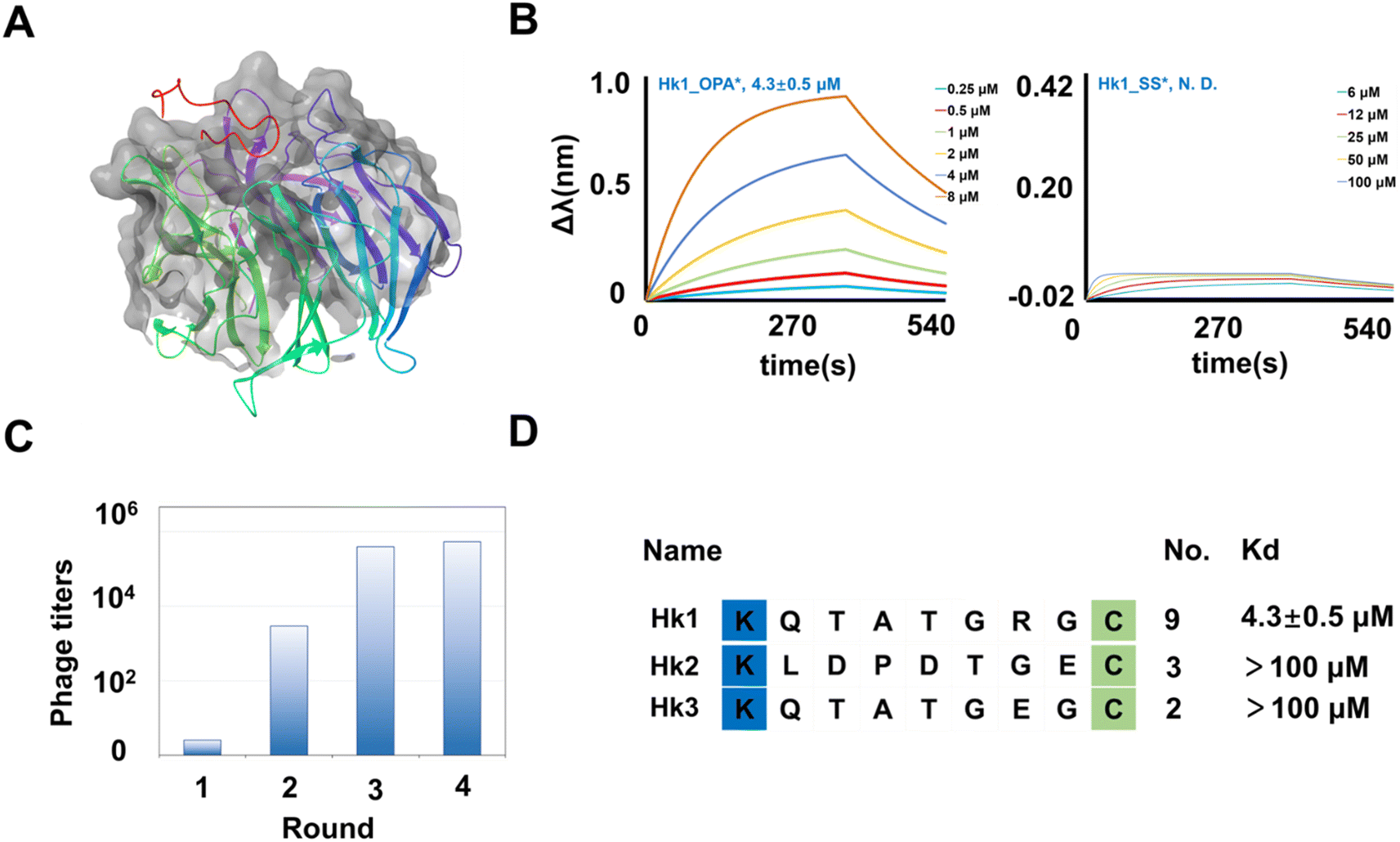
In our pursuit of uncovering the potential of the cyclic library in targeting proteases involved in protein–protein interactions, we conducted phage panning against hKeap1. As a transcriptional regulator, hKeap1 influences the cellular oxidative stress response by upregulating gene expression.48 The binding of hKeap1 to Nrf2 (Fig. 6A) plays a pivotal role in regulating the ubiquitination of this substrate, initiating a cascade of signal responses.49 Interfering with this interaction has been demonstrated to modulate responses to oxidative stress signals,50 offering a promising avenue for discovering inhibitors to treat diseases associated with inflammatory responses and neurodegeneration.51,52 hKeap1, featuring an N-terminal His tag, was expressed (Fig. S15†) and immobilized on a plate for screening the KX7C cyclic peptide library (Fig. 2C). Following four cycles of panning (Fig. 6C), 20 randomly selected clones from the output population underwent sequence identification (Fig. S13†). Similar to the results observed with PTP1B, the panning process enriched several sequences (Fig. 6D), which were then synthesized through solid-phase peptide synthesis. The purified linear precursors were processed using OPA to generate the desired cyclic peptides. A BLI assay was employed to verify the binding affinity of peptides hitting hKeap1 (Fig. S16 and S17†). Among the synthesized peptides, Hk1_OPA demonstrated a low micromolar binding capacity to Keap1 (Kd = 4.3 ± 0.5 μM) (Fig. 6B). Notably, the precursor peptide Hk1_SS showed no interaction with the target protein (Fig. 6B), indicating the success of panning towards the OPA-modified cyclic peptide library in generating the expected cyclic ligands. Because of false positive hits, other tests on the remaining cyclic peptides, Hk2 (Fig. S18†) and Hk3 (Fig. S19†), did not exhibit significant affinity binding to Keap1 (up to 100 μM). Subsequently, the binding basis of Hk1_OPA to Keap1 was further explored through molecular docking studies, revealing that the cyclic peptide ligand adapts to the Nrf2 binding pocket of hKeap1 (Fig. S20†). In previous reports, a variety of potent cyclic peptide ligands targeting hKeap1 had been found by phage display. Most of these peptide molecules contain conserved sequences “ETGE” derived from Nrf2. However, the only active peptide Hk1_OPA, KQTATGRGC is significantly different from the reported active sequence and contains only “TG”. The reasons for weak binding affinities probably resulted from the defects in the method of library construction, which leads to the loss of a large number of affinity sequences during the amplification of random sequences.
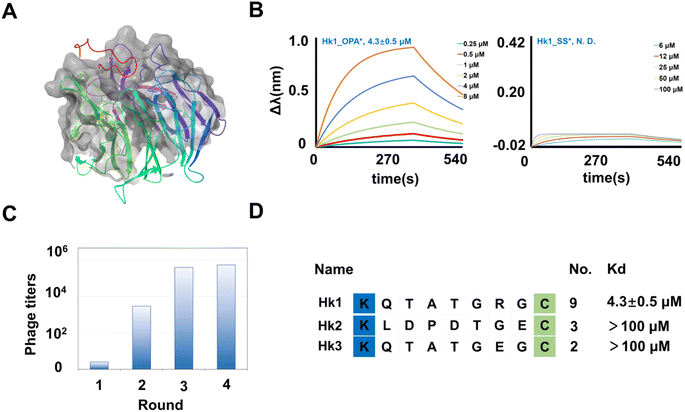 |
| | Fig. 6 Identification of cyclic binders of hKeap1. (A) Crystal structure of hKeap1 interacting with Nrf2 (pdb 2FLU). (B) The fitted curves from BLI assay results of Hk1_OPA and Hk1_SS binding hKeap1. The fitted curves from bio-layer interferometry assay results of Hk1_OPA and Hk1_SS binding hKeap1. (C) The phage titers after each round of screening. The increasing phage titers revealed the continuous enrichment of peptides after each round of panning. (D) Enriched peptide sequences from the cyclic peptide library. The Kd value of the binder with hKeap1 was extracted via fitting. | |
Conclusions
In summary, we have developed a display platform by integrating ortho-phthalaldehyde (OPA) onto phage particles using a three-step strategy, resulting in the generation of genetically encoded cyclic peptides featuring the isoindole skeleton. The OPA-cyclization has been proven to be highly chemoselective under physiological conditions. While OPA cyclization applied to phage-display remains challenging, we successfully loaded OPA into the N-terminal of pIII to gain the KX7C cyclic peptide library without compromising the infectivity of phage particles. The efficacy of our display platform was substantiated by its application in developing ligands for protein targets associated with various diseases. The discovery of a peptide ligand binding to PTP1B with single-digit micromolar dissociation constants (Kd) and inhibiting the dephosphorylation activity of PTP1B with 10.43 ± 1.06 μM showcases the potential of our platform. Molecular docking and mutagenesis studies further revealed that the cyclic peptide engages with the unique B and C sites of PTP1B, providing a distinct approach compared to small molecules. Expanding our exploration, we identified ligands for hKeap1 and NEK7, thereby affirming the versatility and adaptability of the OPA-cyclization strategy for diverse phage display applications. While the short random sequences investigated in this study limited the affinity of the discovered ligands to the single-digit micromolar range, we anticipate that expanding the size of the randomized peptide segment holds promise for achieving higher affinity. In addition, there are some limitations for the OPA-cyclization strategy derived from the necessary proteolytic digestion and CBT blocking steps, due to the inherent regioselectivity of OPA for recognizing other N-terminal amino, Lys, and Cys in pIII proteins or randomized sequences, resulting in operational complexity of this method. Here, our contribution lies in seamlessly integrating the advantages of OPA-cyclization, such as the accessibility of linkers without complex chemical processes and simplicity of crosslinking reaction, into the generation of side-chain cyclized libraries for phage display, thus extending the applications of phage display. We believe that the click cyclization-based high-throughput integration platform holds promise for facilitating the discovery of potent macrocyclic peptides targeting a broader range of challenging biological entities in future research.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
X. Lei designed the study and supervised the project. H. Xiang and L. W. Bai performed the experiments. T. Dan, P. Cheng, X. Q. Yang, H. L. Ai and K. Li collected and analyzed the data. All the authors discussed the results and co-wrote the manuscript.
Conflicts of interest
X. Lei, H. Xiang and L. W. Bai are inventors of the Chinese provisional patent that incorporates the discoveries described in this manuscript.
Acknowledgements
This work was supported by the Science and Technology Major Program of Gansu Province of China (22ZD6FA006 and 23ZDFA015). The MD simulations are supported by the Supercomputing Center of Lanzhou University. We also thank the Analytical Measuring Center, School of Pharmaceutical Sciences, SCMU for their help with ESI-MS and BLI measurements.
Notes and references
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- A. K. Malde, T. A. Hill, A. Iyer and D. P. Fairlie, Chem. Rev., 2019, 119, 9861–9914 CrossRef CAS PubMed.
- C. Sohrabi, A. Foster and A. Tavassoli, Nat. Rev. Chem, 2020, 4, 90–101 CrossRef CAS PubMed.
- T. Passioura, T. Katoh, Y. Goto and H. Suga, Annu. Rev. Biochem., 2014, 83, 727–752 CrossRef CAS PubMed.
- F. J. Chen, N. Pinnette, F. Yang and J. Gao, Angew. Chem., Int. Ed., 2023, 62, e202306813 CrossRef PubMed.
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y. H. Tsai and C. Wu, J. Am. Chem. Soc., 2020, 142, 5097–5103 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- M. Zheng, F. J. Chen, K. Li, R. M. Reja, F. Haeffner and J. Gao, J. Am. Chem. Soc., 2022, 144, 15885–15893 CrossRef CAS PubMed.
- J. Y. Wong, R. Mukherjee, J. Miao, O. Bilyk, V. Triana, M. Miskolzie, A. Henninot, J. J. Dwyer, S. Kharchenko, A. Iampolska, D. M. Volochnyuk, Y. S. Lin, L. M. Postovit and R. Derda, Chem. Sci., 2021, 12, 9694–9703 RSC.
- S. Chen, R. Gopalakrishnan, T. Schaer, F. Marger, R. Hovius, D. Bertrand, F. Pojer and C. Heinis, Nat. Chem., 2014, 6, 1009–1016 CrossRef CAS PubMed.
- X. S. Wang, P. C. Chen, J. T. Hampton, J. M. Tharp, C. A. Reed, S. K. Das, D. S. Wang, H. S. Hayatshahi, Y. Shen, J. Liu and W. R. Liu, Angew. Chem., Int. Ed., 2019, 58, 15904–15909 CrossRef CAS PubMed.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed.
- S. S. Kale, M. Bergeron-Brlek, Y. Wu, M. G. Kumar, M. V. Pham, J. Bortoli, J. Vesin, X.-D. Kong, J. F. Machado, K. Deyle, P. Gonschorek, G. Turcatti, L. Cendron, A. Angelini and C. Heinis, Sci. Adv., 2019, 5, eaaw2851 CrossRef CAS PubMed.
- S. Chen, S. Lovell, S. Lee, M. Fellner, P. D. Mace and M. Bogyo, Nat. Biotechnol., 2021, 39, 490–498 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- X. D. Kong, J. Moriya, V. Carle, F. Pojer, L. A. Abriata, K. Deyle and C. Heinis, Nat. Biomed. Eng., 2020, 4, 560–571 CrossRef CAS PubMed.
- M. Zheng, F. Haeffner and J. Gao, Chem. Sci., 2022, 13, 8349–8354 RSC.
- T. R. Oppewal, I. D. Jansen, J. Hekelaar and C. Mayer, J. Am. Chem. Soc., 2022, 144, 3644–3652 CrossRef CAS PubMed.
- K. S. Lee and D. G. Drescher, Int. J. Biochem., 1978, 9, 457–467 CrossRef CAS PubMed.
- M. Todorovic, K. D. Schwab, J. Zeisler, C. Zhang, F. Bénard and D. M. Perrin, Angew. Chem., Int. Ed., 2019, 58, 14120–14124 CrossRef CAS PubMed.
- B. Le Bourdonnec, R. El Kouhen, M. M. Lunzer, P. Y. Law, H. H. Loh and P. S. Portoghese, J. Med. Chem., 2000, 43, 2489–2492 CrossRef CAS PubMed.
- D. J. Maly, J. A. Allen and K. M. Shokat, J. Am. Chem. Soc., 2004, 126, 9160–9161 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhang, C. T. T. Wong and X. Li, J. Am. Chem. Soc., 2019, 141, 12274–12279 CrossRef CAS PubMed.
- M. Todorovic and D. M. Perrin, Methods Enzymol., 2020, 639, 313–332 CAS.
- T. Wei, D. Li, Y. Zhang, Y. Tang, H. Zhou, H. Liu and X. Li, Small Methods, 2022, 6, e2201164 CrossRef PubMed.
- J. R. Benson and P. E. Hare, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 619–622 CrossRef CAS PubMed.
- J. Liu, D. Machalz, G. Wolber, E. J. Sorensen and M. Bureik, Appl. Biochem. Biotechnol., 2021, 193, 218–237 CrossRef CAS PubMed.
- Z. Y. Zhang, Y. Wang and J. E. Dixon, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 1624–1647 CrossRef CAS PubMed.
- A. Alonso, J. Sasin, N. Bottini, I. Friedberg, I. Friedberg, A. Osterman, A. Godzik, T. Hunter, J. Dixon and T. Mustelin, Cell, 2004, 117, 699–711 CrossRef CAS PubMed.
- A. Ostman, C. Hellberg and F. D. Böhmer, Nat. Rev. Cancer, 2006, 6, 307–320 CrossRef PubMed.
- P. Kefalas, T. R. Brown and P. M. Brickell, Int. J. Biochem. Cell Biol., 1995, 27, 551–663 CrossRef CAS PubMed.
- A. Salmeen, J. N. Andersen, M. P. Myers, N. K. Tonks and D. Barford, Mol. Cell, 2000, 6, 1401–1412 CrossRef CAS PubMed.
- R. A. Haeusler, T. E. McGraw and D. Accili, Nat. Rev. Mol. Cell Biol., 2018, 19, 31–44 CrossRef CAS PubMed.
- M. Elchebly, P. Payette, E. Michaliszyn, W. Cromlish, S. Collins, A. L. Loy, D. Normandin, A. Cheng, J. Himms-Hagen, C. C. Chan, C. Ramachandran, M. J. Gresser, M. L. Tremblay and B. P. Kennedy, Science, 1999, 283, 1544–1548 CrossRef CAS PubMed.
- T. O. Johnson, J. Ermolieff and M. R. Jirousek, Nat. Rev. Drug Discovery, 2002, 1, 696–709 CrossRef CAS PubMed.
- N. Krishnan, C. A. Bonham, I. A. Rus, O. K. Shrestha, C. M. Gauss, A. Haque, A. Tocilj, L. Joshua-Tor and N. K. Tonks, Nat. Commun., 2018, 9, 283 CrossRef PubMed.
- Y. N. Chen, M. J. LaMarche, H. M. Chan, P. Fekkes, J. Garcia-Fortanet, M. G. Acker, B. Antonakos, C. H. Chen, Z. Chen, V. G. Cooke, J. R. Dobson, Z. Deng, F. Fei, B. Firestone, M. Fodor, C. Fridrich, H. Gao, D. Grunenfelder, H. X. Hao, J. Jacob, S. Ho, K. Hsiao, Z. B. Kang, R. Karki, M. Kato, J. Larrow, L. R. La Bonte, F. Lenoir, G. Liu, S. Liu, D. Majumdar, M. J. Meyer, M. Palermo, L. Perez, M. Pu, E. Price, C. Quinn, S. Shakya, M. D. Shultz, J. Slisz, K. Venkatesan, P. Wang, M. Warmuth, S. Williams, G. Yang, J. Yuan, J. H. Zhang, P. Zhu, T. Ramsey, N. J. Keen, W. R. Sellers, T. Stams and P. D. Fortin, Nature, 2016, 535, 148–152 CrossRef CAS PubMed.
- N. Maheshwari, C. Karthikeyan, P. Trivedi and N. Moorthy, Curr. Drug Targets, 2018, 19, 551–575 CrossRef CAS PubMed.
- B. Sharma, L. Xie, F. Yang, W. Wang, Q. Zhou, M. Xiang, S. Zhou, W. Lv, Y. Jia, L. Pokhrel, J. Shen, Q. Xiao, L. Gao and W. Deng, Eur. J. Med. Chem., 2020, 199, 112376 CrossRef CAS PubMed.
- R. Liu, C. Mathieu, J. Berthelet, W. Zhang, J. M. Dupret and F. Rodrigues Lima, Int. J. Mol. Sci., 2022, 23, 7027–7047 CrossRef CAS PubMed.
- Y. He, M. Y. Zeng, D. Yang, B. Motro and G. Nunez, Nature, 2016, 530, 354–357 CrossRef CAS PubMed.
- R. Tan, S. Nakajima, Q. Wang, H. Sun, J. Xue, J. Wu, S. Hellwig, X. Zeng, N. A. Yates, T. E. Smithgall, M. Lei, Y. Jiang, A. S. Levine, B. Su and L. Lan, Mol. Cell, 2017, 65, 818–831 CrossRef CAS PubMed.
- F. Van Hauwermeiren and M. Lamkanfi, Nat. Immunol., 2016, 17, 223–224 CrossRef CAS PubMed.
- H. Sharif, L. Wang, W. L. Wang, V. G. Magupalli, L. Andreeva, Q. Qiao, A. V. Hauenstein, Z. Wu, G. Nunez, Y. Mao and H. Wu, Nature, 2019, 570, 338–343 CrossRef CAS PubMed.
- J. Fu and H. Wu, Annu. Rev. Immunol., 2023, 41, 301–316 CrossRef CAS PubMed.
- X. Chen, G. Liu, Y. Yuan, G. Wu, S. Wang and L. Yuan, Cell Death Dis., 2019, 10, 906–918 CrossRef CAS PubMed.
- S. A. Ejaz, M. Aziz, Z. Zafar, N. Akhtar and H. A. Ogaly, Sci. Rep., 2023, 13, 4304 CrossRef CAS PubMed.
- K. Itoh, N. Wakabayashi, Y. Katoh, T. Ishii, K. Igarashi, J. D. Engel and M. Yamamoto, Genes Dev., 1999, 13, 76–86 CrossRef CAS PubMed.
- S. C. Lo, X. Li, M. T. Henzl, L. J. Beamer and M. Hannink, EMBO J., 2006, 25, 3605–3617 CrossRef CAS PubMed.
- M. Sandberg, J. Patil, B. D'Angelo, S. G. Weber and C. Mallard, Neuropharmacology, 2014, 79, 298–306 CrossRef CAS PubMed.
- A. Bresciani, A. Missineo, M. Gallo, M. Cerretani, P. Fezzardi, L. Tomei, D. O. Cicero, S. Altamura, A. Santoprete, R. Ingenito, E. Bianchi, R. Pacifici, C. Dominguez, I. Munoz-Sanjuan, S. Harper, L. Toledo-Sherman and L. C. Park, Arch. Biochem. Biophys., 2017, 631, 31–41 CrossRef CAS PubMed.
- J. Iegre, S. Krajcovicova, A. Gunnarsson, L. Wissler, H. Kack, A. Luchniak, S. Tangefjord, F. Narjes and D. R. Spring, Chem. Sci., 2023, 14, 10800–10805 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab