
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA carbon-based bifunctional heterogeneous enzyme: toward sustainable pollution control†
Yuting
Sun‡
 a,
Ming
Guo‡
*b,
Shengnan
Hu
b,
Yankun
Jia
b,
Wenkai
Zhu
*b,
Yusuke
Yamauchi
*cde and
Chaohai
Wang
*f
a,
Ming
Guo‡
*b,
Shengnan
Hu
b,
Yankun
Jia
b,
Wenkai
Zhu
*b,
Yusuke
Yamauchi
*cde and
Chaohai
Wang
*f
aCollege of Environmental and Resource Sciences, Zhejiang Agricultural & Forestry University, Hangzhou, Zhejiang 311300, China
bCollege of Chemistry and Materials Engineering, Zhejiang Agricultural & Forestry University, Hangzhou, Zhejiang 311300, China. E-mail: guoming@zafu.edu.cn
cAustralian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: y.yamauchi@uq.edu.au
dDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8601, Japan
eDepartment of Plant & Environmental New Resources, College of Life Sciences, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 17104, South Korea
fHenan International Joint Laboratory of Green Low Carbon Water Treatment Technology and Water Resources Utilization, School of Municipal and Environmental Engineering, Henan University of Urban Construction, Pingdingshan, Henan 467036, China. E-mail: chaohai@huuc.edu.cn
First published on 18th September 2024
Abstract
We present a study on an immobilized functional enzyme (IFE), a novel biomaterial with exceptional sustainability in enzyme utility, widely employed across various fields worldwide. However, conventional carriers are prone to eroding the active functional domain of the IFE, thereby weakening its intrinsic enzyme activity. Consequently, there is a burgeoning interest in developing next-generation IFEs. In this study, we engineered a carbon-based bifunctional heterogeneous enzyme (MIP-AMWCNTs@lipase) for the intelligent recognition of di(2-ethylhexyl)phthalate (DEHP), a common plasticizer. The heterogeneous enzyme contains a bifunctional structural domain that both enriches and degrades DEHP. We investigated its dual-response performance for the enrichment and specific removal of DEHP. The imprinting factor of the carrier for DEHP was 3.4, demonstrating selectivity for DEHP. The removal rate reached up to 94.2% over a short period. The heterogeneous enzyme exhibited robust activity, catalytic efficiency, and excellent stability under harsh environmental conditions, retaining 77.7% of its initial lipase activity after 7 cycles. Furthermore, we proposed a stepwise heterogeneous enzyme reaction kinetic model based on the Michaelis–Menten equation to enhance our understanding of enzyme reaction kinetics. Our study employs a dual-effect recognition strategy of molecular blotting and enzyme immobilization to establish a method for the removal of organic pollutants. These findings hold significant implications for the fields of biomaterials and environmental science.
Introduction
Molecular modification of enzymes,1 mining of functional materials from biological enzymes,2 and deep expansion of enzyme synthetic biology3 are current hot topics. Enzymes are macromolecular catalysts found in nature and are the safest and most environmentally friendly catalysts known. However, they require stringent environmental conditions to perform optimal catalytic functions, and are difficult to recover and expensive to purify. Thus enzymes are unsuitable for direct participation in many important catalytic processes.4 Cost control and improved catalysis can be achieved by establishing cost-effective enzyme processes.5 Some scientists have tried to immobilize enzymes on inert supports to allow for recovery of enzymes and reduce the cost.6 Immobilized enzymes are at the frontier of current research in the field of enzymology.7 However, carriers of immobilized enzymes hinder the diffusion of biomolecules, which decreases enzymatic activity and catalytic potency.8 To enhance the enzyme immobilization technique, we suggest adding bifunctional structural domains at the interface between the carrier and the enzyme. These domains will enrich the target substance and achieve the desired degradation. Usually, free enzymes are only retained in a single phase. To stably retain enzymes in two or more phases, we have designed synthetic enzymes called heterogeneous enzymes. These enzymes can improve the dispersion and catalytic activity of the enzyme in a heterogeneous phase. Compared to traditional chemical catalysis, the reaction conditions are milder and more environmentally friendly. This technology is expected to replace traditional fine chemicals for heterogeneous biocatalysis of organic pollutants and improve the cost-effectiveness of bioenzymes.9Pollution due to microplastics (MPs)10 has become a global problem. It has become a medium for the transfer of chemical pollutants from seawater to living organisms.11 Di(2-ethylhexyl)phthalate (DEHP)12 is one of the most widely used plastic additives in the phthalic acid ester (PAE) class. DEHP is potentially biotoxic and estrogenic. Small molecules that enter the body and bind to receptors can affect normal hormone levels and reproductive development in humans. In the aqueous environment, MPs readily adsorb DEHP and interact with DEHP through non-covalent forces.13 Thus, DEHP and MPs can have synergistic effects.14 They induce immune responses in organisms, leach chemotoxic substances, and have unpredictable toxic effects on organisms.15 However, MPs are difficult to biodegrade in the body and are prone to causing disease.16 It is possible to remove DEHP first and then gradually mitigate and disintegrate the combined effects of MPs and DEHP. The traditional physical adsorption method is efficient but makes it easy to desorb. The photocatalytic method is prone to secondary pollution. Enzymatic degradation is a more environmentally friendly and efficient method, and it shows great promise for the removal of organic pollutants.
Enzymatic degradation is a safe and efficient way to remove organic contaminants. Lipase (E.C. 3.1.1.3) is a biocatalyst that meets the requirements of sustainable chemical production17 and can efficiently catalyze the hydrolysis of esters. Lipase shows great potential for the development of a new generation of heterogeneous enzyme functional materials that can provide sustainable solutions for remediation of pollution.18
Nanocarriers offer high enzyme loading capacities and efficient mass transfer.19 The potential of multi-walled carbon nanotubes (MWCNTs)20 as carriers for enzymes is well recognized because of their large specific surface areas, modifiable surface properties, and availability of numerous adsorption sites.21 The success of these carriers hinges on the development of an optimized microenvironment at the support interface,22 making carrier biocompatibility crucial for maintaining the enzyme structure and activity.23 Ji et al.24 immobilized porcine pancreatic lipase on functionalized carbon nanotubes and maintained 69% of its enzyme activity after seven repeat applications. However, the pure loading function of carriers struggles to meet the application requirements of heterogeneous enzymes for hard-to-degrade organic contaminants. Surface molecular imprinting technology25 is a technique that mimics artificial antibodies that can polymerize on the carrier surface. This is used to modify the carrier for reactive recognition of the target contaminant, which facilitates efficient catalysis of heterogeneous enzymes. A bifunctional spatial domain is formed between the carrier and the enzyme, which simultaneously respond to the same target contaminant. This synergistic effect helps remove persistent organic contaminants.
In this study, we prepared a heterogeneous enzyme (MIP-AMWCNTs@lipase) with a bifunctional structural domain that can enrich and remove pollutants, thus extending the immobilized enzyme technology. MIP-MWCNTs were obtained by surface molecular imprinting technology and served as a carrier for lipase. Using a synergistic cross-linking approach, covalent and non-covalent modification of lipase was achieved, resulting in the formation of a bifunctional structural domain specifically recognizing the target contaminant in the interface space between lipase and the carrier. We investigated the structural characteristics, enzymatic properties, and immobilization effects of MIP-MWCNTs@lipase in detail, including optimizing the conditions for enzymatic activity and studying enzymatic reaction kinetics. Finally, the specific removal of DEHP was achieved by the synergistic adsorption–degradation effect of the bifunctional structural domain. Our work has practical significance and excellent development prospects.
Results and discussion
Synthesis mechanism
Scheme 1 illustrates the synthetic procedure for MIP-AMWCNTs@lipase. Initially, MWCNTs were subjected to a two-step functionalization process to obtain MWCNTs-COOH and AMWCNTs. The molecularly imprinted prepolymer was achieved using DEHP as the template molecule and acrylamide (Am) as the functional monomer. EDGMA was used as a cross-linking agent to bind the prepolymer to the nanotube surface, which produced the carbon-based molecularly imprinted functional units (MIP-AMWCNTs). Activation of the carriers with the glutaraldehyde cross-linking agent followed by bonding with lipase through a cross-linking reaction provided the carbon-based bifunctional heterogeneous enzyme (MIP-AMWCNTs@lipase) with a bifunctional structural domain that was enriched and degraded by DEHP. | ||
| Scheme 1 Schematic illustration of the synthesis of MIP-AMWCNTs@lipase. | ||
Optimization of the pre-polymerization system
Methyl methacrylate (MMA), methacrylic acid (MAA), and acrylamide (AM) are commonly used functional monomers, and DEHP exhibits UV absorption at 275 nm, whereas none of these functional monomers exhibit absorption at this wavelength (Fig. S1a†). After 12 h of pre-polymerization with each of the three functional monomers, UV absorption peaks at 275 nm were observed for all three blends (Fig. S1b†). The strength was in the order of AM > MMA > MAA. AM had the strongest peak after interacting with DEHP, which indicated that it was the most effective functional monomer for this experiment. Then, we prepared mixtures of DEHP with AM at different ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:10) and observed the UV absorption peaks (Fig. S1c†). The intensity was increased significantly with increases in the molar ratio and plateaued at approximately 1:4, which indicated that a molar ratio of 1:4 was sufficient for DEHP to interact effectively with AM. According to these results, and considering both the cost and efficacy, a molar ratio of 1:4 was selected.
:1 to 1:10) and observed the UV absorption peaks (Fig. S1c†). The intensity was increased significantly with increases in the molar ratio and plateaued at approximately 1:4, which indicated that a molar ratio of 1:4 was sufficient for DEHP to interact effectively with AM. According to these results, and considering both the cost and efficacy, a molar ratio of 1:4 was selected.
Characterization of the carrier
To observe the changes in the micromorphology of the material, the topography of each stage was examined by TEM. Initial MWCNTs had a straight tubular structure that was highly wound with obvious hollows, closed ports, and smooth walls, and a sidewall structure that was largely defect-free (Fig. 1a and b). The MWCNTs exhibited strong hydrophobicity. However, their hydrophilicity and dispersibility can be significantly improved by introducing –COOH through surface modification.26 The TEM image also showed a reduction in the winding degree. After oxidation, MWCNTs-COOH had sidewall defects due to impurity removal from the nanotube surface. Reactive groups, such as carboxyl groups, were also attached to the tube walls, which would make it easier to carry out further modifications (Fig. 1c and d). After amination, agglomeration was reduced, the entanglement was alleviated, and the dispersion performance was greatly enhanced (Fig. 1e and f). After attaching the MIP, AMWCNT wall thickness also increased to about 4.5 nm with the presence of the MIP layer (Fig. 1g and h). During template elution, numerous pores were left behind, resulting in a sparsely porous MIP layer. This, in turn, increased the adsorption surface area and binding sites, which was favorable for the subsequent enzyme synthesis step. Additionally, the winding degree of NIP-AMWCNTs was similar to that of MIP-AMWCNTs (Fig. 1i and j), and the NIP layer was flatter than the MIP layer. To determine the elemental and valence states of the materials during the transformation process, the materials were tested by XPS at each stage. XPS spectra of the nanotubes in various chemical states and the percentage of the element are shown in Fig. 1k. A distinctive C 1s peak and secondary O 1s peak were observed in the MWCNTs at 532.7 and 284.8 eV, respectively. The relative contents of elemental carbon and oxygen were 98.7% and 1.3%, respectively. The oxygen content of MWCNTs-COOH was 7.4%, which showed that a number of oxygen-containing functional groups were added by carboxylation. After addition of APTES, the carboxyl groups interacted with the terminal amino groups of APTES to form AMWCNTs, which produced an N 1s peak. AMWCNTs contained 3.3% nitrogen and 2.4% silicon. The elements and valence states in MIP-AMWCNTs and NIP-AMWCNTs were not altered during the imprinting process. The content of elemental nitrogen increased to 3.8% with addition of the functional monomer (acrylamide). Quantitative results for the surface functional groups and their contents were obtained by further element analysis and fine spectral separation of peaks. The MWCNTs-COOH C 1s fine spectral splitting curve showed five distinct peaks (Fig. 1l). The major peak, which was characteristic of the graphitized structure of carbon nanotubes, occurred at 284.8 eV, which corresponded to the sp2 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hybridization. A secondary peak located near 285.5 eV, which corresponded to sp3 C–C hybridization, indicated that there were defects in the carbon nanotube structure. Carboxylation produced peaks at 286.6 and 288.9 eV, which were attributed to carbon atoms in various oxygen-containing functional groups. Finally, a π–π* leap loss peak was observed at 290.7 eV. After the amidation dehydration condensation reaction, two new characteristic peaks for AMWCNTs were observed at 286.4 eV for amine (C–NHx) and 288.8 eV for the amide bond (CON) (Fig. 1m). These results show that the successful introduction of amino groups on the surface of nanotubes improves their dispersion in solution. The contact surface of the nanotube with the solution also improved, which would enhance subsequent adsorption and lipase immobilization. Details for C 1s peaks of MWCNTs-COOH and AMWCNTs are shown in Table S1.†
C hybridization. A secondary peak located near 285.5 eV, which corresponded to sp3 C–C hybridization, indicated that there were defects in the carbon nanotube structure. Carboxylation produced peaks at 286.6 and 288.9 eV, which were attributed to carbon atoms in various oxygen-containing functional groups. Finally, a π–π* leap loss peak was observed at 290.7 eV. After the amidation dehydration condensation reaction, two new characteristic peaks for AMWCNTs were observed at 286.4 eV for amine (C–NHx) and 288.8 eV for the amide bond (CON) (Fig. 1m). These results show that the successful introduction of amino groups on the surface of nanotubes improves their dispersion in solution. The contact surface of the nanotube with the solution also improved, which would enhance subsequent adsorption and lipase immobilization. Details for C 1s peaks of MWCNTs-COOH and AMWCNTs are shown in Table S1.†
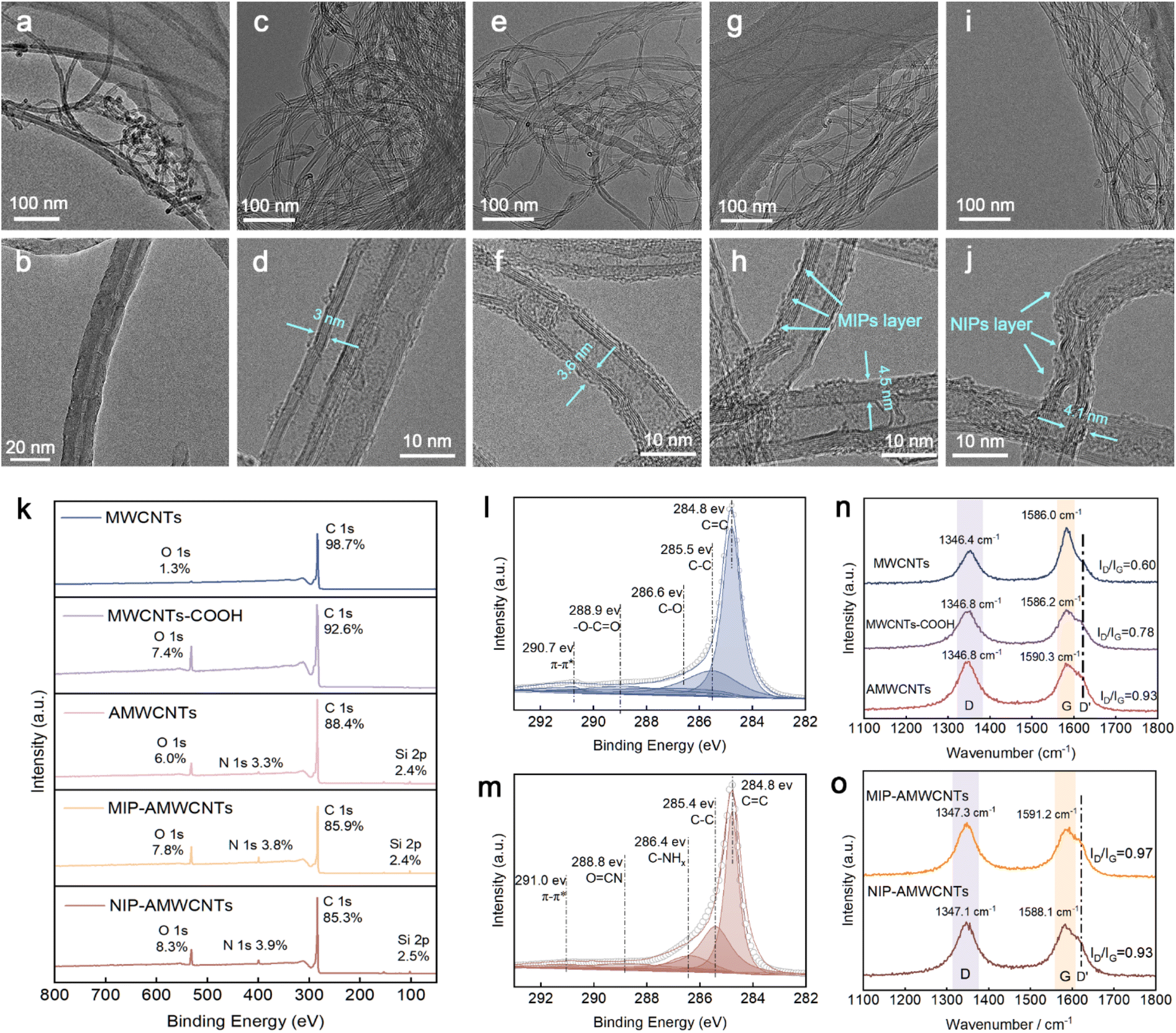 | ||
| Fig. 1 TEM images of MWCNTs (a and b), MWCNTs-COOH (c and d), AMWCNTs (e and f), MIP-AMWCNTs (g and h) and NIP-AMWCNTs (i and j); XPS survey spectra of the intermediate products (k); C 1s split-peak fitting of MWCNTs-COOH (l); C 1s split-peak fitting of AMWCNTs (m); Raman spectra of MWCNTs, MWCNTs-COOH and AMWCNTs (n); Raman spectra of MIP-AMWCNTs and NIP-AMWCNTs (o). | ||
Raman spectroscopy was used to describe the functionalization level of carbon materials. The largest peak (G band) occurred between 1570 and 1600 cm−1, and was characteristic of the graphitized structure of carbon atoms on the nanotubes. The D band, which was the second strongest peak in the 1330–1360 cm−1 range, was characteristic of the severity of flaws in the nanotubes.27 The integrated area ratio of the D band to G band (IG/ID) represents the integrity and orderliness of nanotubes. Three measurements were recorded in different locations for each sample, and the most representative findings were used. Raman spectra of the five samples showed comparable morphologies with clear D and G bands, which were characteristic of MWCNTs (Fig. 1n and o). With deeper functionalization, the D′ band (1620 cm−1) appears as a shoulder of the G band of MWCNTs-COOH, AMWCNTs, MIP-AMWCNTs, and NIP-AMWCNTs. This is similar to the lattice vibration of the G band, which implies that there are significant defects in the functionalized carbon nanotubes.28 The band position shifted slightly as the degree of modification increased, and the IG/ID ratio gradually improved. After acid oxidation, the surface of the MWCNTs showed a considerable improvement in the IG/ID ratio, which increased from 0.60 to 0.78. This indicated a notable increase in the severity of nanotube defects and a decrease in orderliness. Surface polymerization increased the number of flaws in MIP-AMWCNTs, and the IG/ID ratio reached 0.97, which also made it easier to immobilize the lipase.
Characterization of the heterogeneous enzymes
The FITC molecule reacts with the primary amine group of lipase to form a stable thiourea bond. Meanwhile FITC as a fluorescent probe can change the secondary structure and surface charge of the enzyme.29 This allows tracking of the distribution of lipase on the carrier.30 FITC-labeled lipase was clearly uniformly distributed on the carrier surface in the dark field, bright field, and superimposed field (Fig. 2a–c). However, the distribution of lipase was relatively sparse, probably due to the limitations of the carrier structure and enzyme loading. Appropriate density distribution is actually more favorable to improve the efficiency of the adsorption–degradation reaction of heterogeneous enzymes.31 Also, a small percentage of lipases showed agglomeration. Carbon nanotubes tend to clump together, which may explain this phenomenon. A small amount agglomerated when dispersed and was observed under a microscope. The immobilized lipase also agglomerated with the carbon nanotubes. From a microscopic characterization point of view, these results showed that the lipase was successfully immobilized. | ||
| Fig. 2 MIP-AMWCNTs@lipase fluorescence microscope scan image (a–c); immobilization efficiency and specific activity with different materials as carriers (d): A is MWCNTs@lipase, B is MWCNTs-COOH@lipase, C is AMWCNTs@lipase, and D is MIP-AMWCNTs@lipase; N2 adsorption–desorption isotherms of MIP-AMWCNTs and MIP-AMWCNTs@lipase (e and f); XPS survey spectra of MIP-AMWCNTs and MIP-AMWCNTs@lipase (g); schematic diagram of electrochemical sensors (h); cyclic voltammetry curves of different electrode materials (i); CV curves of MIP-AMWCNTs@lipase at different scan rates: 5, 10, 20, 50, 80, 100, 150, 200, 250 and 300 mV s−1 in a 5.0 mM K3[Fe(CN6)] solution of 0.1 M KCl (j); peak current vs. square root of the scan rate (k). | ||
Different degrees and modalities of modification can disrupt the native structure of nanotubes. These changes can improve the enzyme–carrier interaction and increase adhesion of the enzyme. Therefore, we investigated the immobilization effects of MWCNTs, MWCNTs-COOH, AMWCNTs, and MIP-AMWCNTs when used as carriers. The immobilization effect and specific activity improve with increases in the degree of modification (Fig. 2d). This may be because the binding of primitive MWCNTs to the lipase is mainly dependent on hydrophobic interaction between the lipase and the carrier, and lipase is easily released. The functionalized nanotubes have more reactive groups on the surface to bind to lipase. MIP-AMWCNTs are slightly less effective than AMWCNTs when used as carriers, due to the fact that the imprinted polymer occupies some space on the nanotube surface. The immobilization rate reached 72.4% and specific activity reached 7.9 U mg−1 when MIP-AMWCNTs were used as the carrier.
The performance of the heterogeneous enzyme was influenced greatly by spatial constraints and limitations in mass transfer between lipase and the carrier. The development of carriers with high specific surface area, high porosity, and large pore size is necessary to optimize the activity of the heterogeneous enzyme. The N2 adsorption–desorption curves of MIP-AMWCNTs and MIP-AMWCNTs@lipase showed similar patterns in the P/P0 range from 0.0 to 1.0. Both samples exhibited essentially flat curves for both materials with values between 0.0 and 0.9. At P/P0 above 0.9, the isotherms grew rapidly and the adsorption and desorption isotherms did not overlap, showing hysteresis loops. The Barrett–Joyner–Halenda (BJH) model was used to predict the pore size distribution curve. The pore size of carriers directly affects the preparation of the heterogeneous enzyme and the activity and stability of lipase. In this study, the pore size distribution curves of both materials were similar (Fig. 2e and f). This similarity indicates that the process of enzyme immobilization did not change the original pore structure of MWCNTs. Additionally, the average pore size increased from 26.5 nm to 28.3 nm after immobilization. This is because lipase was mostly bound to the carrier by surface cross-linking, and lipase was adsorbed more in micropores or mesopores and less in macropores. The surface area and pore volume were determined using the Brunauer–Emmett–Teller (BET) model and the BJH model, respectively. MIP-AMWCNTs showed a large specific surface area and pore volume (Table S2†), making them an ideal carrier material. According to our measurements, the specific surface area of the material decreased from 204.8 to 134.6 m2 g−1 after lipase loading, and the pore volume decreased from 1.6 to 1.0 cm3 g−1. The more significant changes in the data were caused by lipase occupying the pores on the carrier surface. Taken together, these results demonstrated that lipase was successfully loaded onto the MIP-AMWCNTs. Furthermore, compared with the previous studies (Table S3†), the specific surface area of the carrier possessed a large effect on the immobilization efficiency. The efficiency of MIP-AMWCNTs as a carrier to immobilize lipase was significantly increased. According to XPS elemental analysis, the relative contents of O and N in MIP-AMWCNTs@lipase were higher than in the MIP-AMWCNTs (Fig. 2g). Lipase was the only source of O and N. Therefore, the lipase molecules were adsorbed onto the surface of MIP-AMWCNTs. The amount of Cl increased slightly, which was attributed to the Tris–HCl buffer used during immobilization. The XPS results demonstrated the successful addition of lipase to the surface of MIP-AMWCNTs.
MIP-AMWCNTs/GCE and MIP-AMWCNTs@lipase/GCE electrochemical sensors were prepared (Fig. 2h). The current intensity of the redox peaks of cyclic voltammetry was used as a preliminary comparison of electrode conductivity in order to simply compare the strength of electrode conductivity. Cyclic voltammetry curves were constructed in the range of 0.2 to 0.6 V with a scan rate of 50 mV s−1 using K3Fe(CN)6 solution as a probe. There are obvious redox peaks in the three electrode materials (Fig. 2i). The peak currents for the MIP-AMWCNTs/GCE were much higher than those for the bare electrodes. Significantly increased conductivity was observed compared to that of bare electrodes. This is because the unique structure of the nanotubes provides an efficient channel for electron transport.32 However, the conductivity of MIP-AMWCNTs@lipase/GCE was much weaker than that of MIP-AMWCNTs/GCE. This difference indicates that the addition of non-conductive lipase affects electron transfer on the surface of the material. These results confirm that lipase was successfully immobilized on the surface of MIP-AMWCNTs.
When the scan speed was increased from 5 to 300 mV s−1 under the same experimental conditions, the redox peak current of MIP-AMWCNTs@lipase/GCE increased with the scan rate (Fig. 2j). MIP-AMWCNTs@lipase/GCE showed significant electrochemical performance over a wide range of scan rates. The oxidation peak potential shifted toward the positive pole, while the reduction peak potential shifted toward the negative pole. The oxidation peak was not symmetrical with the reduction peak. Although the linear fit is very good here, the line does not cross the origin. The relationship between the scan rate's square root of the oxidation peak current (Ipa) and the reduction peak current (Ipc) shows deviation (Fig. 2k). The peak current ratios of oxidation and reduction peaks were more than unity.33 The peak potential difference (ΔEp = Epc – Epa) was calculated for different scan speeds (Table S4†). All the results indicate that the reaction is quasi-reversible.34 The current response of the quasi-reversible reaction is closely related to the scanning speed. When the scanning speed is small, it is close to the reversible reaction state. At high scanning speeds, the system approaches an irreversible response state. This suggests that the prepared electrodes exhibit better reversibility at low scanning speeds. This study developed MIP-AMWCNTs@lipase/GCE biosensors that show promise for contaminant analysis and detection in the future.
Enzymatic performance
The optimum catalytic conditions for lipase were systematically investigated. The pH had a large effect on lipase in both the free and bound states (Fig. S2a†). The optimum reaction pH for both types of lipases was 9.0. MIP-AMWCNTs@lipase showed higher catalytic activity than free lipase over a wide pH range (4.0–11.0). As shown in Fig. S2b,† the optimum temperature for MIP-AMWCNTs@lipase was 55 °C, whereas the ideal temperature for free lipase is 40 °C (range of 25 °C to 80 °C). The relative activity of MIP-AMWCNTs@lipase showed a much flatter trend with a wider temperature range than free lipase. In a high temperature environment, the interaction forces between the active group of lipase and the carrier hinder the diffusion of lipase. This limits conformational changes of the lipase.35 This makes it less sensitive to temperature and more resistant to high temperatures.Catalytic reaction mechanism of MIP-AMWCNTs@lipase
First, we modeled the kinetics of the complex enzymatic reaction of MIP-AMWCNTs@lipase with the Michaelis–Menten equation for simplifying the kinetics of the heterogeneous enzyme. The correlation coefficient (R2) for both free lipase and MIP-AMWCNTs@lipase was close to 0.99. The relationship between 1/[S] and 1/V0 was well linear (Fig. 3a) and followed Michaelis–Menten kinetics. The Km and Vmax were obtained by fitting the Michaelis–Menten equation (Table S5†). The Km of MIP-AMWCNTs@lipase decreased from 0.905 to 0.187 mmol L−1. The decrease in Km may be attributed to the strong affinity between MIP-AMWCNTs@lipase and the substrate, as Km reflects the affinity between lipase and the substrate. The Vmax of MIP-AMWCNTs@lipase decreased from 1.970 × 10−3 to 4.846 × 10−4 mmol (L min)−1. The decrease in Vmax is caused by the lipase confinement to the surface space after lipase is cross-linked to the surface of the vector. As a result, the flexibility of lipase decreased, and the substrate's accessibility to the lipase's active site was reduced. Thus, lipase's ability to operate on the substrate was reduced, and its reaction rate also decreased. The catalytic efficiency was represented by the ratio kcat/Km.35 Thus, the kcat/Km of MIP-AMWCNTs@lipase increased 1.2-fold. The immobilization process exposed the lipase catalytic site36 and enhanced the catalytic effectiveness of lipase. Consequently, catalytic activity was greatly increased, and its affinity for the substrate was improved. These findings also showed that the MIP-AMWCNTs@lipase reaction kinetics were impacted by the emergence of novel structural domains between lipase and the carrier. | ||
| Fig. 3 The Lineweaver–Burk plot (a); the substrate–product plot (b); illustration of enzymatic reaction kinetics (c). | ||
The kinetics of the enzymatic reactions of heterogeneous enzymes are complex and the process is influenced by various aspects, such as diffusion,37 spatial steric hindrance,38 and conformational effects.39 The traditional Michaelis–Menten equation is not adequate to describe this process.40 Consumption of substrates and the formation of products occur throughout the reaction, which creates a concentration differential between the solid-phase carrier and the aqueous-phase solution, resulting in a diffusion effect (Fig. 3c). The substrate diffuses to the active site of lipase on the nanotube surface through three mechanisms: direct diffusion, single hindrance diffusion, and multiple hindrance diffusion. Therefore, the enzymatic reaction of heterogeneous enzymes is somewhat limited by the carrier, which produces a spatial steric hindrance effect.41 This effect causes changes in the active center of the lipase–carrier complex, which leads to a decrease in activity over time and affects the overall reaction rate. The reaction kinetics of heterogeneous enzymes can be broken down into three stages. The first stage is substrate diffusion, which involves non-specific adsorption and can be describe by using the Freundlich adsorption model.42 Lipase reacts with the substrate on the carrier, following the Michaelis–Menten equation. As time passes, the activity decreases until it reaches saturation, similar to the zero-level stage in the equation. Eqn (1) simplifies the reaction of the substrate with the heterogeneous enzyme.
 | (1) |
| Pp-NP = k1[S](−n) + Vmax × [S] × t2/(k2 + [S]) | (2) |
Stability
After incubation at pH 9.0 and 60 °C for different periods, the activities of the two types of lipases decreased to different degrees (Fig. 4a). The inactivation rate of free lipase was obviously faster. After 180 min of incubation, only 31.9% of its initial activity was maintained for free lipase, whereas MIP-AMWCNTs@lipase maintained 52.5% of its initial activity. MIP-AMWCNTs@lipase had better tolerance to heating than free lipase.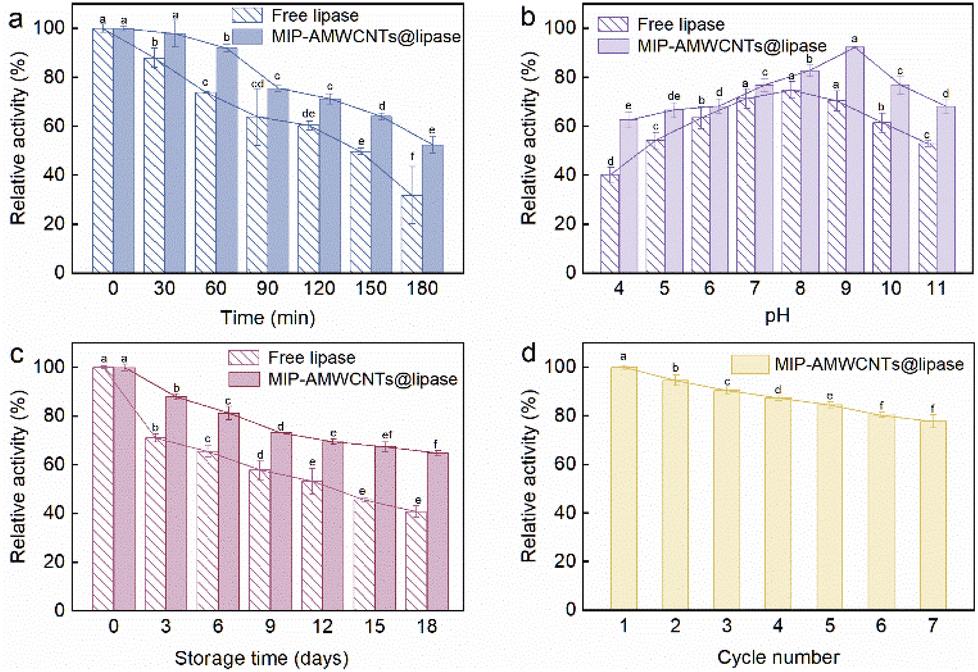 | ||
| Fig. 4 Thermal stability (a); pH stability (b); storage stability (c); effect of repeated use on the activity of MIP-AMWCNTs@lipase (d). (The different English letters show significance between each treatment while p < 0.05.) | ||
Different pH conditions affect the ionization state of the catalytic group of the enzyme molecule. This affects the binding of the lipase to the substrate. The pH stability of free lipase was poor (Fig. 4b). An environment that is too acidic or too alkaline is not conducive to the preservation of free lipase activity. However, MIP-AMWCNTs@lipase showed higher pH tolerance and better catalytic activity over a wide pH range (pH 4.0–11.0).
Storage stability of enzymes has been a hot topic in the field of biocatalysis. When enzymes are immobilized on inert carriers, their resistance against changes in the outside environment is enhanced. Thus, the storage time of the enzyme can be extended, subsequently reducing costs in industry. After 18 days of incubation, MIP-AMWCNTs@lipase maintained 64.9% of its initial activity. Free lipase only maintained 40.8% of its initial activity (Fig. 4c). These showed that the storage stability of MIP-AMWCNTs@lipase is greatly improved and it has a longer shelf life.
The catalysis of free enzymes is very costly and they are difficult to recover as it is very arduous to isolate them from the reaction medium.43 Compared to free lipase, heterogeneous enzymes are easier to isolate from the reaction solution and reuse. This overcomes the problem of the high cost of free lipase, which is difficult to recover. Fig. 4d shows that the activity gradually decreased as the number of operations increased. After 7 cycles, approximately 77.7% of the initial activity was retained. This showed that MIP-AMWCNTs@lipase had good reusability.
Adsorption–degradation
The surface of MIP-AMWCNTs has DEHP-imprinted cavities left by our molecular imprinting technique. This cavity can specifically interact with DEHP in an aqueous environment, i.e., trapping. After being trapped near MIP-AMWCNTs@lipase, DEHP could be directly degraded by lipase, thus improving the removal efficiency of DEHP from water. This is a synergistic adsorption–degradation coordination.42 We first investigated the adsorption behavior of the carrier with 5 mg L−1 DEHP. MIP-AMWCNTs and NIP-AMWCNTs reached adsorption equilibrium around 120 min (Fig. 5a and S3a†) with adsorption rates of 66.3% and 52.1%, respectively. These indicated that MIP-AMWCNTs have higher adsorption capacity than NIP-AMWCNTs. After linear fitting by adsorption kinetics, quasi-secondary kinetics showed a better fit (Fig. 5b and S3b†). | ||
| Fig. 5 The adsorption kinetic curves of MIP-AMWCNTs and NIP-AMWCNTs (a); pseudo-second-order kinetics fit of MIP-AMWCNTs and NIP-AMWCNTs (b); selective identification of adsorption (c); MIP-AMWCNTs@lipase removal efficiency for different initial DEHP concentrations (d); first-order kinetics for degradation (e). | ||
Selective adsorption experiments were performed with DMP, DnOP, and DIDP (Fig. 5c). The large specific surface area and the porous nature of the carrier made the adsorption of other compounds unavoidable. Fig. 5c shows that the presence of DMP and DNOP produced a more interfering effect on the adsorption of DEHP by MIP-AMWCNTs. This is due to the small molecular weights of DMP and DNOP, which are less than and equal to that of DEHP, respectively. They are easily adsorbed by the blotting cavities.44 DEHP had a more significant adsorption effect than the substance due to structural differences. The adsorption effect of MIP-AMWCNTs for DEHP was much higher than that with the other compounds. This could be attributed to the imprinted cavities and exclusive active sites for DEHP on the surface of MIP-AMWCNTs. The imprinting factor (IF) of DEHP was 3.4, that was much higher than those of the other three compounds. Thus, MIP-AMWCNTs have a good imprinting effect for DEHP. The selectivity coefficient (SC) was used to measure the recognition ability of MIP-AMWCNTs for DEHP. The SC of MIP-AMWCNTs for DMP, DnOP, and DIDP was all less than one. Furthermore, the SC of DIDP was close to zero, which indicated that specific selectivity of MIP-AMWCNTs for DEHP was the highest in the presence of DIDP. The results indicate that functional units (MIP-AMWCNTs) have specific selective structural domains on their surfaces.
DEHP (5–40 mg L−1) was used as the target contaminant and the adsorption–degradation synergistic removal ability of MIP-AMWCNTs@lipase was investigated. Removal rates of DEHP (5, 10, 20, 30 and 40 mg L−1) within 240 min reached 94.2%, 87.4%, 80.8%, 77.0%, and 70.6%, respectively (Fig. 5d). These results showed that MIP-AMWCNTs@lipase could be applied to the removal of high concentrations of organic pollutants in the future. Compared with the carrier alone, MIP-AMWCNTs@lipase increased removal of DEHP (5 mg L−1) from 66.3% to 94.2% within 240 min. This difference could be attributed to enzymatic degradation. The DEHP removal ability was significantly improved by MIP-AMWCNTs@lipase. It is assumed that the removal of DEHP in the range of 5–40 mg L−1 by MIP-AMWCNTs@lipase follows first-order kinetics, as shown in Fig. 5e (linear fitting equation) and Table S6† (first-order kinetics parameters). The results indicate that lipase follows a first-order kinetic equation for the adsorption and degradation of MIP-AMWCNTs.
Conclusions
In summary, we have developed a carbon-based bifunctional heterogeneous enzyme (MIP-AMWCNTs@lipase) that exhibits both selectivity and stability. To achieve this, we first synthesized MIP-AMWCNTs using surface molecular imprinting technology. We found that this material exhibited selectivity for the target contaminant (DEHP); however, the removal rate was poor. To address this issue, we immobilized lipase on MIP-AMWCNTs through cross-linking reactions. The study found that the end material (MIP-AMWCNTs@lipase) efficiently removed the target contaminants (94.2% for 5 mg L−1 DEHP) and improved the activity, stability, and reusability of lipase. Additionally, we identified the potential application of MIP-AMWCNTs@lipase for electrochemical biosensors due to its sensitive electrochemical properties, which could be useful for future pollutant analysis and detection. The methodology presented in this study offers a feasible and efficient approach for synthesizing functional biomaterials and removing persistent organic pollutants.Data availability
Data and figures for this manuscript are available within the main manuscript.Author contributions
Y. S. and M. G. contributed equally to this work. M. G., W. Z., Y. Y. and C. W. conceived and supervised the project. M. G. designed the experiments. Y. S. and S. H. analyzed the data and wrote the manuscript. Y. S. carried out most of the experiments. Y. J. provided the theoretical support. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Natural Science Foundation of Zhejiang Province (LTGS23C160003), the ES Program (via Nagoya University until May 2024), the ARC Laureate Fellowship (FL230100095), and Baima Lake Laboratory Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (LBMHZ24B060001). We express our gratitude for English editing software, such as Grammarly and ChatGPT, for refining language and checking grammatical errors in our manuscript. Additionally, this work used the Queensland node of the NCRIS-enabled Australian National Fabrication Facility (ANFF).Notes and references
- P. Giri, A. D. Pagar, M. D. Patil and H. Yun, Biotechnol. Adv., 2021, 53, 107868 CrossRef CAS PubMed.
- (a) J. C. Breger, J. N. Vranish, E. Oh, M. H. Stewart, K. Susumu, G. Lasarte-Aragonés, G. A. Ellis, S. A. Walper, S. A. Díaz, S. L. Hooe, W. P. Klein, M. Thakur, M. G. Ancona and I. L. Medintz, Nat. Commun., 2023, 14, 1757 CrossRef CAS PubMed; (b) L. Li and G. Chen, J. Am. Chem. Soc., 2022, 144, 16232–16251 CrossRef CAS.
- (a) A. F. Ogata, A. M. Rakowski, B. P. Carpenter, D. A. Fishman, J. G. Merham, P. J. Hurst and J. P. Patterson, J. Am. Chem. Soc., 2020, 142, 1433–1442 CrossRef CAS; (b) A. I. Freitas, L. Domingues and T. Q. Aguiar, J. Adv. Res., 2022, 36, 249–264 CrossRef CAS PubMed.
- P. Chandra, Enespa, R. Singh and P. K. Arora, Microb. Cell Fact., 2020, 19, 169 CrossRef CAS.
- L. P. Christopher, H. Kumar and V. P. Zambare, Appl. Energy, 2014, 119, 497–520 CrossRef CAS.
- (a) Y.-P. Cao, G.-Y. Zhi, L. Han, Q. Chen and D.-H. Zhang, Food Chem., 2021, 364, 130428 CrossRef CAS; (b) W. Wei, F. Mazzotta, I. Lieberwirth, K. Landfester, C. T. J. Ferguson and K. A. I. Zhang, J. Am. Chem. Soc., 2022, 144, 7320–7326 CrossRef CAS.
- (a) S. Miri, J. A. E. Perez, S. K. Brar, T. Rouissi and R. Martel, Environ. Pollut., 2021, 285, 117678 CrossRef CAS; (b) G. Awad and E. F. Mohamed, Appl. Catal., B, 2019, 253, 88–95 CrossRef CAS; (c) A. I. Benitez-Mateos, S. Bertella, J. B. de Bueren, J. S. Luterbacher and F. Paradisi, ChemSusChem, 2021, 14, 3198–3207 CrossRef CAS PubMed; (d) Y. J. Chen, P. Li, J. W. Zhou, C. T. Buru, L. Dordevic, P. H. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 1768–1773 CrossRef CAS PubMed.
- L. Feng, Y. Cao, D. Xu, S. You and F. Han, Ultrason. Sonochem., 2016, 32, 224–230 CrossRef CAS.
- J. M. Bolivar and B. Nidetzky, Molecules, 2019, 24, 3460 CrossRef CAS.
- A. D. Vethaak and J. Legler, Science, 2021, 371, 672–674 CrossRef CAS PubMed.
- (a) A. Bakir, I. A. O'Connor, S. J. Rowland, A. J. Hendriks and R. C. Thompson, Environ. Pollut., 2016, 219, 56–65 CrossRef CAS PubMed; (b) A. Mohammadi, M. Malakootian, S. Dobaradaran, M. Hashemi and N. Jaafarzadeh, Sci. Total Environ., 2022, 842, 156838 CrossRef CAS.
- P. Song, J. Gao, X. Li, C. Zhang, L. Zhu, J. Wang and J. Wang, Environ. Int., 2019, 129, 10–17 CrossRef CAS.
- A. Paluselli, V. Fauvelle, F. Galgani and R. Sempere, Environ. Sci. Technol., 2019, 53, 166–175 CrossRef CAS PubMed.
- Y. Deng, Z. Yan, R. Shen, M. Wang, Y. Huang, H. Ren, Y. Zhang and B. Lemos, Environ. Int., 2020, 143, 105916 CrossRef CAS PubMed.
- H. Zhang, Q. Zhou, Z. Xie, Y. Zhou, C. Tu, C. Fu, W. Mi, R. Ebinghaus, P. Christie and Y. Luo, Sci. Total Environ., 2018, 616, 1505–1512 Search PubMed.
- S. Miri, R. Saini, S. M. Davoodi, R. Pulicharla, S. K. Brar and S. Magdouli, Chemosphere, 2022, 286, 131670 CrossRef CAS PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- G. Bijoy, R. Rajeev, L. Benny, S. Jose and A. Varghese, Chemosphere, 2022, 307, 135759 CrossRef CAS PubMed.
- S. A. Ansari and Q. Husain, Biotechnol. Adv., 2012, 30, 512–523 CrossRef CAS PubMed.
- M. Chen, X. S. Qin and G. M. Zeng, Trends Biotechnol., 2017, 35, 836–846 CrossRef CAS PubMed.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- W. Xu, Z. Fu, G. Chen, Z. Wang, Y. Jian, Y. Zhang, G. Jiang, D. Lu, J. Wu and Z. Liu, Nat. Commun., 2019, 10, 2684 CrossRef PubMed.
- E. M. Pelegri-O'Day, N. M. Matsumoto, K. Tamshen, E. D. Raftery, U. Y. Lau and H. D. Maynard, Bioconjugate Chem., 2018, 29, 3739–3745 CrossRef.
- S. Ji, W. Liu, S. Su, C. Gan and C. Jia, LWT--Food Sci. Technol., 2021, 149, 111897 CrossRef CAS.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- Y. Liu, X. Wang, W. Wang, B. Li, P. Wu, M. Ren, Z. Cheng, T. Chen and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 7991–7999 CrossRef CAS.
- A. Jorio and R. Saito, J. Appl. Phys., 2021, 129, 021102 CrossRef CAS.
- A. Rinaldi, J. Zhang, B. Frank, D. S. Su, S. B. Abd Hamid and R. Schlögl, ChemSusChem, 2010, 3, 254–260 CrossRef CAS.
- S. Bingaman, V. H. Huxley and R. E. Rumbaut, Microcirculation, 2015, 10, 221–231 CrossRef.
- B. P. Carpenter, A. R. Talosig, J. T. Mulvey, J. G. Merham, J. Esquivel, B. Rose, A. F. Ogata, D. A. Fishman and J. P. Patterson, Chem. Mater., 2022, 34, 8336–8344 CrossRef CAS PubMed.
- A. I. Benitez-Mateos, C. Huber, B. Nidetzky, J. M. Bolivar and F. Lopez-Gallego, ACS Appl. Mater. Interfaces, 2020, 12, 56027–56038 CrossRef CAS PubMed.
- R. Xu, R. Tang, Q. Zhou, F. Li and B. Zhang, Chem. Eng. J., 2015, 262, 88–95 CrossRef CAS.
- H. A. Kashmery, Int. J. Hydrogen Energy, 2019, 44, 22173–22180 CrossRef.
- K. Hyun, S. W. Han, W.-G. Koh and Y. Kwon, Int. J. Hydrogen Energy, 2015, 40, 2199–2206 CrossRef CAS.
- Y. Li, T. Jing, G. Xu, J. Tian, M. Dong, Q. Shao, B. Wang, Z. Wang, Y. Zheng and C. Yang, Polymer, 2018, 149, 13–22 CrossRef CAS.
- X. Liu, Y. Hu, B. Wei, F. Liu, H. Xu, C. Liu, Y. Li and H. Liang, Process Biochem., 2022, 118, 323–334 CrossRef CAS.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef CAS.
- A. Ledesma-Fernandez, S. Velasco-Lozano, J. Santiago-Arcos, F. López-Gallego and A. L. Cortajarena, Nat. Commun., 2023, 14, 2587 CrossRef CAS PubMed.
- D. F. Kienle, R. M. Falatach, J. L. Kaar and D. K. Schwartz, ACS Nano, 2018, 12, 8091–8103 CrossRef CAS.
- Z. Jin, N. Dridi, G. Palui, V. Palomo, J. V. Jokerst, P. E. Dawson, Q.-X. Amy Sang and H. Mattoussi, J. Am. Chem. Soc., 2023, 145, 4570–4582 CrossRef CAS PubMed.
- (a) Z. Jin, N. Dridi, G. Palui, V. Palomo, J. V. Jokerst, P. E. Dawson, Q.-X. A. Sang and H. Mattoussi, Anal. Chem., 2023, 95, 2713–2722 CrossRef CAS; (b) Z. Jin, C. Ling, Y. Li, J. Zhou, K. Li, W. Yim, J. Yeung, Y.-C. Chang, T. He and Y. Cheng, Nano Lett., 2022, 22, 8932–8940 CrossRef CAS.
- C. Duan, M. A. Alibakhshi, D.-K. Kim, C. M. Brown, C. S. Craik and A. Majumdar, ACS Nano, 2016, 10, 7476–7484 CrossRef CAS.
- B. C. Ekeoma, L. N. Ekeoma, M. Yusuf, A. Haruna, C. K. Ikeogu, Z. M. A. Merican, H. Kamyab, C. Q. Pham, D.-V. N. Vo and S. Chelliapan, J. Biotechnol., 2023, 369, 14–34 CrossRef CAS.
- B. Wu, J. Wan, Y. Zhang, B. Pan and I. M. C. Lo, Environ. Sci. Technol., 2020, 54, 50–66 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03752a |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2024 |