
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomer engineering for deep understanding of aggregation-induced photothermal enhancement in conjugated systems†
Peiyang
Gu
 *a,
Tengfei
He
*c,
Zuoyu
Wang
d,
Shifan
Wang
ab,
Liming
Dong
b,
Hanning
Yao
e,
Tao
Jia
*d,
Guankui
Long
c,
Guangfeng
Liu
a and
Hua
Sun
*ab
*a,
Tengfei
He
*c,
Zuoyu
Wang
d,
Shifan
Wang
ab,
Liming
Dong
b,
Hanning
Yao
e,
Tao
Jia
*d,
Guankui
Long
c,
Guangfeng
Liu
a and
Hua
Sun
*ab
aJiangsu Province Engineering Research Center of Biodegradable Materials, School of Petrochemical Engineering, Changzhou University, Changzhou 213164, P. R. China. E-mail: gupeiyang0714@cczu.edu.cn
bSchool of Material and Chemistry Engineering, Xuzhou University of Technology, 2 Lishui Road, Yunlong District, Xuzhou, 221018, China. E-mail: iamsunhua@xzit.edu.cn
cSchool of Materials Science and Engineering, National Institute for Advanced Materials, Renewable Energy Conversion and Storage Center (RECAST), Nankai University, Tianjin, 300350, China. E-mail: hetf2021@nankai.edu.cn
dKey Laboratory of Forest Plant Ecology, Ministry of Education, Engineering Research Center of Forest Bio-Preparation, Heilongjiang Provincial Key Laboratory of Ecological Utilization of Forestry Based Active Substances, College of Chemistry, Chemical Engineering and Resource Utilization, Northeast Forestry University, 26 Hexing Road, Harbin 150040, P. R. China. E-mail: jiataopolychem@nefu.edu.cn
eCollege of Agronomy, Northeast Agricultural University, 600 Changjiang Road, Harbin 150038, P. R. China
First published on 24th July 2024
Abstract
Organic photothermal materials based on conjugated structures have significant potential applications in areas such as biomedical diagnosis, therapy, and energy conversion. Improving their photothermal conversion efficiency through molecular design is critical to promote their practical applications. Especially in similar structures, understanding how the position of heteroatoms affects the conversion efficiency is highly desirable. Herein, we prepared two isomeric small D–A molecules with different sulfur atom positions (TBP–MPA and i-TBP–MPA), which display strong and broad absorption in the UV-visible region due to their strong intramolecular charge transfer characteristics. Compared to i-TBP–MPA, TBP–MPA demonstrates aggregation-induced photothermal enhancement (AIPE). Under simulated sunlight (1 kW m−2) irradiation, the stable temperature of TBP–MPA powder reached 60 °C, significantly higher than the 50 °C achieved by i-TBP–MPA. Experimental and theoretical results indicate that the S⋯N non-covalent interactions in TBP–MPA impart a more rigid conjugated framework to the molecule, inducing ordered molecular stacking during aggregation. This ordered stacking provides additional non-radiative transition channels between TBP–MPA molecules, enhancing their photothermal performance in the aggregated state. Under 1 sun irradiation, TBP–MPA achieved a water evaporation rate of 1.0 kg m−2 h−1, surpassing i-TBP–MPA's rate of 0.92 kg m−2 h−1.
Introduction
The utilization of organic conjugated molecules as carriers for achieving light-to-light, electric-to-light, and light-to-electric conversions has gradually progressed. Breakthroughs have been witnessed in the fields of organic photoluminescence,1 organic electroluminescence,2 and organic photovoltaics.3 However, the conversion of light to heat is often taken for granted. Therefore, research on light-to-heat (photothermal) conversion based on organic conjugated molecules has been relatively limited. Novel applications of photothermal conversion, such as solar-driven water evaporation,4 photothermal therapy,5 and photoacoustic imaging,6 have sparked enthusiasm for studying the photothermal properties of organic conjugated molecules. Two factors must be addressed to achieve efficient photothermal conversion: broad-spectrum light absorption and sufficient non-radiative transition.By endowing organic conjugated molecules with strong intramolecular charge-transfer (ICT) or radical characteristics,7 the absorption spectra can be effectively broadened. We reason that besides efforts on light-absorption efficiency, understanding the photophysical processes, including structure–efficiency relationships for the absorptive and emissive radiative transitions and radiationless transitions (e.g., R + hv → *R, *R → R + hv and *R → R + heat), is a fundamental issue for designing future conjugated molecular systems with good photothermal properties. In addition to molecular structure, the aggregation behavior of organic conjugated molecules and the molecular motion behind the aggregation significantly impact the photophysical processes of the excited state returning to the ground state.8 The flourishing development of aggregation-induced emission (AIE) has led to in-depth research on the relationship between conjugated molecule aggregation behavior and emissive radiative transitions.9 In contrast to the evident luminescence phenomenon, the study of photothermal conversion dominated by dark processes in conjugated molecular systems is not extensive.10 It has recently been demonstrated that enhancing molecular motion in the aggregated state can facilitate the return of excited-state molecules to the ground state through non-radiative transitions, thereby improving photothermal performance.11 Inspired by the reverse consideration of the AIE phenomenon, it is also reasonable to infer that the π–π interactions, which provide diverse non-radiative channels for decay from excited states back to the ground state in the aggregated state, should also be applicable for enhancing the photothermal properties of organic conjugated systems.
Isomers are compounds with the same molecular formula but different photophysical and electrochemical properties. Many studies have been conducted on isomer effects in organic light-emitting diodes,12 organic field-effect transistors,13 and organic photovoltaics.14 Because of the accurate quantitative variation, the isomers provide an ideal platform for a clear study of the mechanism behind the molecular structures and photothermal performances.15 Recently, Zhao et al. reported on covalent organic frameworks (COFs) based on azulene and its structural isomer naphthalene, namely COF-Azu-TP and COF-Na-TP, respectively, and systematically studied their photothermal properties. In comparison to COF-Na-TP, COF-Azu-TP, based on azulene, exhibits a broader optical absorption range from ultraviolet to infrared light and significantly enhanced photothermal performance. This enhancement is primarily attributed to azulene's unique aromatic structure, which provides a narrow bandgap and a large dipole moment.16 In Pt-bridged dimeric complexes, it has been confirmed that configuration isomers also exhibit distinct photophysical properties, which consequently manifest as differences in photothermal performance.17 However, for metal-free organic conjugated systems, the topic of such isomerism is rarely addressed in photothermal conversion. Therefore, studying the effect of isomers on photothermal performance is necessary, as it may provide new insights for developing good-performance photothermal materials.
Herein, because the electronic and aggregation structures are critical to the photothermal properties of organic conjugated systems, we designed and synthesized two regioisomeric organic conjugated small molecules, TBP–MPA and i-TBP–MPA. The 4-methoxy-N-(4-methoxyphenyl)-N-phenylaniline (MPA) unit was chosen as the electron donor and molecular rotor, while dithieno[2,3-a:3′,2′-c]benzo[i]phenazine (TBP) and dithieno[3,2-a:2′,3′-c]benzo[i]phenazine (i-TBP) could not only act as the acceptor units allowing for high electron delocalization but also provide platforms for intermolecular interactions, leading to strong ICT from the donor to acceptor and the formation of the broad-spectrum light absorption. Furthermore, changes in sulfur atom orientation not only affect the electronic structure of molecules but also significantly influence intra/intermolecular interactions. Recently, in the paper by Tang et al., single-crystal diffraction data on TBP derivatives demonstrated that the rigid planar structure of TBP tends to form tight π–π stacking arrangements.18 The S⋯N non-covalent interactions formed between sulfur and nitrogen atoms in the TBP structural unit enhance the rigidity of the conjugated rings,14 suppressing the vibration of the conjugated ring framework, promoting ordered molecular stacking of TBP–MPA and increasing non-radiative transition channels under aggregated states, thereby exhibiting the phenomenon of aggregation-induced photothermal enhancement (AIPE). Under simulated sunlight irradiation (1 sun, 1 kW m−2), the temperature of TBP–MPA powder reached around 60 °C, while the maximum stable temperature of i-TBP–MPA powder was maintained at approximately 50 °C. Despite only differing in the orientation of sulfur atoms, TBP–MPA and i-TBP–MPA exhibited a 10 °C difference in photothermal conversion under simulated sunlight. For a schematic representation of the mechanism, please refer to Scheme 1. For further application of TBP–MPA and i-TBP–MPA, loaded cellulose papers in solar-driven water evaporation showed promising results.
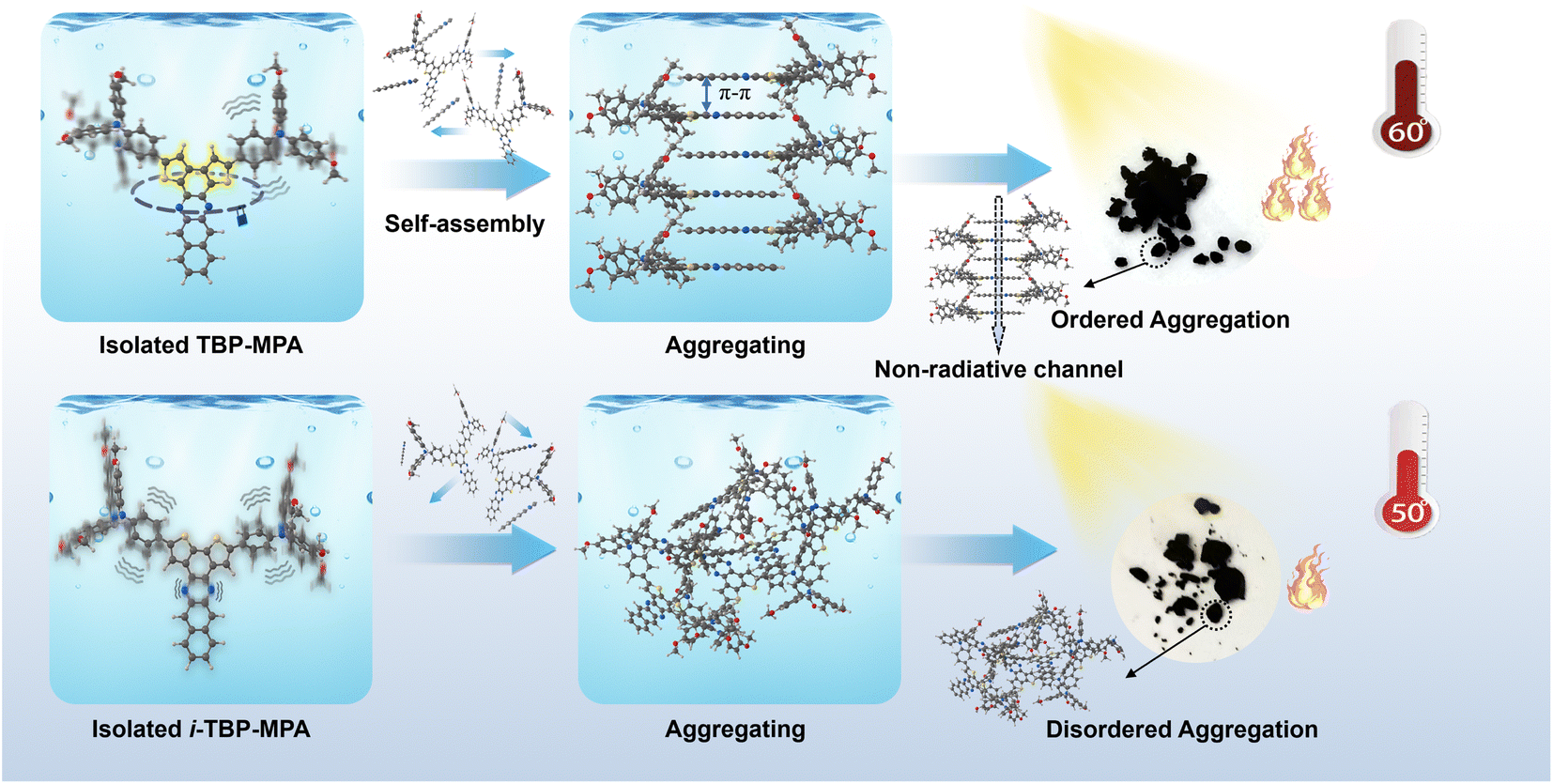 | ||
| Scheme 1 The schematic diagram of AIPE in this work. | ||
Results and discussion
Molecular design and synthesis
The molecular structures of TBP–MPA and i-TBP–MPA are shown in Fig. 1a. The details of synthetic procedures and characterization are provided in the ESI,† following previously reported literature.18 Thermogravimetric analysis revealed excellent thermal stabilities for TBP–MPA and i-TBP–MPA, with decomposition temperatures (Td, 5% weight loss, Fig. S1, ESI†) reaching 443 °C and 453 °C, respectively. As expected, density functional theory (DFT) calculations revealed that the lowest unoccupied molecular orbital (LUMO) electron densities of TBP–MPA and i-TBP–MPA are distributed over the TBP and i-TBP electron-withdrawing structural units. In contrast, the highest occupied molecular orbital (HOMO) electron clouds are primarily localized at the MPA and thiophene structural units (Fig. 1b). The intramolecular push–pull electronic interactions between the donor and acceptor structural units confer ICT characteristics to TBP–MPA and i-TBP–MPA molecules, enhancing their light absorption capability. In the ground state, the optimized molecular conformations of TBP–MPA and i-TBP–MPA (see Fig. 1b) reveal that the TBP and i-TBP units form planar structures in two dimensions, while the MPA end-groups adopt a three-dimensional propeller-like configuration. The dihedral angles between the phenyl ring and the TBP and i-TBP planes are 24° and 23°, respectively. In addition to its electron-donating characteristics, the propeller-shaped MPA units also serve as end rotors to regulate the molecular motion.19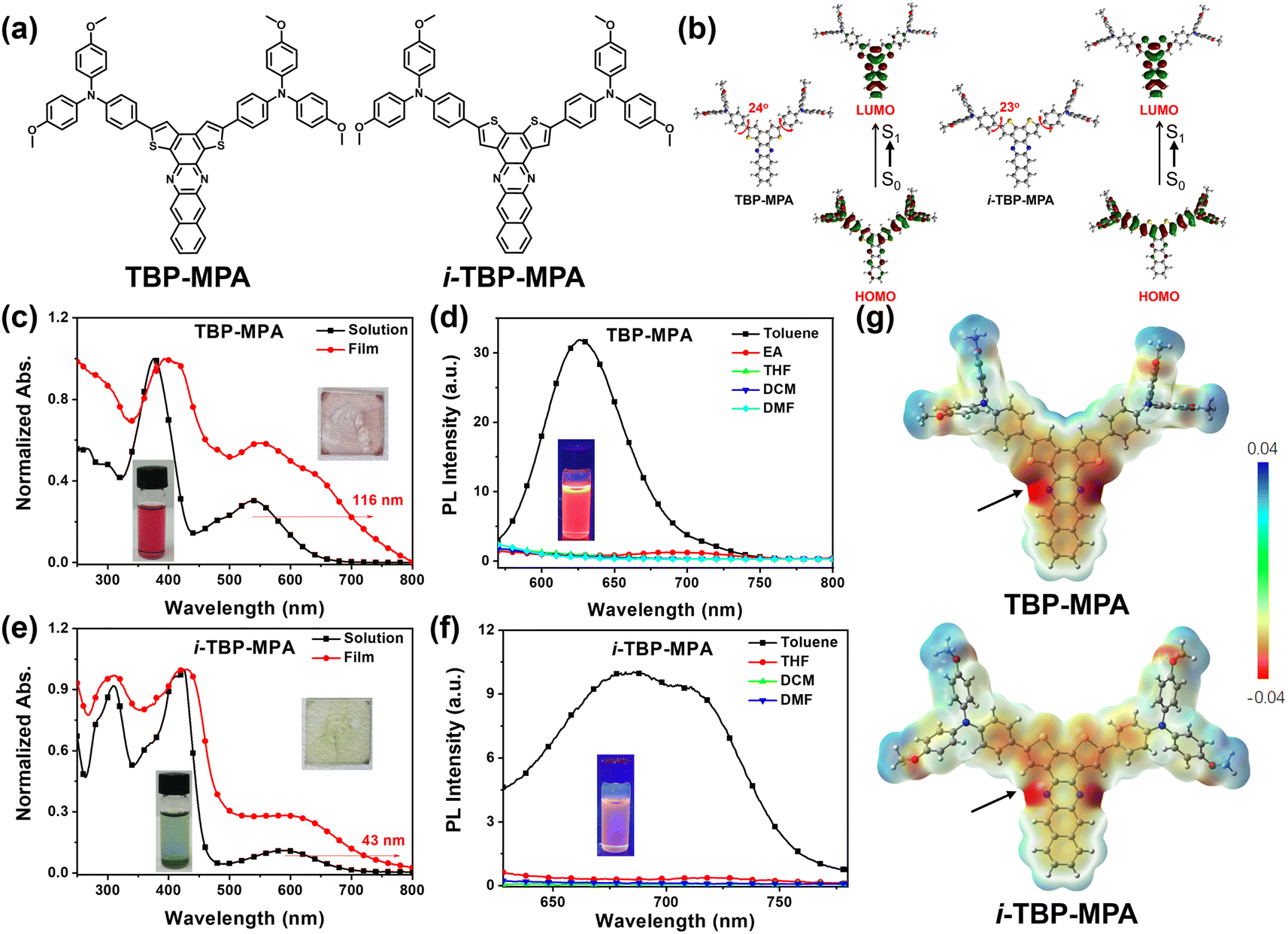 | ||
| Fig. 1 (a) Chemical structures of TBP–MPA and i-TBP–MPA. (b) DFT calculation results for TBP–MPA and i-TBP–MPA. Normalized UV-vis absorption spectra of (c) TBP–MPA and (e) i-TBP–MPA in chloroform solution and as films. PL spectra of (d) TBP–MPA and (f) i-TBP–MPA in solvents of various polarities. (g) ESP surfaces of TBP–MPA and i-TBP–MPA. | ||
Photophysical properties
The UV-vis absorption spectra of TBP–MPA and i-TBP–MPA are shown in Fig. 1c and e, respectively. In the solution state, TBP–MPA exhibits strong light absorption in the range of 250–650 nm, with two prominent absorption peaks at 376 and 539 nm, with molar extinction coefficients of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 037 and 17322 M−1 cm−1 (Fig. S2†), respectively. In comparison, i-TBP–MPA shows a slight redshift in its chloroform solution absorption spectrum, with the onset of absorption around 700 nm. The characteristic absorption peaks appear at 312, 425, and 593 nm, with molar extinction coefficients of 60162, 67003, and 6963 M−1 cm−1 (Fig. S2†), respectively. The TBP–MPA and i-TBP–MPA absorption bands in the low-energy region (539 and 593 nm) are attributed to the ICT transition from the electron-rich MPA units to the electron-deficient TBP and i-TBP units. Compared to i-TBP–MPA, the ICT absorption peak of TBP–MPA is significantly stronger. Upon closer examination, a shoulder peak around 478 nm for TBP–MPA is observed. Theoretical calculations (Fig. S3a and Table S1†) reveal that the absorption of TBP–MPA in the range of 450–650 nm arises from transitions between the ground state (S0) and the first singlet state (S1) and second singlet state (S2), while the absorption band of i-TBP–MPA in the range of 500–700 nm originates from the S0–S1 transition, with a lower oscillator strength. The differences in electronic processes also result in distinct macroscopic characteristics between i-TBP–MPA and TBP–MPA (see the inset in Fig. 1c and e). In the film state, the absorption spectrum of TBP–MPA exhibits a pronounced redshift of 116 nm and a broadening of characteristic peaks compared to its solution state. The shoulder peak of the S0–S2 transition becomes more pronounced. Conversely, the UV-vis absorption profile of the i-TBP–MPA film resembles that of its solution, with a redshift of 43 nm based on the onset. The pronounced redshift indicates strong intermolecular interactions in the aggregated state of TBP–MPA, whereas the intermolecular interactions in the aggregated state of i-TBP–MPA are comparatively weaker. Changes in the UV-vis absorption spectra of TBP–MPA and i-TBP–MPA with increasing solvent polarity were insignificant (see Fig. S3b and c†), suggesting that the polarity of the solvent has minimal effect on the molecules in their ground state.
037 and 17322 M−1 cm−1 (Fig. S2†), respectively. In comparison, i-TBP–MPA shows a slight redshift in its chloroform solution absorption spectrum, with the onset of absorption around 700 nm. The characteristic absorption peaks appear at 312, 425, and 593 nm, with molar extinction coefficients of 60162, 67003, and 6963 M−1 cm−1 (Fig. S2†), respectively. The TBP–MPA and i-TBP–MPA absorption bands in the low-energy region (539 and 593 nm) are attributed to the ICT transition from the electron-rich MPA units to the electron-deficient TBP and i-TBP units. Compared to i-TBP–MPA, the ICT absorption peak of TBP–MPA is significantly stronger. Upon closer examination, a shoulder peak around 478 nm for TBP–MPA is observed. Theoretical calculations (Fig. S3a and Table S1†) reveal that the absorption of TBP–MPA in the range of 450–650 nm arises from transitions between the ground state (S0) and the first singlet state (S1) and second singlet state (S2), while the absorption band of i-TBP–MPA in the range of 500–700 nm originates from the S0–S1 transition, with a lower oscillator strength. The differences in electronic processes also result in distinct macroscopic characteristics between i-TBP–MPA and TBP–MPA (see the inset in Fig. 1c and e). In the film state, the absorption spectrum of TBP–MPA exhibits a pronounced redshift of 116 nm and a broadening of characteristic peaks compared to its solution state. The shoulder peak of the S0–S2 transition becomes more pronounced. Conversely, the UV-vis absorption profile of the i-TBP–MPA film resembles that of its solution, with a redshift of 43 nm based on the onset. The pronounced redshift indicates strong intermolecular interactions in the aggregated state of TBP–MPA, whereas the intermolecular interactions in the aggregated state of i-TBP–MPA are comparatively weaker. Changes in the UV-vis absorption spectra of TBP–MPA and i-TBP–MPA with increasing solvent polarity were insignificant (see Fig. S3b and c†), suggesting that the polarity of the solvent has minimal effect on the molecules in their ground state.
The photoluminescence (PL) spectra of TBP–MPA and i-TBP–MPA in different solvents are shown in Fig. 1d and f, respectively. Due to the twisted intramolecular charge transfer (TICT) effect, TBP–MPA and i-TBP–MPA exhibit obvious fluorescence only in low-polarity toluene solutions. The emission peak of TBP–MPA in toluene is around 627 nm, with a fluorescence quantum yield of approximately 2%. i-TBP–MPA exhibits broad emission in toluene solution with emission peaks at 685 and 710 nm and a fluorescence quantum yield of about 0.5%. In toluene, the fluorescence lifetimes of TBP–MPA and i-TBP–MPA are 1.49 ns and 2.77 ns, respectively (Fig. S4†). The weaker emission and longer fluorescence lifetime of i-TBP–MPA in toluene may result from the more active molecular motion in solution. For the excited state of i-TBP–MPA, the process of returning to the ground state through radiative transition involves a more complex molecular conformational change, resulting in an overall longer emission lifetime and lower emission efficiency. The shoulder peak of i-TBP–MPA may be attributed to the high-frequency vibrations of the molecular skeleton.20 On the other hand, the weak fluorescence emission of i-TBP–MPA enhances the visibility of fine structural vibrations in the spectrum. This could be another reason for the pronounced appearance of the shoulder peak and the spectral broadening. For TBP–MPA, the difference is attributed to the S⋯N non-covalent interactions within the TBP structure,21 where the sulfur atom in the TBP unit interacts with the nitrogen atom to enhance molecular rigidity and suppress non-radiative transitions caused by skeletal vibrations, thus increasing fluorescence quantum yield.
The electrostatic potential (ESP) calculation results (Fig. 1g) support this assertion, demonstrating that in solution, fused negative charge centers form between the sulfur and nitrogen atoms within the TBP structural unit, whereas in the i-TBP structure, the negative charge centers on the sulfur and nitrogen atoms remain separate. These S⋯N non-covalent interactions effectively suppress the vibrations in the TBP structure, enhance the rigidity of conjugated units, and mitigate non-radiative transition losses, resulting in a higher fluorescence quantum yield. TBP–MPA and i-TBP–MPA exhibit nearly identical optical bandgaps. Through photoelectron spectroscopy testing (Fig. S5†) combined with the UV-vis absorption spectra of TBP–MPA and i-TBP–MPA in thin films, we obtained HOMO energy levels of −5.40 and −5.43 eV for TBP–MPA and i-TBP–MPA, respectively, and LUMO energy levels of −3.76 and −3.78 eV, respectively.
Photothermal properties
The UV-vis absorption spectra of TBP–MPA and i-TBP–MPA powders are generally similar (see Fig. S6†), exhibiting good light absorption in the range of 250–800 nm, covering the entire UV-visible region. There is a slight enhancement in light absorption around 700 nm for TBP–MPA powder compared to i-TBP–MPA, corresponding to the higher oscillator strength of TBP–MPA within this range. The photothermal properties of TBP–MPA and i-TBP–MPA were evaluated by investigating the temperature variation of TBP–MPA and i-TBP–MPA powders (5 mg) under simulated sunlight exposure. Under 1 sun irradiation, the temperature of TBP–MPA powder rapidly increased to 55 °C within 350 s and stabilized around 60 °C after approximately 1000 s (Fig. 2a). In contrast, the temperature of i-TBP–MPA powder under sunlight rapidly rose to 50 °C within 350 s and remained at that temperature (Fig. 2b). Compared to the i-TBP–MPA powder, TBP–MPA took longer to reach a stable temperature. This is likely due to the low bulk thermal conductivities of organic semiconductors. During the temperature increase from 55 °C to 60 °C, there is a delay in the conduction of accumulated heat within the material. Both TBP–MPA and i-TBP–MPA demonstrate excellent photothermal stability and photobleaching tolerance. Under prolonged illumination, both TBP–MPA and i-TBP–MPA powders can maintain stable temperatures around 60 and 50 °C, respectively. Through 5 cycles of on–off heating and cooling processes, TBP–MPA and i-TBP–MPA exhibit good photothermal responsiveness (Fig. 2c and d). In each cycle, accompanied by simulated sunlight (1 sun) radiation, TBP–MPA and i-TBP–MPA powders stabilize at approximately 60 and 50 °C, respectively, and rapidly return to room temperature without light. The stability and cycling tests thoroughly confirm that the superior photothermal performance of TBP–MPA over i-TBP–MPA is an intrinsic property of the material. | ||
| Fig. 2 Temperature change curves of (a) TBP–MPA and (b) i-TBP–MPA powders under 1 sun irradiation for 1 h. Photobleaching resistance properties during five cycles of on–off processes of (c) TBP–MPA and (d) i-TBP–MPA powders. | ||
Excitingly, as structural isomers, TBP–MPA and i-TBP–MPA differ only in the orientation of sulfur atoms in their molecular structures, yet TBP–MPA exhibits a significant improvement in photothermal performance compared to i-TBP–MPA. We aim to explain this impressive phenomenon through both experimental and theoretical approaches. PL measurements were conducted by gradually increasing the fraction of poor solvents in solution to study the luminescent properties of TBP–MPA and i-TBP–MPA from their monomeric state to aggregated states (Fig. 3a and d). In the tetrahydrofuran (THF)/water system, water serves as a poor solvent. With the increase in water volume fraction (fwater), the fluorescence of both molecules is weakened (Fig. 3a and d). This is because adding water increases the polarity of the mixed solvent system, leading to the quenching of the fluorescence of the D–A type molecules influenced by the TICT effect (Fig. S7a and b†). However, as the poor solvent increases, TBP–MPA and i-TBP–MPA exhibit significantly different luminescent properties in the aggregated state. For TBP–MPA, when fwater increases to 60%, the fluorescence intensity shows a certain enhancement, further increasing upon adding water. This is because adding a large amount of poor solvent causes TBP–MPA to aggregate, thereby hindering the intramolecular motion necessary to form the TICT state. However, the fluorescence intensity of TBP–MPA in the aggregated state is lower than that in the THF solution (Fig. S7a†), and it does not exhibit the AIE phenomenon. A comparison of PL spectra reveals a significant redshift of 130 nm for TBP–MPA in the aggregated state compared to that in THF, with a noticeable broadening of the spectrum. This indicates strong intermolecular interactions in the aggregated state of TBP–MPA. For i-TBP–MPA, as fwater exceeds 40%, molecules aggregate, and restricted molecular motion induces the AIE effect,22 significantly increasing fluorescence intensity. When fwater reaches 90%, i-TBP–MPA exhibits the strongest emission, with a fluorescence intensity 15-fold higher than that of the pure THF solution (Fig. S7b†). Compared to the THF solution, aggregated i-TBP–MPA undergoes a 35 nm blue-shift. This indicates weaker intermolecular interactions among i-TBP–MPA molecules in the aggregated state. After aggregation, the environment surrounding the molecules is less polar than THF, resulting in the observed blue-shift. TBP–MPA and i-TBP–MPA exhibit significantly different fluorescence properties in various thin film states. As shown in Fig. S7c and e,† poly(methyl methacrylate) (PMMA) films doped with a small amount of TBP–MPA (1 wt%) exhibit significantly enhanced luminescence compared to pure TBP–MPA films, with a spectral blue-shift of up to 79 nm. This indicates strong intermolecular interactions among TBP–MPA molecules in the aggregated state, suppressing molecular radiative transitions. However, i-TBP–MPA demonstrates completely opposite results. Pure i-TBP–MPA films exhibit stronger luminescence than PMMA films doped with a small amount of i-TBP–MPA (Fig. S7d and e†), further confirming the AIE characteristics of i-TBP–MPA molecules. The PL spectrum of the doped i-TBP–MPA film only experiences a 13 nm blue-shift compared to the pure film. The above results clearly demonstrate that, compared to the monomeric state, molecular aggregation suppresses the radiative transitions of TBP–MPA, while the radiative transitions of i-TBP–MPA are enhanced in the aggregated state.
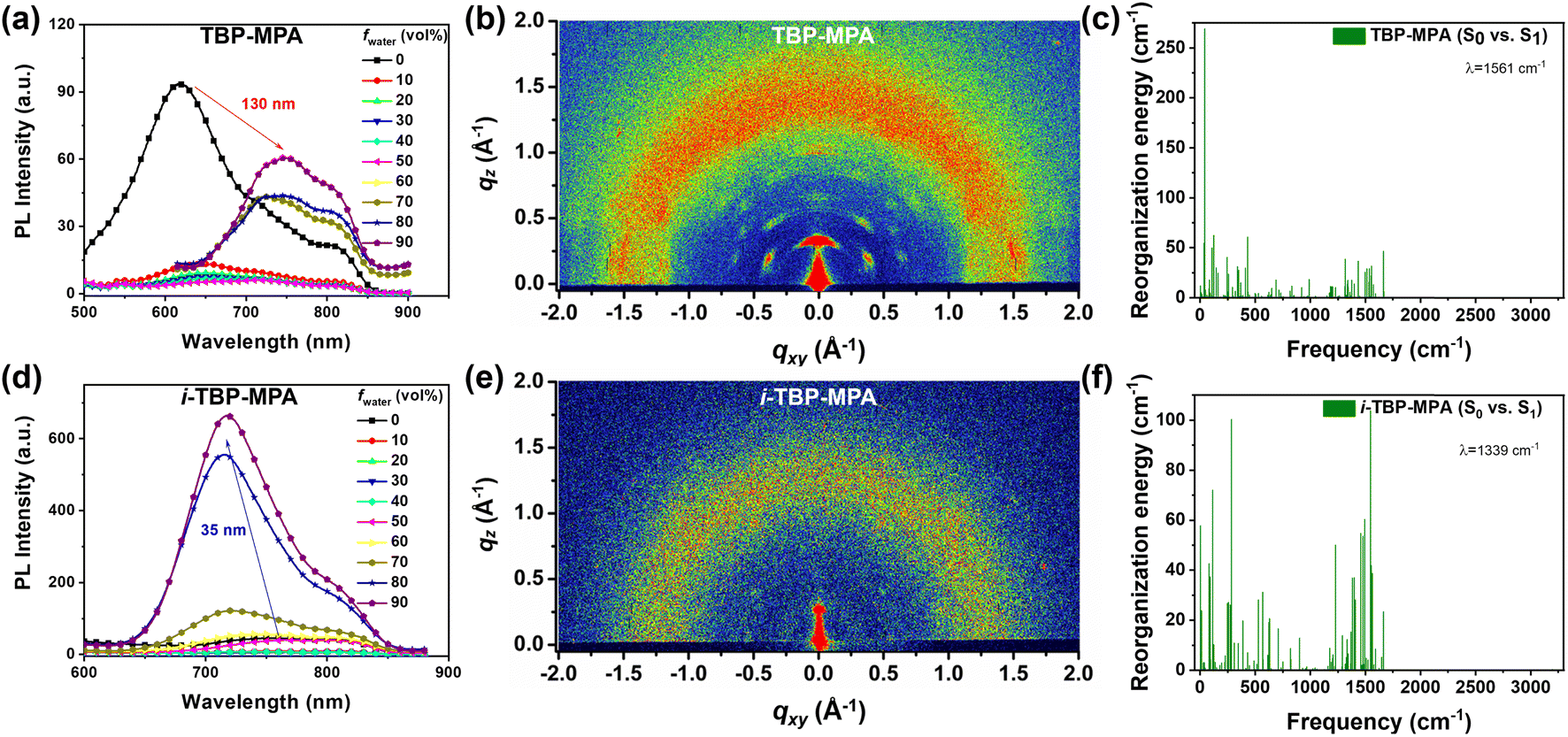 | ||
| Fig. 3 PL spectra of (a) TBP–MPA and (d) i-TBP–MPA in a THF/H2O mixture with different fwater. 2D GIWAXS patterns of (b) TBP–MPA and (e) i-TBP–MPA films. Calculated reorganization energy (environment: toluene) of (c) TBP–MPA and (f) i-TBP–MPA at different wavenumbers. | ||
The intermolecular aggregation behaviors were investigated by conducting grazing-incidence wide-angle X-ray scattering (GIWAXS) tests on the spin-coated TBP–MPA and i-TBP–MPA films on silicon substrates. As shown in Fig. 3b, strong diffraction signals are observed in the two-dimensional (2D) GIWAXS patterns of the TBP–MPA film in both the out-of-plane and in-plane directions. This indicates a preferred growth orientation of TBP–MPA in both directions. The TBP–MPA film exhibits crystal texture characteristics. Near 1.75 Å−1 in the out-of-plane direction, diffraction signals of TBP–MPA can be observed, consistent with the π–π stacking of TBP planar rings, as suggested by recent work by Tang and colleagues on TBP molecules.18 The π–π distance of around 3.6 Å matches closely with the 3.5 Å for similar molecules. The well-crystallized nature of the TBP–MPA film suggests good self-assembly properties between TBP–MPA molecules during film formation, inducing ordered growth and strong intermolecular interactions in the aggregated state. In contrast, the 2D GIWAXS pattern of the i-TBP–MPA film shows no distinct diffraction signals (Fig. 3e), indicating disordered molecular stacking during aggregation, resulting in an amorphous film state.
To further elucidate the intrinsic relationship between molecular structure, molecular aggregation, and photothermal properties, we conducted computational calculations of the recombination energy (λ) using the MOMAP program23 in the single-molecule state (with the simulated environment being toluene) to investigate the geometric distortion and vibrational coupling of TBP–MPA and i-TBP–MPA molecules returning from the excited state to the ground state. As shown in Fig. 3c and f, although both molecules have comparable λ values, 1561 cm−1 for TBP–MPA and 1339 cm−1 for i-TBP–MPA, there are noticeable differences in the vibrational behavior of the molecules. The molecular vibrations of TBP–MPA primarily arise from low-frequency vibrations corresponding to skeletal stretching or structural unit oscillations. By refining the contributions of each component to the recombination energy (Fig. S8 and S9†), it can be clearly observed that the dihedral angles in the MPA structural unit mainly contribute to the molecular vibrations of TBP–MPA. In contrast, the recombination energy of i-TBP–MPA is widely distributed in both low and high-frequency regions. The Huang–Rhys factor of i-TBP–MPA is significantly larger than that of TBP–MPA molecules (Fig. S8 and S9†). This indicates that, compared to TBP–MPA, the high-frequency vibrations from the i-TBP unit contribute significantly to the overall vibration of the molecule. This result effectively explains the low fluorescence quantum yield of i-TBP–MPA in toluene. The non-radiative transition losses due to the active molecular motion of i-TBP in the monomeric state are suppressed in the aggregates due to the restricted intramolecular motion, leading to enhanced radiative transitions and exhibition of AIE behavior. Meanwhile, the rigid conjugated structure of TBP facilitates the formation of intermolecular π–π interactions in aggregates, increasing the channels for non-radiative transitions and exhibiting the AIPE phenomenon.
To further elucidate the photophysical properties of TBP–MPA and i-TBP–MPA molecules, we investigated the electronic processes of TBP–MPA and i-TBP–MPA in solution and thin films using femtosecond transient absorption spectroscopy (fs-TA) measurements. Fig. 4 shows strong positive signals of the differential absorption (ΔA) were observed in the three-dimensional fs-TA spectra of both TBP–MPA and i-TBP–MPA in both solution and thin films. The positive ΔA signals are distributed over a wide energy range, consistent with the characteristics of excited-state absorption (ESA), originating from S1–Sn photo-induced absorption. In solution, TBP–MPA exhibits a broader energy distribution of ESA signals, suggesting the presence of richer singlet-to-singlet internal conversion processes within TBP–MPA molecules. TBP–MPA and i-TBP–MPA show no long-lived signals in solution and thin film states, indicating a simple singlet decay process back to the ground state, consistent with theoretical calculations (Table S1†). Fitting the dynamics curves of TBP–MPA reveals a decay time of 115.7 ps in solution (650 nm, Fig. S10a†) and 30.2 ps in thin film (680 nm, Fig. S10b†), while i-TBP–MPA exhibits decay times of 390.4 ps in solution (635 nm, Fig. S10c†) and 34.1 ps in thin film (650 nm, Fig. S10d†). In solution, the singlet decay lifetime of i-TBP–MPA is much longer than that of TBP–MPA, possibly due to diverse molecular conformation changes induced by the active skeletal vibrations of the i-TBP conjugated unit. In thin films, the decay lifetimes of TBP–MPA and i-TBP–MPA are comparable, which is attributed to restricted molecular motion in the aggregated state. Notably, TBP–MPA exhibits a significant redshift in ESA signals in thin films compared to solution, further confirming strong intermolecular interactions in the aggregated state of TBP–MPA.
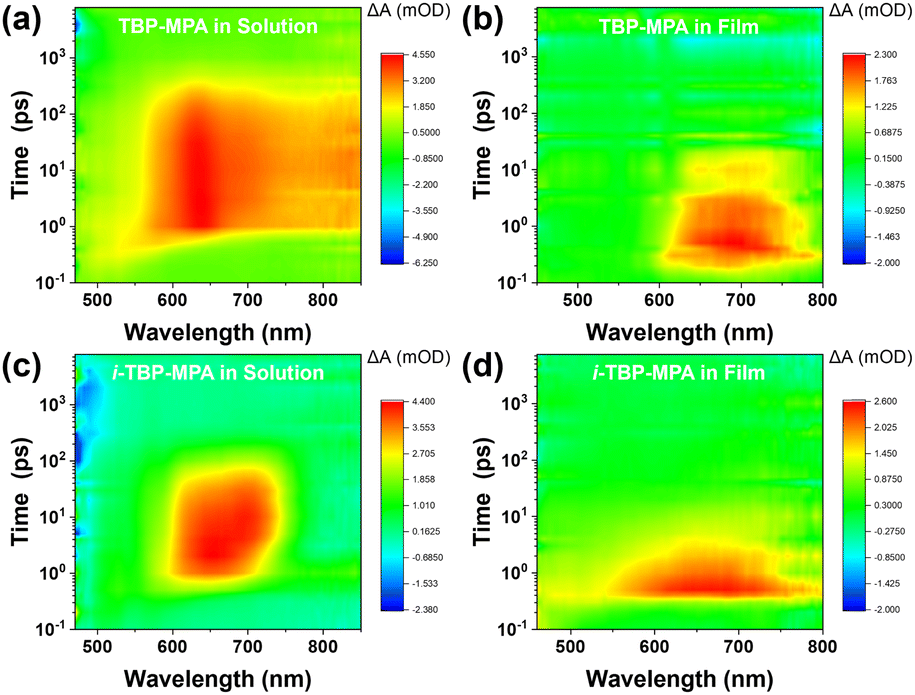 | ||
| Fig. 4 Three-dimensional fs-TA spectra of (a and b) TBP–MPA and (c and d) i-TBP–MPA in solution (a and c) and as films (b and d). | ||
Based on targeted experiments and computational results, we provide the following mechanistic explanation for the AIPE exhibited by TBP–MPA compared to i-TBP–MPA (also refer to Scheme 1). In the single-molecule state, the S⋯N noncovalent interactions within TBP–MPA molecules endow the conjugated units of TBP with a rigid structure, effectively suppressing the vibration of the conjugated backbone at the single-molecule level. During aggregation, the planar rigid backbone is endowed with self-assembly properties through intermolecular π–π interactions, forming ordered molecular stacking and strong intermolecular interactions within the aggregate. These interactions not only enhance light absorption but also significantly promote non-radiative transitions, resulting in enhanced photothermal performance. In contrast, due to the absence of S⋯N non-covalent interactions, vibrations of the i-TBP conjugated unit are active in the single-molecule state. As aggregation occurs, the weak interactions between adjacent molecules lead to the formation of disordered aggregates. Moreover, in the aggregated state, vibrations of the i-TBP structural unit are constrained, inhibiting non-radiative transitions and favoring luminescence, consistent with the AIE phenomenon observed in i-TBP–MPA molecules. Additionally, as a Y-shaped molecule, TBP–MPA may form asymmetric molecular stacking,18 providing more excellent space for the motion of rotor units, facilitating the movement of rotor structures, and contributing to enhancing the photothermal properties.
Benefiting from their excellent photothermal properties, we employed TBP–MPA and i-TBP–MPA as light absorbers to achieve solar-driven water evaporation. Due to their conjugated bicyclic structure, both TBP–MPA and i-TBP–MPA exhibit hydrophobic characteristics, with contact angles measured at 90° and 92.3°, respectively (Fig. S11†). To enhance solar-driven water evaporation efficiency, we have loaded TBP–MPA and i-TBP–MPA onto cellulose papers. The cellulose papers serve as floating carriers supporting organic photothermal small molecules and utilize their own capillary pore structure to quickly transport water to the air/liquid interface, facilitating solar-driven interfacial steam generation.24 Under simulated sunlight (1 sun, 1 kW m−2) exposure, cellulose paper loaded with TBP–MPA (5 mg on 1.57 cm2) rapidly heats up and stabilizes at around 60 °C (Fig. 5a), whereas cellulose paper loaded with i-TBP–MPA (5 mg on 1.57 cm2) stabilizes at around 50 °C (Fig. 5c), a significantly higher temperature than the 37 °C for blank cellulose paper. For water evaporation performance, TBP–MPA showed a water evaporation rate of 1.0 kg m−2 h−1 with a solar-to-vapor conversion efficiency of 69.3% (Fig. 5b), while i-TBP–MPA exhibited a water evaporation rate of 0.92 kg m−2 h−1 and a solar-to-vapor conversion efficiency of 63.7% (Fig. 5d). The superior water evaporation performance of TBP–MPA is attributed to its enhanced photothermal conversion efficiency, which results from its AIPE characteristic. It's inspiring that these results suggest further derivatization of TBP–MPA molecules to extend their light absorption range into the infrared region. This could leverage the AIPE properties of TBP–MPA-like molecules for efficient photothermal tumor therapy and photoacoustic imaging. Additionally, exploring their potential application in infrared photothermal imaging for anti-counterfeiting purposes would be worthwhile.
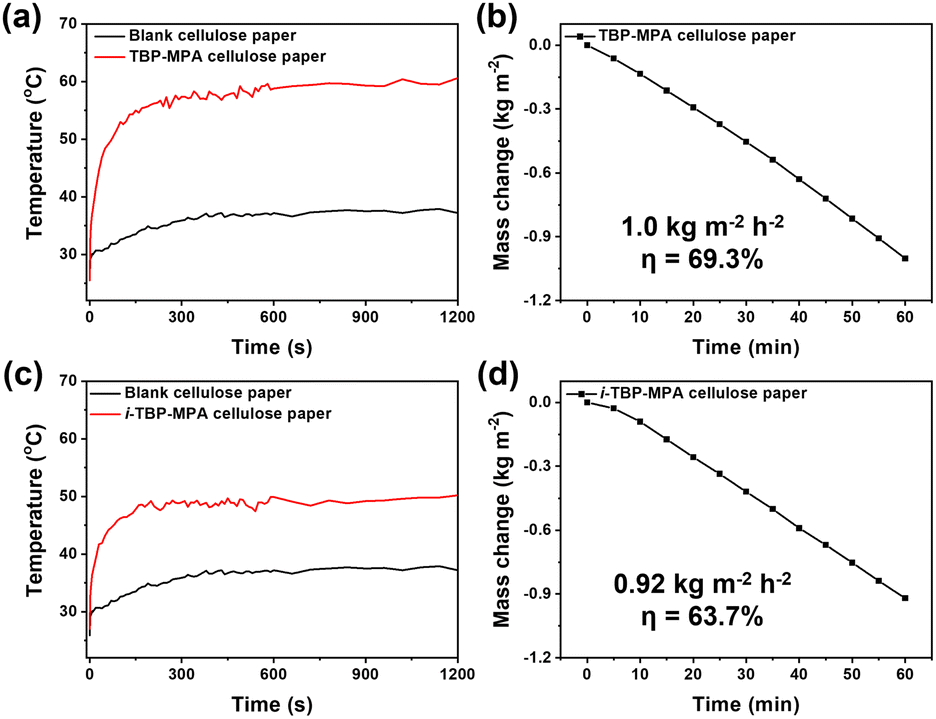 | ||
| Fig. 5 Temperature change curves of (a) TBP–MPA cellulose paper and (c) i-TBP–MPA cellulose paper and blank cellulose papers under a power density of 1 kW m−2 upon 20 min irradiation. Water evaporation curves of (b) TBP–MPA cellulose paper and (d) i-TBP–MPA cellulose paper. | ||
Conclusions
We have designed and synthesized two isomeric conjugated small molecules with a D–A structure, TBP–MPA and i-TBP–MPA. Utilizing isomerism as an ideal platform for structure–property relationship studies, we systematically investigated the relationship between molecular structure, molecular aggregation behavior, and photothermal performance. Compared to i-TBP–MPA, TBP–MPA exhibited the AIPE phenomenon. As expected, TBP–MPA also demonstrated superior solar-driven water evaporation performance. Under 1 sun, TBP–MPA achieved a water evaporation rate of 1.0 kg m−2 h−1 with a solar-to-vapor conversion efficiency of 69.3%. Meanwhile, i-TBP–MPA showed a water evaporation rate of 0.92 kg m−2 h−1 and a solar-to-vapor conversion efficiency of 63.7%. We elucidated the underlying mechanisms behind the AIPE phenomenon through experimental and theoretical calculations. We hope that the research on AIPE mechanisms in this work can provide instructive guidelines for designing organic conjugated molecules with high photothermal conversion efficiency in the aggregated state. Targeted molecular design can reconcile seemingly contradictory molecular motion and ordered molecular stacking. For instance, considering the TBP–MPA molecule, it can be viewed as a Y-shaped molecule. The upper end of the Y-shape can introduce rotors as a motion center (similar to the MPA unit), while the lower end's rigid π-conjugated TBP unit can establish strong intermolecular interactions between adjacent molecules, constructing non-radiative transition channels, thereby enhancing photothermal performance in the aggregated state.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
P. G., G. L., and H. S. conceived the work. H. S. synthesized the compounds with help from S. W. and L. D. H. S. and P. G. conducted tests on optical physics and analyzed the data. T. J. supervised the photothermal characterization and data analysis. Z. W. and H. Y. conducted tests on the photothermal performance. G. L. performed the GIWAXS measurements and corresponding analysis. T. H. carried out the calculations with help from G. L. This manuscript was mainly prepared by H. S., T. H. and P. G., and all authors participated in the manuscript preparation and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Basic Research Program of Xuzhou Science and Technology Bureau (KC22009). This work was also supported by the Changzhou Introduction Program of Innovative Leading Talents (No. CQ20220111), Applied Basic Research Programs of Science and Technology Commission Foundation of Jiangsu Province (No. BK20231340), and Jiangsu Province Engineering Research Center of Biodegradable Materials (KF2302).Notes and references
- Y. Zhang, S. Zhang, C. Liang, J. Shi and L. Ji, Adv. Mater., 2023, 35, 2302732 CrossRef CAS.
- (a) H.-H. Cho, D. G. Congrave, A. J. Gillett, S. Montanaro, H. E. Francis, V. Riesgo-Gonzalez, J. Ye, R. Chowdury, W. Zeng, M. K. Etherington, J. Royakkers, O. Millington, A. D. Bond, F. Plasser, J. M. Frost, C. P. Grey, A. Rao, R. H. Friend, N. C. Greenham and H. Bronstein, Nat. Mater., 2024, 23, 519–526 CrossRef CAS; (b) X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photonics, 2023, 17, 280–285 CrossRef CAS; (c) T. Huang, Q. Wang, H. Zhang, Y. Zhang, G. Zhan, D. Zhang and L. Duan, Nat. Photonics, 2024, 18, 516–523 CrossRef CAS.
- (a) K. Liu, Y. Jiang, G. Ran, F. Liu, W. Zhang and X. Zhu, Joule, 2024, 8, 835–851 CrossRef CAS; (b) H. Sun, X. Song, J. Xie, P. Sun, P. Gu, C. Liu, F. Chen, Q. Zhang, Z.-K. Chen and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 29924–29931 CrossRef CAS; (c) H. Sun, F. Chen and Z.-K. Chen, Mater. Today, 2019, 24, 94–118 CrossRef CAS; (d) J. Wang, Y. Xie, K. Chen, H. Wu, J. M. Hodgkiss and X. Zhan, Nat. Rev. Phys., 2024, 6, 365–381 CrossRef CAS.
- (a) J. Liu, Y. Cui, Y. Pan, Z. Chen, T. Jia, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117087 CrossRef CAS; (b) J. Zhu, Y. Wang, X. Qiu, H. Kong, Y. Li, J. Yan, J. Zheng, S. Chen, Y. Wang and Y. Wang, Adv. Funct. Mater., 2023, 33, 2306604 CrossRef CAS; (c) D. Wang, S. Qi, J. Dong, X. Wang, Y. Zhang, S. Zhou, P. Gu, T. Jia and Q. Zhang, Org. Lett., 2023, 25, 5730–5734 CrossRef CAS.
- (a) Z. Yang and X. Chen, Acc. Chem. Res., 2019, 52, 1245–1254 CrossRef CAS; (b) Z. Jiang, C. Zhang, X. Wang, M. Yan, Z. Ling, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 22376–22384 CrossRef CAS; (c) H. S. Jung, P. Verwilst, A. Sharma, J. Shin, J. L. Sessler and J. S. Kim, Chem. Soc. Rev., 2018, 47, 2280–2297 RSC; (d) H. Sun and Q. Zhang, ChemPhotoChem, 2024, 8, e202300213 CrossRef CAS.
- (a) Z. Jiang, Z. Liang, Y. Cui, C. Zhang, J. Wang, H. Wang, T. Wang, Y. Chen, W. He, Z. Liu and Z. Guo, J. Am. Chem. Soc., 2023, 145, 7952–7961 CrossRef CAS; (b) Y. Cui, X. Wang, Z. Jiang, C. Zhang, Z. Liang, Y. Chen, Z. Liu and Z. Guo, Angew. Chem., Int. Ed., 2023, 62, e202214505 CrossRef CAS.
- (a) G. Chen, J. Sun, Q. Peng, Q. Sun, G. Wang, Y. Cai, X. Gu, Z. Shuai and B. Z. Tang, Adv. Mater., 2020, 32, 1908537 CrossRef CAS PubMed; (b) Y. Cui, J. Liu, Z. Li, M. Ji, M. Zhao, M. Shen, X. Han, T. Jia, C. Li and Y. Wang, Adv. Funct. Mater., 2021, 31, 2106247 CrossRef CAS; (c) L. Yuan, J. Yang, S. Qi, Y. Liu, X. Tian, T. Jia, Y. Wang and C. Dou, Angew. Chem., Int. Ed., 2023, 62, e202314982 CrossRef CAS PubMed; (d) Z. Wang, J. Zhou, Y. Zhang, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202113653 CrossRef CAS PubMed; (e) L. Cheng, Z. Ju, X. Ji, Y. Ma, J. Fan, P. Sun, S. Wang, L. Dong, H. Sun and J. Zhang, Synth. Met., 2023, 293, 117278 CrossRef CAS.
- (a) R. Xu, Natl. Sci. Rev., 2018, 5, 1 CrossRef; (b) F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 14192–14196 CrossRef PubMed.
- (a) Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS; (c) H. Sun, T. He, C. Zhang, S. Wang, L. Dong, Z. Li, P.-Y. Gu, Z. Wang, G. Long and Q. Zhang, Chem.–Eur. J., 2023, 29, e202300029 CrossRef CAS; (d) H. Sun, J. Jin, Q. Wang, S. Wang, W. Na, Z. Li, B. Yao, P. Sun, L. Dong and X.-C. Hang, Dyes Pigm., 2022, 200, 110169 CrossRef CAS.
- G. Feng, G.-Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- (a) Z. Zhao, C. Chen, W. Wu, F. Wang, L. Du, X. Zhang, Y. Xiong, X. He, Y. Cai, R. T. K. Kwok, J. W. Y. Lam, X. Gao, P. Sun, D. L. Phillips, D. Ding and B. Z. Tang, Nat. Commun., 2019, 10, 768 CrossRef CAS PubMed; (b) C. Xu, R. Ye, H. Shen, J. W. Y. Lam, Z. Zhao and B. Zhong Tang, Angew. Chem., Int. Ed., 2022, 61, e202204604 CrossRef CAS.
- P. Han, C. Lin, H. Xu, E. Xia, D. Yang, A. Qin, D. Ma and B. Z. Tang, ACS Mater. Lett., 2022, 4, 1087–1093 CrossRef CAS.
- H.-Y. Chen, G. Schweicher, M. Planells, S. M. Ryno, K. Broch, A. J. P. White, D. Simatos, M. Little, C. Jellett, S. J. Cryer, A. Marks, M. Hurhangee, J.-L. Brédas, H. Sirringhaus and I. McCulloch, Chem. Mater., 2018, 30, 7587–7592 CrossRef CAS.
- Z. Chen, J. Zhu, D. Yang, W. Song, J. Shi, J. Ge, Y. Guo, X. Tong, F. Chen and Z. Ge, Energy Environ. Sci., 2023, 16, 3119–3127 RSC.
- S. N. Intorp, M. Hodecker, M. Müller, O. Tverskoy, M. Rosenkranz, E. Dmitrieva, A. A. Popov, F. Rominger, J. Freudenberg, A. Dreuw and U. H. F. Bunz, Angew. Chem., Int. Ed., 2020, 59, 12396–12401 CrossRef CAS.
- P.-J. Tian, X.-H. Han, Q.-Y. Qi and X. Zhao, Small, 2024, 20, 2307635 CrossRef CAS.
- S. Lv, B. Wang, Y. Wu, R. Zhang, E. Feng, T. Liu, X. Xie, J. Jiang, X. Hou, D. Liu and F. Song, Acta Biomater., 2024, 174, 400–411 CrossRef CAS.
- Y. Li, S. Liu, H. Ni, H. Zhang, H. Zhang, C. Chuah, C. Ma, K. S. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian, X. Lu and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 12822–12826 CrossRef CAS PubMed.
- C. Zhang, T. Zhang, Z. Qiu, Z. Wang, T. Huang, C. Luo, C. Zhang, Y. Li, Q. Wang, X.-C. Hang, H. Sun and Q. Zhang, Adv. Opt. Mater., 2024, 12, 2302952 CrossRef CAS.
- Y. Xie, W. Liu, W. Deng, H. Wu, W. Wang, Y. Si, X. Zhan, C. Gao, X.-K. Chen, H. Wu, J. Peng and Y. Cao, Nat. Photonics, 2022, 16, 752–761 CrossRef CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS.
- Y. Tu, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Natl. Sci. Rev., 2021, 8, nwaa260 CrossRef.
- Z. Shuai, Chin. J. Chem., 2020, 38, 1223–1232 CrossRef CAS.
- S. Cao, P. Rathi, X. Wu, D. Ghim, Y.-S. Jun and S. Singamaneni, Adv. Mater., 2021, 33, 2000922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03542a |
| This journal is © The Royal Society of Chemistry 2024 |