
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpotting d-band centers of single-atom catalysts by oxygen intermediate-boosted electrochemiluminescence†
Ruyu
Xie
a,
Kaitao
Li
ac,
Rui
Tian
*ac and
Chao
Lu
 *abc
*abc
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: tianrui@mail.buct.edu.cn; luchao@mail.buct.edu.cn
bPingyuan Laboratory, College of Chemistry, Zhengzhou University, Zhengzhou, 450001, China
cQuzhou Institute for Innovation in Resource Chemical Engineering, Quzhou, 324000, China
First published on 7th October 2024
Abstract
Catalytic activities of single-atom catalysts are strongly dependent on their d-band centers. However, it is a long-standing challenge to provide a cost-effective and accurate evaluation for the positions of d-band centers of these catalysts due to the fact that the widely applicable photoelectron spectroscopy methodologies require complicated sampling and spectral unfolding processes. In this contribution, we have proposed oxygen intermediate-boosted electrochemiluminescence (ECL) for rapid spotting of the d-band centers of single-atom catalysts, involving single atomic Au, Ag, Cu and Fe. It was disclosed that the d-band centers of single-atom catalysts closer to the Fermi level could facilitate the interaction between catalysts and oxygen intermediates, leading to higher luminol ECL intensities as a result of the promoted adsorption and reduction ability towards oxygen intermediates. Moreover, this correlation was also adapted for other metal catalysts such as Au and Ag nanoparticles. This correspondence could be utilized for an accurate identification of d-band centers of single-atom catalysts. It is anticipated that the proposed strategy could be beneficial for a deep understanding of microstructure studies of single-atom catalysts to achieve advanced catalytic performances.
Introduction
The d-band model for single-atom catalysts highlights the role of the electronic states in the valence band in influencing the reaction activity.1 To unravel the factors that govern the catalytic activities of single-atom catalysts, the positions of their d-band centers have been studied as a descriptor for the modulation of the electronic structure.2 The position of d-band centers could determine the hybridization energy between the bonding and antibonding states of oxygen intermediates and d-bands of single atoms.1 As the d-band center shifts up, more antibonding states are pulled above the Fermi level,3 lowering the energy barrier and facilitating the oxygen reduction reaction.4,5 To achieve high-efficiency catalytic performances, increasing efforts have been devoted to engineering the d-band centers of single-atom catalysts.6–8 For example, the single-atom catalysts could be anchored on a specific support through the coordination interaction, and the electronic metal–support interactions (EMSI) contributed to the redistributed electrons, modulated d-band centers and thereby boosted catalytic activities.9,10 Therefore, it is critically important to identify the positions of d-band centers of single-atom catalysts to reveal the electronic structure–activity relationship.To evaluate the positions of the d-band centers of single-atom catalysts, the general strategies relied on photoelectron spectroscopy methodologies, such as X-ray photoelectron spectroscopy (XPS)11,12 and ultraviolet photoelectron spectroscopy (UPS).13 However, the detection depth of XPS measurements is on the nanometer scale, failing to provide comprehensive information on the electronic structure of catalysts.14,15 UPS showed high requirements for samples, of which the surface should be fresh and free from contamination. In addition, the density functional theory calculations to study the structure of catalysts are time-consuming and demand extensive theoretical knowledge.16 These strategies are hardly accessible as widely applicable methodologies for the studies on single-atom catalysts. Therefore, it is highly desirable to explore a sensitive and feasible strategy to identify the d-band centers of single-atom catalysts and provide an in-depth recognition of the variations of the electronic structure during the reactions.
Electrochemiluminescence (ECL), a kind of chemiluminescence triggered by electrochemical reactions, has been known for its inherent advantages of low background signal, cost-effectiveness and high sensitivity.17,18 Specifically, the electrochemical oxidation of luminol involves the reaction of reactive oxygen species (ROS) and contributes to the oxygen intermediate-boosted ECL signals.19,20 These features make ECL a promising strategy for identifying both the quantities and reaction behaviors of oxygen intermediates, thereby allowing the evaluation of the d-band centers of single-atom catalysts. In this work, we have localized the Ag, Au, Cu and Fe single atoms on a two-dimensional layered double hydroxide (LDH) support on ITO glass (referred to as Ags/LDH/ITO, Aus/LDH/ITO, Cus/LDH/ITO, and Fes/LDH/ITO), and the varied d-band centers of these single-atom catalysts were identified through the luminol ECL intensities (Scheme 1). This is because the upshifted d-band centers towards the Fermi levels of these single-atom catalysts are apt to create favorable binding interactions between catalysts and oxygen intermediates, resulting in the facilitated adsorption and reduction kinetics of oxygen intermediates. Accordingly, the oxygen intermediate-boosted ECL signals of these single-atom catalysts conformed to the same tendency as the positions of d-band centers relative to the Fermi level: Ags/LDH/ITO > Aus/LDH/ITO > Cus/LDH/ITO > Fes/LDH/ITO. Moreover, this study was extended to Ag and Au nanoparticles. The results showed the higher catalytic performance of Ag species than Au species, and the promoted atomic utilization of single atoms compared to that of nanoparticles was also validated. Therefore, we have proposed intermediate-boosted ECL as a sensitive and cost-effective approach to spot the d-band centers of single-atomic and metallic catalysts. It is anticipated that our strategy would provide valuable information for the study and modulation of the electronic structures of catalysts.
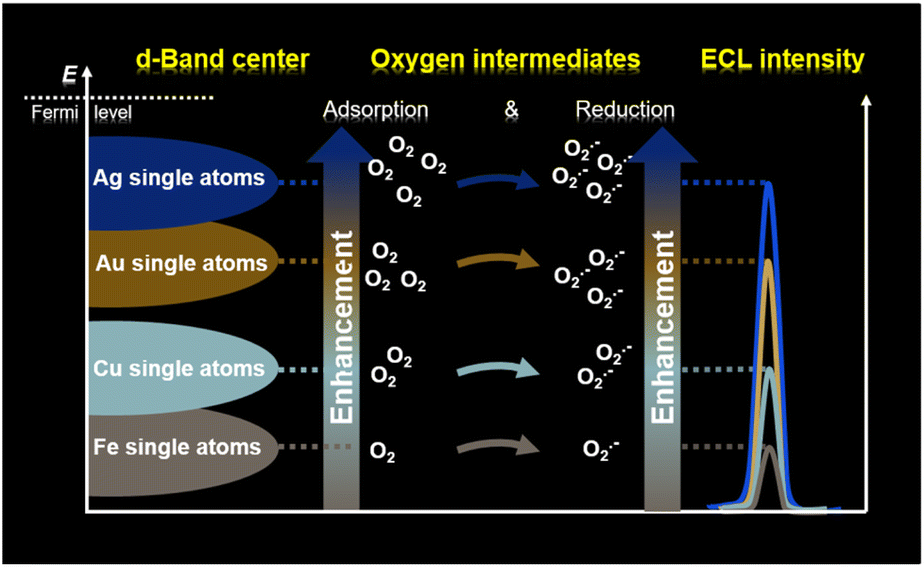 | ||
| Scheme 1 Schematic illustration of ECL to spot the positions of d-band centers of single-atom catalysts. | ||
Results and discussion
Correlation between the d-band centers and ECL intensities
CoAl/LDH, featuring the two-dimensional structural characteristics of LDHs and the catalytically active Co element, was employed as a support to localize different kinds of catalysts.21,22 CoAl/LDH was prepared on an ITO electrode through a hydrothermal method. To localize the different metal species with varied d-band centers onto LDH/ITO, electrochemical deposition was employed. The structure of the localized metal was highly dependent on the mass loadings of the metal species during the electrochemical deposition, corresponding to the minimum supersaturation on the support.23 In detail, single atomic Au and Ag were anchored onto the LDH/ITO electrodes through the electrochemical deposition for 2 cycles (denoted as Ags/LDH/ITO and Aus/LDH/ITO), and the contents of Au and Ag were determined to be 0.071 mol% and 0.077 mol% by inductively coupled plasma optical emission spectrometry. The referenced Au and Ag nanoparticles were electrochemically deposited on the LDH/ITO electrodes after 30 cycles (labelled as Aunp/LDH/ITO and Agnp/LDH/ITO), with the contents of Au and Ag of 0.214 mol% and 0.226 mol%. The d-band centers relative to the Fermi level of these catalysts were studied by implementing the highly surface-sensitive ultraviolet photoelectron spectroscopy (UPS). The UPS valence-band spectra showed that the energy of d-band centers of Ags/LDH/ITO was located at −4.24 eV, which was higher than that of −4.33 eV for Aus/LDH/ITO (Fig. 1a). Furthermore, the values of d-band centers of Agnp/LDH/ITO and Aunp/LDH/ITO were determined to be −4.45 eV and −4.54 eV, respectively. It could be concluded that more shifts towards the Fermi level could be acquired for the d-band centers of single atoms than those of the nanoparticles, which could be attributed to the larger proportion of low coordinated atoms in the nanoparticles.24 Moreover, Ag species shared the higher d-band center values than Au did, and this phenomenon could be explained by the electron distribution of different metal species.2 Concretely, the values of the d-band centers of these modified electrodes followed the trend of Ags/LDH/ITO > Aus/LDH/ITO > Agnp/LDH/ITO > Aunp/LDH/ITO. According to the d-band theory proposed by Nørskov, the up-shifts of d-band centers would result in the variations of anti-bonding states,25 leading to the strong adsorption strength towards intermediates and facilitating the rate-limiting reactions for the catalytic reactions.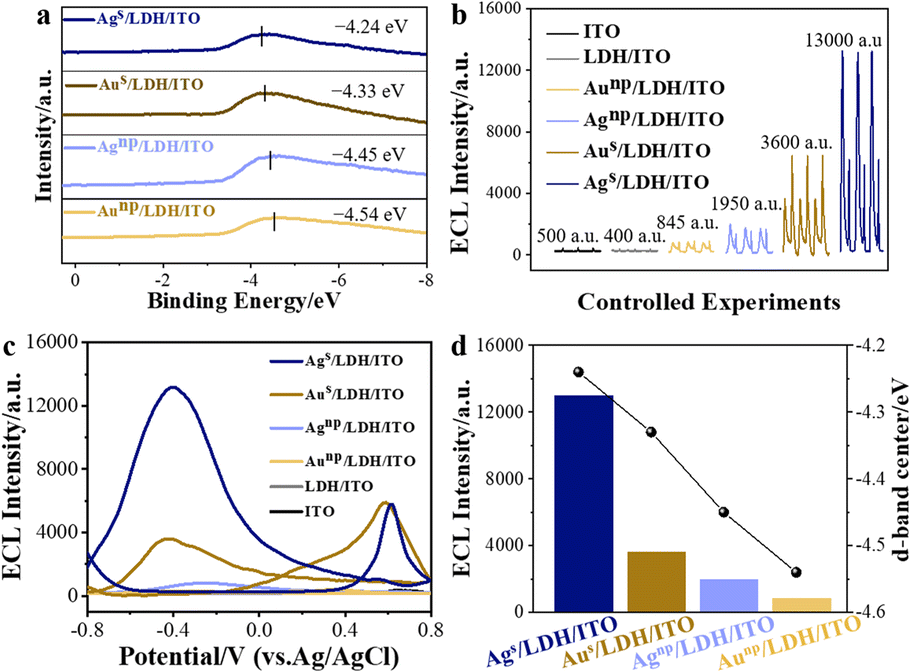 | ||
| Fig. 1 (a) UPS valence-band spectra of Ags/LDH/ITO, Aus/LDH/ITO, Agnp/LDH/ITO, and Aunp/LDH/ITO relative to the Fermi level; the black solid lines represent the positions of the d-band centers; (b) ECL intensities and (c) IECL–potential curves for ITO, LDH/ITO, Aunp/LDH/ITO, Agnp/LDH/ITO, Aus/LDH/ITO and Ags/LDH/ITO in 0.01 mmol per L luminol (PBS, pH = 7.5); (d) correlation between the d-band centers and ECL intensities of Ags/LDH/ITO, Aus/LDH/ITO, Agnp/LDH/ITO, and Aunp/LDH/ITO electrodes (the values of d-band centers were measured by UPS as shown in (a). | ||
The ECL performances of these modified electrodes were recorded in a 0.01 mmol L−1 luminol solution (0.1 mol L−1 phosphate buffered saline, pH = 7.5) over a voltage range of −0.8 to 0.8 V (Fig. 1b). No obvious ECL signals could be observed on the bare ITO or LDH/ITO electrodes. Upon the deposition of single atomic Au or Ag, the modified electrodes showed the prominently promoted ECL intensities. The cathodic luminol ECL intensity of Ags/LDH/ITO was about 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 a.u., while that of Aus/LDH/ITO was only 3600 a.u. In comparison with the localized single atoms, Agnp/LDH/ITO and Aunp/LDH/ITO with downshifted d-band centers showed less effect on promoting the ECL signals, with the ECL intensities of 1950 a.u. and 845 a.u., respectively. The IECL–potential and synchronous cyclic voltammetry (CV) curves of different modified electrodes were recorded for validation. An anodic ECL peak at +0.6 V appeared for both Ags/LDH/ITO and Aus/LDH/ITO electrodes during the positive scan, and the cathodic ECL peak at −0.4 V was noted during the negative scan for these electrodes (Fig. 1c). The ECL spectra recorded at the cathode showed a maximum emission wavelength of around 425 nm (Fig. S1†), validating the emissive species as the excited 3-aminophthalate dianion from luminol.26 The CV curve of Ags/LDH/ITO showed a broad oxidation peak at +0.45 V during the positive scan (Fig. S2†), which was correlated to the oxidation of Agδ+ and luminol.27 During the reversed potential scan, the reduction reactions of Ag+ and dissolved oxygen were found at −0.005 V and −0.55 V, respectively.28 For Aus/LDH/ITO, the reduction of the oxidized gold could be observed at +0.03 V, and the oxidation process of luminol was identified at +0.58 V.29,30 In comparison, the peaks at the cathode during the negative scanning potential, which were associated with the reduction of the oxidized metal species and the dissolved oxygen,31 were more pronounced for Ags/LDH/ITO than for the Aus/LDH/ITO electrode. These results demonstrated that single atomic Ag showed a stronger oxygen adsorption capacity to reduce oxygen to generate intermediate radical species. These superior behaviors of Ags/LDH/ITO could be reflected by the ECL due to its ultrasensitive features to interpret the subtle changes during an electrochemical reaction.32
000 a.u., while that of Aus/LDH/ITO was only 3600 a.u. In comparison with the localized single atoms, Agnp/LDH/ITO and Aunp/LDH/ITO with downshifted d-band centers showed less effect on promoting the ECL signals, with the ECL intensities of 1950 a.u. and 845 a.u., respectively. The IECL–potential and synchronous cyclic voltammetry (CV) curves of different modified electrodes were recorded for validation. An anodic ECL peak at +0.6 V appeared for both Ags/LDH/ITO and Aus/LDH/ITO electrodes during the positive scan, and the cathodic ECL peak at −0.4 V was noted during the negative scan for these electrodes (Fig. 1c). The ECL spectra recorded at the cathode showed a maximum emission wavelength of around 425 nm (Fig. S1†), validating the emissive species as the excited 3-aminophthalate dianion from luminol.26 The CV curve of Ags/LDH/ITO showed a broad oxidation peak at +0.45 V during the positive scan (Fig. S2†), which was correlated to the oxidation of Agδ+ and luminol.27 During the reversed potential scan, the reduction reactions of Ag+ and dissolved oxygen were found at −0.005 V and −0.55 V, respectively.28 For Aus/LDH/ITO, the reduction of the oxidized gold could be observed at +0.03 V, and the oxidation process of luminol was identified at +0.58 V.29,30 In comparison, the peaks at the cathode during the negative scanning potential, which were associated with the reduction of the oxidized metal species and the dissolved oxygen,31 were more pronounced for Ags/LDH/ITO than for the Aus/LDH/ITO electrode. These results demonstrated that single atomic Ag showed a stronger oxygen adsorption capacity to reduce oxygen to generate intermediate radical species. These superior behaviors of Ags/LDH/ITO could be reflected by the ECL due to its ultrasensitive features to interpret the subtle changes during an electrochemical reaction.32
It should be noted that the ECL intensities were highly correlated with the positions of d-band centers of the modified electrodes. The trends for the d-band centers relative to the Fermi level of Ags/LDH/ITO, Aus/LDH/ITO, Agnp/LDH/ITO, and Aunp/LDH/ITO are in good accordance with the tendencies of their ECL performances: Ags/LDH/ITO > Aus/LDH/ITO > Agnp/LDH/ITO > Aunp/LDH/ITO (Fig. 1d). In other words, the larger upshift of d-band centers towards the Fermi level was associated with the higher ECL activity of the modified electrodes. The varied positions of the d-band center of metal species could be induced by the electronic metal–support interaction (EMSI),33,34 which could regulate the electronic structure and the electron transfer ability of Ag or Au species. These results demonstrated the strength of ECL in identifying and screening the position of d-band centers of single-atomic and metallic catalysts.
Structural and morphological studies
To unveil the reason for this correlation, the structural and morphological features of these modified electrodes were studied. Taking the Ag species as an example, the typical characteristic diffraction peaks of LDH at 11.56°, 23.21° and 35.26° could be observed for Ags/LDH/ITO and Agnp/LDH/ITO (Fig. S3†), demonstrating the presence of the periodically layered LDHs.35 The characteristic diffraction peak of Ag nanoparticles at 38.28° could be observed for Agnp/LDH/ITO and Agnp/ITO. In contrast, no such peak could be observed for Ags/LDH/ITO, suggesting the absence of Ag nanoparticles. UV-vis spectra showed the surface plasma resonance peak at ∼460 nm for Agnp/ITO, while this peak was absent for Ags/LDH/ITO (Fig. S4†). These results ruled out the aggregation of Ag in Ags/LDH/ITO and demonstrated the successfull preparation of monodispersed Ag single atoms on the LDH/ITO electrodes. Similarly, the localization of the single atomic Au on the LDH/ITO electrodes could be confirmed by the absence of the characteristic diffraction peak of Au nanoparticles in XRD patterns (Fig. S5†) and the surface plasma resonance peak in UV-vis spectra (Fig. S6†). The morphological characterization studies were performed to verify the presence of Ag and Au species in the as-prepared samples. The SEM images showed the vertically arranged CoAl/LDH arrays on the ITO substrate, with 1 μm in length and ∼20 nm in thickness (Fig. S7†). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Ags/LDH/ITO showed a number of isolated bright spots located on the CoAl/LDH, which can be attributed to the single atomic Ag (Fig. 2a). Furthermore, the mapping images showed that the Ag, Co and Al elements were uniformly distributed on the CoAl/LDH (Fig. 2b), validating the localization of single atomic Ag onto the LDH support. Similarly, bright spots ascribed to the single atomic Au could be observed on the surface of the CoAl/LDH (Fig. 2c and d), demonstrating the successful preparation of Aus/LDH/ITO. Moreover, Au and Ag nanoparticles were mono-dispersed on the LDH support in Aunp/LDH/ITO and Agnp/LDH/ITO, as observed from TEM images (Fig. S8†). | ||
| Fig. 2 (a) HAADF-STEM image and (b) STEM-EDS mapping images of Co, Al, and Ag for Ags/LDH/ITO; (c) HAADF-STEM image and (d) STEM-EDS mapping images of Co, Al, and Au for Aus/LDH/ITO. | ||
Electronic structure studies
The electronic structure and local coordination of these modified electrodes were investigated by X-ray photoelectron spectroscopy (XPS). It can be observed that the binding energies of Ag 3d5/2 and Ag 3d3/2 were located at 367.5 and 373.5 eV for Agnp/ITO (Fig. 3a), demonstrating the presence of metallic Ag0.36 These peaks shifted to higher binding energies of 367.8 and 373.9 eV in Agnp/LDH/ITO. Furthermore, the peaks of Ag 3d5/2 for Ags/LDH/ITO shifted to 368.2 eV, demonstrating an intermediate valence between the metallic Ag0 and Ag+ (0 < Agδ+ < +1). Notably, the peaks were more inclined to Ag+ in Ags/LDH/ITO, suggesting the strong interaction between single atomic Ag and LDHs, which may be responsible for the significant upshift of d-band centers to the Fermi level.37 The O 1s spectra of the electrodes modified with Ag species were deconvoluted into three peaks at 533.8, 531.8, and 530.6 eV (Fig. 3b), ascribed to the oxygen in adsorbed molecular water (OH), adsorbed oxygen (Oo), and lattice oxygen (Olat), respectively.38,39 The peaks indexed to O 1s in CoAl/LDH were shifted to a lower binding energy upon localization of Ag nanoparticles or single atoms. A new peak at 529.5 eV could be observed for Ags/LDH/ITO, suggesting the formation of Ag–O bonds.40 Moreover, XPS spectra for Co 2p were studied to reveal the changes in the electronic structure of CoAl/LDH, and the core level Co 2p spectra were split into two peaks at 781.4/797.2 eV for Co2+ and 783.2/798.7 eV for Co3+ (Fig. 3c). These peaks of CoAl/LDH shifted to lower binding energies upon localization of Agnp and Ags, accompanied by a significant decrease in the content of high-valent Co3+. These variations indicated a possible electron transfer from Ags and Agnp to CoAl/LDH through the O-bridge bimetal structure. Similar results could be observed for Aus/LDH/ITO and Aunp/LDH/ITO electrodes. The peaks of Au 4f7/2 for Aus/LDH/ITO were located at 84.6 eV (Fig. 3d), as an intermediate valence between the metallic Au0 and Au+.41 In contrast, Aunp/ITO and Aunp/LDH/ITO showed peaks around 83.7 and 83.8 eV, respectively. The presence of the Au–O bond42 could be observed as a new peak around 532.0 eV in the O 1s spectra for Aus/LDH/ITO (Fig. 3e). Shifts to lower binding energies could also be observed in O 1s and Co 2p spectra for Aus/LDH/ITO and Aunp/LDH/ITO, in comparison with CoAl/LDH (Fig. 3e and f). These findings suggested the partial electron transfer from Aus and Aunp to the adjacent coordination due to the EMSI effect. In addition, Al 2p core-level spectra were deconvoluted into two peaks at 74.7 and 74.1 eV.43 No obvious changes could be observed in the Al 2p spectra for all the modified electrodes (Fig. S9 and S10†), suggesting that Al atoms in LDH showed negligible effects on the interfacial electronic interactions.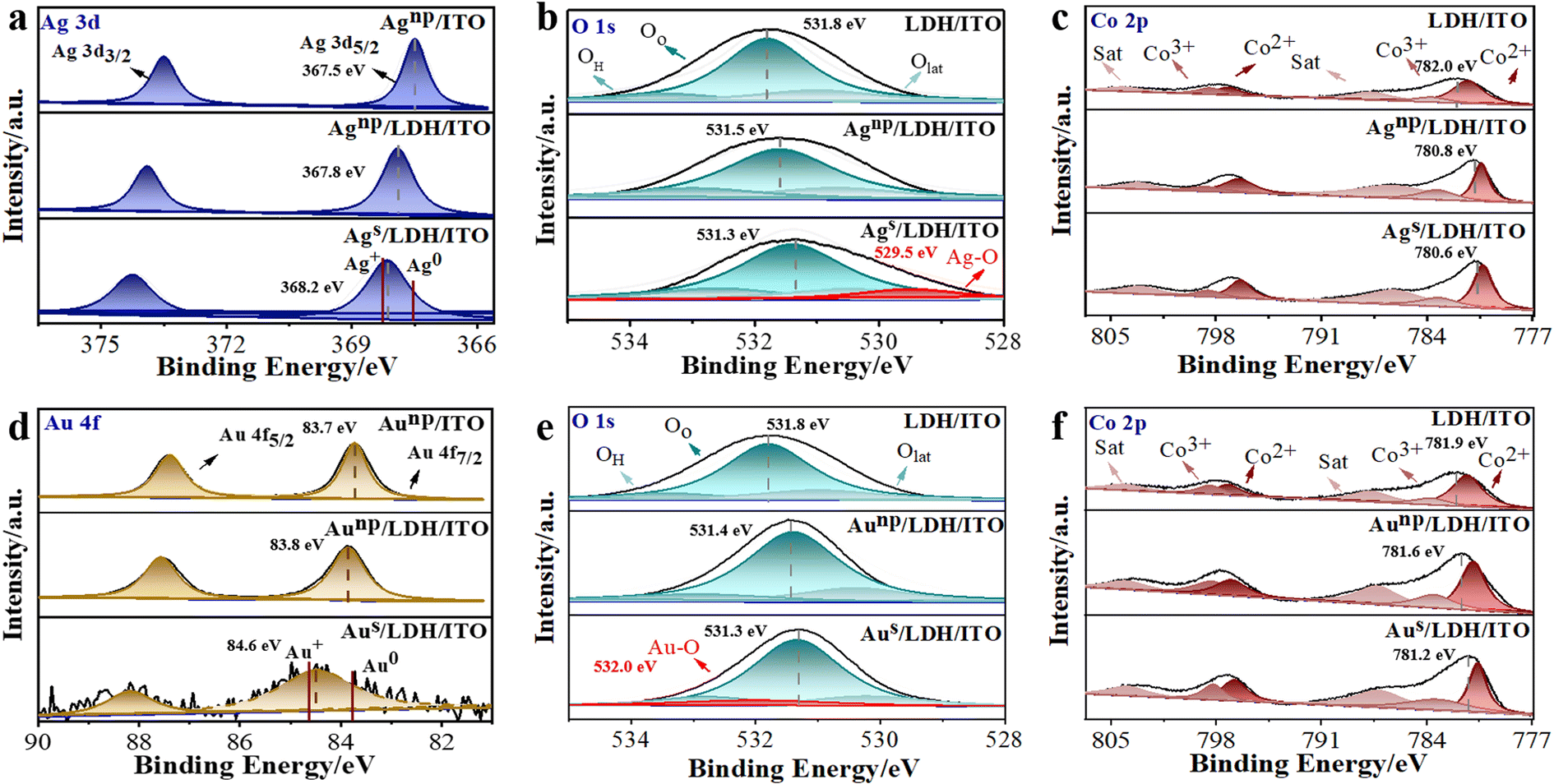 | ||
| Fig. 3 High-resolution XPS spectra of (a) Ag 3d, (b) O 1s and (c) Co 2p for LDH/ITO, Agnp/ITO, Agnp/LDH/ITO and Ags/LDH/ITO; high-resolution XPS spectra of (d) Au 4f, (e) O 1s and (f) Co 2p for LDH/ITO, Aunp/ITO, Aunp/LDH/ITO and Aus/LDH/ITO (the positions of the dotted line are marked in the diagram). | ||
Quantitative results from XPS spectra have addressed the differences between these modified electrodes. The ratios of Co3+/Co2+ showed a slight decrease from 71.9% in CoAl/LDH to 70.3% in Agnp/LDH/ITO and 68.4% in Aunp/LDH/ITO, while more significant reduction could be observed for 40.6% in Ags/LDH/ITO and 65.4% in Aus/LDH/ITO (Table S1†). Such a phenomenon could be ascribed to the unique electronic structure and well-defined active sites of single atoms, exhibiting great potentials in enhancing the catalytic activity during an electrochemical reaction.44,45 Moreover, the electronic structure of metal catalysts could be effectively regulated through the electronic metal–support interaction (EMSI).46 It has been known that the EMSI effect is associated with the orbital rehybridization of metal catalysts and charge transfer across the metal–support interface.47 Note that the rearrangement of electrons with a considerable EMSI effect is only confined to a couple of atomic layers at the metal–support interface.48 Accordingly, the stronger EMSI effect could be induced for single-atom catalysts than for the nanoparticles, leading to the modulated d state and the facilitated electron transfer from single-atom catalysts to LDH/ITO. Therefore, we will focus on the microstructural variations of these single atoms in the following discussions.
The average oxidation states of single atomic Ag and Au on the LDH/ITO have been calculated through the quantitative peak deconvolution and integration, and the oxidation states of single atomic Ag and Au on the LDH/ITO supports were estimated to be +0.9 and +0.7 eV, respectively (please see Table S2† for the detailed information and calculation process). As expected, electrons were transferred from Ag or Au single atoms to the LDH/ITO support due to the EMSI effect, and more electron transfer occurred for Ags/LDH/ITO with upshifted d-band centers. It is thus concluded that Ag single atoms with a higher oxidation state were energetically favorable for the adsorption and activation of oxygen species for the ECL reactions. Moreover, O 1s spectra showed that the contents of oxygen vacancies (Oo) for Ags/LDH/ITO and Aus/LDH/ITO were 71.2% and 70.3%, respectively (Table S3†). The higher contents of oxygen vacancies in Ags/LDH/ITO indicated the stronger oxygen adsorption capacity, in accordance with the positions of the d-band centers. The Ag–O and Au–O bonds accounted for 7.5% and 6.0% in O 1s spectra for Ags/LDH/ITO and Aus/LDH/ITO, suggesting the stronger metal–oxygen bonds in Ags/LDH/ITO for the electronic redistribution. Therefore, the data from XPS confirmed the upshifted d-band center of single atomic Ag and Au towards the Fermi level, in correlation with adsorption and activation abilities toward oxygen intermediates.
Ability to absorb, activate, and utilize the oxygen species
The redistributed electrons and oxidation state of metal species may affect their abilities to absorb, activate, and utilize the oxygen species during reaction. The oxygen adsorption ability at the modified electrodes was studied by O2-temperature programmed desorption (O2-TPD). It has been reported that the adsorbed oxygen changed from the physically adsorbed O2 (desorbed below 200 °C) to the chemically adsorbed oxygen species (O2−/O−, in the range of 200–400 °C) and eventually to the O2− in the lattice of the catalysts (higher than 400 °C).49,50 LDH/ITO showed two oxygen desorption peaks at about 291 °C and 736 °C (Fig. 4a), corresponding to the chemically adsorbed oxygen species and lattice oxygen species, respectively. Upon localization of single atomic Ag and Au, the temperatures of these two peaks decreased to 270/634 °C for Aus/LDH/ITO and 244/602 °C for Ags/LDH/ITO, suggesting the improved oxygen adsorption capacity of the modified electrodes. Note that compared to Aus/LDH/ITO, Ags/LDH/ITO with upshifted d-band centers required a lower temperature to absorb oxygen species and showed stronger potential to produce active oxygen species.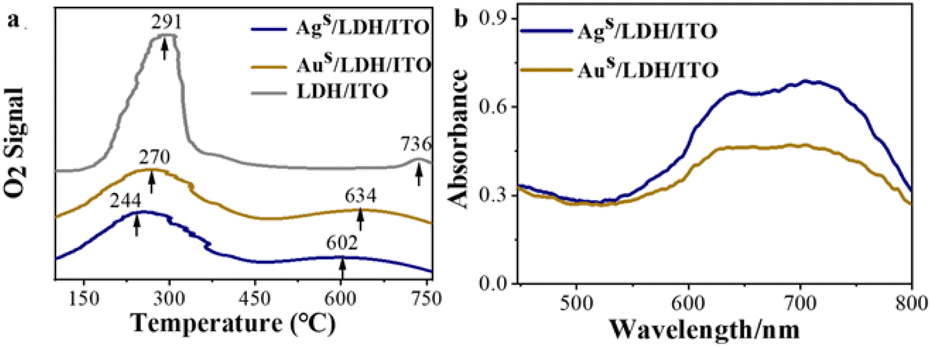 | ||
| Fig. 4 (a) O2-TPD pattern of LDH/ITO, Aus/LDH/ITO and Ags/LDH/ITO electrodes; (b) UV-vis spectra of methyl hydrazone for the Ags/LDH/ITO and Aus/LDH/ITO electrodes in KOH/DMSO solution. | ||
These adsorbed oxygen species could be converted to ROS during the negative scan, such as superoxide (O2˙−), singlet oxygen (1O2), and hydroxyl radicals (OH˙).51 To validate the presence of these radicals, radical scavengers, such as p-benzoquinone (BQ, O2˙− radical scavenger), isopropyl alcohol (IPA, OH˙ radical scavenger) and NaN3 (1O2 radical scavenger),52 have been added to the luminol solution in the presence of different electrodes. It can be observed that the ECL signals during the scan from −0.8 to 0 V were quenched by BQ, IPA and NaN3 at Ags/LDH/ITO and Aus/LDH/ITO electrodes (Fig. S11 and S12†), indicating the significance of O2˙−, OH˙ and 1O2 for the ECL emission. Particularly, the ECL signals were significantly quenched for 99% at Ags/LDH/ITO and 97% at Aus/LDH/ITO when the O2˙− was scavenged by BQ (Table S4†). These results suggested that O2˙− was the dominant ROS that contributed to the impressive cathodic emission of the luminol-dissolved O2 ECL system. The quantity of O2˙− was further evaluated by nitro blue tetrazolium chloride (NBT),53 and the amount of oxidation product methyl hydrazone was recorded by UV-vis spectroscopy (Fig. 4b). Notably, the absorbance of the oxidized product was higher for Ags/LDH/ITO than for Aus/LDH/ITO, illustrating the excellent ability of Ags/LDH/ITO to catalyse the generation of O2˙− than Aus/LDH/ITO did. These results demonstrate that O2˙− played vital roles in determining the ECL signals,47 due to its functionality to oxidize the luminol anions (L˙−) to form excited-state 3-aminophthalate (AP22−*) for the ECL emissions (Scheme S1†). Furthermore, we measured the ECL in the range from 0 to +0.8 V and quantified the generated O2˙− in the anodic ECL reaction for luminol. Upon the addition of BQ, most of the ECL signals at Ags/LDH/ITO and Aus/LDH/ITO were quenched, and the absorbance values of the oxidation products of NBT were almost the same at the Aus/LDH/ITO and Ags/LDH/ITO electrodes (Fig. S13†). These results suggested the generation of similar amounts of O2˙− during the anodic ECL reaction at the Aus/LDH/ITO and Ags/LDH/ITO electrodes, responsible for the similar amounts of AP2−* and ECL emissions (Scheme S1†). Therefore, we have employed the cathodic ECL as the indicator, and Ags/LDH/ITO showed superior ability in absorbing and activating the ROS to achieve the enhanced catalytic performances and cathodic ECL intensities.
Mechanisms to spot the d-band centers by the cathodic luminol ECL
Electrochemical impedance spectroscopy (EIS) was performed on the modified electrodes in 5.0 mmol L−1 K3[Fe(CN)6]/K4[Fe(CN)6] (0.1 mol L−1 KCl) from 0.1 Hz to 100 kHz. An equipped equivalence circuit is shown as the inset in Fig. S14,† where Rs, Rct, Cdl, and W represent the resistance of solution, charge transfer resistance, double layer capacitance, and the Warburg constant, respectively. The charge transfer resistance (Rct) from the Nyquist plot followed the trend: LDH/ITO (82.4 Ω) > ITO (69.6 Ω) > Aus/LDH/ITO (54.5 Ω) > Ags/LDH/ITO (46.9 Ω), indicating the higher electron transfer ability of Ags/LDH/ITO than that of Aus/LDH/ITO (Fig. S14†). The oxygen reduction reaction (ORR) is a critical step to generate ROS through electrochemical reactions and is highly correlated with the efficiency of the luminol ECL emission.51 The linear sweep voltammetry (LSV) curves showed that Ags/LDH/ITO possessed more positive onset potential and a higher ORR current than Aus/LDH/ITO did (Fig. 5a), indicating the effectively activated catalytic activity of Ags/LDH/ITO. This result was consistent with the ECL performance of the modified electrodes. Tafel slope curves further showed the values of 164.4 mV dec−1 for Ags/LDH/ITO and 208.4 mV dec−1 for Aus/LDH/ITO (Fig. 5b). The smaller Tafel slope of Ags/LDH/ITO indicated the faster electron transport and superior kinetics for the ORR.54 It has been known that the ORR kinetics process can proceed via either a 2e− (O2 + H2O + 2e− HO2− + OH−) or 4e− (O2 + 2H2O + 4e− 4OH−) pathway, which is dependent on the propensity to break the O–O bond during the ORR process.55 To figure out the pathways of the ORR process, the electron transfer number (n) involved in the ORR process was studied according to the Koutecky–Levich (K–L) plots (Fig. S15 and S16†). The number of electrons transferred during the ORR was different for different modified electrodes: the calculated n was 3.53 for Ags/LDH/ITO and 2.46 for Aus/LDH/ITO (Fig. 5c). Such a result suggested that the 4e− pathway of the ORR process was preferred in the presence of Ags/LDH/ITO, and Ags/LDH/ITO exhibited better ORR electrocatalytic activity and reaction kinetics than Aus/LDH/ITO did. Note that the 4e− reaction pathway involves more ROS, beneficial for the ECL reaction and emission enhancement.56 In consequence, these findings well explained the significantly enhanced ECL signals by Ags/LDH/ITO.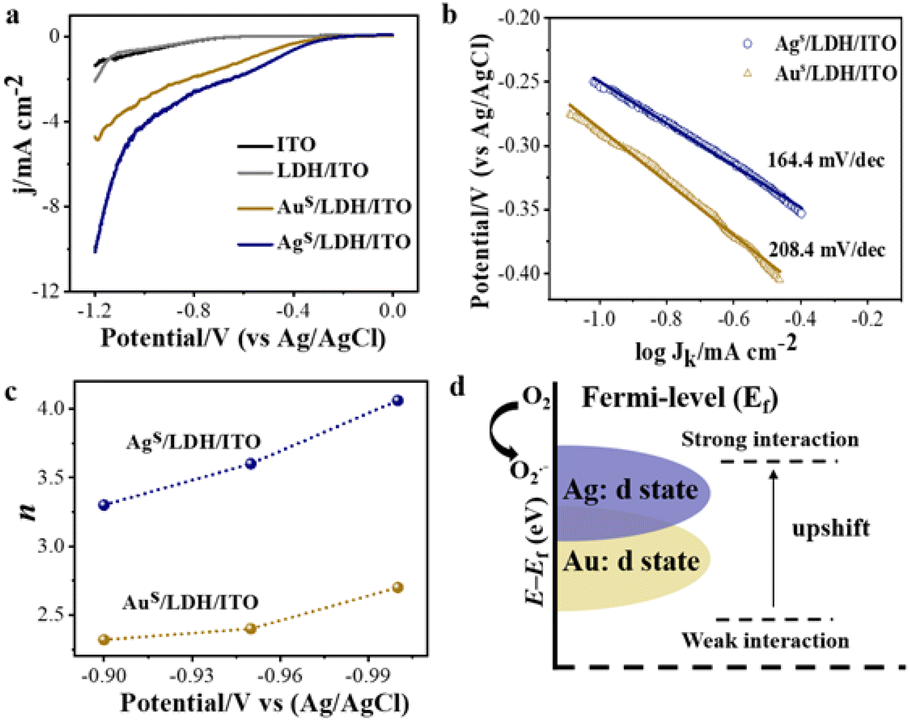 | ||
| Fig. 5 (a) Current density (j)–voltage curves in O2-saturated PBS (pH = 7.5) at ITO, LDH/ITO, Aus/LDH/ITO and Ags/LDH/ITO electrodes; (b) Tafel slope curves of Ags/LDH/ITO and Aus/LDH/ITO electrodes; (c) average electron transfer numbers (n) calculated from the K–L equation during the oxygen reduction reaction; (d) schematic illustration of the d-band center shift from the Fermi level for single atomic Ag and Au. | ||
The d-band model proposed by Nørskov was used to explain the electronic structure and thus the catalytic performances for these modified electrodes. Upon adsorption of O2 on the surface of the modified electrodes, bonding and antibonding states were formed between the p-band of adsorbate and d-band of metal species.1 The d-band centers of the metal species were shifted as a consequence. The closer location of d-band centers of Ags/LDH/ITO to the Fermi level than that of Aus/LDH/ITO resulted in the strengthened bonding interaction between Ag single atoms and oxygen intermediates (Fig. 5d). Therefore, much stronger oxygen reduction ability could be acquired for Ags/LDH/ITO, facilitating the generation of ROS for the ORR process and the promoted ECL emission of luminol. Therefore, the ECL signals showed the strength and accuracy to evaluate the d-band centers of single-atom catalysts in a sensitive approach.
Finally, we explored the universality of the proposed ECL strategy by other single-atom catalysts. First, Cu, which is listed in the same main group of Au and Ag, was prepared as single atomic Cu on LDH/ITO (labelled as Cus/LDH/ITO). XRD and UV-vis spectra have excluded the appearance of Cu nanoparticles in Cus/LDH/ITO (Fig. S17 and S18†). The HAADF-STEM image showed the uniform distribution of bright spots, which were ascribed to the single atomic Cu (Fig. S19†). The UPS valence-band spectra showed that the energy of d-band centers of Cus/LDH/ITO was located at −4.46 eV (Fig. S20†), and the ECL measurements showed that the cathodic ECL intensity of Cus/LDH/ITO was about 1200 a.u. (Fig. S21†). Second, single atomic Fe was prepared, as validated using the XRD spectra, UV-vis spectra and HAADF-STEM images (Fig. S22–S24†). The as-prepared Fes/LDH/ITO exhibited the energy of d-band centers at −4.97 eV (Fig. S25†) and the cathodic ECL intensity of 400 a.u. (Fig. S26†). As expected, both the ECL intensities and the positions of d-band centers for these single-atom catalysts followed the same trend: Ags/LDH/ITO > Aus/LDH/ITO > Cus/LDH/ITO > Fes/LDH/ITO (Fig. S27†). Therefore, we validated the universality of the proposed intermediate-boosted ECL in spotting the d-band centers of single-atom catalysts.
Conclusions
In summary, we have proposed oxygen intermediate-boosted ECL signals for an accurate and sensitive evaluation of the d-band centers of single-atom catalysts. Ag, Au, Cu and Fe single atoms with different d-band centers were localized onto CoAl/LDH supports as catalysts. In accordance with the largest upshift of d-band centers towards the Fermi level, Ags/LDH/ITO displayed a notable oxygen adsorption capacity and remarkable catalytic activity to generate ROS, leading to the strongest ECL emissions from luminol reactions. The variation tendency of the ECL signals and the positions of d-band centers were highly correlated: Ags/LDH/ITO > Aus/LDH/ITO > Cus/LDH/ITO > Fes/LDH/ITO. Such a correlation was also adapted for the nanoparticle catalysts such as Ag and Au nanoparticles, validating the higher catalytic efficiency of single atoms than that of nanoparticles. The proposed ECL strategy featured some advantages, such as high sensitivity, good accuracy, and simple sampling and analysing processes. Distinguishable signals with large variations could be acquired for the metal catalysts through an ECL cell for the luminol reaction. These advantages revealed that the proposed ECL strategy is an applicable and versatile method to evaluate the d-band centers for the metal catalysts. Moreover, the proposed strategy has also deepened the understanding of the d-band centers in determining the catalytic efficiencies for catalysts. This prospective methodology could be further applied to study and tailor the electronic structures for catalysts.Data availability
The data supporting the findings of this study are available within the article and ESI.† All other relevant source data are available from the corresponding authors upon reasonable request.Author contributions
Chao Lu and Rui Tian conceived the experiments. Ruyu Xie and Kaitao Li carried out the experiments. Ruyu Xie, Rui Tian and Chao Lu contributed to the data analysis and writing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (U22A20397 and 22374008) and Beijing Natural Science Foundation (2222018).Notes and references
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- Z. Li, Z. L. Tian, H. Cheng, T. Wang, W. Zhang, Y. Lu, Y. Q. Lai and G. J. He, Energy Storage Mater., 2023, 59, 102764 CrossRef.
- H. Sun, M. F. Wang, X. C. Du, Y. Jiao, S. S. Liu, T. Qian, Y. C. Yan, C. Liu, M. Liao, Q.-H. Zhang, L. X. Meng, L. Gu, J. Xiong and C. L. Yan, J. Mater. Chem. A, 2019, 7, 20952–20957 RSC.
- L. L. Li, Y.-F. Jiang, T. H. Zhang, H. F. Cai, Y. L. Zhou, B. Y. Lin, X. Y. Lin, Y. Zheng, L. R. Zheng, X. Y. Wang, C.-Q. Xu, C. Au, L. L. Jiang and J. Li, Chem, 2022, 8, 749–768 CAS.
- H. Liu, C. Wang, C. Liu, X. Zong, Y. F. Wang, X. X. Huang, Z. Z. Hu and Z. Q. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 29110–29119 CrossRef CAS.
- X. Li, J. Liu, Q. Cai, Z. Kan, S. Liu and J. Zhao, J. Colloid Interface Sci., 2022, 628, 331–342 CrossRef CAS PubMed.
- X. Y. Tian, R. J. Ren, F. Y. Wei, J. J. Pei, Z. B. Zhuang, L. Zhuang and W. C. Sheng, Nat. Commun., 2024, 15, 76 CrossRef CAS.
- Y. Shi, Z.-R. Ma, Y.-Y. Xiao, Y.-C. Yin, W.-M. Huang, Z.-C. Huang, Y.-Z. Zheng, F.-Y. Mu, R. Huang, G.-Y. Shi, Y.-Y. Sun, X.-H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef.
- X. Wu, F. Chen, N. Zhang, Y. Lei, Y. Jin, A. Qaseem and R. L. Johnston, Small, 2017, 13, 1603387 CrossRef PubMed.
- Q. Yuan, J. Zhao, D. H. Mok, Z. Zheng, Y. Ye, C. Liang, L. Zhou, S. Back and K. Jiang, Nano Lett., 2022, 22, 1257–1264 CrossRef CAS PubMed.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS.
- K. Artyushkova, A. Serov, H. Doan, N. Danilovic, C. B. Capuano, T. Sakamoto, H. Kishi, S. Yamaguchi, S. Mukerjee and P. Atanassov, J. Electron Spectrosc. Relat. Phenom., 2019, 231, 127–139 CrossRef CAS.
- J. Mahoney, C. Monroe, A. M. Swartley, M. G. Ucak-Astarlioglu and C. A. Zoto, Spectrosc. Lett., 2020, 53, 726–736 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. P. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS.
- S. F. Douman, D. Collins, L. R. Cumba, S. Beirne, G. G. Wallace, Z. L. Yue, E. I. Iwuoha, F. Melinato, Y. Pellegrin and R. J. Forster, Chem. Commun., 2021, 57, 4642–4645 RSC.
- M. Feng, A. L. Dauphin, L. Bouffier, F. Zhang, Z. Wang and N. Sojic, Anal. Chem., 2021, 93, 16425–16431 CrossRef CAS PubMed.
- Y. H. Huang, R. Y. Xie, K. T. Li, R. Tian, Y. Lin and C. Lu, ACS Appl. Mater. Interfaces, 2023, 15, 1610–1618 CrossRef CAS.
- Y. P. Dong, J. Wang, Y. Peng and J. J. Zhu, J. Electroanal. Chem., 2016, 781, 109–113 CrossRef CAS.
- D. Wei, L. Chen, H. Zhao, L. Tian, S. Ramakrishna and D. Ji, ACS Appl. Nano Mater., 2023, 6, 11061–11069 CrossRef CAS.
- X. Zhou, J. J. Gao, Y. X. Hu, Z. Y. Jin, K. L. Hu, K. M. Reddy, Q. H. Yuan, X. Lin and H.-J. Qiu, Nano Lett., 2022, 22, 3392–3399 CrossRef CAS.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- E. D. Boyes, A. P. LaGrow, M. R. Ward, R. W. Mitchell and P. L. Gai, Acc. Chem. Res., 2020, 53, 390–399 CrossRef CAS.
- N. Acerbi, S. C. E. Tsang, G. Jones, S. Golunski and P. Collier, Angew. Chem., Int. Ed., 2013, 52, 7737–7741 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS.
- A. J. Marenco, D. B. Pedersen, S. Wang, M. W. P. Petryk and H. B. Kraatz, Analyst, 2009, 134, 2021–2027 RSC.
- S. A. Chala, M.-C. Tsai, W.-N. Su, K. B. Ibrahim, B. Thirumalraj, T.-S. Chan, J.-F. Lee, H. J. Dai and B.-J. Hwang, ACS Catal., 2019, 9, 117–129 CrossRef CAS.
- R. Zou, Y. J. Lin and C. Lu, Anal. Chem., 2021, 93, 2678–2686 CrossRef CAS.
- H. Cui, Y. Xu and Z. F. Zhang, Anal. Chem., 2004, 76, 4002–4010 CrossRef CAS PubMed.
- Y. H. Huang, R. Zou, Y. J. Lin and C. Lu, Anal. Chem., 2021, 93, 11291–11297 CrossRef CAS PubMed.
- X. L. Huo, H. Yang, M.-X. Li, W. Zhao, J.-J. Xu, Y. Wang, X.-L. Luo and H.-Y. Chen, Nanoscale, 2018, 10, 19224–19230 RSC.
- J. Wei, K. Xiao, Y. Chen, X. P. Guo, B. Huang and Z. Q. Liu, Energy Environ. Sci., 2022, 15, 4592–4600 RSC.
- J. Li, Q. Q. Guan, H. Wu, W. Liu, Y. Lin, Z. H. Sun, X. X. Ye, X. S. Zheng, H. B. Pan, J. F. Zhu, S. Chen, W. H. Zhang, S. Q. Wei and J. L. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- Y. Hou, J. Feng, R. Tian, C. Lu and X. Duan, Angew. Chem., Int. Ed., 2023, 62, e202307573 CrossRef CAS PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS.
- X. Wang, S. Li, Z. Yuan, Y. Sun, Z. Tang, X. Gao, H. Zhang, J. Li, S. Wang, D. Yang, J. Xie, Z. Yang and Y.-M. Yan, Angew. Chem., Int. Ed., 2023, 62, e202303794 CrossRef CAS PubMed.
- H. Zeng, L. Deng, H. Zhang, C. Zhou and Z. Shi, J. Hazard. Mater., 2020, 400, 123297 CrossRef CAS PubMed.
- L. Wu, J. Hong, Q. Zhang, B. Y. Chen, J. Wang and Z. Dong, Chem. Eng. J., 2020, 385, 123620 CrossRef CAS.
- F.-F. Zhang, C.-Q. Cheng, J.-Q. Wang, L. Shang, Y. Feng, Y. Zhang, J. Mao, Q.-J. Guo, Y.-M. Xie, C.-K. Dong, Y.-H. Cheng, H. Liu and X.-W. Du, ACS Energy Lett., 2021, 6, 1588–1595 CrossRef CAS.
- L. J. Lei, H. Liu, Z. W. Wu, Z. F. Qin, G. F. Wang, J. Y. Ma, L. Luo, W. B. Fan and J. G. Wang, ACS Appl. Nano Mater., 2019, 2, 5214–5223 CrossRef CAS.
- P. Kundu, N. Singhania, G. Madras and N. Ravishankar, Dalton Trans., 2012, 41, 8762–8766 RSC.
- L. Rosenberger, R. Baird, E. McCullen, G. Auner and G. Shreve, Surf. Interface Anal., 2008, 40, 1254–1261 CrossRef CAS.
- C. P. Wang, S. J. Mao, Z. Wang, Y. Z. Chen, W. T. Yuan, Y. Ou, H. Zhang, Y. T. Gong, Y. Wang, B. B. Mei, Z. Jiang and Y. Wang, Chem, 2020, 6, 752–765 CAS.
- J. Li, M. F. Stephanopoulos and Y. Xia, Chem. Rev., 2020, 120, 11699–11702 CrossRef CAS PubMed.
- A. Bruix, J. A. Rodriguez, P. J. Ramírez, S. D. Senanayake, J. Evans, J. B. Park, D. Stacchiola, P. Liu, J. Hrbek and F. Illas, J. Am. Chem. Soc., 2012, 134, 8968–8974 CrossRef CAS PubMed.
- G. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas and K. Prince, Nat. Mater., 2011, 10, 310–315 CrossRef CAS.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- P. Li, C. He, J. Cheng, C. Y. Ma, B. J. Dou and Z. P. Hao, Appl. Catal., B, 2011, 101, 570–579 CrossRef CAS.
- W. Deng, Q. Tang, S. Huang, L. Zhang, Z. Jia and L. Guo, Appl. Catal., B, 2020, 278, 119336 CrossRef CAS.
- H. Xia, X. L. Zheng, J. Li, L. G. Wan, Y. Xue, C. Peng, Y. C. Han, Y. Wang, S. J. Guo, J. Wang and E. K. Wang, J. Am. Chem. Soc., 2022, 144, 7741–7749 CrossRef CAS.
- W. Gu, H. J. Wang, L. Jiao, Y. Wu, Y. X. Chen, L. Y. Hu, J. M. Gong, D. Du and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 3534–3538 CrossRef CAS PubMed.
- R.-H. Liu, S.-Y. Fu, H.-Y. Zhan and L. A. Lucia, Ind. Eng. Chem. Res., 2009, 48, 9331–9334 CrossRef CAS.
- W. Wang, Y. Liu, J. Li, J. Luo, L. Fu and S. Chen, J. Mater. Chem. A, 2018, 6, 14299–14306 RSC.
- J. Hu, W. Liu, C. Xin, J. Guo, X. Cheng, J. Weai, C. Hao, G. Zhang and Y. Shi, J. Mater. Chem. A, 2021, 9, 24803–24829 RSC.
- W. Gu, X. S. Wang, J. Wen, S. Y. Cao, L. Jiao, Y. Wu, X. Q. Wei, L. R. Zheng, L. Y. Hu, L. Z. Zhang and C. Z. Zhu, Anal. Chem., 2021, 93, 8663–8670 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03763d |
| This journal is © The Royal Society of Chemistry 2024 |