
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCCC-NHC Au(III) pincer complexes as a reliable platform for isolating elusive species†
Hugo
Valdés
*ab,
Nora
Alpuente
b,
Pedro
Salvador
 b,
A. Stephen K.
Hashmi
*cd and
Xavi
Ribas
*b
b,
A. Stephen K.
Hashmi
*cd and
Xavi
Ribas
*b
aDepartamento de Química Orgánica y Química Inorgánica, Instituto de Investigación Química “Andrés M. del Río” (IQAR), Facultad de Farmacia, Universidad de Alcalá, Alcalá de Henares, 28805 Madrid, Spain. E-mail: hugo.valdes@uah.es
bInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, Girona E-17003, Catalonia, Spain. E-mail: xavi.ribas@udg.edu
cOrganisch-Chemisches Institut, Heidelberg University, 69120 Heidelberg, Germany. E-mail: hashmi@hashmi.de
dChemistry Department, Faculty of Science, King Abdulaziz University (KAU), Jeddah 21589, Saudi Arabia
First published on 2nd October 2024
Abstract
The reactivity of unprecedented CCC-NHC Au(III) pincer complexes has been investigated, employing a novel methodology for their preparation. Notably, this marks the inaugural case of CCC-NHC Au(III) pincer complexes with a central aryl moiety where the two arms of the pincer ligand consist of N-heterocyclic carbenes (NHC). The stability conferred by the CCC-NHC ligand facilitated the isolation of elusive Au(III) species, encompassing Au(III)–formate, Au(III)–F, Au(III)–Me, and Au(III)–alkynyl. Our study also unveiled the elusive Au(III)–H species, offering valuable insights into its formation, stability, and reactivity. While the CCC-NHC Au(III)–H complex remains stable at room temperature, its decomposition becomes conspicuous at elevated temperatures (>60 °C), exhibiting a more pronounced tendency under acidic conditions compared to basic ones. Through comprehensive experiments, we indirectly demonstrated the potential of Au(III)–formate to undergo β-hydride elimination, becoming a key step in the dehydrogenation of formic acid. Theoretical calculations revealed variations in the reactivity of Au(III)–H species towards sodium hydride and formic acid, highlighting a link between σ-donation from the pincer ligand and reaction energetics. Pincers with lower electron donation favored the reaction with sodium hydride but impeded the reaction with formic acid, whereas those with higher electron donation exhibited the opposite behavior. Additionally, the CCC-NHC Au(III) pincer complex exhibited Lewis acid behavior, catalyzing the synthesis of phenols. In summary, the CCC-NHC Au(III) pincer complex emerges as a versatile platform for isolating reactive species and unraveling elementary catalytic steps.
1 Introduction
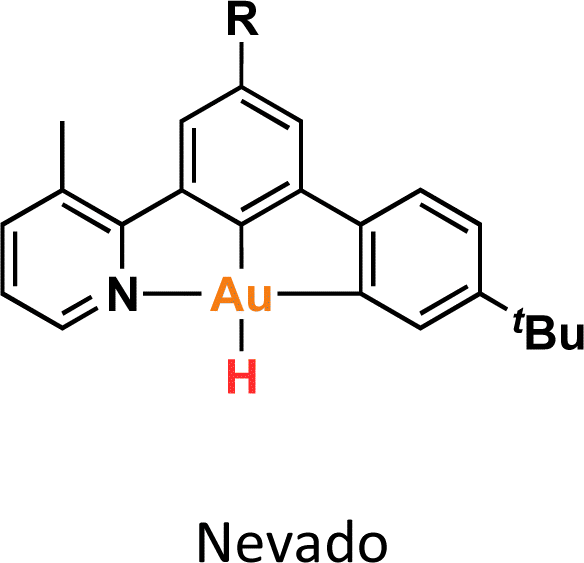
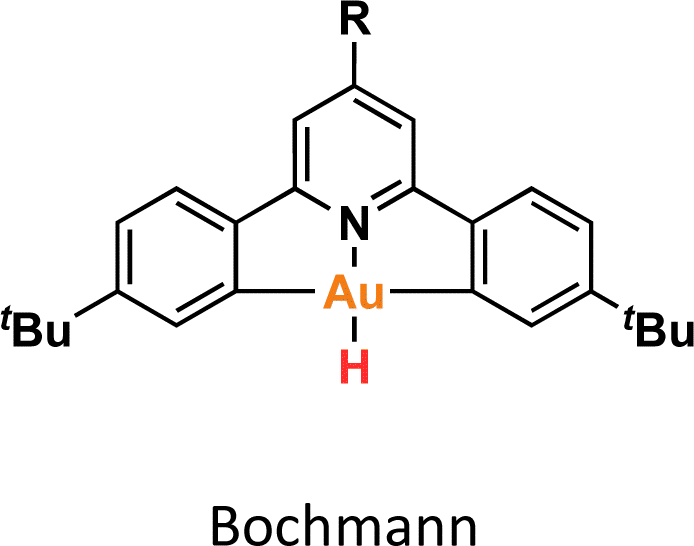
One crucial facet of metal-based catalysis lies in unraveling the intermediate species intricately involved in catalytic cycles. This comprehension not only facilitates the enhancement and design of novel catalysts but also directly impacts the efficiency of the catalytic process. In the domain of gold catalysis, elucidating these species proves challenging owing to the inherent reactivity of gold. Its notably high oxidation potential (EAu(III)/Au(I)red = +1.41 V, EAu(III)/Au(0)red = +1.36 V) designates gold(III) as a potent oxidizing metal, rendering its coordination with highly electron-rich ligands generally incompatible.1–3 An illustrative example is provided by Au(III)–H species,4–6 which remains elusive, with few examples in the literature (Fig. 1a).1,7–12 Stable examples typically involve the use of multidentated ligands. In separate studies, Bochmann and collaborators,8,11 as well as Benzuidenhout and co-workers,12 employed CNC pincer ligands to isolate and characterize Au(III)–H species. Very recently, Nevado's group also employed PNC to stabilize Au(III)–H complexes.13 The heightened stability of these complexes can be attributed to the weak trans effect of the N-containing ligand positioned opposite the hydride group, thereby augmenting the Au–H bond dissociation enthalpy. In contrast, Au(III)–H featuring a robust trans-carbon σ-donor exhibits significantly higher lability.14 While composing this manuscript, Goldberg and colleagues reported the oxygen insertion into the Au(III)–H bond of a PCP pincer complex. This process led to the formation of a PCP Au(III)–OOH species.15 Moreover, the use of bidentated CC-ligands enabled the isolation of an air-sensitive Au(III)–H complex.16 The introduction of a PPh3 ligand provided additional stabilization, enabling its characterization through X-ray diffraction. However, Au(III) phosphane complexes with organic substituents might decompose via reductive elimination to form a P–C bond. Utilizing N-heterocyclic carbene (NHC) ligands could prevent this undesired reaction due to their strong σ-donor character.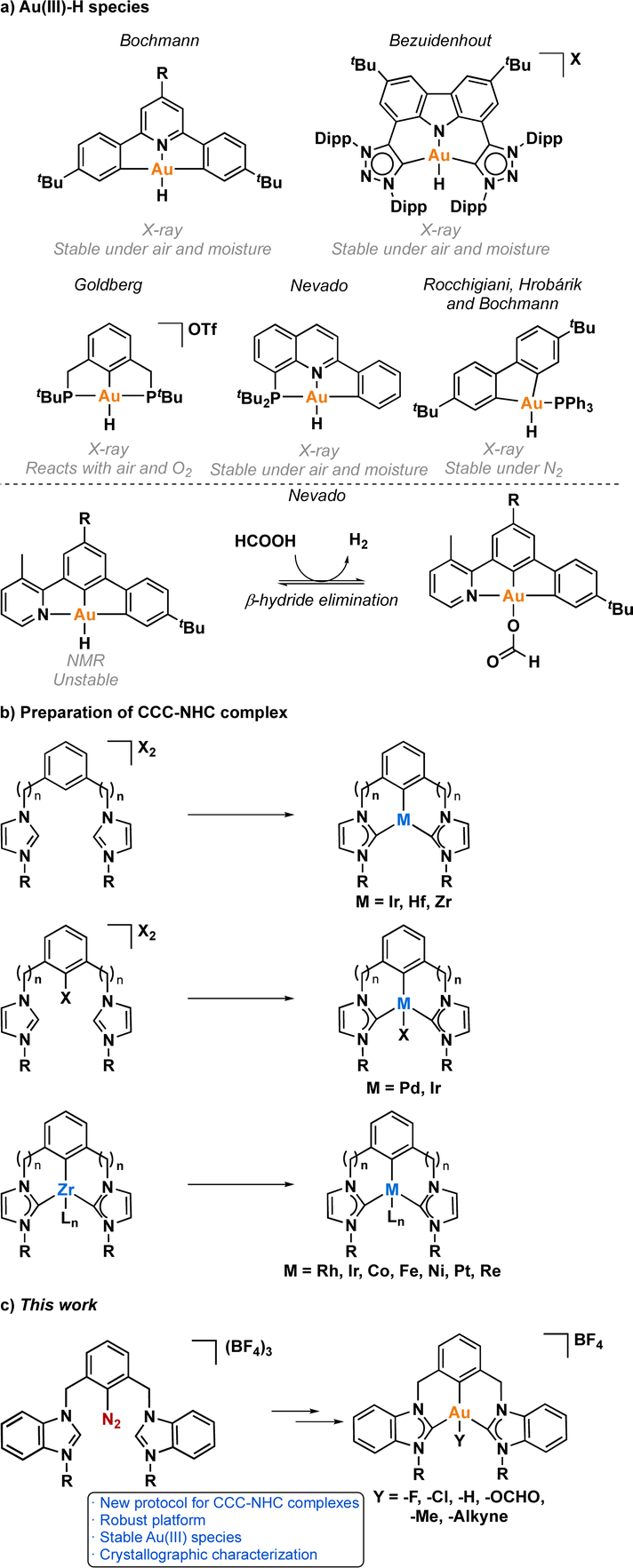 | ||
| Fig. 1 (a) Overview of reported Au(III)–H species to date and their significance in the dehydrogenation of formic acid. (b) Overview of methodologies for synthesizing CCC-NHC transition metal complexes. (c) Our work describing the innovative synthesis of CCC-Au(III) pincer complexes. | ||
Au(III)–H species, alongside Au(III)–carboxylates17,18 and -formates, play a pivotal role as proposed intermediates in the dehydrogenation of formic acid19–22 and Water-Gas Shift (WGS) processes.23 In 2017, Nevado and colleagues elucidated the formic acid dehydrogenation employing a CCN–Au(III) pincer complex (Fig. 1a).24 They outlined the reaction of a CCN Au(III)–F complex with formic acid, yielding the formate complex in quantitative yield. The Au(III)–formate complex, in principle, can undergo β-hydride elimination, leading to the formation of an Au(III)–H species. Subsequently, this species reacts with another molecule of formic acid, generating hydrogen gas and regenerating the Au(III)–formate. The latter process was observed at high temperatures due to the highly energetic cost of β-hydride elimination. It is noteworthy that the CCN Au(III)–H species was exclusively identified through in situ cryo-NMR experiments,16 and its reactivity was probed through trapping experiments conducted in the presence of alkynes.24 Additionally, the Au(III)–formate complex exhibited stability in the solid state, but it decomposed in solution over time under heating at 100 °C.
Motivated by the limited structural diversity observed in stable gold(III)–hydrides and recognizing their importance and potential roles in hydrofunctionalization, water-gas shift, formic acid dehydrogenation reactions, and β-hydride elimination processes, we focused our efforts in employing a CCC-NHC pincer ligand with a central aryl moiety to stabilize elusive Au(III) species, where the two arms of the pincer ligand consist of NHC. The synthesis of CCC-NHC pincer complexes is presently limited to very few methodologies (Fig. 1b).25,26 Coordinating the metal atom with the central aryl fragment poses a notable synthetic challenge. For instance, initiating with a bis(azolium)–aryl precursor necessitates the activation of the central aryl's C–H bond to prepare the pincer complex derivative. This C–H activation is predominantly achievable with a subset of transition metals, including Ir,27–36 Hf37 and Zr.38–40 Functionalizing the central aryl fragment with a halogen group facilitates coordination with certain transition metals capable of oxidative addition, such as Pd(0).41 In 2005, Hollis and colleagues introduced a general synthetic method involving a transmetallation reaction employing Zr derivatives,37,39,40,42,43 proven effective for synthesizing Rh,44,45 Ir,46 Co,47,48 Fe,49,50 Ni,51 Pt,52–54 Re.55 Despite the success of these strategies in obtaining pincer complexes with various transition metals, the synthesis of CCC-NHC Au-pincer (one central aryl and two NHC-based arms) complexes remains undocumented. So far, all attempts reported to obtain CCC-NHC Au-pincer complexes have resulted in the formation of linear Au(I) complexes56 or bimetallic species.57,58
A widely utilized approach for synthesizing CCD-Au(III) pincer complexes, where D is a N atom or a NHC carbene ligand, involves a transmetalation reaction utilizing Sn,59 Hg,60,61 Si,62 or Au(I)63 species, as well as acidic conditions.64–66 Notably, in 2020, Breher and Klopper innovatively detailed the synthesis of a non-palindromic CCC-NHC complex, where two aryl groups and one NHC arm were coordinated to Au(III).59 The complex was prepared via a transmetalation reaction employing Sn(IV). However, a notable drawback of these methodologies is, in some cases, the generation of toxic waste owing to the formation of Sn and Hg species. In contrast, Hashmi and co-workers have circumvented the need for transmetalation agents by employing diazonium salts to create pincer Au(III) complexes.67,68 This approach hinges on the use of light to facilitate the C–N2 activation by Au(I), a key step requiring the presence of a donating heteroatom in the pincer framework. The initial phase of this reaction involves the coordination of the heteroatom, which promotes close interaction between Au(I) and the diazonium salt. Subsequently, the application of light triggers the oxidative addition of C–N2 to Au(I), yielding an Au(III) species capable of activating aromatic C–H bonds. It is noteworthy that this specific strategy has not been previously employed for the synthesis of CCC-NHC pincer complexes. As such, we envisioned that the synthesis of CCC-NHC Au(III) pincer complexes should be attainable through this innovative approach.
Based on the above-mentioned, we present an innovative methodology for synthesizing CCC-NHC Au(III) complexes, featuring NHC ligands as both arms of the pincer ligand (Fig. 1c). The pivotal non-photoirradiated activation of a diazonium salt using sodium ascorbate played a crucial role in the success of this strategy.69 Utilizing this reliable platform, we successfully isolated otherwise elusive Au(III) species, including Au(III)–H, Au(III)–F, Au(III)–alkynyl, Au(III)–Me, and Au(III)–formate. The reactivity of the Au(III)–H complex towards both acids and bases was investigated, shedding light on its stability under varying conditions. Additionally, we delved into the mechanism of β-hydride elimination in the Au(III)–formate complex, enhancing our understanding of this crucial reaction. Finally we explored the Lewis acid catalysis of the CCC-NHC Au(III)–Cl complex in the synthesis of phenol derivatives.
2 Results and discussion
The synthesis of our targeted CCC-NHC pincer ligand (Fig. 1c) precursors began with the reflux of 1,3-dimethylnitrobenzene (1) in the presence of N-bromo succinimide (NBS) and dibenzoyl peroxide (BPO) as a radical initiator in 1,2-dichloroethane (Scheme 1). Following purification, the desired product (2) was obtained in a 20% yield. Subsequently, this product was reacted with 1-substituted benzimidazole to yield the targeted bis(azolium) salts 3a and 3b. Reduction of the nitro group was achieved using zinc and NH4Cl in methanol under reflux conditions to afford 4a and 4b. The diazonium-bis(azolium) salts, 5a or 5b, were then synthesized by reacting with tBu-ONO in the presence of HBF4. An important advantage of this entire pincer ligand precursor synthesis process is its simplicity, eliminating the need for chromatographic columns or laborious procedures (see ESI†). Additionally, the synthetic process for the diazonium-bis–azolium salt offers the benefit of producing all resulting products on a gram-scale, with the added advantage of excellent bench stability (stable for months in solid state). Subsequently, we reacted [Au(SMe2)Cl] with the diazonium-bis(azolium) salt in the presence of KOtBu and blue LEDs, which led to a very complex reaction mixture. Thus, we resorted to a different approach. The reaction of the diazonium-bis(azolium) salt 5b with [Au(SMe2)Cl] in the presence of LiCl (2.7 equivalents) and sodium ascorbate (0.3 equivalents) resulted in the formation of the Au(III) complex 7b–Au.69–72 We observed immediately the formation of N2 gas bubbles in the reaction mixture, which suggested the reaction with gold. At this stage of the reaction, ESI-MS monitoring showed a peak at 755.2 m/z that matched with aryl-Au(III) complex 7b–Au (See ESI, Fig. S1†), in which the central aryl fragment is coordinate to Au(III)Cl3 and the azolium salts remained uncoordinated. Interestingly, LiCl not only acted as an accelerator for C–N2 bond activation by forming a chloride diazonium-bis(azolium) but also promoted the formation of the [AuCl2]− species (See ESI, Scheme S12†).73 To complete the assembling of the pincer complex, we added a DMSO solution of potassium tert-butoxide to the reaction mixture, allowing the deprotonation of the azolium and forming the pincer CCC-Au(III)–Cl complex. It is worth noting that 6a and 6b were isolated as their BF4− salts, as confirmed by X-ray diffraction analyses and subsequent reactivity studies. During the work-up and purification process of the pincers, water was employed, and the selective formation of these pincer salts was likely due to the limited solubility of BF4− species in water.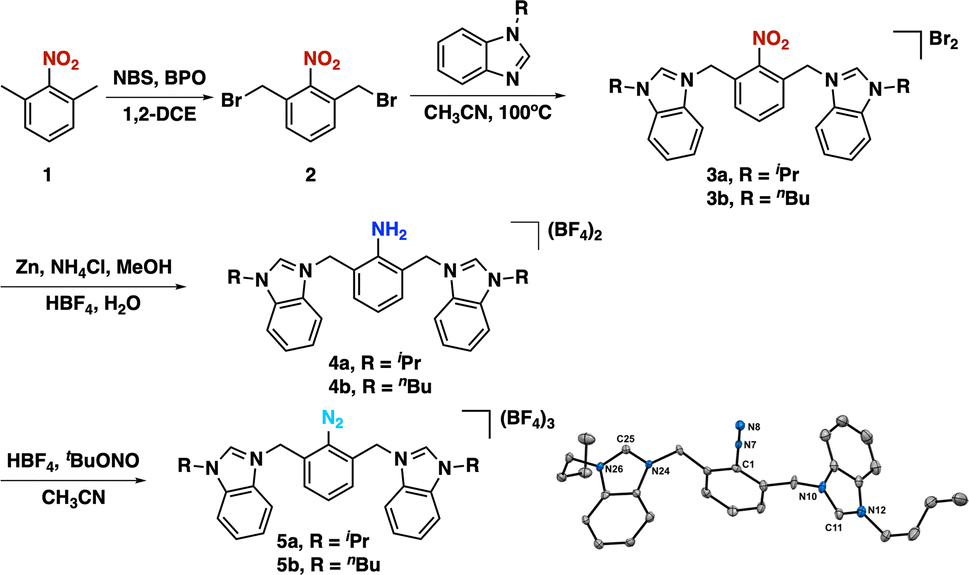 | ||
| Scheme 1 Synthesis of the diazonium–bis–azolium salt. | ||
Based on earlier studies,67,69–71 it is proposed that sodium ascorbate can activate the diazonium salt by generating an aryl radical (See ESI, Scheme S12†). This aryl radical may subsequently react with the [AuCl2]− species, leading to the formation of an Au(II) species. This species may proceed along two distinct pathways. Firstly, it could transfer an electron to another molecule of the diazonium–bis–azolium salt, thereby propagating the radical reaction.74 Alternatively, it may undergo disproportionation with another molecule of the Au(II) complex, resulting in the formation of Au(I) and Au(III) compounds. The identification of these compounds was confirmed through ESI-MS analysis (See ESI†). Ultimately, the introduction of a base promotes the formation of the pincer complex by deprotonating the azolium salts.
The pincer complexes 6a and 6b were obtained in 40% yield, and were thoroughly characterized by various analytical techniques (NMR, HRMS, and crystallographic analysis). In the 1H NMR spectra, the absence of the acidic proton of the azolium salts served as a strong indication of pincer formation, along with the presence of two pairs of diastereotopic signals corresponding to the –CH2- bridges (see Fig. S46–55†). Meanwhile, the 13C NMR spectra exhibited a characteristic signal for the carbene carbon atom at 170.3 ppm and 168.8 ppm for complex 6a and complex 6b, respectively. This small difference is probably due to the more electron-donating character of the nBu derivative.75,76
Fortunately, we were able to obtain suitable crystals of both CCC–Au(III)–Cl pincer complexes, enabling their analysis by X-ray diffraction of a monocrystal (Scheme 2). The resulting structures of both complexes were found to be isostructural, with Au(III) coordinated to one pincer ligand and one chloride ligand completing its coordination sphere. The metal center exhibited a distorted square planar geometry, with C(NHC)–Au–C(NHC) angles lower than the ideal for a perfect square, measuring 171.4(3)° for 6a (iPr) and 172.44(18)° for 6b (nBu). Interestingly, despite having different N-substituents, the average length of the Au-Ccarbene bond was very similar between the two complexes, measuring 2.044 Å for iPr and 2.042 Å for nBu.
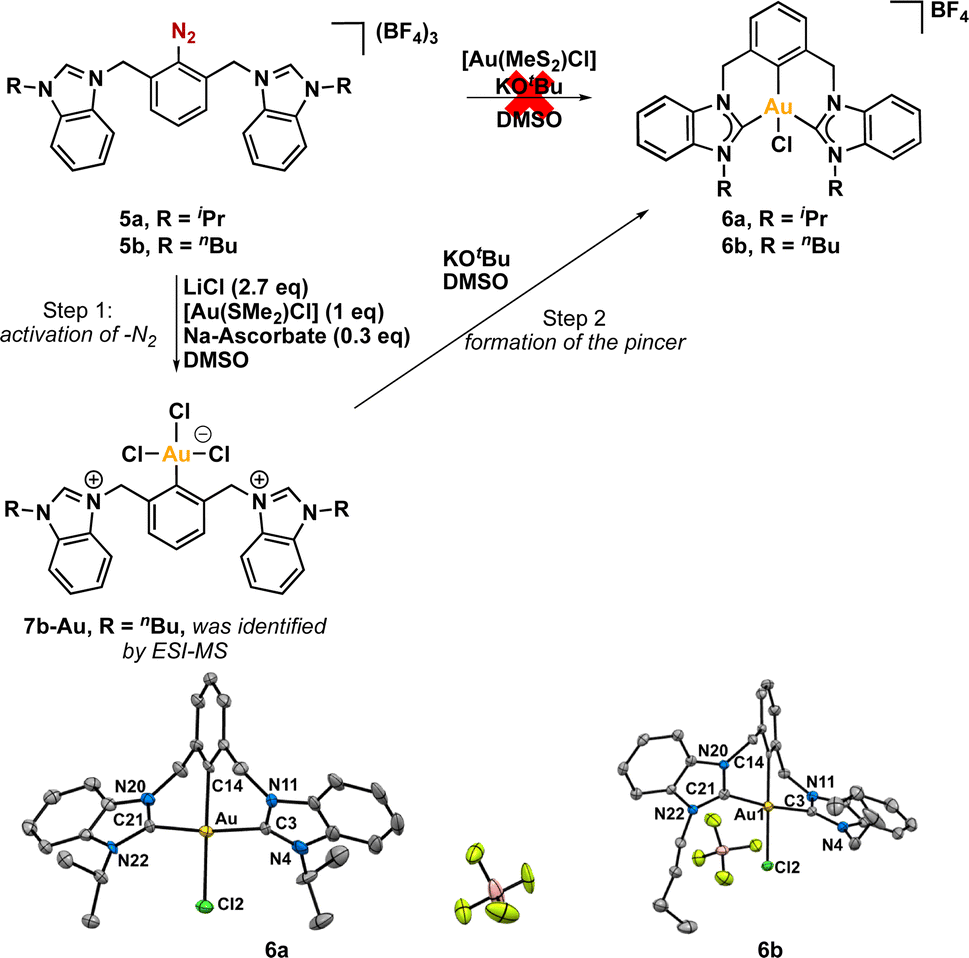 | ||
| Scheme 2 Synthesis of gold(III) pincer complexes through C–N2 bond activation promoted by sodium ascorbate. | ||
Moreover, crystal structures of both complexes 6a and 6b consist of a racemic mixture of two helical conformations of the pincer complexes. This phenomenon arises from hindered rotation of the methylene bridges (–CH2- fragments), leading to the formation of two atropisomers. A similar behavior was previously reported by Hahn and co-workers in the context of related Pd(II) pincer complexes.41,77 The observed axial chirality may potentially play a pivotal role in the realm of gold catalysis,78–81 provided these racemic mixtures could be separated and used for asymmetric gold catalysis.
2.1 Reactivity studies of CCC–Au(III)–Cl pincer complexes
We embarked on a study to explore the reactivity of both complexes towards various nucleophiles (Scheme 3). Our initial focus was on substituting the chlorine atom in 6a/6b with different functional groups. To accomplish this, we conducted reactions between both complexes with methyl magnesium chloride in dichloromethane at room temperature. The outcome was a successful exchange reaction between the chloride and methyl ligand. Both complexes underwent thorough characterization using spectroscopic techniques and X-ray diffraction analyses. Notably, the Heteronuclear Multiple Bond Correlation (1H, 13C-HMBC) analysis revealed a correlation between the proton atoms of the methyl group and the carbon atoms coordinated to the gold atom (Fig. S56–65†). This observation strongly suggests that the methyl group is indeed coordinated to the metal center. In the mass spectra, we observed a single peak corresponding to the CCC–Au(III)–Me complex at 633.2 m/z for 8a and 661.3 for 8b. To obtain high-quality crystals for further analysis, we employed the slow diffusion method, where n-hexane was diffused into a concentrated solution of the complexes in dichloromethane. The resulting crystals were suitable for X-ray diffraction analysis. Both complexes 8a and 8b, clearly show the coordination of the methyl moiety, with similar Au–CH3 bond lengths, measuring 2.108(9) Å for iPr (8a) and 2.14(3) Å for nBu (8b).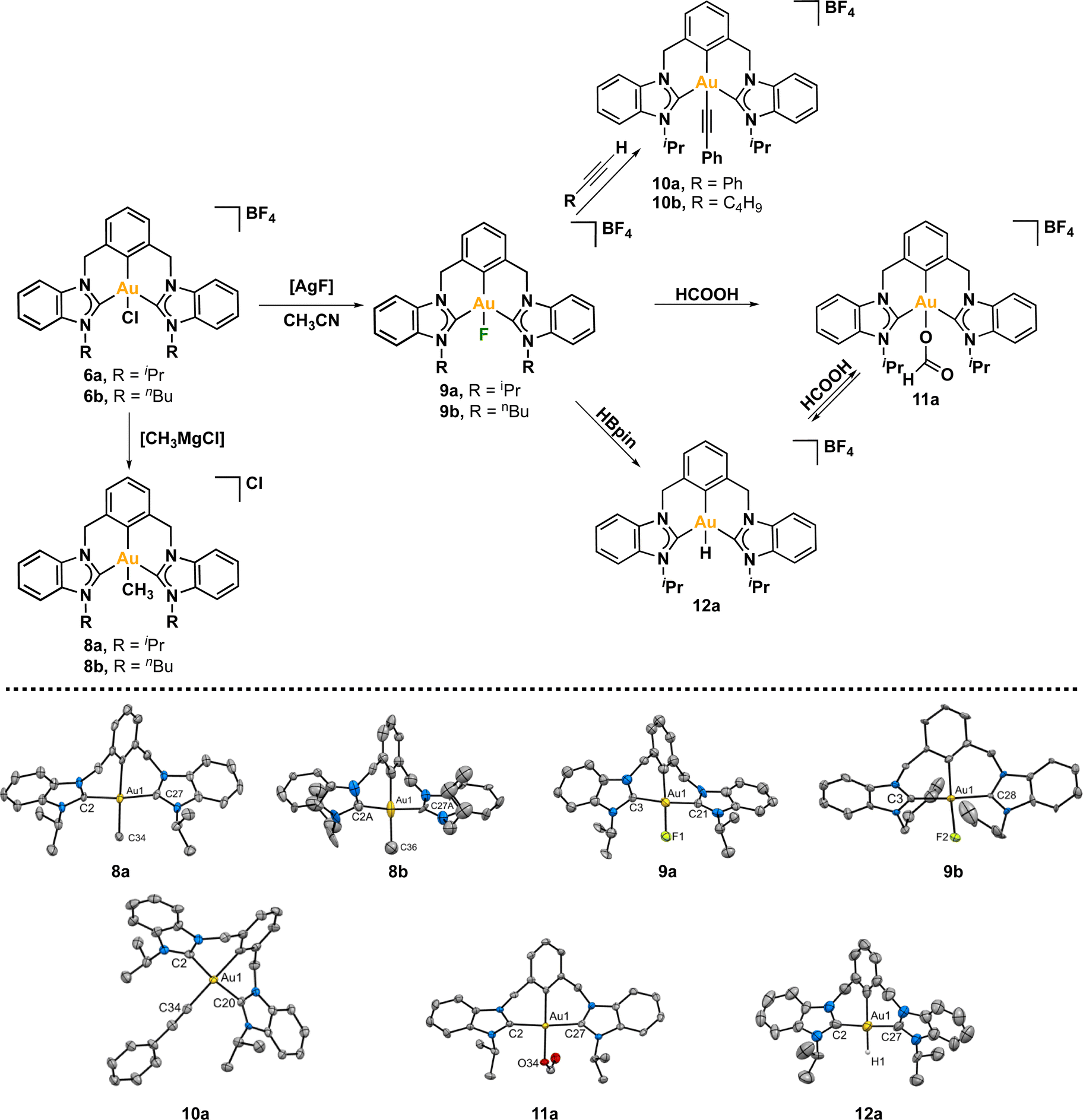 | ||
| Scheme 3 Reactivity studies of Au(III) complexes. | ||
It is worth noting that both complexes were remarkably stable. First, we heated a solution of complex 8b in 1,2-DCE at 100 °C overnight, with no apparent decomposition observed by 1H NMR. When a solution of complex 8b in CD2Cl2 was heated at 60 °C in the presence of formic acid, no significative changes were observed in the 1H NMR. Furthermore, the complex was stable towards NaH at room temperature and at 60 °C. Hence, complex 8b is thermally stable, and resistant to the presence of soft acid and strong base.
Next, we reacted the CCC–Au(III)–Cl pincer complexes 6a and 6b with AgF in CH3CN at room temperature, seeking to exchange the chloride ligand by a fluorine atom. The 1H NMR of the complexes did not show significant changes. Interestingly, in the 13C{1H} NMR spectra the signals of the carbene carbon atom and the metalated aryl carbon appeared as multiplets due to the coupling with the fluorine atom. In complex 9a, the carbenic carbon appeared as a doublet at 170.0 ppm with a 2JC–F of 2.6 HZ, while the metalated aryl carbon appeared at lower frequencies 124.1 ppm and a larger 2JC–F of 37 Hz. In the case of complex 9b the signal of the carbenic carbon appeared as singlet at 169.2 ppm, and the metalated aryl carbon was showed as doublet at 125.7 with a 2JC–F of 26 Hz. Finally, in the 19F NMR spectra appeared a signal at −256.1 ppm for 9a and at −260.1 ppm for 9b, which is within the typical chemical shift range for Au(III)–F complexes.24,82,83
With complex Au(III)–F 9a in hand, our focus turned to investigating its reactivity. The nucleophilic nature of the fluorine ligand, characterized by a negative charge, enables it to act as a base, reacting with acidic species. To illustrate this concept, we conducted a reaction between complex Au(III)–F 9a and phenylacetylene at room temperature for 2 h. The relatively acidic character of the C(sp)–H proved sufficient for a quantitative reaction with complex 9a, considering the pKa of phenylacetylene is 28.7.84 The fluorine ligand acted as a base, deprotonating the alkyne, leading to the formation of HF and generating a negatively charged C(sp) that coordinates to Au(III). This reactivity aligns with observations made previously by Nevado.82 The CCC-Au(III)–alkynyl complex 10a was thoroughly characterized, including structural elucidation through crystallographic analysis. A similar behavior was observed with 1-hexyne, which resulted in the formation of complex 10b.
Having established this proof of concept, it becomes evident that complex 9a may react with stronger acids, such as formic acid. While formate Au(III) complexes are infrequently reported in the literature,18,24 their significance lies in their pivotal role in dehydrogenation processes.85,86 Thus, we proceeded to react 9a with formic acid in CD2Cl2 at room temperature. Gratifyingly, the reaction progressed quantitatively in just a few minutes. The initial indication of the exchange of the fluorine atom by a formate ligand was revealed by 19F NMR, as the signal of the Au–F bond disappeared. The crystal structure of the resulting CCC–Au(III)–OOCH complex 11a unveiled an Au–O distance of 2.081(3) Å, comparable to that reported for gold(III)–OOCR complexes.17,18,24 Remarkably, the complex remained stable in CD2Cl2 at room temperature for several days. Additionally, the complex showed no apparent signs of decomposition when heated in CD2Cl2 at 60 °C for 24 h.
In contrast, heating a 1,2-DCE solution of the complex 11a at 100 °C for 24 h resulted in significant changes in the 1H NMR spectra. First, the signal corresponding to the formate disappeared. This observation, in principle, should be an indication that the Au(III)–formate complex may undergo β-hydride elimination, forming an Au(III)–H complex and CO2 as a side-product. Unfortunately, no hydride complex was observed by 1H NMR; instead, the signals matched those of complex Au(III)–Cl 6a. We then wondered whether a simple ligand exchange between formate and chloride occurs, or if the formation of the Au(III)–H takes place, this species is unstable at high temperatures.1 Regardless, the presence of complex 6a was further confirmed by ESI-MS, which exhibited a peak at 653.2 m/z corresponding to the molecular weight of the complex minus the counter ion ([6a–BF4]+). The ubiquity of chlorides in most solvents, including chlorinated ones, may facilitate the formation of the chlorinated complex 6a. More importantly, a dicharged peak at 619.2 m/z was observed, suggesting the possibility of a dimetallic species in which each gold atom has an oxidation state of +1, with each metal linearly coordinated to two NHC fragments, and the central aryl moiety has been converted to arene.
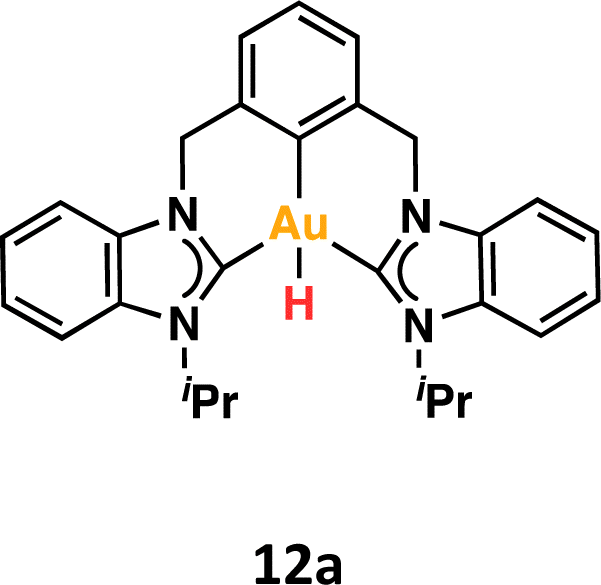
To shed light on the possible β-hydride elimination reaction, we embarked on the preparation of the Au(III)–H derivative. We reacted Au(III)–F complex with HBpin in CD2Cl2. To our delight, the CCC-Au(III)–H species 12a was formed in quantitative yield (99%) within a few minutes. The formation of the stable B–F bond is likely the driving force behind this reaction. The complex 12a exhibited stability in the solid state, allowing for a comprehensive characterization, including the elucidation of its crystal structure through X-ray diffraction analysis. To our knowledge, this constitutes the first X-ray structure of a CCC–Au(III)–H species. In the 1H NMR spectrum, the signal of the Au–H appeared at 1.43 ppm. This value is similar to that reported for Au(III)–H complexes featuring bidentate C^C ligands, where an aryl carbon atom is positioned trans to the hydrogen ligand.16 Recently, Goldberg and co-workers previously reported a chemical shift of 3.50 ppm for a PCP Au(III)–H complex.15
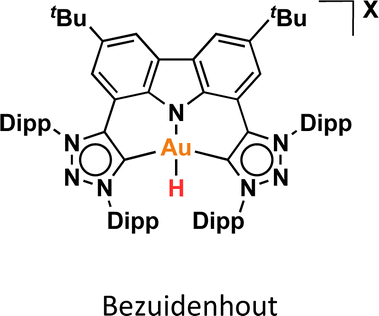
The reactivity of Au(III)–H species with organic acids is uncommon. For instance, the CNC Au(III)–H pincer, as reported by Bezuidenhout and co-workers, exhibited no reactivity towards TfOH, CF3COOH, or HCl.12 A similar unreactive trend was observed for the CNC Au(III)–H pincer reported by Bochmann, which showed no response to acetic acid.1,6,11 In contrast, Nevado and co-workers proposed that a CCN–Au(III)–formate complex undergoes β-hydride elimination to in situ form a CCN–Au(III)–H and releasing CO2, and allowing the CCN–Au(III)–H with hydridic character to react with formic acid, affording H2 gas and CCN–Au(III)–formate to restart the catalytic cycle. With these precedents, we sought to explore the reactivity of CCC–Au(III)–H 12a with formic acid. Therefore, we prepared a CD2Cl2 solution of the Au(III)–H complex 12a and added a substantial excess of formic acid. Surprisingly, 12a demonstrated stability towards formic acid at room temperature for 24 h, as revealed by the 1H NMR. However, after 24 hours at 60 °C, significant changes were observed in the 1H NMR of the reaction mixture, only small amounts of the hydride derivative remained, and a new complex was formed, but no H2 was generated. Also, the presence of the Au(III)–Cl and Au(III)–formate species was further corroborated by ESI-MS (see ESI†). The new species was crystallographically identified as a dimetallic Au(I) species 13a, bearing two non-metalated arene moieties and each metal coordinated to two NHC fragments in a linear arrangement.
Since no H2 evolution was observed with formic acid, we wondered whether the hydrogen atom coordinated to gold possesses a “protic” instead of “hydridic” character. Notably, Bezuidenhout and co-workers12 reported the stoichiometric formation of T-shaped CNC Au(I) pincer complex and H2 gas upon the reaction of the CNC Au(III)–H complex with protic character with NaH. To discern this, we reacted the Au(III)–H complex 12a with a strong base NaH in CD2Cl2, with no significant changes at 60 °C for 24 h. However, upon heating the solution at 60 °C for 5 days, we again observed the formation of the dimetallic Au(I) species 13a.
From the above reactivity of CCC–Au(III)–H 12a with formic acid or NaH several conclusions can be drawn: (a) Au(III)–H complex 12a displays greater stability in the presence of NaH than in the presence of formic acid; (b) the formation of the arene C–H bond suggests reductive elimination of a CCC–Au(III)–H species. We propose that the formation of the dimetallic species 13a might follow the mechanism depicted in Scheme 4. Initially, one arm of the pincer is decoordinated, followed by hydride cis–trans isomerization promoted by the strong trans effect of the NHC ligand. Subsequently, the Au(III) atom undergoes reductive elimination between the aryl fragment and the hydrogen atom. Finally, two of these molecules dimerize to form 13a. The decoordination of the NHC ligand is likely to be more favorable under acidic conditions via the formation of the corresponding azolium salt. Conversely, under basic conditions, the free NHC ligand may dissociate more sluggishly and a transient bis-hydride species might drive the formation of 13a.
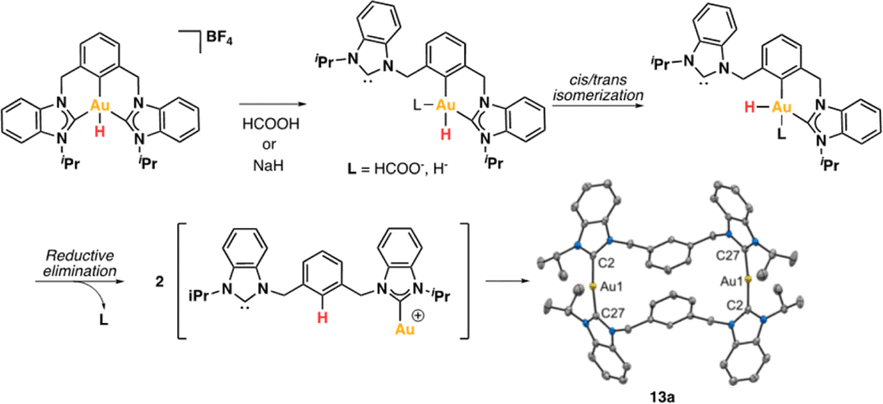 | ||
| Scheme 4 Convergent decomposition route of Au(III)–H complex (12a) towards 13a with HCOOH and NaH. | ||
Finally, we were interested in evaluating the possibility of performing the dehydrogenation of formic acid. We reacted the Au(III)–F complex 9a with a large excess of formic acid in CD2Cl2 at 90 °C. After 24 h, we observed neither the formation of H2 gas nor the Au(III)–H complex; instead, we detected the formation of the Au(III)–formate complex. According to some reports, the dehydrogenation of formic acid proceeds under neat conditions.24,86 Therefore, we heated a solution of the Au(III)–F complex in DCOOH at 100 °C for 168 h. By 1H NMR and ESI-MS studies, we detected the formation of the dimetallic complex (major species by 1H NMR), Au(III)–formate, and Au(III)–Cl complex, but the formation of HD or H2 gas was not observed by 1H NMR or GC (see ESI†).
With all the above information, we propose that the formation of the dimetallic complex should occur through β-hydride elimination at the formed Au(III)–formate 11a to form Au(III)–H 12a (Scheme 3), followed by successive decomposition steps as in Scheme 4: de-coordination of NHC, hydride cis–trans isomerization, and reductive elimination. Thus, thermal decomposition of the Au(III)–H leads to the formation of the dimetallic complex 13a, so its formation is a good indication of the intermediacy of 12a in this reaction. Furthermore, the reaction of 12a with triflic acid at room temperature in CD2Cl2 leads to the formation of 13a within minutes.
DFT calculations were conducted to elucidate the observed reactivity of the Au(III)–H species from Bochmann,11 Bezuidenhout,12 and Nevado24 (Fig. 1a) and our 12a towards sodium hydride and formic acid for the generation of H2. Details of the calculations are provided in the ESI,† and Table 1 summarizes the thermodynamics of all the reactions. According to our calculations, Bezuidenhout's Au(III)–H species is the only one with a favorable thermodynamic value of −2.2 kcal mol−1 for the reaction with NaH, and this agrees with the H2 evolution observed in THF upon reaction with NaH.
| Entry | Au(III)–H species |
|
|
Occ. EFO pincer |
|---|---|---|---|---|
| 1 |
|
+4.7 | +0.8 | 0.449 |
| 2 |
|
+37.2 | −8.2 | 0.414 |
| 3 |
|
+14.4 | +9.7 | 0.451 |
| 4 |
|
−2.2 | +33.2 | 0.502 |

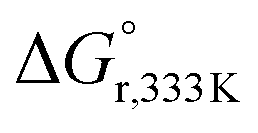
On the contrary, unfavorable endergonic reaction is found for the other Au(III)–H species. On the other hand, the calculated thermodynamics for the formation of the Au(III)–formate species and liberation of H2 are highly unfavorable, particularly for the sterically hindered Bezuidenhout's species. Remarkably, the reaction is thermodynamically accessible for Nevado's species, which indeed showed reactivity with formic acid for the generation of H2 and the formation of the intermediate Au(III)–formate.24 In the case of 12a, we observed degradation upon prolonged exposure to formic acid at high temperatures (which ultimately leads to the formation of the bimetallic species 13a). In this case, the reaction is almost isoenergetic, accounting for the detection of the Au(III)–formate species by ESI-MS.
We endeavored to explain the energetic trends by scrutinizing the electronic structure of the Au(III)–species. Specifically, we quantified the amount of σ-donation87 from the pincer ligand to the metal using a method known as effective fragment orbitals (EFOs). The shape and occupations of the relevant EFOs for the pincer, Au and H fragments are depicted in Fig. S116 and S117.† The main σ-donation channel occurs via the in phase interaction of the NHC lone pairs in cis position with respect to the H atom. As the occupancy of this EFO decreases, the σ-donation from the pincer increases. We observed a significant correlation between the σ-donation (Table 1 and Fig. S118†) and the energetics of the reaction with NaH and formic acid. A less electron-donating pincer (i.e. Bezuidenhout's) results in a more electrophilic H moiety, allowing reaction with hydride while inhibiting reaction with formic acid. Conversely, the more electron-donating pincer of Nevado's species induces the opposite reactivity. The intermediate situation of our CCC scaffold suggests that the reactivity of 12a could potentially be fine-tuned by adding the correct substituent to our pincer ligand.
2.2 Catalytic studies of CCC-NHC Au(III) pincer complex 6a
We further delved into the capabilities of the Au(III) pincer complex as a Lewis acid catalyst.88,89 It is well-established that gold can coordinate to multiple bonds, activating them and rendering them susceptible to nucleophilic attacks. Building on this understanding, we chose to assess the catalytic activity of complex 6a in the synthesis of phenols. To achieve this, we reacted ω-alkynylfurans90 with 5 mol% of 6a, along with a halogen scavenger, as a model reaction for Lewis acid catalysis of gold(III) to trigger the cyclization-mediated production of the corresponding phenol derivative (Scheme 5).91–95 The halogen scavenger traps the chlorine ligand, generating a vacant coordination site within the Au(III) coordination sphere.96,97 This enables the coordination of the alkyne moiety to Au(III), resulting in the formation of an η2 adduct. Following this, subsequent rearrangements occur, ultimately leading to the production of the desired phenol product.92,93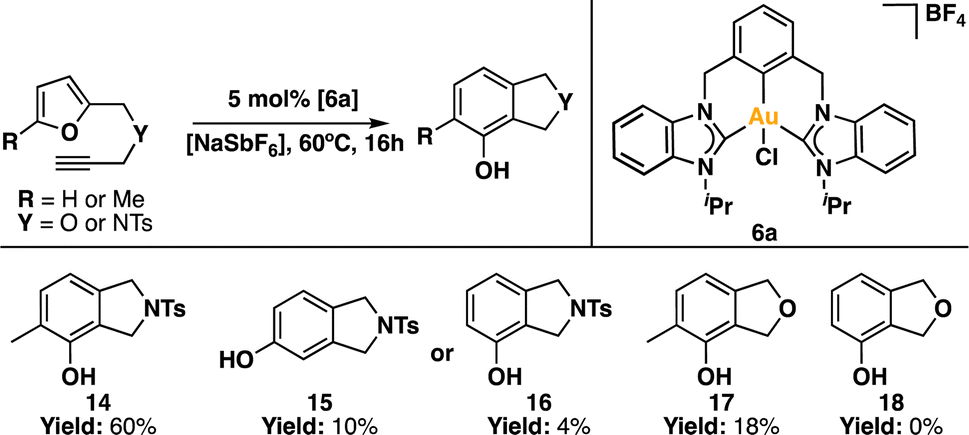 | ||
| Scheme 5 Synthesis of phenol products catalyzed by 6a. | ||
In our initial attempts, we used [NaSbF6] in CDCl3 as the reaction medium at room temperature. Unfortunately, the reaction yielded products in low quantities (<5%) after 24 h. Consequently, we decided to carry out the catalytic reactions at a higher temperature. To our delight, when the reaction was performed at 60 °C for 16 h, we detected the formation of the desired product. Using 4-methyl-N-((5-methylfuran-2-yl)methyl)-N-(prop-2-yn-1-yl)benzenesulfonamide as the substrate, we achieved a yield of 60% for compound 14, with a TON of 12.
When the analogous compound with a hydrogen atom replacing the methyl group on the furan fragment (N-(furan-2-ylmethyl)-4-methyl-N-(prop-2-yn-1-yl)benzenesulfonamide) was used, we observed a mixture of isomers 15 and 16, with yields of 10% and 4%, respectively, resulting in a TON of ∼3. When the benzenesulfonamide group was replaced by an ether group, the yields were significantly lower. Specifically, using 2-methyl-5-((prop-2-yn-1-yloxy)methyl)furan as the substrate, the yield was 18% (TON ∼4), while with 2-((prop-2-yn-1-yloxy)methyl)furan, no product formation was observed.
The differences in reactivity among the substrates can likely be attributed to the Thorpe-Ingold effect.98–100 The bulkier benzenesulfonamide group, compared to the ether fragment, brings the furan and alkyne fragments into closer proximity, facilitating their interaction and, consequently, the formation of the phenol product. This catalytic behaviour has been described previously.91,101 Additionally, the presence of the methyl group on the furan fragment likely enhances the reaction due to an electronic effect.
Our complex 6a demonstrated catalytic activity comparable to other Au(III)-based catalysts.91,94,101–104 For instance, using 2 mol% of AuCl3 at 20 °C, compound 14 was obtained in 97% yield after 30 h.91 Another example involves the use of bidentate N^O ligands;94 however, in that case, an induction period was observed, suggesting that the well-defined complex functions as a pre-catalyst. Nevertheless, this preliminary study serves as compelling evidence of the Lewis acid role that our CCC-NHC Au(III) pincer complex can play, opening avenues for further exploration and applications in catalysis.
3 Conclusion
Our investigation into the reactivity and catalytic potential of the CCC-NHC Au(III) pincer complexes has revealed intriguing insights into their behavior and applications. The successful synthesis of the pincer complex through innovative approaches, such as non-photoactivated aryl-diazonium bond activation mediated by sodium ascorbate, marks a significant advancement in the field. The obtained CCC-Au(III) pincer complexes demonstrated stability and resistance to various conditions, showcasing their robust nature. Ligand exchange reactivity of the CCC-Au(III)–L pincer complex using nucleophiles allowed the coordination of a methyl, fluorine, alkyne and formate ligands, all of them fully characterized spectroscopically and crystallographically. The study also sheds light on the elusive Au(III)–H species, offering valuable insights into its formation, stability, and reactivity. The successful preparation of the Au(III)–H derivative and its reactivity in the presence of formic acid present opportunities for further exploration in catalytic processes in the context of H2 generation. The DFT calculations shed light on the reactivity differences observed among various Au(III)–H species, including those from Bochmann, Bezuidenhout, and Nevado, as well as our own 12a, towards sodium hydride and formic acid for H2 generation. Correlation between reaction thermodynamics and the σ-donating character of the pincer ligand was found, showing that less electron-donating pincers favored reaction with NaH but hindered the reaction with formic acid, while more electron-donating pincers induced the opposite reactivity. These findings offer valuable insights for designing and understanding gold-mediated catalytic processes. Furthermore, the CCC-Au(III)–Cl pincer complex shows very good Lewis acid reactivity as catalyst for the synthesis of phenols, underscoring its potential as a Lewis acid catalyst.In summary, our findings not only contribute to the understanding of gold(III) chemistry but also highlight the CCC-NHC Au(III) pincer complex as a versatile and promising candidate for catalytic applications. The demonstrated stability, reactivity, and Lewis acid behavior pave the way for future studies aimed at unraveling additional facets of gold catalysis and expanding the utility of CCC-NHC Au(III) pincer complexes in various synthetic transformations.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. V. and N. A. performed the experiments. P. S. performed DFT calculations. All authors were involved in the conceptual design of the study, discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank MINECO Spain (PID2022–136970NB–I00 to X.R.) and Generalitat de Catalunya (2021SGR00475 and Beatriu de Pinós contract 2019–BP–0080 to H.V.). X.R. is grateful for an ICREA Acadèmia award. The authors would like to acknowledge the STR–UdG for technical support. A.S.K.H. is grateful for support by the Deutsche Forschungsgemeinschaft (DFG).Notes and references
- A. S. K. Hashmi, Angew. Chem., Int. Ed., 2012, 51, 12935–12936 CrossRef CAS.
- N. Ibrahim, M. H. Vilhelmsen, M. Pernpointner, F. Rominger and A. S. K. Hashmi, Organometallics, 2013, 32, 2576–2583 CrossRef CAS.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- A. Comas-Vives, C. González-Arellano, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Am. Chem. Soc., 2006, 128, 4756–4765 CrossRef CAS.
- A. Comas-Vives, C. González-Arellano, M. Boronat, A. Corma, M. Iglesias, F. Sánchez and G. Ujaque, J. Catal., 2008, 254, 226–237 CrossRef CAS.
- D.-A. Roşca, J. A. Wright and M. Bochmann, Dalton Trans., 2015, 44, 20785–20807 RSC.
- D.-A. Roşca, J. A. Wright, D. L. Hughes and M. Bochmann, Nat. Commun., 2013, 4, 2167 CrossRef.
- A. Pintus, L. Rocchigiani, J. Fernandez-Cestau, P. H. M. Budzelaar and M. Bochmann, Angew. Chem., Int. Ed., 2016, 55, 12321–12324 CrossRef CAS.
- F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920–11929 CrossRef CAS PubMed.
- J. Jiang, B. Cao, Y. Chen, H. Luo, J. Xue, X. Xiong and T. Zou, Angew. Chem., Int. Ed., 2022, 61, e202201103 CrossRef CAS.
- D.-A. Roşca, D. A. Smith, D. L. Hughes and M. Bochmann, Angew. Chem., Int. Ed., 2012, 51, 10643–10646 CrossRef.
- G. Kleinhans, M. M. Hansmann, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, J. Am. Chem. Soc., 2016, 138, 15873–15876 CrossRef CAS PubMed.
- J. Martín, J. Schörgenhumer, M. Biedrzycki and C. Nevado, Inorg. Chem., 2024, 63, 8390–8396 CrossRef PubMed.
- M. S. M. Holmsen, A. Nova and M. Tilset, Acc. Chem. Res., 2023, 56, 3654–3664 CrossRef CAS.
- A. S. Phearman, Y. Ardon and K. I. Goldberg, J. Am. Chem. Soc., 2024, 1446, 4045–4059 CrossRef PubMed.
- L. Rocchigiani, J. Fernandez-Cestau, I. Chambrier, P. Hrobárik and M. Bochmann, J. Am. Chem. Soc., 2018, 140, 8287–8302 CrossRef CAS PubMed.
- D.-A. Roşca, D. A. Smith and M. Bochmann, Chem. Commun., 2012, 48, 7247–7249 RSC.
- D. A. Smith, D.-A. Roşca and M. Bochmann, Organometallics, 2012, 31, 5998–6000 CrossRef CAS.
- Q.-Y. Bi, X.-L. Du, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS.
- X. Liu, L. He, Y.-M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793–804 CrossRef CAS PubMed.
- S. Mousavi-Salehi, S. Keshipour and F. Ahour, J. Phys. Chem. Solids, 2023, 176, 111239 CrossRef CAS.
- B. W. J. Chen, M. Stamatakis and M. Mavrikakis, ACS Catal., 2019, 9, 9446–9457 CrossRef CAS.
- G. Bond, Gold Bull., 2009, 42, 337–342 CrossRef CAS.
- R. Kumar, J.-P. Krieger, E. Gómez-Bengoa, T. Fox, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 12862–12865 CrossRef CAS PubMed.
- J. A. Denny, G. M. Lang and T. K. Hollis, in Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018, pp. 251–272 Search PubMed.
- R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC.
- M. Raynal, C. S. J. Cazin, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Chem. Commun., 2008, 3983–3985 RSC.
- M. Raynal, R. Pattacini, C. S. J. Cazin, C. Vallée, H. Olivier-Bourbigou and P. Braunstein, Organometallics, 2009, 28, 4028–4047 CrossRef CAS.
- M. Jagenbrein, A. A. Danopoulos and P. Braunstein, J. Organomet. Chem., 2015, 775, 169–172 CrossRef CAS.
- L. González-Sebastián and A. B. Chaplin, Inorg. Chim. Acta, 2017, 460, 22–28 CrossRef.
- A. R. Chianese, A. Mo, N. L. Lampland, R. L. Swartz and P. T. Bremer, Organometallics, 2010, 29, 3019–3026 CrossRef CAS.
- A. R. Chianese, S. E. Shaner, J. A. Tendler, D. M. Pudalov, D. Y. Shopov, D. Kim, S. L. Rogers and A. Mo, Organometallics, 2012, 31, 7359–7367 CrossRef CAS.
- W. Zuo and P. Braunstein, Dalton Trans., 2012, 41, 636–643 RSC.
- W. Zuo and P. Braunstein, Organometallics, 2012, 31, 2606–2615 CrossRef CAS.
- L.-H. Chung, H.-S. Lo, S.-W. Ng, D.-L. Ma, C.-H. Leung and C.-Y. Wong, Sci. Rep., 2015, 5, 15394 CrossRef CAS PubMed.
- C.-Y. Kuei, S.-H. Liu, P.-T. Chou, G.-H. Lee and Y. Chi, Dalton Trans., 2016, 45, 15364–15373 RSC.
- J. Cho, T. K. Hollis, E. J. Valente and J. M. Trate, J. Organomet. Chem., 2011, 696, 373–377 CrossRef CAS.
- J. Cho, T. K. Hollis, T. R. Helgert and E. J. Valente, Chem. Commun., 2008, 5001–5003 RSC.
- W. D. Clark, J. Cho, H. U. Valle, T. K. Hollis and E. J. Valente, J. Organomet. Chem., 2014, 751, 534–540 CrossRef CAS.
- H. U. Valle, G. Akurathi, J. Cho, W. D. Clark, A. Chakraborty and T. K. Hollis, Aust. J. Chem., 2016, 69, 565–572 CrossRef CAS.
- F. E. Hahn, M. C. Jahnke and T. Pape, Organometallics, 2007, 26, 150–154 CrossRef CAS.
- R. J. Rubio, G. T. S. Andavan, E. B. Bauer, T. K. Hollis, J. Cho, F. S. Tham and B. Donnadieu, J. Organomet. Chem., 2005, 690, 5353–5364 CrossRef CAS.
- W. D. Clark, K. N. Leigh, C. E. Webster and T. K. Hollis, Aust. J. Chem., 2016, 69, 573–582 CrossRef CAS.
- S. W. Reilly, G. Akurathi, H. K. Box, H. U. Valle, T. K. Hollis and C. E. Webster, J. Organomet. Chem., 2016, 802, 32–38 CrossRef CAS.
- E. D. Amoateng, J. Zamora-Moreno, G. Kuchenbeiser, B. Donnadieu, F. Tham, V. Montiel-Palma and T. Keith Hollis, J. Organomet. Chem., 2022, 979, 122499 CrossRef CAS.
- E. B. Bauer, G. T. S. Andavan, T. K. Hollis, R. J. Rubio, J. Cho, G. R. Kuchenbeiser, T. R. Helgert, C. S. Letko and F. S. Tham, Org. Lett., 2008, 10, 1175–1178 CrossRef CAS PubMed.
- J. A. Denny, R. W. Lamb, S. W. Reilly, B. Donnadieu, C. E. Webster and T. K. Hollis, Polyhedron, 2018, 151, 568–574 CrossRef CAS.
- S. W. Reilly, C. E. Webster, T. K. Hollis and H. U. Valle, Dalton Trans., 2016, 45, 2823–2828 RSC.
- J. Steube, A. Kruse, O. S. Bokareva, T. Reuter, S. Demeshko, R. Schoch, M. A. Argüello Cordero, A. Krishna, S. Hohloch, F. Meyer, K. Heinze, O. Kühn, S. Lochbrunner and M. Bauer, Nat. Chem., 2023, 15, 468–474 CrossRef CAS PubMed.
- J. Mensah, V. K. Adiraju, J. D. Cope, R. W. Lamb, X. X. Li, B. Donnadieu, I. V. Rubtsov, C. E. Webster and T. K. Hollis, Organometallics, 2024, 43, 273–283 CrossRef CAS.
- J. D. Cope, J. A. Denny, R. W. Lamb, L. E. McNamara, N. I. Hammer, C. E. Webster and T. K. Hollis, J. Organomet. Chem., 2017, 845, 258–265 CrossRef CAS.
- X. Zhang, A. M. Wright, N. J. DeYonker, T. K. Hollis, N. I. Hammer, C. E. Webster and E. J. Valente, Organometallics, 2012, 31, 1664–1672 CrossRef CAS.
- A. J. Huckaba, B. Cao, T. K. Hollis, H. U. Valle, J. T. Kelly, N. I. Hammer, A. G. Oliver and C. E. Webster, Dalton Trans., 2013, 42, 8820–8826 RSC.
- X. Zhang, B. Cao, E. J. Valente and T. K. Hollis, Organometallics, 2013, 32, 752–761 CrossRef CAS.
- H. H. Pham, B. Donnadieu and T. K. Hollis, Appl. Organomet. Chem., 2022, e6789 Search PubMed.
- W. Feuerstein and F. Breher, Dalton Trans., 2021, 50, 9754–9767 RSC.
- M. Monticelli, C. Tubaro, M. Baron, M. Basato, P. Sgarbossa, C. Graiff, G. Accorsi, T. P. Pell, D. J. D. Wilson and P. J. Barnard, Dalton Trans., 2016, 45, 9540–9552 RSC.
- A. Herbst, C. Bronner, P. Dechambenoit and O. S. Wenger, Organometallics, 2013, 32, 1807–1814 CrossRef CAS.
- W. Feuerstein, C. Holzer, X. Gui, L. Neumeier, W. Klopper and F. Breher, Chem.–Euro. J., 2020, 26, 17156–17164 CrossRef CAS.
- P. A. Bonnardel, R. V. Parish and R. G. Pritchard, J. Chem. Soc., Dalton Trans., 1996, 3185–3193 RSC.
- G. Alesso, M. A. Cinellu, S. Stoccoro, A. Zucca, G. Minghetti, C. Manassero, S. Rizzato, O. Swang and M. K. Ghosh, Dalton Trans., 2010, 39, 10293–10304 RSC.
- A. Ahrens, L. F. P. Karger, F. Rominger, M. Rudolph and A. S. K. Hashmi, Organometallics, 2023, 42, 1561–1566 CrossRef CAS.
- M. Contel, M. Stol, M. A. Casado, G. P. M. van Klink, D. D. Ellis, A. L. Spek and G. van Koten, Organometallics, 2002, 21, 4556–4559 CrossRef CAS.
- S. Stoccoro, G. Alesso, M. A. Cinellu, G. Minghetti, A. Zucca, M. Manassero and C. Manassero, Dalton Trans., 2009, 3467–3477 RSC.
- L. Tabrizi and H. Chiniforoshan, Sens. Actuators, B, 2017, 245, 815–820 CrossRef CAS.
- L. Tabrizi and H. Chiniforoshan, Dalton Trans., 2017, 46, 14164–14173 RSC.
- D. Eppel, P. Penert, J. Stemmer, C. Bauer, M. Rudolph, M. Brückner, F. Rominger and A. S. K. Hashmi, Chem.–Euro. J., 2021, 27, 8673–8677 CrossRef CAS.
- D. Eppel, M. Rudolph, F. Rominger and A. S. K. Hashmi, ChemSusChem, 2020, 13, 1986–1990 CrossRef CAS PubMed.
- I. Medina-Mercado, E. O. Asomoza-Solís, E. Martínez-González, V. M. Ugalde-Saldívar, L. G. Ledesma-Olvera, J. E. Barquera-Lozada, V. Gómez-Vidales, J. Barroso-Flores, B. A. Frontana-Uribe and S. Porcel, Chem.–Euro. J., 2020, 26, 634–642 CrossRef CAS.
- U. Costas-Costas, E. Gonzalez-Romero and C. Bravo-Diaz, Helv. Chim. Acta, 2001, 84, 632–648 CrossRef CAS.
- F. P. Crisostomo, T. Martin and R. Carrillo, Angew. Chem., Int. Ed., 2014, 53, 2181–2185 CrossRef CAS PubMed.
- C. Cai, M.-j. Bu and G.-p. Lu, Synlett, 2015, 26, 1841–1846 CrossRef.
- R. Visbal, A. Laguna and M. C. Gimeno, Chem. Commun., 2013, 49, 5642–5644 RSC.
- D. Zhu, S. V. Lindeman and J. K. Kochi, Organometallics, 1999, 18, 2241–2248 CrossRef CAS.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- F. E. Hahn, M. C. Jahnke, V. Gomez-Benitez, D. Morales-Morales and T. Pape, Organometallics, 2005, 24, 6458–6463 CrossRef CAS.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 CAS.
- B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981–2988 CrossRef CAS.
- H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS PubMed.
- R. Kumar, A. Linden and C. Nevado, Angew. Chem., Int. Ed., 2015, 54, 14287–14290 CrossRef CAS PubMed.
- R. Kumar, A. Linden and C. Nevado, J. Am. Chem. Soc., 2016, 138, 13790–13793 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- J. J. A. Celaje, Z. Lu, E. A. Kedzie, N. J. Terrile, J. N. Lo and T. J. Williams, Nat. Commun., 2016, 7, 11308 CrossRef CAS PubMed.
- G. Comas-Vilà and P. Salvador, ChemPhysChem, 2024, 25, e202400582 CrossRef.
- L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- S. Carrettin, M. C. Blanco, A. Corma and A. S. K. Hashmi, Adv. Synth. Catal., 2006, 348, 1283–1288 CrossRef CAS.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- A. S. K. Hashmi, M. Rudolph, J. P. Weyrauch, M. Wölfle, W. Frey and J. W. Bats, Angew. Chem., Int. Ed., 2005, 44, 2798–2801 CrossRef CAS PubMed.
- H. Rabaâ, B. Engels, T. Hupp and A. S. K. Hashmi, Int. J. Quantum Chem., 2007, 107, 359–365 CrossRef.
- A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejović, Angew. Chem., Int. Ed., 2004, 43, 6545–6547 CrossRef CAS PubMed.
- A. S. K. Hashmi, M. Rudolph, H.-U. Siehl, M. Tanaka, J. W. Bats and W. Frey, Chem.–Euro. J., 2008, 14, 3703–3708 CrossRef CAS PubMed.
- J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 3949–3959 CrossRef.
- J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 2493–2502 CrossRef.
- B. L. Shaw, J. Am. Chem. Soc., 1975, 97, 3856–3857 CrossRef CAS.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 RSC.
- M. J. O'Neill, T. Riesebeck and J. Cornella, Angew. Chem., Int. Ed., 2018, 57, 9103–9107 CrossRef.
- A. S. K. Hashmi, T. Hengst, C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315–1337 CrossRef CAS.
- Y. Chen, W. Yan, N. G. Akhmedov and X. Shi, Org. Lett., 2010, 12, 344–347 CrossRef CAS.
- A. S. K. Hashmi, M. C. Blanco, E. Kurpejović, W. Frey and J. W. Bats, Adv. Synth. Catal., 2006, 348, 709–713 CrossRef CAS.
- A. S. K. Hashmi, A. Loos, A. Littmann, I. Braun, J. Knight, S. Doherty and F. Rominger, Adv. Synth. Catal., 2009, 351, 576–582 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For materials, instrumentation, experimental procedures and spectroscopic characterization of all compounds. CCDC 2334004-2334014. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02999b |
| This journal is © The Royal Society of Chemistry 2024 |