
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnleashing the potential of Li–O2 batteries with electronic modulation and lattice strain in pre-lithiated electrocatalysts†
Zhengcai
Zhang‡
a,
Dulin
Huang‡
a,
Shuochao
Xing
a,
Minghui
Li
a,
Jing
Wu
a,
Zhang
Zhang
a,
Yaying
Dou
*ab and
Zhen
Zhou
 *a
*a
aInterdisciplinary Research Center for Sustainable Energy Science and Engineering (IRC4SE2), School of Chemical Engineering, Zhengzhou University, Zhengzhou 450001, China. E-mail: yydou@zzu.edu.cn; zhenzhou@zzu.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China
First published on 22nd July 2024
Abstract
Efficient catalysts are indispensable for overcoming the sluggish reaction kinetics and high overpotentials inherent in Li–O2 batteries. However, the lack of precise control over catalyst structures at the atomic level and limited understanding of the underlying catalytic mechanisms pose significant challenges to advancing catalyst technology. In this study, we propose the concept of precisely controlled pre-lithiated electrocatalysts, drawing inspiration from lithium electrochemistry. Our results demonstrate that Li+ intercalation induces lattice strain in RuO2 and modulates its electronic structure. These modifications promote electron transfer between catalysts and reaction intermediates, optimizing the adsorption behavior of Li–O intermediates. As a result, Li–O2 batteries employing Li0.52RuO2 exhibit ultrahigh energy efficiency, long lifespan, high discharge capacity, and excellent rate performance. This research offers valuable insights for the design and optimization of efficient electrocatalysts at the atomic level, paving the way for further advancements in Li–O2 battery technology.
Introduction
Lithium–oxygen (Li–O2) batteries have garnered significant attention as a promising “beyond lithium-ion battery” technology for next-generation energy storage systems. By capitalizing on the lightweight properties of lithium metal and the abundant availability of atmospheric oxygen, Li–O2 batteries offer an exceptional theoretical energy density of up to 5220 W h kg−1.1–3 This, in conjunction with their cost-effectiveness and low pollution characteristics, positions Li–O2 batteries as an enticing solution in the realm of electrochemical energy storage. Nevertheless, the practical implementation encounters many hurdles primarily arising from the insulating nature of Li2O2, resulting in diminished energy efficiency, rapid capacity decay, and sluggish reaction kinetics.4–6Addressing these challenges, extensive research efforts have been directed towards both solid and liquid catalysts. However, the utilization of liquid catalysts often introduces the issue of the “shuttle effect”, resulting in the corrosion of lithium metal and subsequent reduction in battery durability.7 Alternatively, various solid catalysts, including metal oxides,8,9 alloys,5 and high-entropy catalysts,10 have been extensively investigated in Li–O2 batteries. Previous studies emphasized the importance of modulating the adsorption strength between LiO2 and catalyst surfaces to facilitate the formation and decomposition of Li2O2, a pivotal process in Li–O2 batteries.10–13
To further enhance the catalytic activity of these candidates, surface engineering techniques are commonly employed to modulate atom arrangement and electronic structure. Established methods such as crystal facet engineering, defect engineering, and surface/interface modification are widely utilized towards achieving these objectives.14–17 Among these strategies, doping with heteroatoms has shown promising electrocatalytic activity for boosting the performance of Li–O2 batteries. By introducing heteroatoms with varying valence states and electronegativity, the charge and spin density of materials can be redistributed, thereby influencing the adsorption of oxygen-containing intermediates at active sites.
For instance, research conducted by Lu et al. demonstrated that incorporating excess Co into the (101) plane of RuO2 results in abundant Ru/Co dual-atom sites on the RuO2 (110) surface. This approach effectively optimizes both charge transfer and the accessibility of the intermediate *OOH species in zinc–air batteries.18 However, traditional doping methods often entail complex preparation procedures, impeding precise control over the foreign element concentration and the rational design of catalysts. Moreover, the structure–activity relationship of catalysts prepared via traditional chemical methods, especially at the atomic level, remains elusive for oxygen electrochemical processes in Li–O2 batteries.
Therefore, it is imperative to develop an efficient and controllable preparation method that strikes a delicate balance between cost-effectiveness and the precise preparation of catalysts. This will provide a solid foundation for the development of high-performance Li–O2 batteries.
Electron-ion coupled transfer in electrochemistry offers a promising alternative for modifying the electronic or crystal structure of host materials. Unlike conventional chemical synthesis, electrochemical techniques operate at lower temperatures and pressures, leading to reduced energy consumption and waste generation. Furthermore, these methods afford greater control over impurity concentration through adjustable electrochemical parameters.19–21 This controllability allows for increased freedom in modulating the atom arrangement and electronic properties of catalytic materials, thereby facilitating the design and synthesis of tailored catalysts.
Electrochemical methods, including galvanic replacement, electrochemical exfoliation, and electrochemical insertion/extraction, have found wide application in the synthesis of energy catalytic materials, demonstrating promising outcomes in various electrocatalytic applications such as water splitting and carbon dioxide reduction.22–24 For example, the electrochemical treatment of Li2Co2O4 spinel facilitates the formation of amorphous active layers, thereby enhancing the oxygen evolution reaction (OER) due to the presence of Co4+ ions and oxygen sites with electronic holes.25 Similarly, treating MnO2 with lithium exhibited improved catalytic performance in Li–O2 batteries, indicating the promising recycling of depleted Li–MnO2 batteries.26 Although these examples underscore the distinct advantages of electrochemical treatments in fabricating efficient catalysts, the specific catalytic mechanisms still need to be further revealed, especially in Li–O2 batteries.
Drawing inspiration from this perspective, we propose a simple lithium electrochemical tuning method to enhance the catalytic activity of RuO2, the most commonly used representative in Li–O2 batteries. This method allows for the quantitative adjustment of Li+ concentration (x). The findings reveal that Li+ not only induces lattice strain by embedding into the lattice interstitial of RuO2 but also functions as an electron donor, directly modulating the electronic structure of RuO2. Specifically, the valence state of Ru decreases with Li+ intercalation, accompanied by the formation of oxygen vacancies. These modifications facilitate efficient electron transfer from the catalyst to the reaction intermediates while optimizing the adsorption behavior of the Li–O intermediates, particularly LiO2, on the electrode surface. Consequently, Li–O2 batteries employing Li0.52RuO2 as a catalyst demonstrate ultrahigh energy conversion efficiency and long-term reversibility. The elucidation of the atomic-level catalytic mechanism provides valuable insights into the rational design and optimization of advanced electrocatalysts for Li–O2 batteries.
Experimental
Fabrication of LixRuO2 cathodes
Firstly, RuO2@CNT (carbon nanotubes) was prepared using the method described previously.8 Then, a mixture comprising 90 wt% of RuO2@CNT and 10 wt% polyvinylidene difluoride (PVDF) binder was prepared by mixing them with N-methylpyrrolidone (NMP) in a mortar. After ultrasonic dispersion, the mixture was evenly spread onto a carbon paper with a diameter of 12 mm. Subsequently, the resulting cathode was dried at 110 °C under vacuum for 12 hours before use.To prepare LixRuO2, a Li+ intercalation process was employed. Specifically, the RuO2@CNT cathode was assembled into CR2032 coin cells, with Li foil as the counter-electrode. The electrolyte was a 1 M bis(trifluoromethane)sulfonamide lithium salt (LiTFSI) dissolved in tetraethylene glycol dimethyl ether (TEGDME). The Li+ intercalation into RuO2 was achieved by discharging the cell at a constant current density of 10 mA g−1 in an argon atmosphere. While the amount of Li+ intercalation was controlled by the discharge time. The obtained LixRuO2 was used as the as-prepared cathode for Li–O2 batteries.
Battery assembly and tests
The Li–O2 batteries were assembled inside an Ar-filled glovebox, in which the as-prepared electrode, Li foil and glass fiber paper (Whatman, GF/D) were used as the cathode, anode and separator, respectively. 100 μL of electrolyte (1 M LiTFSI/TEGDME) was added to the battery. Before electrochemical tests, the batteries were purged with 99.995% O2 for 1 h. Galvanostatic discharge–charge tests were conducted under a LAND CT2001A battery testing system. The current and specific capacity were calculated based on the active mass of the cathode. Linear sweep voltammetry (LSV) curves were conducted with an electrochemical workstation (Solartron 1470E).Experimental characterization
After discharge/charge tests, the batteries were disassembled in a glovebox. The cathode was then removed and washed with anhydrous acetonitrile solvent, followed by vacuum drying before undergoing a series of characterizations. X-ray diffraction (XRD) analysis was performed using an Ultima IV X-ray diffractometer equipped with a graphite monochromatized Cu-Kα radiation source, operating at 40 kV and 40 mA. The morphology of the samples was examined using a Hitachi S4800 field emission scanning electron microscope (FESEM), while the crystal structure was analyzed using a JEM-2800 transmission electron microscope (TEM). Surface elemental properties were analyzed by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific K-Alpha+ instrument. All spectra were calibrated using the C 1s peak at 284.8 eV.Differential electrochemical mass spectrometry (DEMS) measurements of Li–O2 batteries. Quantitative DEMS was employed to investigate the stability and reversibility of Li–O2 batteries. A custom-designed Li–O2 battery, equipped with two securely attached poly(ether-ether-ketone) (PEEK) capillary tubes for gas inlet and outlet, was connected to a commercially available magnetic sector mass spectrometer (Thermo Fisher) using a specially engineered gas purging system. The flow rate of the purging gas was precisely regulated by a digital mass flow meter (Bronkhorst). During discharging, a gas mixture of Ar/O2 (mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) with a controlled flux of 0.4 sccm served as the carrier gas to accurately measure the consumption of O2. For charging Li–O2 batteries, high-purity (99.999%) Ar was utilized as the carrier gas to quantify O2 evolution. The DEMS battery assembly and testing followed procedures similar to those outlined in the section on electrochemical measurements.
:4) with a controlled flux of 0.4 sccm served as the carrier gas to accurately measure the consumption of O2. For charging Li–O2 batteries, high-purity (99.999%) Ar was utilized as the carrier gas to quantify O2 evolution. The DEMS battery assembly and testing followed procedures similar to those outlined in the section on electrochemical measurements.
Computational methods
All the computations were carried out by the DFT method including van der Waals (vdW) corrections, as implemented in the Vienna Ab Initio Simulation Package (VASP).27 The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was used to describe the exchange–correlation interaction.28 Projector augmented wave (PAW) methods are used for pseudopotentials.29 An energy cutoff of 400 eV is adopted for the plane-wave basis. The vacuum layers are set to ∼30 Å to decouple the interaction between periodic images. The Brillouin zones are sampled using Gamma-centered k-mesh of 3 × 3 × 1. The energy convergence criterion of geometry relaxation is set to 10−5 eV. The rest atomic layers and adsorbates are free to relax until the net force per atom is less than 0.05 eV Å−1. The DFT-D3 method is used to describe the van der Waals interaction.30 The VASPKIT code is used for the post-processing of the VASP computational data.31 The structures were visualized using the VESTA package.32The differential charge density is calculated according to Δρ = ρAB − ρA − ρB, where ρAB, ρA, and ρB represent the charge densities of LixRuO2 (x = 0, 0.5) covered by Li2O2 with or without adsorbed LiO2, and isolated LiO2, respectively. Yellow and blue colors indicate the charge accumulation and depletion, respectively.
The adsorption energy is calculated according to the equation Eads = EAB − EA − EB, where EAB is the total energy of LixOy (x = 1, 2, 4, y = 2, 4) molecules adsorbed on the LixRuO2 (x = 0, 0.5), EA is the energy of isolated LixOy (x = 1, 2, 4, y = 2, 4) molecule, EB is the energy of LixRuO2 substrate.
Results and discussion
Catalyst characterization
The Li+-intercalated RuO2 (LixRuO2) with adjustable Li+ concentration was prepared via an electrochemical lithiation process involving coupled ion-electron transfer. Linear sweep voltammetry (LSV) was conducted to investigate the Li+ intercalation process (Fig. S1a†). Notably, a distinct reduction peak at approximately 2 V was observed, indicating the Li+ intercalation into RuO2, followed by the transformation of LixRuO2 to Ru/Li2O,33,34 as illustrated in Fig. 1a. Furthermore, a constant-current discharge was conducted to verify this phenomenon (Fig. S1b†). Remarkably, a prominent discharge plateau was observed around 2 V, followed by a gradual voltage decrease towards 1 V, consistent with the LSV results. These findings confirm the feasibility of synthesizing LixRuO2via the electrochemical approach. The precise control of Li+ content (x) in LixRuO2 is of utmost importance as it allows for the accurate adjustment of the atomic structure and electronic properties of the catalyst. By constant current density discharge, a linear relationship between the Li+ concentration and time is established. Further details regarding the estimation of the nominal lithium concentration can be found in Fig. S2.†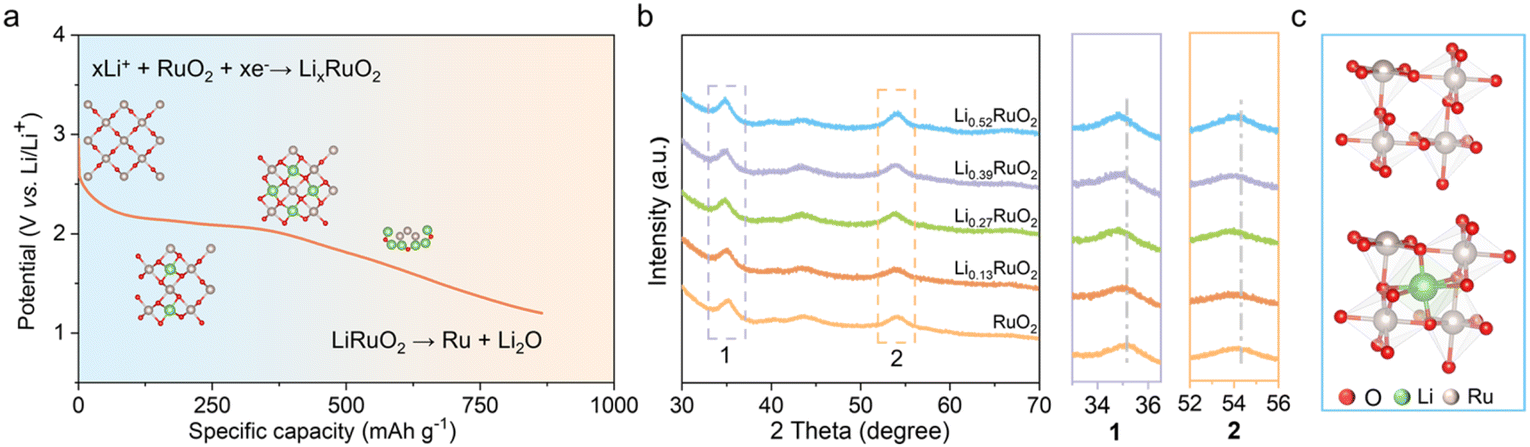 | ||
| Fig. 1 (a) Schematic diagram illustrating Li+ intercalation into RuO2 under a constant current density of 10 mA g−1. (b) XRD patterns of LixRuO2 with Li+ concentration x from 0 to 0.52 and the corresponding zoom-in images. (c) RuO6 octahedron before (up) and after (down) Li+ intercalation. | ||
XRD was conducted to investigate the influence of Li+ intercalation on the crystal structure of RuO2. Fig. S3† illustrates the XRD patterns of pristine RuO2, displaying three distinct diffraction peaks at approximately 28.1°, 35.1°, and 54.4°, in accordance with the characteristic diffraction pattern of rutile RuO2. The analysis of LixRuO2 primarily focused on the peaks at 35.1° and 54.4°, to circumvent the diffraction interference of carbon at 28.1°. Evidently, LixRuO2 retain the overall diffraction characteristics of RuO2. However, as the Li+ concentration increases, these peaks' positions gradually shift slightly towards lower angles, indicating expansion of the RuO2 lattice due to Li+ intercalation (Fig. 1b). The simulations, as depicted in Fig. S4,† further confirm this phenomenon. The lithium intercalation levels used were 0.08, 0.25, 0.33, and 0.5, based on experimental data and computational feasibility. The calculations illustrate that Li+ intercalates into the octahedral interstice formed by six adjacent O atoms, rather than replacing the Ru cations, thus leading to the expansion of the RuO2 lattice (Fig. 1c). The fitted lattice parameters of RuO2 before and after Li+ intercalation, along with the corresponding dilatation strains, are presented in Table S1.† Specifically, with increasing the lithiation degree, the expansion strains along a, b, and c-axis increase by 3.73%, 3.89%, and 0.26%, respectively.
The morphology and crystal structure of the-thus prepared samples were analyzed using SEM and TEM. As shown in Fig. S5,† the initial cathode exhibited a uniform distribution of RuO2 and CNT. After 16 h electrochemical treatment, no significant changes were observed, except for a slight reduction in pore size. Selected area electron diffraction (SAED) analyses shown in Fig. 2a reveal a series of lattice fringes in pristine RuO2, corresponding to the (110), (101), and (200) planes of the rutile RuO2. With Li+ intercalation, the lattice spacing gradually increases, as demonstrated in Fig. 2b–f. Specifically, in the case of Li0.52RuO2, the lattice spacings of the (110), (101), and (200) planes increase to 0.2427 nm, 0.2768 nm, and 0.3157 nm, respectively. This indicates that Li+ intercalation causes lattice expansion, aligning with the observed shift of characteristic peak positions towards lower angles in XRD.
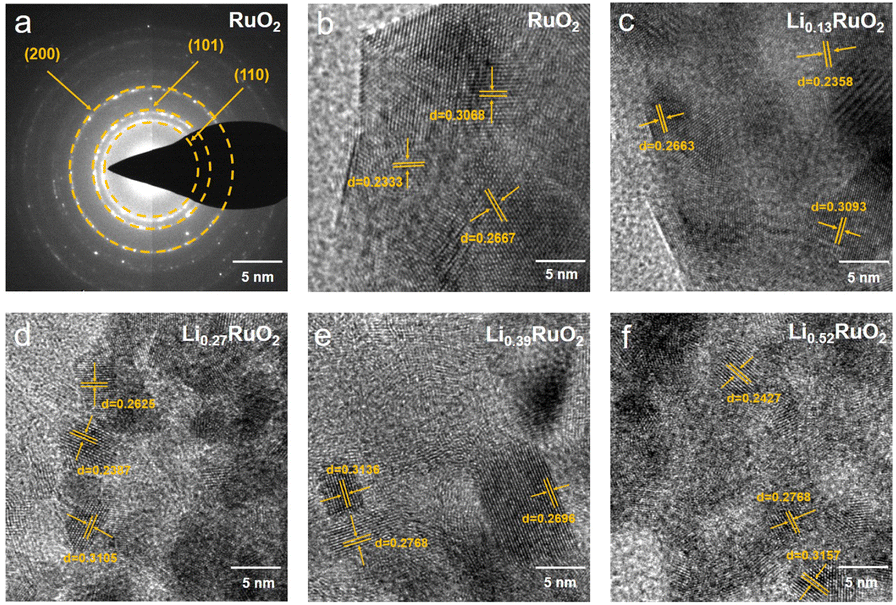 | ||
| Fig. 2 The SAED patterns (a) of pristine RuO2 and HRTEM images (b–f) of the RuO2 with different Li+ concentrations. | ||
To investigate the influence of electrochemical treatment on surface chemical states and electronic structure of LixRuO2, XPS was employed. Fig. 3a illustrates the Ru 3d spectra of pristine RuO2 and a series of LixRuO2 samples. The Ru 3d5/2 spectrum exhibits peaks at 281 and 282 eV, corresponding to Ru(III) and Ru(IV), respectively. Two satellite peaks associated with Ru 3d3/2 are also observed. Pristine RuO2 exclusively displays the Ru(IV) peak at 282 eV, consistent with previous reports.35,36 However, upon Li+ intercalation, a characteristic peak of Ru(III) at 281 eV emerges. The content of Ru(IV) and Ru(III) in LixRuO2 samples is summarized in Fig. 3c. With increasing Li+ concentration, the proportion of Ru(IV) decreases to 93.5%, 87.3%, 76.2%, and 71.4%. Moreover, both the Ru(IV) and Ru(III) peaks exhibit a slight shift towards higher binding energies, indicating electron transfer and a decrease in electron density around the Ru sites. Furthermore, an analysis of O 1s spectra was conducted, as presented in Fig. 3b. The peak around 529.4 eV in RuO2, attributed to lattice oxygen, gradually shifts to lower binding energies with Li+ intercalation. This shift suggests an increased electron density surrounding the oxygen sites, indicating partial electron transfer from the Ru sites to the O sites facilitated by Li+ intercalation.37,38 Notably, after 8 h intercalation, a new peak appears around 530.5 eV, corresponding to oxygen vacancy, whose proportion gradually increases with prolonged intercalation time, as statistically demonstrated in Fig. 3d. This increase might provide additional active sites for oxygen electrochemical reactions.
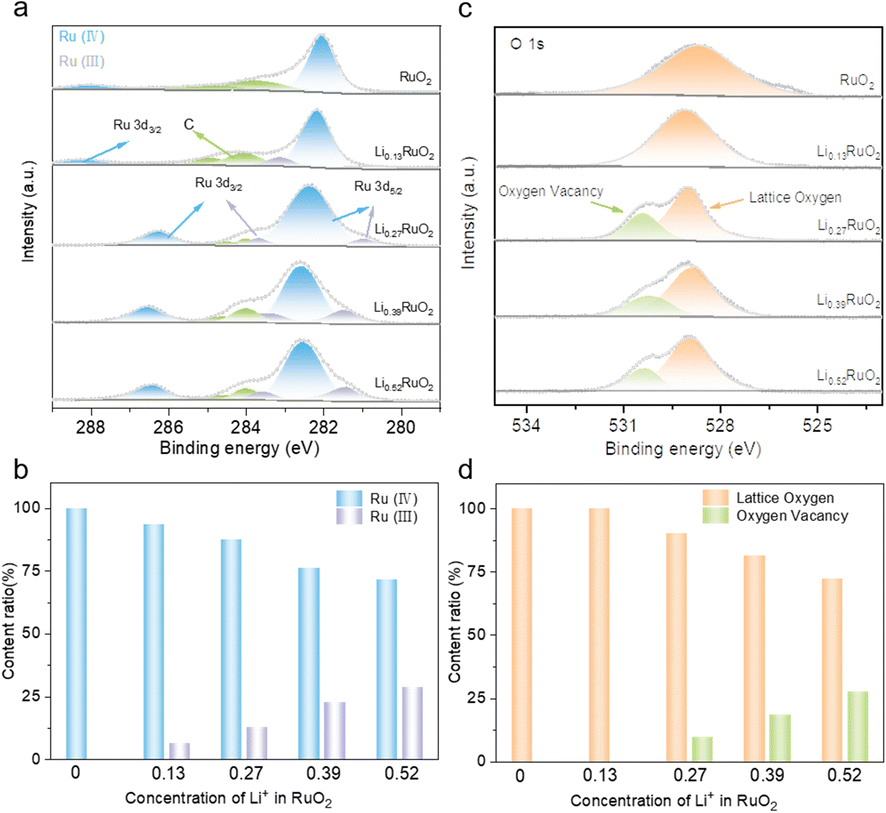 | ||
| Fig. 3 XPS of Ru 3d (a) and O 1s (b) of RuO2 and LixRuO2, and (c and d) content comparison of different chemical species calculated from the fitted XPS spectra. | ||
Li–O2 battery performance
A series of electrochemical tests were conducted to evaluate the unique electrocatalytic capability of the elaborately designed LixRuO2 catalyst for Li–O2 batteries. The discharge profiles at different current densities reveal that the electrochemically treated LixRuO2 exhibits enhanced discharge capacity compared with pristine RuO2 (Fig. 4a and b). Moreover, with an increase in Li+ intercalation, the capacity demonstrates corresponding enhancement. This can be attributed to the enhanced adsorption of intermediate LiO2 on LixRuO2, resulting in full utilization of inner space, which will be discussed in detail below.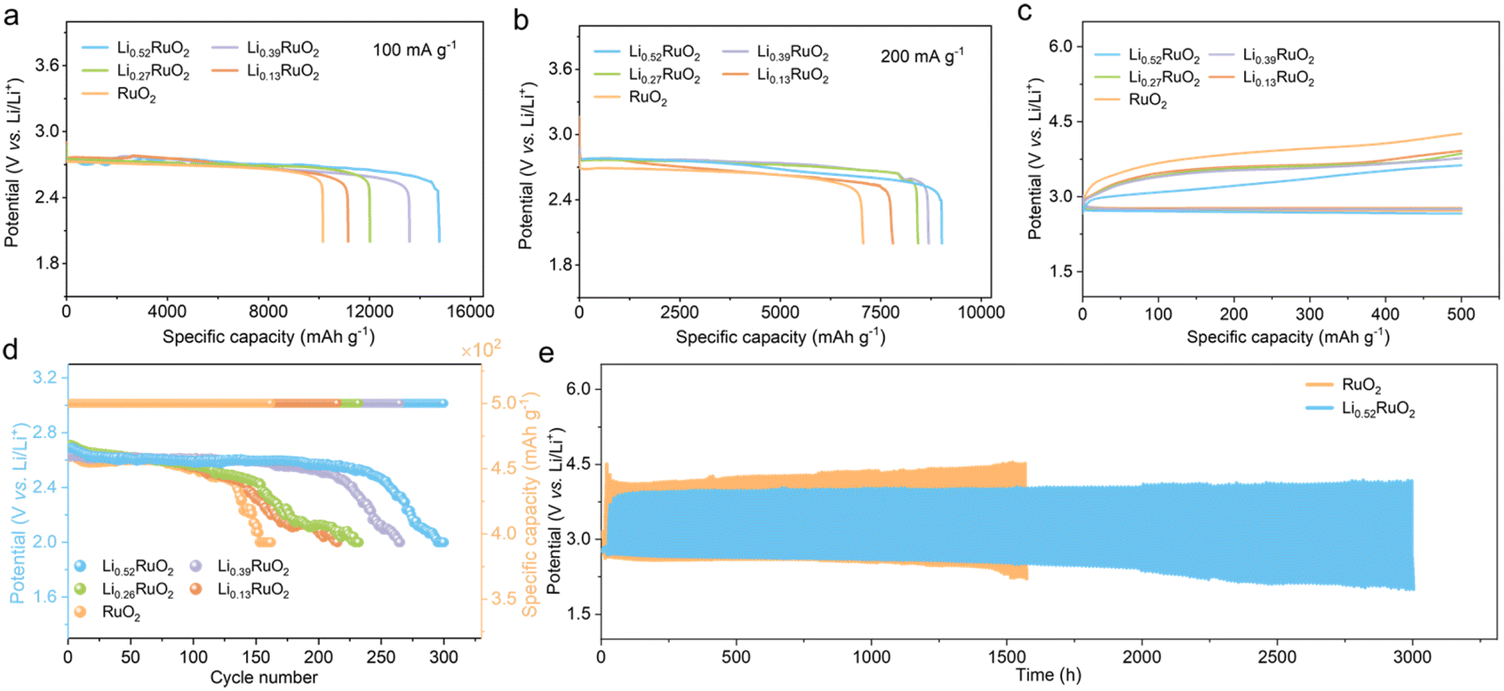 | ||
| Fig. 4 Discharge profiles of Li–O2 batteries with RuO2 or LixRuO2 cathode at a current density of (a) 100 mA g−1 and (b) 200 mA g−1. The first-cycle charge–discharge curve (c), terminal discharge voltages and corresponding capacity (d) and cycling stability (e) of Li–O2 batteries with RuO2 or LixRuO2 cathode at 100 mA g−1 with a limited capacity of 500 mA h g−1. | ||
Fig. 4c shows the first-cycle charge–discharge curve of Li–O2 batteries with RuO2 or LixRuO2 cathode, which is another crucial criterion for evaluating the catalytic activity of materials. Compared with the RuO2 cathode, Li–O2 batteries based on LixRuO2 demonstrated smaller charge overpotentials, which is negatively correlated with the Li+ concentration. That is, the higher the Li+ intercalation level, the lower the reaction overpotential. Notably, the first charge voltage of Li0.52RuO2-based Li–O2 batteries decreased to approximately 3.41 V, which could effectively mitigate parasitic reactions at higher voltages. Moreover, Li–O2 batteries based on RuO2 displayed a limited cycle life of 150 cycles, while the batteries incorporating LixRuO2 demonstrated significantly improved cycling performance (Fig. 4d). Among them, the Li–O2 batteries utilizing Li0.52RuO2 exhibited the best cycling stability, with a remarkable cycle life of 300 cycles and stable operation exceeding 3000 hours (Fig. 4e). These results indicate that the Li+ intercalation significantly enhances the catalytic activity of LixRuO2 towards Li2O2 decomposition, which can be attributed to additional vacancy oxygen and the modulated electronic structure, providing additional active sites and enhancing reaction kinetics.
The practical feasibility of Li0.52RuO2-based Li–O2 batteries was evaluated at an increased current density of 500 mA g−1. Fig. 5a and b illustrate that the Li–O2 batteries with Li0.52RuO2 demonstrated improved cycling life of 321 cycles, surpassing that with untreated RuO2. Even at a higher cutoff capacity of 1000 mA h g−1, as displayed in Fig. 5c, Li–O2 batteries with Li0.52RuO2 demonstrated remarkable cycling stability for 153 cycles, whereas those with RuO2 showed limited cycle life of 111 cycles due to rapid voltage increase (Fig. 5d). These results highlight the exceptional ability of LixRuO2 to mitigate charging voltage and enhance cycling stability, demonstrating its practical potential. The rate performance of Li–O2 batteries utilizing Li0.52RuO2 is depicted in Fig. 5k. In comparison with pristine RuO2, the Li0.52RuO2-incorporating battery exhibits minimal discharge and charge voltage fluctuation, even under a high current density of 1000 mA g−1, which can be attributed to enhanced kinetics of oxygen electrochemical reactions. These findings underscore the paramount significance of modulating the atom structure and electronic feature of the catalyst in enhancing the performance of Li–O2 batteries.
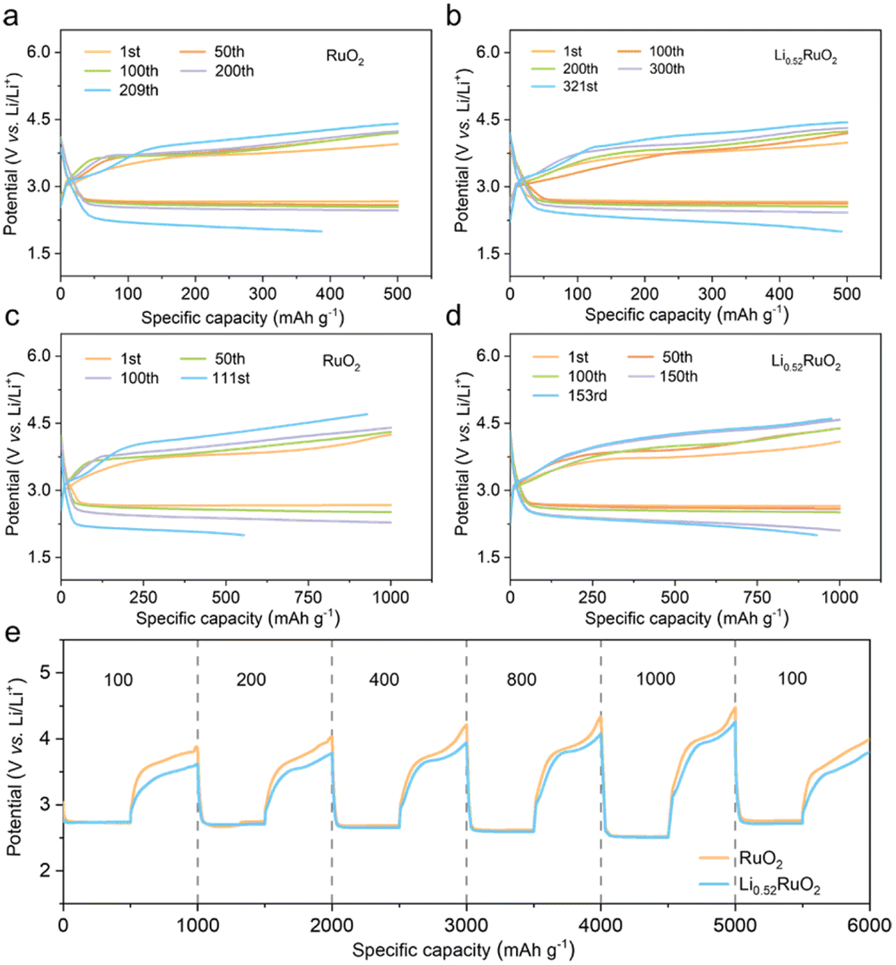 | ||
| Fig. 5 Cycling performance of RuO2 and LixRuO2 in Li–O2 batteries with a cutoff capacity of (a and b) 500 mA h g−1 or (c and d) 1000 mA h g−1 at a current density of 500 mA g−1. (e) Rate performance of RuO2 and Li0.52RuO2 under current density changing from 100 to 1000 mA g−1. | ||
To gain deeper insights into the underlying catalytic mechanism of LixRuO2, which is closely linked to the component and morphology of Li–O2 battery products and their electrochemical performance, XRD and SEM were employed to examine the cathodes in different discharge/charge states. As demonstrated in Fig. S6,† both the discharged RuO2 and LixRuO2 cathodes exhibited diffraction peaks at 32.9° and 35.0°, corresponding to the (100) and (101) crystal planes of Li2O2 (PDF#09-0355), indicating Li2O2 as the primary discharge product. Upon charging completion, the Li2O2 diffraction peak vanished, while the cathode peak reappeared, suggesting complete decomposition of discharge products for both RuO2 and LixRuO2 cathodes. Considering that XRD analysis provides information solely on the crystalline components, it is crucial to employ quantitative techniques like DEMS to evaluate the reversibility of Li–O2 batteries.39 The amount of O2 consumption/evolution during battery operation can be monitored using DEMS, which is imperative for the assessment of truly rechargeable Li–O2 batteries.
For an ideally reversible Li–O2 battery, the ratio of electrons to O2 molecule (e−/O2) shall be 2.0, and O2 is the only gaseous species involved in the discharge/recharge cycle. The typical galvanostatic discharge/charge profiles and the corresponding gas consumption/evolution rate are shown in Fig. 6. For the RuO2 based Li–O2 battery (Fig. 6a), a significantly deviated value of 2.39 e−/O2 was obtained upon discharge, with an ORR efficiency of only 80.5%, suggesting much undesired parasitic reaction. However, the e−/O2 ratio was quantified to be 2.08 (≈2.0 e−/O2) for the Li0.52RuO2-based Li–O2 battery, as depicted in Fig. 6b, with a slight deviation of 4% from the theoretical value. This negligible discrepancy could be attributed to the inevitable shuttle effect of oxygen and Li–O intermediates. The results indicate that the discharge reaction, catalyzed by Li0.52RuO2, primarily involved Li2O2 formation, which is consistent with the XRD result. Furthermore, the catalytic activity of Li0.52RuO2 and RuO2 during recharge was also evaluated using DEMS. As exhibited in Fig. 6c, the RuO2 based Li–O2 battery displays a high charge potential and a widely observed OER profile with a dip in the middle of charge, which usually is accompanied by a hydrogen evolution reaction (HER) resulted from the 1O2 attack, mirroring the missing O2 in the OER profile.40 Additionally, a significant amount of CO2 was observed when the charging voltage reached approximately 4.0 V. Actually, the appearance of gaseous CO2 during recharge is an indicator that the Li–O2 batteries are not ideally reversible, and the amount of CO2 generated directly reflects the extent of undesired parasitic reactions, which has been suggested to originate from the decomposition of carbon cathodes or electrolytes. On the contrary, the battery charged with Li0.52RuO2 (Fig. 6d) does not display a dip in its OER profile, which demonstrated a continuous, stable release of O2, with negligible CO2 generation. As a result, the ratio of charge passed to O2 evolved with Li0.52RuO2 cathode was quantified to be 2.11, which is much lower than 2.26 of the RuO2-based Li–O2 battery. Based on these findings, the disappearance of these three features (OER dip, CO2 release and ratio of e−/O2), the Li0.52RuO2 further confirms its superior catalytic activity. Besides, the parasitic products also were investigated through XPS. After the first cycle, the RuO2 cathode exhibited undecomposed Li2O2 and significant amounts of Li2CO3 byproducts (Fig. S7a†), likely due to elevated charging voltage. These byproducts, due to their wide band gap, are difficult to decompose during cycling, leading to increased charging voltage and eventual cathode passivation. As shown in Fig. S7b,† more Li2CO3 accumulated in the RuO2 cathode surface after the 10th cycle. In contrast, pre-lithiated cathodes exhibited significantly lower Li2CO3 levels after cycling. The Li2CO3 content decreased progressively with increasing Li+ concentration. Notably, the Li0.52RuO2 cathode showed almost no Li2CO3 byproducts, consistent with DEMS results. Even after the 10th cycle, no significant Li2CO3 was observed, indicating the system's exceptional capability in suppressing side reactions. Therefore, the incorporation of Li0.52RuO2 in Li–O2 batteries not only improves reaction kinetics but also reduces charging voltage, leading to reduced side reactions and enhanced reversibility, thereby improving the overall cycle stability.
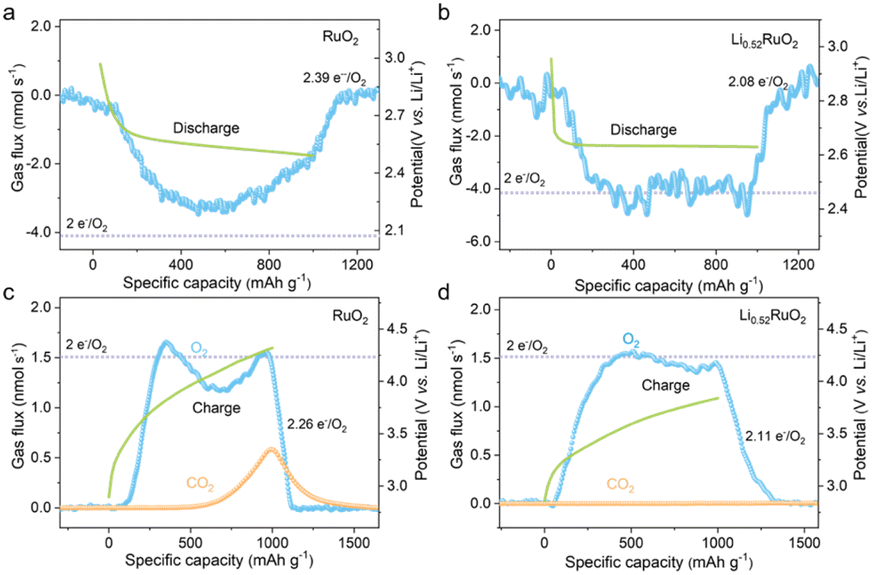 | ||
| Fig. 6 DEMS analyses of gas consumption (a and b) and evolution (c and d) during discharge/charge of Li–O2 batteries based on (a and c) RuO2 and (b and d) Li0.52RuO2 cathodes. | ||
SEM was conducted to investigate the morphological features of discharge products. As depicted in Fig. S4a,† the pristine RuO2 electrode exhibited a homogeneous mixture of RuO2 particles and CNTs. Upon discharge, the electrode was covered by a dense film-like discharge product (Fig. S8a†), which may be the potential reason for limited discharge capacity. However, in addition to the dense film-like discharge product, LixRuO2 also exhibited some rod-like products, as presented in Fig. S8b–e.† Furthermore, increasing Li+ concentration promoted the growth of these products, corresponding to higher discharge capacity. This result is closely linked to the adsorption behavior of reaction intermediates on the catalyst surface, which is modulated by the electronic structure resulting from Li+ intercalation. It may optimize the adsorption strength of LixRuO2 cathodes toward the superoxide intermediates, promoting different oxygen reduction reaction (ORR) routes. Notably, the Li0.52RuO2 cathode exhibited the highest discharge product yield and capacity. Upon charging, residual discharge products were observed on RuO2 cathodes (Fig. S8f†), which severely reduce the availability of active sites and impede electron transfer. Conversely, the LixRuO2 cathode displayed complete products decomposition, showcasing excellent reversibility (Fig. S8g–k†). These results emphasize the importance of electrochemically modulating the electronic structure of materials, optimizing the adsorption characteristics of catalysts towards intermediate species in Li–O2 batteries. Further detailed explanations will be provided in DFT calculations sections.
Catalytic mechanism of LixRuO2 catalyst
DFT calculations were employed to investigate the catalytic mechanisms of pre-lithiation RuO2 in Li–O2 batteries. The calculations focused on the (110) planes of RuO2 and LixRuO2, which were predominantly observed in HRTEM images. The work function (Φ), a crucial descriptor of the electron-donating capability of a solid electrocatalyst, was depicted in Fig. S9a–e.† As the Li+ concentration increases, the work function of the LixRuO2 (110) plane significantly decrease, which highlights the effective modification of electronic structure of RuO2 through Li+ insertion. Such modification facilitates electron transfer from the catalyst to the reaction intermediates, thereby enhancing the ORR and OER kinetics. Fig. S9f† shows the Bader charges of Ru and O, with increasing Li+ concentration, the acquired electron of O increases, while the donated electrons of Ru gradually reduce. It indicates a decline in the valence state of Ru cations, which aligns with the XPS results. The shift in the surface electronic structure can be attributed to the strain effect, which plays a crucial role in modulating electrocatalytic activity.41–44Furthermore, the PDOS in RuO2 and LixRuO2 were recorded to reveal the regulating effect of Li+ insertion on the d-band center of RuO2. As shown in Fig. 7a, the d-band center of LixRuO2 exhibits a significantly upshift from −1.86 (x = 0) to 1.62 eV (x = 0.5), approaching the Fermi level. Meanwhile, the adsorption energy of LixRuO2 toward the key LiO2 intermediate gradually increase −3.02 eV to −3.85 eV, as depicted in Fig. 7b. Moreover, the adsorption profiles of other Li–O intermediates on RuO2 and Li0.5RuO2 were calculated, as illustrated in Fig. S10.† It reveals that the adsorption energy of all Li–O intermediates on the RuO2 (110) plane is significantly lower than that on the Li0.5RuO2 (110) plane, indicating that the incorporation of Li+ strengthens the interaction between Li–O intermediates and the catalyst. Notably, the strong binding interaction, particularly between LiO2 and Li0.5RuO2 cathodes, assumes a pivotal role in determining the growth route of discharge products and facilitating the OER catalytic activities.6,45,46 Visualizations of the differential charge density distributions (Fig. S11†) provide further support for the enhanced adsorption of catalysts towards the reaction species following Li+ intercalation. The electron donation and accumulation between O and the catalyst surface was presented by color of cyan and yellow, respectively. Remarkably, there are fewer electrons transferred from Ru to O on the RuO2 surface compared to the Li0.5RuO2 (110) surface. Based on the aforementioned calculation results, Fig. 7c presents a schematic illustrating the enhanced catalytic performance of RuO2 with Li+ insertion. Specifically, the remarkable improvement in electron transfer ability and adsorption functionality could synergistically optimize the reaction pathways and kinetics of the ORR and OER in Li–O2 batteries.
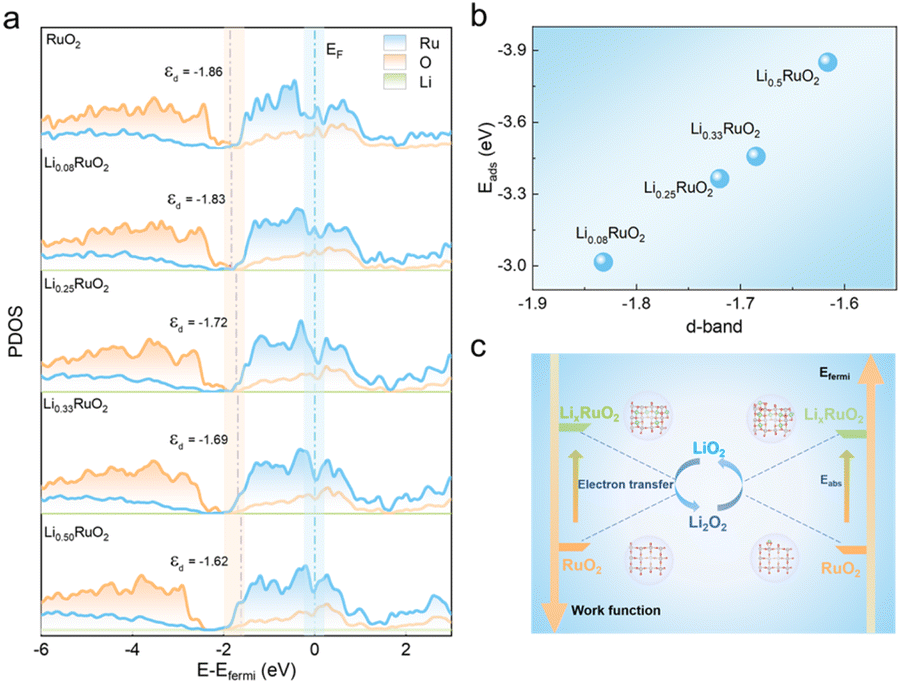 | ||
| Fig. 7 (a) The partial density of states (PDOS) of the LixRuO2 (x = 0, 0.08, 0.25, 0.33, 0.5) and the corresponding d-band center of Ru atom. (b) The variation in the adsorption energy of LiO2 on different LixRuO2 surfaces as a function of the d-band center. (c) Schematic illustration for the improved catalytic performance of RuO2 by Li+ insertion. | ||
Integrating computational calculation with experimental results, we have elucidated the crucial role of the pre-lithiation RuO2 catalyst in promoting the nucleation and growth of Li2O2. Typically, upon ORR, dissolved oxygen initially undergoes a one-electron reduction process, forming LiO2 intermediate. For the RuO2 cathode with weak adsorption, a large number of soluble intermediates were formed at the initial stage of discharge and captured by porous electrodes. As the discharge process advanced, the intermediates distributed uniform crystal seeds into the porous structure, and finally induced the growth of film-shaped Li2O2 in accordance with SEM observation.47 However, for the LixRuO2 cathode, Li2O2 growth occurs through dual growth pathways with distinct morphologies. Specifically, a film-like Li2O2 similar to that on the RuO2 cathode is formed on the CNT surface. Additionally, due to the high affinity between LiO2 and LixRuO2 configurations, a significant confinement effect leads to the formation of rod-like Li2O2 products.11 Furthermore, the charge density distribution shown in Fig. S12† indicates that even when the Li0.5RuO2 surface is covered by Li2O2, Li0.5RuO2 still exhibits strong interactions with LiO2. Consequently, Li0.5RuO2-based batteries can sustain discharge, resulting in a larger discharge capacity. During the subsequent charging process, the enhanced interaction between Li0.5RuO2 and LiO2 intermediates, as well as Li2O2 products, facilitate the charge transfer between oxygen-containing species and oxygen electrode, thereby the OER kinetics. As a result, the discharge products can be decomposed at an ultra-low charging potential, while preventing the accumulation of residue from the discharge process and ensuring the ultralong cycle life for Li–O2 batteries.
Conclusions
In this study, we synthesized a series of LixRuO2 catalysts with tunable Li+ concentrations via electrochemical methods. Results demonstrated that Li+ is inserted into the octahedral interstices of RuO2, inducing lattice strain effect. Furthermore, the Li+ intercalation precisely customized the surface electronic structure of LixRuO2. Specifically, Li+ ions acted as potent electron donors, effectively reducing the valence state of Ru cations, which also results in the formation of oxygen vacancies. Benefitting from these characteristics, the charge transfer and adsorption strength between LixRuO2 and oxygen-containing intermediates were synergistically strengthened, which dramatically enhanced the electrochemical performance Li–O2 batteries. Particularly, the Li0.52RuO2-based Li–O2 battery exhibited an energy conversion efficiency of up to 80%, a long-term lifespan of 321 cycles, a high discharge capacity of 14760 mA h g−1, and desirable rate performance. This study not only presents a facile and controllable method for synthesizing highly efficient catalysts for Li–O2 batteries with atomic-level precision but also offers profound insights into the fundamental understanding of catalytic mechanisms, which demonstrates a promising avenue for the practical implementation of advanced energy conversion and storage systems.
Data availability
The original data supporting this article are available in the main context and ESI.†Author contributions
Zhengcai Zhang: writing – original draft, investigation, visualization. Dulin Huang: investigation, validation, visualization. Shuochao Xing: investigation, methodology, validation, visualization. Minghui Li: investigation. Jing Wu: investigation. Zhang Zhang: writing – review & editing, supervision. Yaying Dou: writing – review & editing, supervision, funding acquisition, conceptualization. Zhen Zhou: writing – review & editing, supervision, conceptualization.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22202182), the Key R&D and Promotion Special (Scientific Problem Tackling) Project of Henan Province (242102240088) and the China postdoctoral science foundation (2023M733211).Notes and references
- X. Chi, M. Li, J. Di, P. Bai, L. Song, X. Wang, F. Li, S. Liang, J. Xu and J. Yu, A highly stable and flexible zeolite electrolyte solid-state Li–air battery, Nature, 2021, 592, 551–557 CrossRef CAS PubMed.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Lithium–Oxygen Batteries and Related Systems: Potential, Status, and Future, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, B. Sun, Y. Xie, A. Tkacheva, F. Qiu, P. He, H. Zhou, K. Yan, X. Guo, S. Wang, A. M. McDonagh, Z. Peng, J. Lu and G. Wang, A long-life lithium-oxygen battery via a molecular quenching/mediating mechanism, Sci. Adv., 2022, 8, eabm1899 CrossRef PubMed.
- X. Han, L. Zhao, J. Wang, Y. Liang and J. Zhang, Delocalized Electronic Engineering of Ni5P4 Nanoroses for Durable Li–O2 Batteries, Adv. Mater., 2023, 35, 2301897 CrossRef CAS PubMed.
- Y. Zhou, Q. Gu, K. Yin, L. Tao, Y. Li, H. Tan, Y. Yang and S. Guo, Cascaded orbital–oriented hybridization of intermetallic Pd3Pb boosts electrocatalysis of Li-O2 battery, Proc. Natl. Acad. Sci. U.S.A., 2023, 120, e2301439120 CrossRef CAS PubMed.
- P. Wang, Y. Ren, R. Wang, P. Zhang, M. Ding, C. Li, D. Zhao, Z. Qian, Z. Zhang, L. Zhang and L. Yin, Atomically dispersed cobalt catalyst anchored on nitrogen-doped carbon nanosheets for lithium-oxygen batteries, Nat. Commun., 2020, 11, 1576 CrossRef CAS PubMed.
- Y. He, L. Ding, J. Cheng, S. Mei, X. Xie, Z. Zheng, W. Pan, Y. Qin, F. Huang, Y. Peng and Z. Deng, A “Trinity” Design of Li-O2 Battery Engaging the Slow-Release Capsule of Redox Mediators, Adv. Mater., 2023, 35, 2308134 CrossRef CAS PubMed.
- Y. Dou, X.-G. Wang, D. Wang, Q. Zhang, C. Wang, G. Chen, Y. Wei and Z. Zhou, Tuning the structure and morphology of Li2O2 by controlling the crystallinity of catalysts for Li-O2 batteries, Chem. Eng. J., 2021, 409, 128145 CrossRef CAS.
- B. Sun, X. Huang, S. Chen, P. Munroe and G. Wang, Porous Graphene Nanoarchitectures: An Efficient Catalyst for Low Charge-Overpotential, Long Life, and High Capacity Lithium–Oxygen Batteries, Nano Lett., 2014, 14, 3145–3152 CrossRef CAS PubMed.
- J. Tian, Y. Rao, W. Shi, J. Yang, W. Ning, H. Li, Y. Yao, H. Zhou and S. Guo, Sabatier Relations in Electrocatalysts Based on High-entropy Alloys with Wide-distributed d-band Centers for Li-O2 Batteries, Angew. Chem., Int. Ed., 2023, 62, e202310894 CrossRef CAS PubMed.
- Z. Lian, Y. Lu, S. Zhao, Z. Li and Q. Liu, Engineering the Electronic Interaction between Atomically Dispersed Fe and RuO2 Attaining High Catalytic Activity and Durability Catalyst for Li-O2 Battery, Adv. Sci., 2023, 10, 2205975 CrossRef CAS PubMed.
- Y. Dou, S. Xing, Z. Zhang and Z. Zhou, Solving the Singlet Oxygen Puzzle in Metal-O2 Batteries: Current Progress and Future Directions, Electrochem. Energy Rev., 2024, 7, 6 CrossRef CAS.
- Q. Xia, D. Li, L. Zhao, J. Wang, Y. Long, X. Han, Z. Zhou, Y. Liu, Y. Zhang, Y. Li, A. A. A. Adam and S. Chou, Recent advances in heterostructured cathodic electrocatalysts for non-aqueous Li–O2 batteries, Chem. Sci., 2022, 13, 2841–2856 RSC.
- Y. Zhou, Q. Gu, Y. Xin, X. Tang, H. Wu and S. Guo, Orbital Coupling of PbO7 Node in Single-Crystal Metal–Organic Framework Enhances Li-O2 Battery Electrocatalysis, Nano Lett., 2023, 23, 10600–10607 CrossRef CAS PubMed.
- Z. Sun, X. Zhao, W. Qiu, B. Sun, F. Bai, J. Liu and T. Zhang, Unlock Restricted Capacity via O-Ce Hybridization for Li-Oxygen Batteries, Adv. Mater., 2023, 35, 2210867 CrossRef CAS PubMed.
- R. Zheng, D. Du, Y. Yan, S. Liu, X. Wang and C. Shu, Cation Vacancy Modulated Interfacial Electronic Interactions for Enhanced Electrocatalysis in Lithium–Oxygen Batteries, Adv. Funct. Mater., 2024, 2316440, DOI:10.1002/adfm.202316440.
- Z. Shen, W. Yu, A. Aziz, K. Chida, T. Yoshii and H. Nishihara, Sequential Catalysis of Defected-Carbon and Solid Catalyst in Li–O2 Batteries, J. Phys. Chem. C, 2023, 127, 6239–6247 CrossRef CAS.
- Q. Lu, X. Zou, X. Wang, L. An, Z. Shao and Y. Bu, Simultaneous reactant accessibility and charge transfer engineering in Co-doped RuO2-supported OCNT for robust rechargeable zinc-air batteries, Appl. Catal., B, 2023, 325, 122323 CrossRef CAS.
- S. Wang, M. Lu, X. Xia, F. Wang, X. Xiong, K. Ding, Z. Pang, G. Li, Q. Xu, H.-Y. Hsu, S. Hu, L. Ji, Y. Zhao, J. Wang, X. Zou and X. Lu, A universal and scalable transformation of bulk metals into single-atom catalysts in ionic liquids, Proc. Natl. Acad. Sci. U.S.A., 2024, 121, e2319136121 CrossRef CAS PubMed.
- H. Li, X. Han, W. Zhao, A. Azhar, S. Jeong, D. Jeong, J. Na, S. Wang, J. Yu and Y. Yamauchi, Electrochemical preparation of nano/micron structure transition metal-based catalysts for the oxygen evolution reaction, Mater. Horiz., 2022, 9, 1788–1824 RSC.
- D. Gao, H. Li, P. Wei, Y. Wang, G. Wang and X. Bao, Electrochemical synthesis of catalytic materials for energy catalysis, Chin. J. Catal., 2022, 43, 1001–1016 CrossRef CAS.
- S. Jiang, H. Suo, X. Zheng, T. Zhang, Y. Lei, Y.-X. Wang, W.-H. Lai and G. Wang, Lightest Metal Leads to Big Change: Lithium-Mediated Metal Oxides for Oxygen Evolution Reaction, Adv. Energy Mater., 2022, 12, 2201934 CrossRef CAS.
- J. Park, H. Kim, K. Jin, B. J. Lee, Y.-S. Park, H. Kim, I. Park, K. D. Yang, H.-Y. Jeong, J. Kim, K. T. Hong, H. W. Jang, K. Kang and K. T. Nam, A New Water Oxidation Catalyst: Lithium Manganese Pyrophosphate with Tunable Mn Valency, J. Am. Chem. Soc., 2014, 136, 4201–4211 CrossRef CAS PubMed.
- K. Jiang, H. Wang, W.-B. Cai and H. Wang, Li Electrochemical Tuning of Metal Oxide for Highly Selective CO2 Reduction, ACS Nano, 2017, 11, 6451–6458 CrossRef CAS PubMed.
- S. Zhang, S. Gu, Y. Wang, C. Liang, Y. Yu, L. Han, S. Zheng, N. Zhang, X. Liu, J. Zhou and J. Li, Spontaneous Delithiation under Operando Condition Triggers Formation of an Amorphous Active Layer in Spinel Cobalt Oxides Electrocatalyst toward Oxygen Evolution, ACS Catal., 2019, 9, 7389–7397 CrossRef CAS.
- Y. Hu, T. Zhang, F. Cheng, Q. Zhao, X. Han and J. Chen, Recycling Application of Li–MnO2 Batteries as Rechargeable Lithium–Air Batteries, Angew. Chem., Int. Ed., 2015, 54, 4338–4343 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- P. Balaya, H. Li, L. Kienle and J. Maier, Fully Reversible Homogeneous and Heterogeneous Li Storage in RuO2 with High Capacity, Adv. Funct. Mater., 2003, 13, 621–625 CrossRef CAS.
- A. S. Hassan, A. Navulla, L. Meda, B. R. Ramachandran and C. D. Wick, Molecular Mechanisms for the Lithiation of Ruthenium Oxide Nanoplates as Lithium-Ion Battery Anode Materials: An Experimentally Motivated Computational Study, J. Phys. Chem. C, 2015, 119, 9705–9713 CrossRef CAS.
- D. J. Morgan, Resolving ruthenium: XPS studies of common ruthenium materials, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- M. Hu, T. Yu, K. Tan, A. Zhou, L. Luo and S. Yin, Ultralow Ru loading RuO2-TiO2 with strong oxide-support interaction for efficient chlorine evolution and ammonia-nitrogen-elimination, Chem. Eng. J., 2023, 465, 143001 CrossRef CAS.
- G. Li, T. Sun, H.-J. Niu, Y. Yan, T. Liu, S. Jiang, Q. Yang, W. Zhou and L. Guo, Triple Interface Optimization of Ru-based Electrocatalyst with Enhanced Activity and Stability for Hydrogen Evolution Reaction, Adv. Funct. Mater., 2023, 33, 2212514 CrossRef CAS.
- Y. Li, T. Xu, Q. Huang, L. Zhu, Y. Yan, P. Peng and F.-F. Li, C60 Fullerenol to Stabilize and Activate Ru Nanoparticles for Highly Efficient Hydrogen Evolution Reaction in Alkaline Media, ACS Catal., 2023, 13, 7597–7605 CrossRef CAS.
- B. Sun, L. Guo, Y. Ju, P. Munroe, E. Wang, Z. Peng and G. Wang, Unraveling the catalytic activities of ruthenium nanocrystals in high performance aprotic Li–O2 batteries, Nano Energy, 2016, 28, 486–494 CrossRef CAS.
- Z. Liang, Q. Zou, J. Xie and Y.-C. Lu, Suppressing singlet oxygen generation in lithium–oxygen batteries with redox mediators, Energy Environ. Sci., 2020, 13, 2870–2877 RSC.
- S. Schnur and A. Groß, Strain and coordination effects in the adsorption properties of early transition metals: A density-functional theory study, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 033402 CrossRef.
- X. Wang, Y. Orikasa, Y. Takesue, H. Inoue, M. Nakamura, T. Minato, N. Hoshi and Y. Uchimoto, Quantitating the Lattice Strain Dependence of Monolayer Pt Shell Activity toward Oxygen Reduction, J. Am. Chem. Soc., 2013, 135, 5938–5941 CrossRef CAS PubMed.
- A. M. Smith, A. M. Mohs and S. Nie, Tuning the optical and electronic properties of colloidal nanocrystals by lattice strain, Nat. Nanotechnol., 2009, 4, 56–63 CrossRef CAS PubMed.
- K. Wang, K. Yu, S. Xu, S. Yuan, L. Xiang, B. Pang, J. Zheng and N. Li, Synergizing lattice strain and electron transfer in TMSs@1T-MoS2 in-plane heterostructures for efficient hydrogen evolution reaction, Appl. Catal., B, 2023, 328, 122445 CrossRef CAS.
- Q. Lv, Z. Zhu, Y. Ni, J. Geng and F. Li, Spin-State Manipulation of Two-Dimensional Metal–Organic Framework with Enhanced Metal–Oxygen Covalency for Lithium-Oxygen Batteries, Angew. Chem., Int. Ed., 2022, 61, e202114293 CrossRef CAS PubMed.
- L.-N. Song, W. Zhang, Y. Wang, X. Ge, L.-C. Zou, H.-F. Wang, X.-X. Wang, Q.-C. Liu, F. Li and J.-J. Xu, Tuning lithium-peroxide formation and decomposition routes with single-atom catalysts for lithium–oxygen batteries, Nat. Commun., 2020, 11, 2191 CrossRef CAS PubMed.
- Z. Gou, Y. Yao, X. Geng, F. Yang, X. Hu, Z. Chen, L. Zheng, Y. Su, F. Wu and J. Lu, Dual Redox Mediators Assisted Hierarchically Porous Hollow Carbon Shell Cathode for Enhanced Performance Li-O2 Battery, Adv. Energy Mater., 2024, 2304272, DOI:10.1002/aenm.202304272.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03242j |
| ‡ Zhengcai Zhang and Dulin Huang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |