
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecond-shell modulation on porphyrin-like Pt single atom catalysts for boosting oxygen reduction reaction†
Tayyaba
Najam
 ab,
Syed Shoaib Ahmad
Shah
c,
Hanqing
Yin
d,
Xin
Xiao
e,
Shamraiz
Talib
f,
Qianqian
Ji
a,
Yonggui
Deng
g,
Muhammad Sufyan
Javed
a,
Jie
Hu
a,
Ruo
Zhao
a,
Aijun
Du
d,
Xingke
Cai
*a and
Qiang
Xu
*eh
ab,
Syed Shoaib Ahmad
Shah
c,
Hanqing
Yin
d,
Xin
Xiao
e,
Shamraiz
Talib
f,
Qianqian
Ji
a,
Yonggui
Deng
g,
Muhammad Sufyan
Javed
a,
Jie
Hu
a,
Ruo
Zhao
a,
Aijun
Du
d,
Xingke
Cai
*a and
Qiang
Xu
*eh
aInstitute for Advanced Study, Shenzhen University, Shenzhen 518060, China. E-mail: cai.xingke@szu.edu.cn
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
cDepartment of Chemistry, School of Natural Sciences, National University of Sciences and Technology, Islamabad 44000, Pakistan
dQUT Centre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, 4000, Australia
eShenzhen Key Laboratory of Micro/Nano-Porous Functional Materials (SKLPM), SUSTech-Kyoto University Advanced Energy Materials Joint Innovation Laboratory (SKAEM-JIL), Department of Chemistry and Department of Materials Science and Engineering, Southern University of Science and Technology (SUSTech), Shenzhen 518055, China. E-mail: xuq@sustech.edu.cn
fAdvanced Materials Chemistry Centre (AMCC), SAN Campus, Khalifa University, Abu Dhabi, P. O. Box 127788, United Arab Emirates
gCollege of Mechatronics and Control Engineering, Shenzhen University, Shenzhen 518060, PR China
hInstitute for Integrated Cell-Material Sciences (WPI-iCeMS), Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 9th October 2024
Abstract
The first coordination shell is considered crucial in determining the performance of single atom catalysts (SACs), but the significance of the second coordination shell has been overlooked. In this study, we developed a post-doping strategy to realize predictable and controlled modulation on the second coordination shell. By incorporating a P atom into the second coordination shell of a porphyrin-like Pt SAC, the charge density at the Fermi level of Pt single atom increases, enhancing its intrinsic activity. Moreover, the P atom shows stronger adsorption towards large size anions (ClO4−) than Pt atoms, preventing the Pt site poisoning in acid. As a result, the Pt–N4P–C catalyst exhibits significantly higher activity than the Pt–N4–C catalyst. It even outperforms commercial Pt/C (20 wt% Pt) with a Pt content of only 0.22 wt% in both alkaline and acidic solutions. This work indicates the second coordination shell modulation also greatly impacts the performance of SACs.
Introduction
Single atom catalysts (SACs) have gained significant attention in catalysis due to their high atomic utilization efficiency and excellent catalytic performance.1–6 The intrinsic activity of SACs is typically attributed to the catalytic centers and their first coordination environment.7 However, the influence of the second coordination shell, which is further from the active centers, is often overlooked. Understanding the extent to which the second coordination atoms can impact the performance of single atom catalysts is a topic that requires further exploration. If the second coordination shell has a significant effect, it could provide an additional avenue for optimizing the performance of SACs, in addition to the regulation of catalytic centers and first coordination shell.To investigate the impact of the secondary coordination shell on catalytic performance, the first coordination shell should be relatively stable. In such cases, modulation of the secondary coordination shell would not alter the first coordination environment. However, most studies have focused on regulating the metal catalytic centers and the first coordination shell,1,8 while accurate modulation of the second coordination shell poses more challenges. When two or more metallic atoms are doped, it is relatively easy to form a bimetallic single atom catalyst.9–11 Similarly, when two or more non-metallic atoms are doped, a monometallic single atom catalyst with heterogeneous non-metallic coordination in the first shell can be easily formed.12–14 There are a few experimental and theoretical studies that have investigated the simultaneous regulation of the first and second coordination shells, making it difficult to separate the individual impacts of these environments on catalyst performance.12,15 So far, the full extent of the importance of the second coordination shell has yet to be fully elucidated and warrants further exploration.
Compared to the MN4 (M = Fe, Co, Ni, Mn, Cu) structured transition metal single atoms, noble metal atoms such as Pt, Ir, and Ru can form a more stable porphyrin-like MN4 structured SACs, where four nitrogen atoms stably bond with the metal atoms.16 Additionally, compared to transition metal atoms, single noble metal atoms in the M–N–C structure have higher intrinsic catalytic activity and can effectively prevent the Fenton reaction during the oxygen reduction reaction (ORR).7,16 Furthermore, the noble metal content in this type of catalyst is extremely low, minimizing its impact on cost.17 Therefore, MN4 structured catalysts consisting of single noble metal atoms are highly significant as ORR catalysts. The regulation of the second coordination environment in these catalysts holds crucial practical importance for the development of energy devices related to ORR.
Among all noble metals, Pt has been widely recognized as the best catalyst for ORR, exhibiting higher catalytic activity compared to other noble metal-based catalysts with porphyrin-like MN4 (M = Ir, Ru, Pd) structure.16 In this study, we focused on regulating the second coordination shell of porphyrin-like PtN4-based catalysts and investigating its influence on ORR performance. Notably, among all reported PtNx structured catalysts, the PtN4 coordination structure has demonstrated the highest ORR activity in basic conditions. With a 3.8 wt% Pt single atom loading, it shows a half-wave potential (E1/2) 40 mV greater than that of Pt/C (20 wt%).17–19 However, under acidic conditions, the catalytic activity of PtN4 catalysts is as low as nitrogen-doped carbon.18 In other words, PtN4 structured single atom catalysts are completely deactivated under acidic conditions. In our study, we discovered that the doping of P atoms in the second coordination shell of PtN4 catalysts further improved their catalytic activity in basic solutions, and remarkably, the catalytic activity of Pt single atoms was restored under acidic conditions. With a mere 0.22 wt% Pt content, the half-wave potential E1/2 of the catalyst was 0.91 V and 0.81 V in basic and acidic conditions, respectively, which significantly surpassed that of PtN4 catalysts (0.88 V, 0.68 V). Our findings demonstrate that the regulation of the second coordination shell plays a critical role in the catalytic performance of Pt single atoms, particularly under acidic conditions.
Results and discussion
Theoretical simulations were conducted to predict the modulation of the second coordination shell of PtN4 by P atoms. It was observed that when P atoms were doped into the first coordination shell of the PtN4 structure, the resulting PtN4−xPx structures exhibited binding energies greater than 0 eV (Fig. 1). This indicates that these structures were not thermodynamically stable. In other words, the P atoms did not replace the nitrogen atoms in the first coordination shell of Pt single atoms. This is in contrast to the behavior observed in transition metal single atoms (such as Fe, Co, Mo, etc.), where the first coordination shell can be easily modulated.12,20 The theoretically optimized structures for the Pt-SACs are revealed as PtC4 and PtN4 without disturbing the first coordination spheres. Therefore, if P atoms were doped into the sample, they would not disrupt the first coordination shell but rather surround the porphyrin-like PtN4 structure.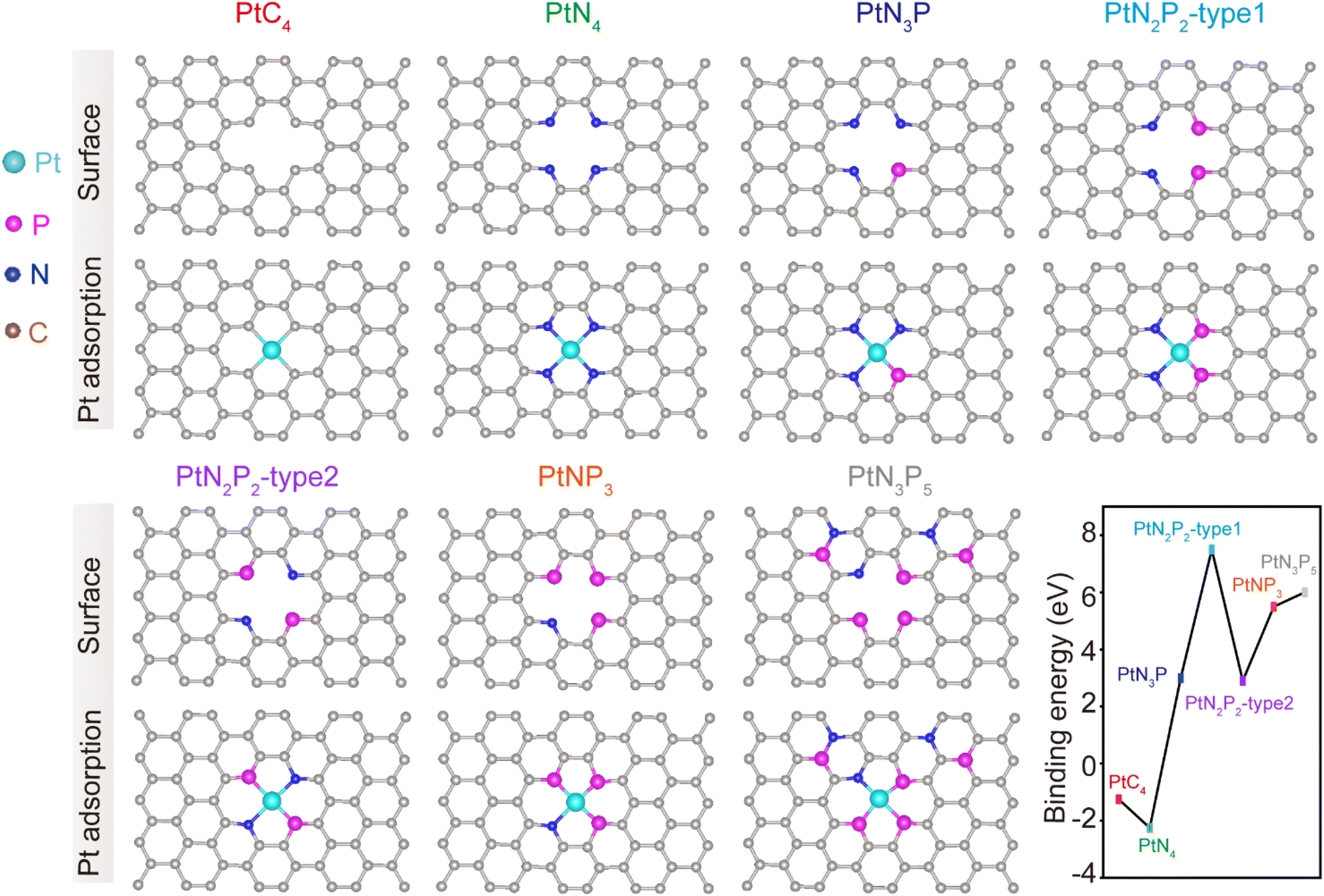 | ||
| Fig. 1 Prediction of the structural stability of Pt single atoms with different first coordination environment and calculated binding energy of Pt atom anchored on the doped-graphene (PtC4, PtN4, PtN3P, PtN2P2-type 1, PtN2P2-type 2, PtNP3, PtN3P5) monolayer. All energies are given in eV. | ||
In order to experimentally demonstrate the feasibility of modulating the second coordination shell of PtN4 with P atoms, we synthesized a P doped PtN4 catalyst using the following procedure (Fig. 2a). First, a solution containing Pt salt, Zn salt, and 2-methyl imidazole (2-MeIM) was prepared with methanol. This solution was then used to synthesize ZIF-8 with Pt atoms trapped inside (referred to as Pt@ZIF-8). Next, the Pt@ZIF-8 material was carbonized to form porphyrin-like Pt single atoms embedded in a nitrogen doped carbon substrate (Pt-NC). Finally, the Pt-NC sample was washed with phytic acid and subjected to a carbonization process to create the Pt-PNC catalyst with P atoms doped into the second coordination shell. As a control, Pt-NC and PNC were also prepared via the same process except that no P and Pt were added, respectively.
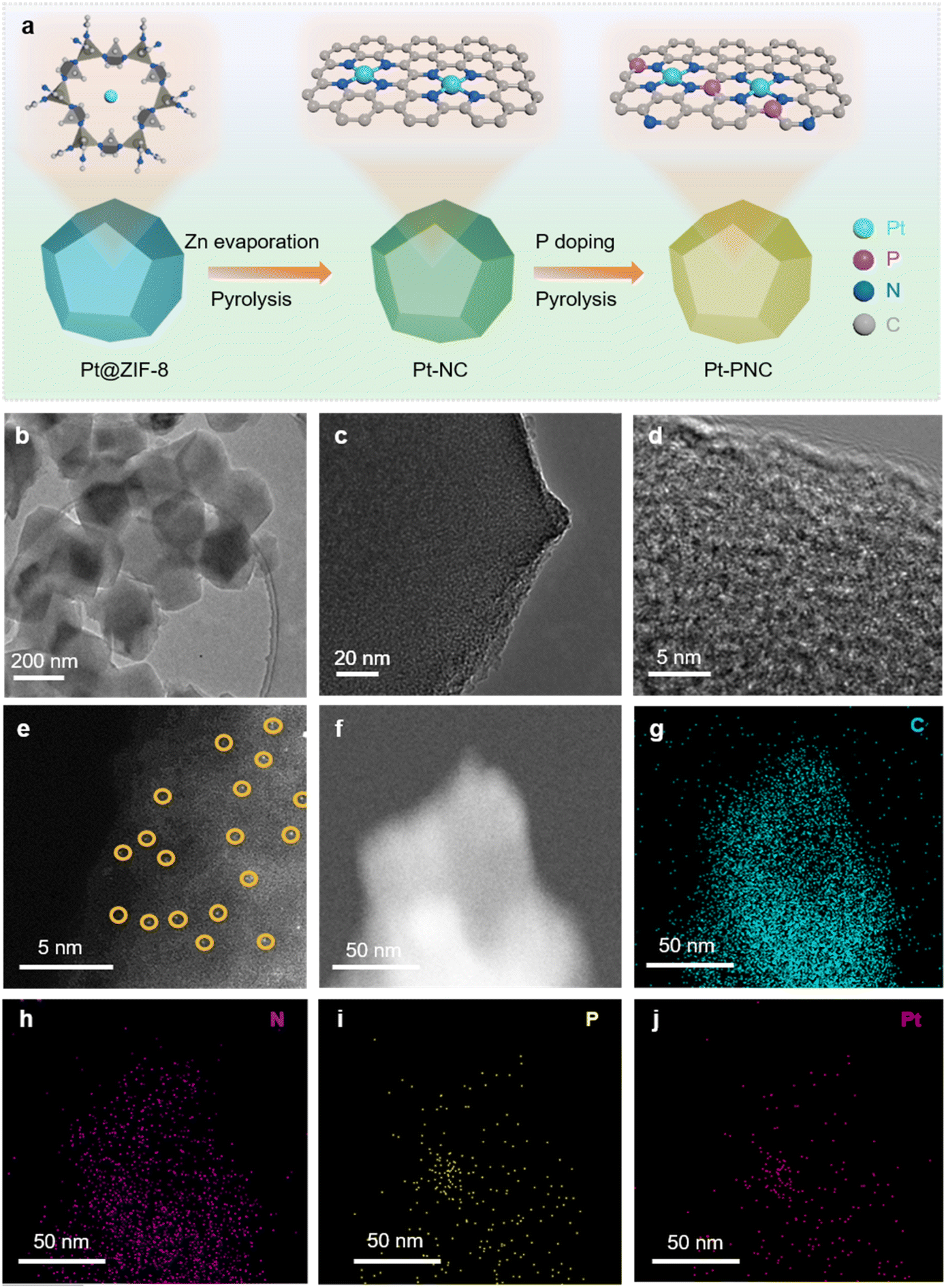 | ||
| Fig. 2 TEM characterization of Pt-PNC. (a) Graphical illustration for the synthesis of Pt-PNC. (b–d) TEM and HRTEM images of Pt-PNC. (e) AC-STEM image of Pt-PNC, the encircled white dots represent Pt single atoms. (f–j) HAADF-STEM and the corresponding EDX elemental mapping images of Pt-PNC. | ||
To optimize the Pt content, Pt@ZIF-8 samples with different Pt loadings were prepared. X-ray diffraction (XRD) analysis of the as-synthesized Pt-NC and Pt-PNC samples (Fig. S1 and S2†) confirmed the absence of peaks corresponding to metallic Pt nanoparticles. The broad peaks observed in the XRD patterns of Pt-NC and Pt-PNC can be attributed to the carbon substrates.
Inductively coupled plasma (ICP) analysis was performed to determine the Pt contents in the catalysts with different loadings (Table S1†). The results showed that the Pt content in the Pt-PNC sample gradually increased from 0.09 wt% to 0.33 wt% with an increase in the Pt precursor from 1 wt% to 5 wt%. These samples exhibited a similar morphology to the original Pt@ZIF-8, indicating that the synthetic process of Pt-PNC did not disrupt the integrated structure of ZIF-8 (Fig. S3 and S4†). An initial assessment of the ORR performance of the samples with different Pt contents in alkaline medium (Fig. S5†) revealed that the Pt-PNC sample with 0.22 wt% Pt exhibited the highest E1/2. However, increasing the Pt content from 0.22 wt% to 0.33 wt% resulted in a decrease in ORR performance, which is consistent with some previously reported results. This decrease in performance can be attributed to the increased defects in the carbon framework, leading to reduced electrical conductivity of the carbon substrates (Fig. S6†).21,22 In the following sections, we focused on the Pt-PNC sample with the optimal Pt concentration of 0.22 wt%.
Based on the transmission electron microscopy (TEM) image in Fig. 2b, the Pt-PNC sample exhibited a similar morphology to the original ZIF-8 particles. The high-resolution TEM (HRTEM) image (Fig. 2c) revealed the absence of Pt nanoparticles in the Pt-PNC sample. Instead, amorphous structures were observed. This result was consistent with the XRD results, indicating the destruction of the original crystal structure of ZIF-8 in Pt-PNC (Fig. 2c and d). The dark-field TEM image showed white spots, indicating the mono-dispersion of Pt atoms throughout the Pt-PNC sample (Fig. 2e). Energy dispersive X-ray (EDX) mapping confirmed the homogeneous distribution of C, N, P, and Pt elements in the entire sample (Fig. 2f–j). This suggests that the P, N, and C elements within the supports reacted with the Pt atoms, resulting in their homogenization within the framework during the carbonization process.
To further understand the exact coordination environment of Pt single atoms in Pt-PNC, X-ray photoelectron spectroscopy (XPS) was used to characterize the bonding environment of each element. Narrow scans of each element in Pt-NC and PNC catalysts were also studied for comparison. The survey scans of the three samples revealed the atomic ratio within them (Table S2†). Fig. 3a shows the N 1s spectra, which exhibit four main peaks corresponding to oxidized graphitic-N, graphitic-N, pyrrolic-N, and pyridinic-N.23,24 Compared to PNC, both Pt-PNC and Pt-NC samples exhibit an additional peak at 399.2 eV, which is typically attributed to Pt–N bonds.25 The slightly higher ratio of graphitic-N in Pt-PNC and PNC compared to Pt-NC can be attributed to the high annealing temperature during the P doping process. The P 1s spectra (Fig. S7†) do not show any peaks corresponding to Pt–P bonds around 129.8 eV, indicating that the P atoms probably not be directly connected to the Pt atoms.26,27 However, P–N and P–C bonds can be observed, indicating that the P atoms are directly connected to the N atoms and located in the second coordination shell. The intensity ratio of P–N bonds to P–C bonds is approximately 1/3, suggesting that the P atoms are connected to one N atom and three C atoms. It is important to note that XPS is a qualitative analysis method, and further evidence is required to confirm this conclusion. The high-resolution Pt 4f analysis (Fig. 3b) indicates the absence of metallic Pt single atoms in both Pt-PNC and Pt-NC samples, and Pt2+ species were mainly present. This further supports the presence of a predominately four coordination PtN(P)4 structure.18,28 Compared to Pt-NC, the Pt-PNC sample exhibits a shift in peak position towards a higher binding energy, indicating that the presence of P atoms can influence the electronic structure of the Pt atoms.13
 | ||
| Fig. 3 Spectroscopic characterization of Pt-PNC. (a) High-resolution N 1s spectra for PNC, Pt-NC, and Pt-PNC. (b) High-resolution Pt 4f spectra for Pt-NC and Pt-PNC. (c) Pt-L3 edge XANES spectra of Pt-PNC, Pt-NC, PtO2, and Pt-foil. (d–g) Wavelet transforms for the k3-weighted Pt K-edge XAFS signals of PtO2, Pt-foil, Pt-NC, and Pt-PNC. (h) k2-weighted FT-XAFS of Pt-PNC, Pt-NC, PtO2, and Pt-foil. (i) Corresponding EXAFS fitting in k space foil for Pt-PNC. | ||
The X-ray absorption near edge structure (XANES) analysis of Pt L3-edge was conducted to determine the structure of the first coordination shell for Pt single atoms (Fig. 3c). The peak heights of Pt-NC and Pt-PNC are similar to the average peak heights of PtO2 and Pt foil, indicating that the Pt atoms exist in a 2+ charge state in both Pt-NC and Pt-PNC. This result suggests that the P doping does not alter the coordination number of the Pt atoms. Wavelet transform (WT) analysis was performed to discriminate the backscattering atoms and provide resolution in both R and k spaces (Fig. 3d and h). The WT contour plots of Pt-PNC and Pt-NC show intensity maxima at approximately 4.0 Å, which is similar to the intensity maximum of PtO2. This suggests that the intensity in Pt-PNC and Pt-NC arises from the backscattering between Pt and light atoms, such as P, N, and O. In contrast, the intensity maximum for Pt foil is observed at approximately 9.5 Å, corresponding to Pt–Pt bonds, which are not observed in Pt-PNC and Pt-NC. These results confirm that the Pt species are atomically dispersed in Pt-PNC and Pt-NC.
X-ray absorption fine structure (XAFS) spectroscopy was utilized to identify the exact coordination environment of Pt single atoms in Pt-PNC and Pt-NC (Fig. 3h). It shows only one prominent peak at 1.56 Å, corresponding to the Pt–N bond length, and no peaks corresponding to Pt–Pt (2.5 Å) and Pt–P (2.25 Å) bonds are observed.29,30 This confirms that Pt exists in the form of single atoms and only coordinates with N atoms. The Pt–N bond length in Pt-PNC is slightly smaller compared to Pt-NC, indicating that P doping can slightly modulate the coordination environment of Pt single atom. The fitting of the XAFS spectra in R space (Fig. 3i) shows a coordination number of 3.6 and 3.7 for Pt-NC and Pt-PNC, respectively (Table S3†). This confirms that the Pt single atoms in both materials are coordinated with four N atoms, forming a porphyrin-like PtN4 structure. The slight influence of P atoms on the coordination environment suggests that they are located in the second coordination sphere of Pt single atoms. However, the exact location of P atoms needs further investigation.
Electrochemical performance
The electrochemical performance of the synthesized samples, Pt-PNC and Pt-NC, was evaluated using rotating disk electrode (RDE) tests for the ORR in both basic (0.1 M KOH) and acidic (0.1 M HClO4) solutions. In 0.1 M KOH solution, the onset potential (Eonset) and half-wave potential (E1/2) for Pt-PNC are 1.04 V and 0.91 V vs. reversible hydrogen electrode (RHE), respectively, which are higher than those of the commercial Pt/C (Pt 20 wt%) with an Eonset and E1/2 of 1.03 V and 0.88 V (Fig. 4a and b). In contrast, Pt-NC and PNC show an E1/2 of 0.88 V and 0.76 V in 0.1 M KOH. The kinetic current density (Jk) of each catalyst at 0.9 V vs. RHE follows the same order: Pt-PNC > Pt/C > Pt-NC > PNC, in terms of specific activity (Fig. 4b).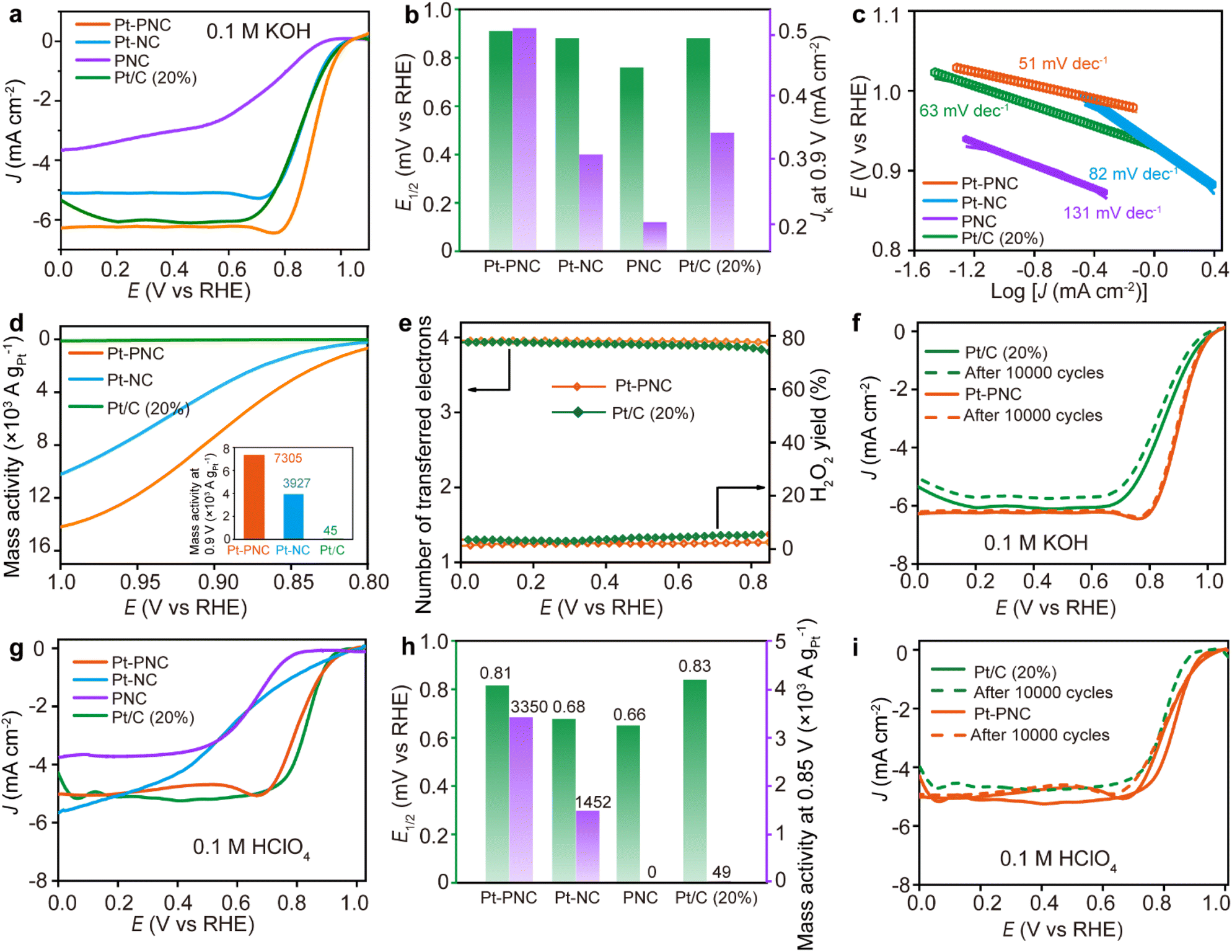 | ||
Fig. 4 ORR performance of Pt-PNC in alkaline and acidic media. (a) LSV curves of Pt-PNC, Pt-NC, PNC, and Pt/C in 0.1 M KOH. (b–d) Comparative study of half-wave-potential and specific activity, Tafel plots, mass activity calculations for Pt-PNC, Pt-NC and Pt/C (inset: their mass activity at 0.9 V vs. RHE). (e) Electron transferred number calculated from RRDE test and corresponding peroxide yield. (f) Stability test carried out for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 CV cycles in 0.1 M KOH. (g) LSV curves of Pt-PNC, Pt-NC, PNC, and Pt/C in 0.1 M HClO4. (h) Comparative study of half-wave-potential and mass activity. (i) Stability test carried out for 10000 CV cycles in 0.1 M HClO4. 000 CV cycles in 0.1 M KOH. (g) LSV curves of Pt-PNC, Pt-NC, PNC, and Pt/C in 0.1 M HClO4. (h) Comparative study of half-wave-potential and mass activity. (i) Stability test carried out for 10000 CV cycles in 0.1 M HClO4. | ||
The Pt-PNC exhibits a Tafel slope of 51 mV dec−1, which is much smaller than that of Pt/C (63 mV dec−1), Pt-NC (82 mV dec−1), and PNC (131 mV dec−1) based on the linear scanning voltammetric (LSV) curves (Fig. 4c). This indicates that the doping of a small amount of P atoms greatly enhances the ORR reaction kinetics of Pt-PNC. It is important to note that the mass loading of Pt in Pt-PNC and Pt-NC is only 0.22 wt% according to ICP measurement, which is approximately 1% of the Pt content in Pt/C, but shows comparable or even better ORR performance. Therefore, we compared the mass activity of the Pt-PNC and Pt-NC samples with the Pt/C. In Fig. 4d, we can easily find the Pt-PNC and Pt-NC show much higher mass activity than the Pt/C at the whole range of 0.8–1.0 V vs. RHE. Specifically, the mass activity of Pt-PNC and Pt-NC at 0.9 V vs. RHE is 7305 and 3927 A gPt−1, which is 161 and 86 times higher than that of Pt/C (45 A gPt−1). This highlights the superior performance of Pt-PNC in terms of mass activity (Fig. 4c). N2 adsorption–desorption measurements reveal high specific surface areas (BET) of 984.1 m2 g−1, 1126.2 m2 g−1, and 1057.4 m2 g−1 for Pt-PNC, Pt-NC, and PNC, respectively (Fig. S8†). The pore size distribution for all three catalysts is centered at 1–5 nm. This suggests that the improved activity of Pt-PNC compared to Pt-NC is not solely attributed to a larger number of Pt catalytic sites, but rather to the higher activity of each Pt site.23 The turnover frequency (TOF) values obtained at 0.9 V for Pt-PNC, Pt-NC, and Pt/C are 15.0, 8.1, and 0.092 e−1 per site s−1, respectively, much higher than the previously reported catalysts (Table S4†). This indicates that P doping greatly enhances the intrinsic ORR activity of Pt single atoms in Pt-PNC.
The ORR pathway on the highly efficient Pt-PNC was firstly investigated by calculating the electron transfer number per oxygen molecule (n) using the RDE technique (Fig. S9†). The Koutecký–Levich (K–L) plots displayed linear relationships at different applied voltages for Pt-PNC (vs. RHE), from which n was determined to be approximately 3.9. Additionally, the ORR was also studied by coating the Pt-PNC catalyst on a rotating ring-disk electrode (RRDE) to study the ratio of each reaction pathway. The Pt-PNC electrocatalyst exhibited an electron transfer number of 3.95–3.98 in the potential range of 0–0.8 V vs. RHE (Fig. 4e and S9†). Therefore, both the RDE and RRDE results indicate the efficient reduction of oxygen to OH− through a four-electron pathway.31 The Pt-PNC displayed a low percentage of peroxide production (<3%), which is a production of two electron reduction pathway, in the potential range of 0–0.8 V vs. RHE, whereas Pt/C (20 wt% Pt) produced peroxide levels exceeding 4% in the same potential range (Fig. 4e). These findings suggest that the superior ORR selectivity of Pt-PNC compared to Pt/C, enabling the efficient progression of the four-electron pathway. Considering the low loading of Pt and its superior overall performance, Pt-PNC can be considered as an efficient ORR catalyst in an alkaline medium compared to other state-of-the-art catalysts reported in the literature (Table S5†).
The stability of Pt-PNC in an alkaline medium is investigated by cycling the electrode between 0.6–1.2 V vs. RHE at a scanning rate of 50 mV s−1 in 0.1 M KOH solution (Fig. 4f). The results demonstrate the good stability of Pt-PNC, as it only exhibits a 5 mV decrease in E1/2 after 10000 cyclic voltammetry (CV) cycles, which is significantly smaller than the 20 mV decrease observed for Pt/C. The XRD pattern and Raman spectrum of Pt-PNC after stability test revealed that the structure were maintained with no significant changes (Fig. S10†).
This improved stability of Pt-PNC compared to Pt/C can be attributed to the better structural stability of the PtN4 configuration in the carbon substrate compared to Pt nanoparticles.32
The ORR performance of Pt-PNC in acidic medium was also investigated in 0.1 M HClO4. Both Pt-PNC and Pt-NC exhibited an onset potential (Eonset) of 0.98 V vs. RHE, which is higher than that of Pt/C (0.94 V). However, the E1/2 for Pt/C, Pt-PNC, and Pt-NC was measured to be 0.83 V, 0.81 V, and 0.68 V vs. RHE, respectively. Notably, the E1/2 value for Pt-NC dropped to a value close to that of PNC (0.66 V). This is consistent with previous literature reports (Table S6†),33–35 where porphyrin-like Pt single atoms anchored onto NC demonstrated better ORR performance than Pt/C in alkaline medium but exhibited a similar E1/2 value to NC in acidic medium.18 These results suggest that the Pt catalytic sites in Pt-NC have been deactivated in acidic medium.
In contrast, the introduction of P atoms to the second coordination shell in Pt-PNC helps to prevent the complete deactivation of Pt catalytic sites in acidic conditions. However, unlike the situation in alkaline medium where Pt-PNC exhibited a 30 mV higher E1/2 than Pt/C, the E1/2 for Pt-PNC (0.81 V) is slightly lower than that of Pt/C (0.83 V) in acidic medium. This indicates that the presence of P atoms cannot fully counteract the detrimental effect of the acid on the ORR activity of the PtN4 structure.
However, despite the slight decrease in E1/2 compared to Pt/C in acidic medium, the mass activity at 0.85 V for Pt-PNC is still 67 times higher than that of Pt/C (Fig. 4h). The RRDE test revealed an electron transfer number of 3.8–3.93 for the Pt-PNC electrocatalyst at 0.2–0.8 V vs. RHE, slightly higher than that of Pt/C in the low potential range. Additionally, the H2O2 yield for Pt-PNC is consistently below 5% across the entire potential range, which is lower than that of Pt/C. This indicates that Pt-PNC still proceeds the ORR through the four-electron pathway, forming H2O molecules rather than H2O2 (Fig. S11†).
Furthermore, Pt-PNC exhibits excellent cycling stability in the acidic solution. After 10000 CV cycles in 0.1 M HClO4, Pt-PNC shows no change in E1/2 (0.81 V), while the E1/2 of Pt/C decreases by 34 mV to 0.80 V (Fig. 4i). Therefore, the P doping in Pt-NC provides it with good stability, making Pt-PNC a superior catalyst to Pt-NC and Pt/C in both alkaline and acidic solutions.
In order to elucidate the influence of the P atom on the activity and anti-toxicity of PtN4, we conducted DFT simulations to analyze the ORR process on the PtN4P structure. Initially, we calculated the energy diagram by placing the P atom at six different sites beyond the first coordination shell in alkaline medium (Fig. 5a and c and S12†). These configurations are denoted as PtN4Px (x = 1, 2, 3, 4, 5, 6). We observed that only the three configurations with the P atom on the second coordination shell exhibit P–N bonds, which is consistent with the structural characterization. Moreover, only PtN4P1 and PtN4P2 demonstrate lower energy barriers compared to PtN4 (Fig. 5d and S12†), indicating that the real structure for PtN4P arises from these configurations.
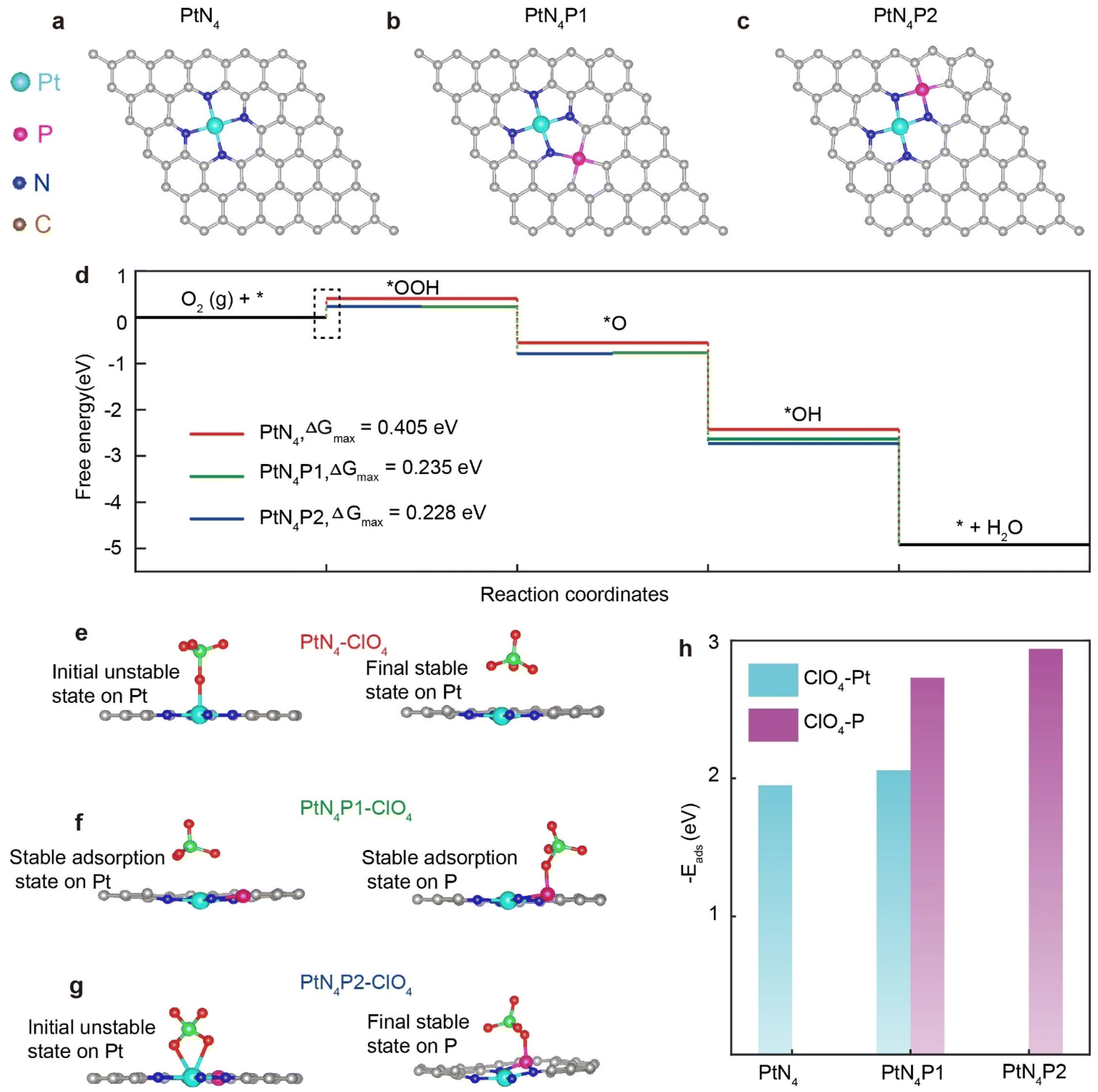 | ||
| Fig. 5 Theoretical calculation to identify the P location in the second coordination shell and its influence on the activity and anti-toxicity of PtN4 structure. (a–c) Structural models of PtN4, PtN4P1 and PtN4P2. (d) Energy diagrams of the three models for ORR in the alkaline medium. (e–g) Adsorption model of the ClO4− on the PtN4, PtN4P1, and PtN4P2 structures. (h) The adsorption energy of ClO4− on stable atomic sites of PtN4, PtN4P1 and PtN4P2. | ||
To further understand the enhanced ORR activity of PtN4P1 and PtN4P2 compared to PtN4, we conducted calculations of the charge density difference (CDD) for these structures. The CDD analysis (Fig. S13a–c†) reveals significant changes in the charge density distribution surrounding the Pt atom upon the introduction of the P atom. In the PtN4P structure, the presence of P atom leads to the delocalization of charges on the N atoms in the Pt–N bonds. This results in an increased charge density on the Pt atoms, facilitating proton adsorption on the Pt single atoms. As a consequence, the energy barrier for the rate-determining step (from *O2 to *OOH) is reduced from 0.405 eV for PtN4 to 0.235/0.228 eV for PtN4P1/PtN4P2, respectively. These findings indicate that the incorporation of the P atom in the second coordination shell of PtN4 significantly influences the charge distribution and facilitates proton adsorption, ultimately leading to a lower energy barrier for the rate-determining step of ORR process.
In order to investigate the influence of the P atom on the electronic structure of PtN4, we calculated the spin-polarized partial density of states (DOS) projected on Pt-5d, 6s, C-2p, N-2p, and P-3p orbitals in both models. The results, shown in Fig. S13d–f,† indicate strong hybridization between the Pt-5d, 6s orbitals and the N-2p and P-3p orbitals. This demonstrates that all these orbitals can affect the electronic structure of the Pt atoms. By introducing the P atom into the second coordination shell, the d band center of PtN4 structure only slightly shifted. However, the charge density at the Fermi level of the carbon atoms is significantly higher in PtN4P compared to PtN4, leading to the much easier adsorption of O2 molecules on the PtN4P, thereby facilitating the ORR kinetics. Therefore, although the P atom is located in the second coordination shell of the Pt single atom, it still significantly promotes the intrinsic activity of PtN4 by increasing the adsorption of O2 molecules and reducing the energy barrier for the proton adsorption.
The reason why porphyrin-like Pt single atom catalysts are prone to full poisoning in acidic solutions but exhibit good activity in basic solutions is still not fully understood.18 In this study, we found that the modulation of the second coordination shell by the P atom can prevent the complete poisoning of Pt single atoms and achieve comparable performance to Pt/C (20 wt% Pt) catalysts. To gain insight into the anti-toxicity mechanism of P modulation in acidic solutions, we first analyzed the differences between basic and acidic solutions. Acidic solutions contain large-sized anions such as ClO4−, HSO4−, and NO3− in addition to protons. On the other hand, basic solutions only contain small-sized alkaline metal ions (Li+, Na+, K+) and OH− anions. Based on this observation, we deduce that the poisoning of PtN4 structure in acid is likely related to the presence of large anions in the solution. These anions may interact strongly with the Pt single atoms, leading to the deactivation of the catalyst. In contrast, the absence of such large anions in basic solutions may contribute to the higher stability and activity of the Pt–N4–C structure.17
To substantiate the above inference, we simulated the interaction between Pt single atoms and large anions in acidic solutions (Fig. 5e–g). Indeed, the presence of large anions from acid solvation, such as ClO4−, can compete with ORR intermediates (*OOH and *OH) for adsorption sites on the catalyst. To further understand the excellent performance of PtN4P1 and PtN4P2 in acidic environments, we investigated the adsorption behavior of ClO4− on pristine PtN4, PtN4P1, and PtN4P2 and proposed a possible mechanism. As shown in Fig. 5e, when ClO4− is adsorbed on PtN4, it tends to occupy the position right above the Pt atom, effectively blocking these active sites from other ORR intermediates. However, the introduction of the P atom in the second coordination sphere breaks the symmetry of the PtN4 site. This results in a preference for ClO4− adsorption on the P site (Fig. 5f and g).
Specifically, the adsorption energy of ClO4− on the P site is significantly higher than that on the Pt site in PtN4P1 (Fig. 5h). In the case of PtN4P2, even if ClO4− is pre-adsorbed on the Pt site, it will spontaneously move to the P site (Fig. 5g). Consequently, the free Pt sites that are not covered by ClO4− remain available for ORR. Therefore, both the PtN4P1 and PtN4P2 can help to avoid the coverage of the Pt catalytic sites by ClO4−, but the PtN4P1 can only partially avoid this coverage and the PtN4P2 can fully avoid this coverage. It should be noted that there is a partial decrease in activity in our experiments, which indicates the performance of PtN4P1 is more consistent with the real catalyst. This explanation should also be extended to other scenarios where there is competition between active sites for ORR intermediates and anions from the acidic environment. Further, the adsorption energies for the *OH were also calculated for the respective active sites i.e., Pt and P (Fig. S14†). The negative value of the binding energy (−3.4 eV) at site A suggests that the *OH adsorption on Pt is more energetically favourable compared to adsorption on the P-atom at site B. Pt exhibits strong OH binding, essential for catalytic activity, while P-modification helps in fine-tuning this interaction for enhanced performance in ORR applications.
Zn–air battery performance
This new Pt-PNC catalyst exhibits excellent ORR performance, good stability, and low noble metal content, and its practical application potential lies in ORR-related devices. In this study, we utilized the Pt-PNC catalyst as the cathode catalyst in a Zn–air battery (ZAB) setup (Fig. 6a). The Pt-PNC-based ZAB demonstrated an open circuit voltage of 1.45 V, higher than that of the Pt/C catalyst (1.39 V) (Fig. 6b and c). Additionally, the peak power density of the Pt-PNC-based ZAB reached 246 mW cm−2, significantly outperforming the Pt/C-based ZAB (192 mW cm−2 (Fig. 6d)).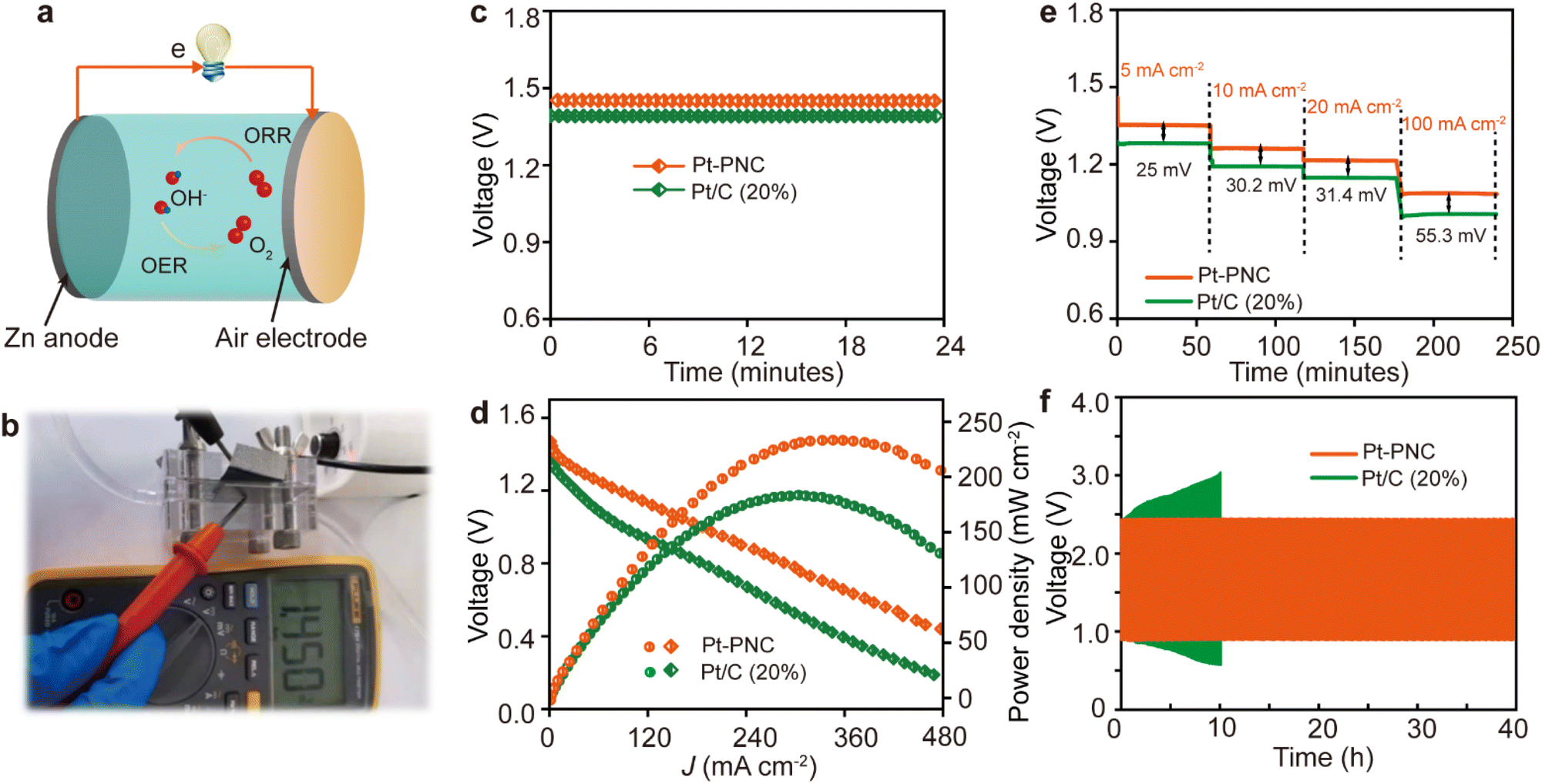 | ||
| Fig. 6 ZAB application of Pt-PNC. (a) Schematic illustration of ZAB. (b) OCP photograph of Pt-PNC-based ZAB. (c) Open circuit voltage curves. (d) Discharge curves and corresponding peak power density plots. (e and f) Galvanostatic discharge curves at varying current densities (5, 10, 20, and 100 mA cm−2) and cycling stability test at 10 mA cm−2 for the ZABs with different catalysts. | ||
To evaluate the performance at different discharge current densities, we analyzed the voltage gap. As the current densities increased from 5, 10, 20, to 100 mA cm−2, the voltage gap increased from 25, 30.2, and 31.4 to 55.3 mV for the Pt-PNC-based ZAB, indicating its smaller polarity and higher reaction kinetics compared to the Pt/C-based ZAB (Fig. 6e). Furthermore, we assessed the stability performance of the Pt-PNC-based ZAB by cycling it at a current density of 10 mA cm−2 (Fig. 6f). After 40 hours of operation, the Pt-PNC-based ZAB showed only a 14 mV potential increase from the original voltage. In contrast, the Pt/C-based ZAB experienced a significant increase in operation potential, highlighting the superior stability of the Pt-PNC-based ZAB. Overall, although the performance of the Pt-PNC catalyst in the ZAB is impressive, further optimization and investigation are necessary to fully exploit its potential in practical applications.
Experimental
Synthesis of Pt-NC
3 g of Zn(NO3)2·6H2O was added into 80 ml of methanol solution containing 6.5 g of 2-methyl imidazole (2-MeIM). Then, acetylacetonate platinum ((C5H7O2)Pt) with varying concentration (1%, 2%, 3%, 4%, 5%) was added into the solution, followed by 24 h stirring at room temperature. The samples were collected after washing with methanol and dried in an oven at 60 °C overnight. The Pt-NC-x (x = 1 to 5) was obtained after carbonization the samples in Ar/H2 (5 at% H2) atmosphere for 2 h at 950 °C.Synthesis of Pt-PNC
0.1 g of Pt-NC was added into 20 ml mixed solution (water:ethanol) and sonicated for 15 minutes. Then, 4 ml phytic acid was added into the solution and stirred for 12 h at room temperature. After that, the suspension was washed with water and ethanol, and dried in an oven at 60 °C overnight. The collected sample was carbonized in an Ar/H2 atmosphere for 1 h at 950 °C to produce Pt-PNC.
Synthesis of NC
3 g of Zn(NO3)2·6H2O was added into 80 ml of methanolic solution containing 6.5 g of 2-MeIM, followed by stirring at room temperature for 24 h. The sample was collected after washing with methanol and dried in an oven at 60 °C overnight. The NC was obtained after carbonization in an Ar/H2 atmosphere for 2 h at 950 °C.Synthesis of PNC
0.1 g of NC was added into 20 ml mixed solution (water:ethanol), and sonicated for 15 minutes. Then, 4 ml phytic acid was added into the solution and stirred for 12 h at room temperature, followed by washing with water and ethanol, dried in an oven at 60 °C overnight. The collected sample was carbonized in an Ar/H2 atmosphere for 1 h at 950 °C to produced PNC.
Electrochemical measurements
Autolab electrochemical work station was used to carry out all the tests for ORR. The electrochemical measurements were taken by three electrode method, where working electrode was glassy carbon rotating disk, counter electrode was graphitic carbon rod and reference electrode was Ag/AgCl electrode. The electrolyte used was 0.1 M KOH/HClO4 purged with N2/O2. For all the catalytic electrodes, the catalyst loading was 0.2 mg cm−2. The cyclic voltammetry (CV) and linear sweep voltammetry (LSV) curves were taken at a scanning rate of 50 and 10 mV s−1, respectively.Zinc–air battery (ZAB) performance measurement
Home-built electrochemical cell was used to measure the ZAB performance, which comprises of two electrodes. A catalyst-loaded tri-layer composite (carbon paper, PTFE waterproof layer, and Ni foam) works as the cathode, Zn foil works as the anode and 6 M KOH was used as the electrolyte (galvanostatic discharge tests). The area of the electrodes exposed to the electrolyte is 1.0 cm2. While electrolytic solution of 6 M KOH + 0.2 M zinc acetate used for cycling test.The specific capacity of ZAB was calculated based on the equation below:
| Capacity = (service hours × current)/(weight of consumed zinc). |
The energy density of ZAB was calculated based on the equation below:
| Energy density = (service hours × current × average discharge voltage)/(weight of consumed zinc). |
Conclusions
This work successfully demonstrated the modulation of the second coordination shell of the porphyrin-like Pt single atom by introducing a P atom. This P modulation enhances the ORR activity of the Pt single atom and prevents its complete poisoning in acidic media. The PtN4P catalyst, with an ultralow Pt content of 0.22 wt%, exhibits significantly improved ORR performance compared to the PtN4 structure with the same Pt content. In both alkaline and acidic media, the PtN4P catalyst shows higher E1/2 values for ORR compared to the PtN4 structure. Furthermore, despite its ultralow Pt content, the PtN4P catalyst demonstrates comparable E1/2 values to the Pt/C catalyst and exhibits superior cycling stability. The enhanced ORR activity of the PtN4P catalyst can be attributed to the delocalization of electrons on the N atom and the increased charge density on the Pt and C atoms due to the presence of the P atom. This facilitates the adsorption of O2 molecules and lowers the energy barrier for the rate-determining step of ORR, leading to improved reaction kinetics and thermodynamics. Moreover, the P atom can also partially avoid the covering of Pt single atoms by large size anions, such as ClO4−, making the PtN4 catalyst show decent activity in acidic medium. Overall, this work highlights the second coordination shell modulation of single atom catalysts, which could be another feasible route to greatly enhance the comprehensive performance besides the change of metal catalytic centers and the first coordination shell.Data availability
All data associated with this work are available in the article and ESI.†Author contributions
X. C. and Q. X. conceived the project. X. C. provided scientific guidance throughout. T. N., S. S. A. S. and M. S. J. conducted the experiments. H. Y., S. T. and A. D. performed the theoretical modelling. Q. J. helped the synchrotron experiments. T. N., Q. J. synthesized and characterized the samples. T. N., S. S. A. S., Y. D., R. Z., X. C. analyzed the data. T. N., X. C. and Q. X. wrote the manuscript. X. X., S. S. A. S, H. Y. gave inputs during the writing process.Conflicts of interest
The authors declared no conflict of interest.Acknowledgements
The work has been supported by the funding from Natural Science Foundation of China (No. 52373266, 52003163 and 22105129), Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515010670 and 2022A1515011048), Science and Technology Innovation Commission of Shenzhen (No. KQTD20170810105439418, No. 20200812112006001 and No. GJHZ20220913142610020), the National Key Research and Development Project (2023YFA1506601) and Guangdong Grants (2021ZT09C064). T. N. thanks the support from China post-doctoral Science Foundation (2020M672790). The authors appreciate the help from the test center in Shenzhen University to do the TEM characterization for us by using JEOL F200. They also appreciate the help from electron microscopy center in Shenzhen University for the testing the aberration corrected HAADF-STEM.References
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS.
- N. Cheng, L. Zhang, K. Doyle-Davis and X. Sun, Electrochem. Energy Rev., 2019, 2, 539–573 CrossRef.
- S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- L. Hu, W. Li, L. Wang and B. Wang, J. Energy Chem., 2021, 3, 100056 CAS.
- J. Qin, H. Liu, P. Zou, R. Zhang, C. Wang and H. L. Xin, J. Am. Chem. Soc., 2022, 144, 2197–2207 CrossRef CAS.
- Q. An, S. Bo, J. Jiang, C. Gong, H. Su, W. Cheng and Q. Liu, Adv. Sci., 2023, 10, 2205031 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS.
- J. Wang, K. Li, R. Xu, S. Li, Y. Li, M. Xia, J. S. Tse and Z. Wu, Appl. Surf. Sci., 2022, 605, 154832 CrossRef CAS.
- Q. Liu, Y. Li, L. Zheng, J. Shang, X. Liu, R. Yu and J. Shui, Adv. Energy Mater., 2020, 10, 2000689 CrossRef CAS.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-j. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS.
- T. Li, J. Liu, Y. Song and F. Wang, ACS Catal., 2018, 8, 8450–8458 CrossRef CAS.
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Adv. Mater., 2021, 33, 2008599 CrossRef CAS.
- Y. Zhao, Z. Zhang, L. Liu, Y. Wang, T. Wu, W. Qin, S. Liu, B. Jia, H. Wu, D. Zhang, X. Qu, G. Qi, E. P. Giannelis, M. Qin and S. Guo, J. Am. Chem. Soc., 2022, 144, 20571–20581 CrossRef CAS.
- Z. R. Ismagilov, A. E. Shalagina, O. Y. Podyacheva, A. V. Ischenko, L. S. Kibis, A. I. Boronin, Y. A. Chesalov, D. I. Kochubey, A. I. Romanenko, O. B. Anikeeva, T. I. Buryakov and E. N. Tkachev, Carbon, 2009, 47, 1922–1929 CrossRef CAS.
- H. Niwa, M. Kobayashi, K. Horiba, Y. Harada, M. Oshima, K. Terakura, T. Ikeda, Y. Koshigoe, J.-i. Ozaki, S. Miyata, S. Ueda, Y. Yamashita, H. Yoshikawa and K. Kobayashi, J. Power Sources, 2011, 196, 1006–1011 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS.
- H. Kiuchi, T. Kondo, M. Sakurai, D. Guo, J. Nakamura, H. Niwa, J. Miyawaki, M. Kawai, M. Oshima and Y. Harada, Phys. Chem. Chem. Phys., 2016, 18, 458–465 RSC.
- K. Kamiya, R. Kamai, K. Hashimoto and S. Nakanishi, Nat. Commun., 2014, 5, 5040 CrossRef CAS.
- W.-Z. Li, B.-A. Lu, L. Gan, N. Tian, P.-Y. Zhang, W. Yan, W.-X. Chen, Y.-H. Chen, Z.-Y. Zhou and S.-G. Sun, Chin. J. Catal., 2021, 42, 2173–2180 CrossRef CAS.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS.
- J. Liu, M. Jiao, B. Mei, Y. Tong, Y. Li, M. Ruan, P. Song, G. Sun, L. Jiang, Y. Wang, Z. Jiang, L. Gu, Z. Zhou and W. Xu, Angew. Chem., Int. Ed., 2019, 58, 1163–1167 CrossRef CAS.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS.
- S. Zaman, Y.-Q. Su, C.-L. Dong, R. Qi, L. Huang, Y. Qin, Y.-C. Huang, F.-M. Li, B. You, W. Guo, Q. Li, S. Ding and B. Yu Xia, Angew. Chem., Int. Ed., 2022, 61, e202115835 CrossRef CAS.
- T. Najam, S. S. A. Shah, W. Ding, J. Jiang, L. Jia, W. Yao, L. Li and Z. Wei, Angew. Chem., Int. Ed., 2018, 57, 15101–15106 CrossRef CAS.
- M. Xiao, J. Zhu, G. Li, N. Li, S. Li, Z. P. Cano, L. Ma, P. Cui, P. Xu, G. Jiang, H. Jin, S. Wang, T. Wu, J. Lu, A. Yu, D. Su and Z. Chen, Angew. Chem., Int. Ed., 2019, 58, 9640–9696 CrossRef CAS.
- Y. Zhou, R. Lu, X. Tao, Z. Qiu, G. Chen, J. Yang, Y. Zhao, X. Feng and K. Müllen, J. Am. Chem. Soc., 2023, 145, 3647–3655 CrossRef CAS.
- H. Yang, R. Shi, L. Shang and T. Zhang, Small Struct., 2021, 2, 2100007 CrossRef CAS.
- J. Yu, C. Su, L. Shang and T. Zhang, ACS Nano, 2023, 17, 19514–19525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and XRD characterization. Details of XAFS measurements and computational methods. Part of electrochemical characterization. See DOI: https://doi.org/10.1039/d4sc03369h |
| This journal is © The Royal Society of Chemistry 2024 |