DOI:
10.1039/D4SC03873H
(Edge Article)
Chem. Sci., 2024,
15, 13405-13414
Tuning the steric hindrance of alkylamines: a predictive model of steric editing of planar amines†
Received
13th June 2024
, Accepted 10th July 2024
First published on 11th July 2024
Abstract
Amines are one of the most prevalent functional groups in chemistry. Perhaps even more importantly, amines represent one of the most ubiquitous moieties within the realm of bioactive natural products and life-saving pharmaceuticals. The archetypal geometrical property of amines is their sp3 hybridization with the lone pair of nitrogen occupying the apex of the pyramid. Herein, we present a blueprint for quantifying the properties of extremely sterically hindered alkylamines. These amines reach planarity around the nitrogen atom due to the excessive steric hindrance, which results in a conformational re-modeling of the amine moiety. Crucially, the steric properties of amines are characterized by the %VBur index, which we show is a general predictive parameter for evaluating the properties of sterically hindered amines. Computational studies on the acidic nature and the reactivity of organometallic Au and Pd complexes are outlined. Density functional theory calculations permit for predictive catalysis, ordering the mapping of extremely hindered tertiary amines by employing artificial intelligence via machine learning. Overall, the study outlines the correlation between the unusual geometry and the key thermodynamic and kinetic properties of extremely hindered alkylamines. The steric hindrance, as quantified by %VBur, is the crucial factor influencing the observed trends and the space required to accommodate sterically hindered tertiary amines.
1. Introduction
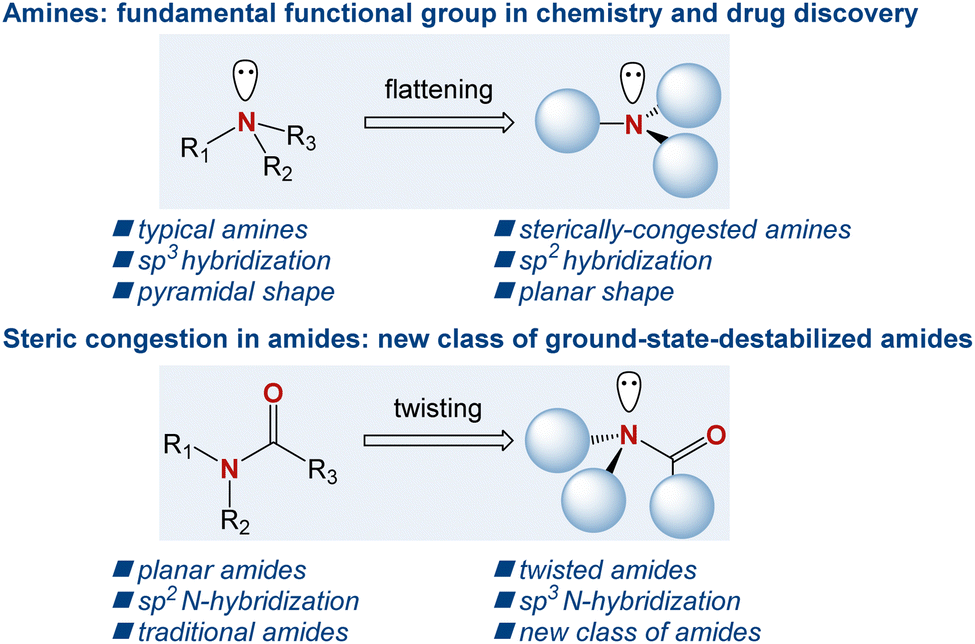
The amine motif is one of the most fundamental functional groups within the realm of organic chemistry and drug discovery.1 In organic synthesis, in addition to being a fundamental part of the target products ranging from bioactive natural products to organometallic chemistry applications, amines serve as key bases and nucleophiles, where the accessibility of the free electron pair at the nitrogen atom enables their common role in synthetic reactions on a daily basis.2 In drug discovery, >80% of small-molecule drugs approved by the FDA contain an amino group, where the nitrogen atom serves as a key pharmacophore in drug–receptor interactions.3 Consequently, over the years, numerous generations of chemists have devised a wide array of synthetic methods to access the amine motif.4 In this context, sterically hindered amines, where the nitrogen atom significantly deviates from the sp3 pyramidalization, are of major synthetic and theoretical interest (Scheme 1).5 In a broader sense, sterically hindered amines are conceptually related to sterically hindered amides, where the typical planarity of the amide bond is geometrically transformed into a non-planar ground-state conformation by N–C(O) bond rotation and nitrogen pyramidalization.6 Recent years have witnessed a surge in applications of such twisted amides owing to their significantly different properties compared to their planar counterparts.7
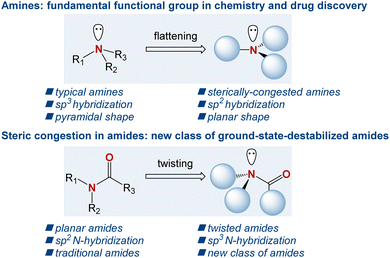 |
| | Scheme 1 The effect of steric hindrance in amines, and sterically destabilized twisted amides. | |
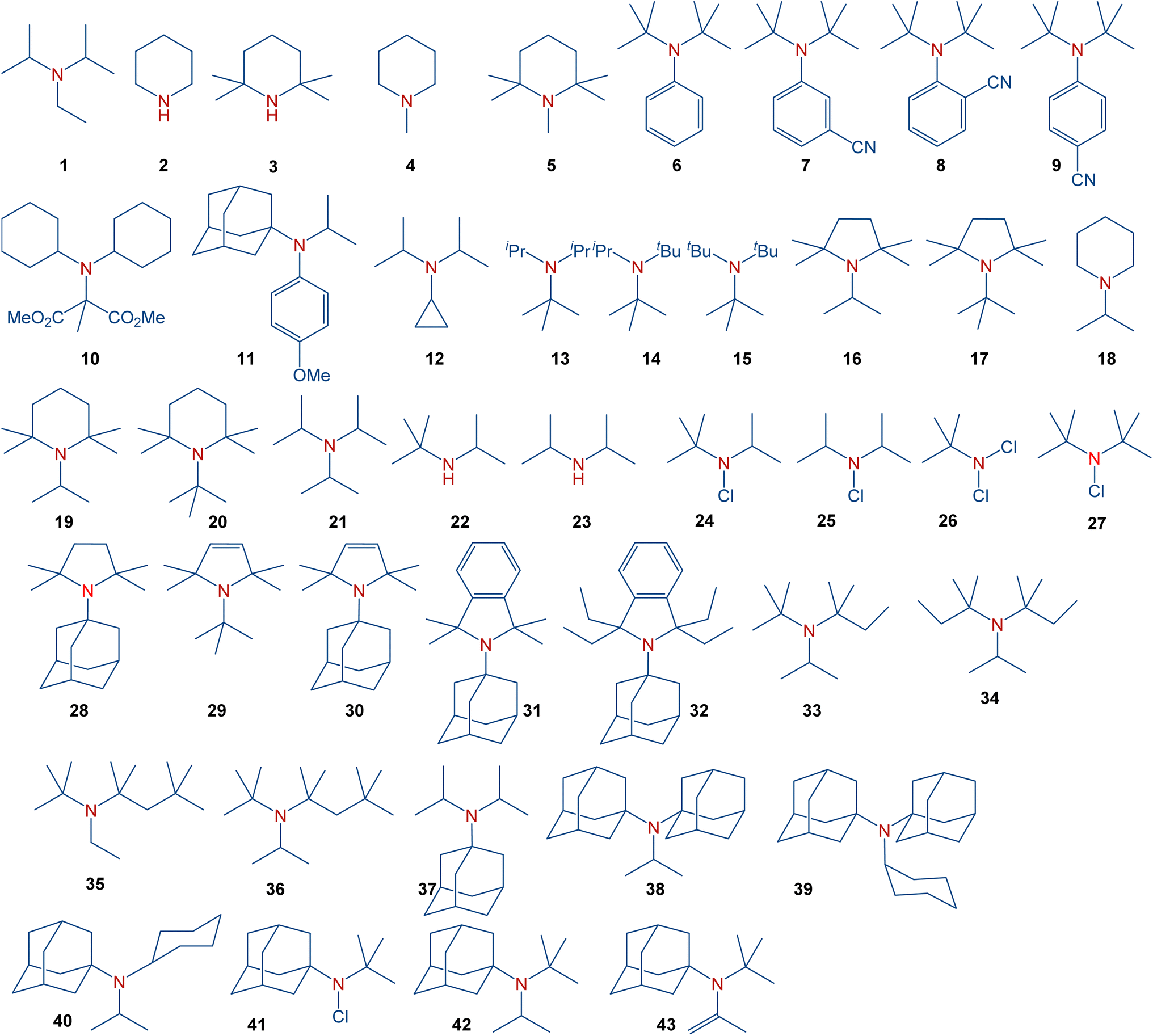
In general, sterically hindered amines have already found diverse applications. Amines discussed in the present manuscript are summarized in Scheme 2. These amines include simple aliphatic amines, such as Hünig's base (1), sterically hindered piperidines (4–5), sterically hindered anilines (6–9), cyclopropylamines (12), aliphatic amines with gradually increasing steric hindrance (13–15), sterically hindered pyrrolidines (16–17), representative dialkylamines (18–20), formylamines (25), chloroamines (22–23), as well as amines with the most steric hindrance prepared or proposed to date (35–42). These species serve, for instance, as bases with low nucleophilicity (1 (ref. 8) and 2 (ref. 9)), precursors for persistent nitroxyl radicals (3 (ref. 10 and 11)), inhibitors in polymerization, and stabilizers (4).12 Moreover, sterically hindered amines have found applications in gas-treating processes,13 and as pharmacological agents.14
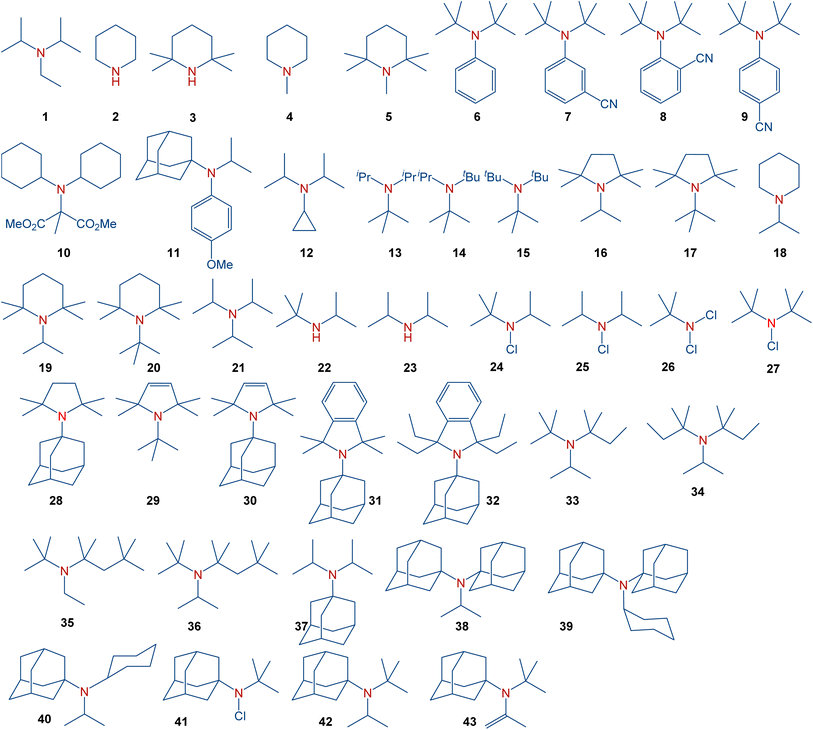 |
| | Scheme 2 Full scope of amines in the present study. | |
It should be noted that several tertiary amines with a notably high degree of steric congestion, e.g., 7–9,1510,1611,17 encompass additional functional groups that significantly influence the molecular structure surrounding the amine nitrogen atom and, consequently, its reactivity. For example, amine 12 has been claimed to be the most hindered tertiary amine known.18 However, this amine derives its properties from the unique electronics and sterics of the cyclopropyl group. For years, triisopropylamine 21 appeared to set the limit for the achievable level of steric congestion in simple trialkylamines.16 This amine was predicted to exhibit nearly planar geometry around the nitrogen atom based on electron diffraction19 and NMR studies,20 while low-temperature crystallographic analysis21 indicated a flattened pyramid shape instead of a perfect planarity. Furthermore, despite claims in organic chemistry textbooks comparing bulky amines to alcohols,22 sterically hindered amines, such as 15 (ref. 23) typically avoid planar structures, while steric congestion in bulky amines should take into account the different bond lengths involved. Specifically, the average C–N bond is considerably shorter than C–C bonds,24 leading to a shortening of the distance between the groups around the nitrogen atom in sterically hindered amines. Synthetically, the stability of hindered amines should also be considered since amines with significant steric hindrance may undergo β-elimination.25 A recent study demonstrated the synthesis of some of the most sterically hindered amines reported to date, including 13–14, 16–18, 30–42 and 44.26 Among the strategies used,27 synthetic methods to access such sterically hindered amines include ammonium salts,28 alkyl29,30 or aryl31 Grignard reagents,32 Bruylants reaction,19,33 or the SN1 alkylation.34 These approaches yielded trialkylamines with exceptionally high steric hindrance, leading to restricted rotation around the C–N bond.26
Given the fundamental importance of extremely sterically hindered amines in various areas of chemistry and the critical role of geometry around the nitrogen atom in amines in drug discovery, herein we report a blueprint to quantify the properties of extremely sterically hindered alkylamines and correlate them with the geometry ranging from low35 to furthermost36 steric hindrance, leading to predictive catalysis.37,38
2. Computational details
DFT calculations were performed with the Gaussian16 set of programs,39 using the hybrid GGA functional of Becke-Lee, Parr, and Yang, i.e., B3LYP,40 and the def2TZVP basis set for all atoms applied,41 except for Au and Pt, which were treated with the quasi-relativistic Stuttgart/Dresden effective core potential with an associated valence basis set (standard SDD keywords in Gaussian16).42 Moreover, we also included the D3 Grimme pairwise scheme to account for dispersion corrections in the geometry optimizations. Geometry optimizations were performed without symmetry constraints, and the characterization of the stationary points was performed by analytical frequency calculations. These frequencies were used to calculate unscaled zero-point energies (ZPEs) as well as thermal corrections and entropy effects at 298.15 K and 1 atm by using the standard statistical mechanics relationships for an ideal gas. We also included the solvent effects of THF solution estimated with the polarizable continuous solvation model (PCM) as implemented in Gaussian16.43
3. Results and discussion
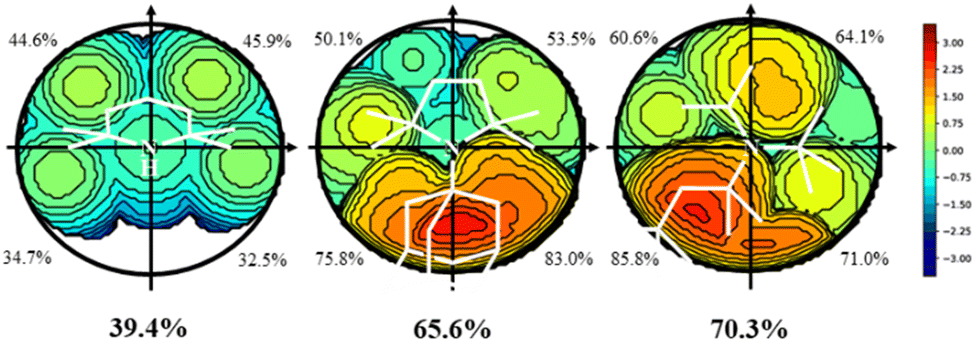
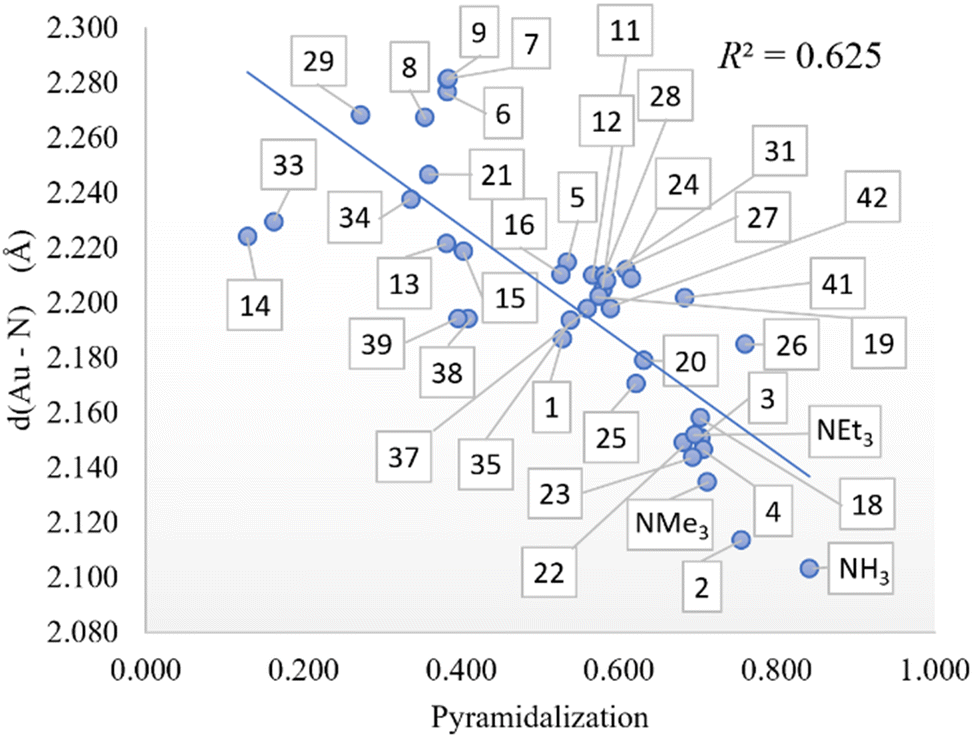
All amines included in Scheme 2 were optimized by screening all potential isomers. To study the sterics, the high flexibility was overcome by means of the coordination of amines to the Au–Cl moiety, screening all isomers again. Then, the geometry of amine ligands in amine–Au–Cl complexes was evaluated by means of the %VBur, developed by Cavallo and coworkers.44 The sterics were evaluated at 2.0 Å from the N atom, since this is the average value for the coordination of amines to metals.38 Further, only the first sphere around the metal was studied since this is where the reactivity takes place. The properties were calculated for the 45 tertiary amines, as well as NH3, NMe3, NEt3, and pyridine (Py) for comparison. Table 1 summarizes the overall %VBur (for additional details, see Table S1† divided into four quadrants to infer any asymmetry that would compensate, partially or completely, for a high overall %VBur value). Fig. 1 includes the odd cases 3 and 36, with a lower (39.4%) and higher occupation (70.3%), respectively, and also a third one, 28, with an intermediate value of 65.6%.
Table 1 Acidity and binding energies (Gibbs energies in kcal mol−1) for 45 amines and NH3, NMe3, NEt3, and Py with a proton, AuCl(P(CH3)3), Pd(P(CH3)3), and PdCl2(P(CH3)3). Total %VBur values of the amines
|
|
H+a |
H+b |
Au+ |
Pd0 |
Pd2+ |
%VBur |
|
The proton is added as a free atom.
The proton comes from acetic acid.
Not located.
|
|
1
|
−0.3 |
22.8 |
24.0 |
13.8 |
3.9 |
49.8 |
|
2
|
−1.3 |
21.7 |
18.4 |
11.8 |
−12.7 |
25.2 |
|
3
|
−3.9 |
19.1 |
17.6 |
8.5 |
−9.1 |
39.4 |
|
4
|
−1.8 |
21.2 |
20.0 |
10.8 |
—c |
36.5 |
|
5
|
−5.1 |
18.0 |
24.1 |
12.7 |
—c |
48.0 |
|
6
|
−0.8 |
22.2 |
33.6 |
17.2 |
—c |
55.8 |
|
7
|
4.2 |
27.2 |
35.0 |
17.4 |
—c |
55.8 |
|
8
|
6.3 |
29.3 |
45.9 |
—c |
—c |
68.5 |
|
9
|
4.5 |
27.5 |
35.8 |
—c |
—c |
55.8 |
|
10
|
2.3 |
25.3 |
—c |
—c |
—c |
63.3 |
|
11
|
−0.6 |
22.5 |
30.7 |
16.4 |
11.1 |
59.0 |
|
12
|
−5.3 |
17.8 |
23.5 |
13.4 |
—c |
55.6 |
|
13
|
1.0 |
24.1 |
33.3 |
21.1 |
—c |
55.0 |
|
14
|
−4.5 |
18.6 |
30.1 |
17.6 |
—c |
61.6 |
|
15
|
−9.1 |
13.9 |
27.2 |
15.1 |
—c |
56.2 |
|
16
|
−6.3 |
16.7 |
24.7 |
13.6 |
—c |
54.0 |
|
17
|
−4.9 |
18.1 |
18.7 |
17.7 |
—c |
59.3 |
|
18
|
−3.1 |
20.0 |
20.7 |
11.1 |
−5.8 |
43.3 |
|
19
|
−8.6 |
14.5 |
21.3 |
10.8 |
—c |
51.9 |
|
20
|
−4.2 |
18.8 |
34.7 |
12.9 |
−2.4 |
46.2 |
|
21
|
−2.4 |
20.6 |
18.1 |
21.2 |
11.6 |
53.8 |
|
22
|
−3.3 |
19.8 |
19.4 |
10.5 |
−7.9 |
38.8 |
|
23
|
−2.1 |
20.9 |
31.8 |
9.7 |
−9.7 |
33.9 |
|
24
|
12.1 |
35.1 |
27.2 |
15.3 |
—c |
43.8 |
|
25
|
13.2 |
36.2 |
34.8 |
13.1 |
−0.1 |
42.0 |
|
26
|
30.7 |
53.7 |
28.7 |
16.0 |
—c |
38.6 |
|
27
|
10.0 |
33.0 |
27.0 |
13.5 |
—c |
50.1 |
|
28
|
−8.4 |
14.6 |
32.9 |
14.1 |
—c |
65.6 |
|
29
|
−4.5 |
18.6 |
27.5 |
18.1 |
—c |
58.4 |
|
30
|
−5.5 |
17.5 |
30.7 |
—c |
—c |
61.0 |
|
31
|
−5.1 |
18.0 |
23.5 |
—c |
—c |
61.4 |
|
32
|
−6.5 |
16.5 |
—c |
—c |
—c |
61.9 |
|
33
|
−3.7 |
19.4 |
32.2 |
19.5 |
—c |
65.1 |
|
34
|
−7.0 |
16.1 |
31.0 |
—c |
—c |
68.2 |
|
35
|
−7.1 |
15.9 |
24.4 |
14.5 |
6.2 |
67.8 |
|
36
|
−6.2 |
16.8 |
—c |
17.3 |
—c |
70.3 |
|
37
|
−5.3 |
17.7 |
28.7 |
16.1 |
—c |
63.6 |
|
38
|
−6.5 |
16.5 |
26.3 |
—c |
8.0 |
67.4 |
|
39
|
−5.8 |
17.2 |
26.0 |
13.6 |
8.7 |
67.9 |
|
40
|
−5.6 |
17.4 |
—c |
—c |
—c |
61.3 |
|
41
|
9.0 |
32.0 |
29.7 |
—c |
—c |
54.5 |
|
42
|
−7.9 |
15.2 |
25.4 |
13.3 |
—c |
61.6 |
|
43
|
−3.4 |
19.6 |
—c |
16.0 |
—c |
63.1 |
| NH3 |
8.4 |
31.4 |
20.7 |
11.0 |
−11.4 |
15.2 |
| NMe3 |
−0.2 |
22.9 |
21.5 |
12.3 |
−7.0 |
29.7 |
| NEt3 |
−3.2 |
19.8 |
21.0 |
11.2 |
0.4 |
44.8 |
| Py |
5.3 |
28.4 |
—c |
—c |
—c |
21.3 |
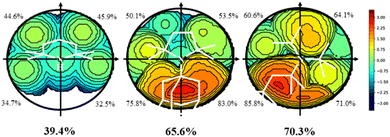 |
| | Fig. 1 Topographic steric maps (xy plane) and %VBur of the amines 3 (left), 28 (middle), and 36 (right). The center is on the Z axis, where the Au–N bond would be, specifically at 2.0 Å from N, and the XY plane contains the plane that best defines the 3 N-coordinated atoms. The isocontour curves of the steric maps are given in Å. The radius of the sphere around the center was set to 3.5 Å, while for the atoms we adopted the Bondi radii scaled by 1.17, and a mesh of 0.1 Å was used to scan the sphere for buried voxels (the isocontour curves of the steric maps are given in Å). | |
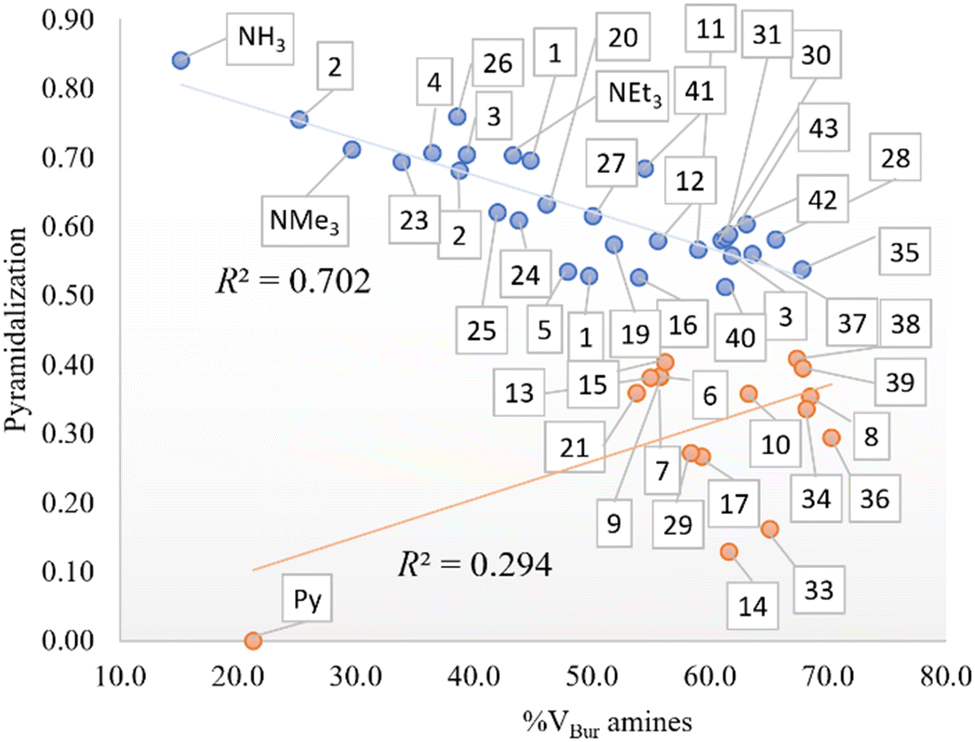
In addition, the role of sterics in tuning amine pyramidalization and the influence of C–N bond lengths were also analyzed. First, the pyramidalization was calculated according to the scheme of Radhakrishnan and Agranat.45 As aforementioned, in sterically hindered amines, the nitrogen atom deviates from sp3 hybridization to assume a planar shape in bulky amines. In the plot of pyramidalization vs. amines %VBur, no correlation was found for all amines, but as shown in Fig. 2, the pyramidalization-%VBur trend deviates from linearity for pyramidalization values below 0.450. On the other hand, for greater values, the pyramidalization is almost directly proportional to amine %VBur, with a Pearson coefficient (R2) of 0.702. The rest of the amines do not show any trend, but except for pyridine, with a pyramidalization index equal to zero, they have in common a tert-butyl moiety, which somehow in the presence of two other bulky groups favors the flattering of the amine to contrast the steric repulsion.
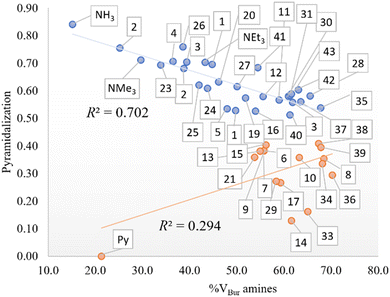 |
| | Fig. 2 Pyramidalization vs. %VBur of the amines (in blue for a pyramidalization index higher than 0.450 and in red for lower values). | |
Apart from the sterics, amines were evaluated according to their capacity to trap protons, a fundamental property of the amine functional group, and thus acidity is also included in Table 1. Two approaches were used: either the addition of a proton (eqn (1)) or the deprotonation of an acetic acid molecule (eqn (2)). It should be noted that for the first approach for the aqueous solvation free energy of the proton, we assumed a value of −272.2 kcal mol−1 from the literature,46 to be compared with values of −250 kcal mol−1 in organic media.47 Our aim was to establish trends among the series of amines; however, it should be noted that this value is probably underestimated. We analyzed in detail how strongly the amines bond to metals using eqn (3)–(5). Next, we analyzed in detail how strongly the amines are bonded to the metals. As representative systems, the aforementioned amine–Au–Cl complexes were selected (eqn (3)) as well as palladium complexes. For the latter, the study included the oxidation state 0 as well as +2 (eqn (4) and (5)), to evaluate how the oxidation state on the metal center affects the affinity for the coordination of amines and to assist in future studies involving amine–metal(0) complexes.48 We must acknowledge that we omitted other relevant models, particularly the R3N–Ni(CO)3 standard in phosphine chemistry, which can be considered congeners of amines.49 Instead, we selected models from eqn (2)–(5), which are more typical of NHCs. Despite the significance of Ni(CO)3 systems, which also possess C3V symmetry and could align with the three substituents on nitrogen, our goal was to focus on the models that reflect current research interests, especially those from the past decade.50
| | | NR3 + (CH3)COOH → (NR3)H+ + (CH3)COO− | (2) |
| | | AuCl(P(CH3)3) + NR3 → AuCl(NR3) + P(CH3)3 | (3) |
| | | Pd(P(CH3)3)2 + NR3 → Pd(P(CH3)3)(NR3) + P(CH3)3 | (4) |
| | | PdCl2(P(CH3)3) + NR3 → Pd(P(CH3)3)Cl2(NR3) | (5) |
Faced with such a large set of similar species, in order to find trends, we had to select the key parameters. In the context of parameters, they must be robust and selective. In a systematic process, a series of reactions were carried out to evaluate the role of amines at the reactivity level.
N-Protonation
Emphasizing the fundamental organic protonation of amines in eqn (2) and analyzing the thermodynamics of the process, the best correlation found with one variable was reached with the Mayer Bond Order (MBO) of the N–H bond, but the fitting is very modest (R2 = 0.527). By increasing the number of variables up to three, the correlations improve. In detail, the addition of the amine LUMO energy and the amine molecular area leads to better correlations with two (R2 = 0.731) and three (R2 = 0.785) variables. In general, the thermodynamics results are thus influenced by electronic factors since both amine LUMO energy and the MBO of the N–H bond are intrinsically linked to amine molecular orbitals. On the other hand, the kinetics of the proton transfer to the amine is more influenced by steric factors. The kinetics appears to be moderately correlated (R2 = 0.502) with the amine %VBur of the least occupied quadrant (Low %VBur amine) with one variable. In contrast, Table 2 shows that the steric-dependent descriptors as well as the pyramidalization on the nitrogen atom and pure steric descriptors such as amine %VBur and ammonium %VBur have some influence on kinetics when using two and three variables. The fit improved up to 0.869 (with two variables) and to 0.884 (with three variables) if the amine pKa was also taken into account.
Table 2 Best correlations for the 43 tertiary amines and NH3, NMe3, NEt3, and Py (R2 = square of the Pearson correlation coefficient; RMSE = root mean squared error)
|
|
Variables number |
|
Filter |
Systems number |
Variables |
Coefficients |
Intercept |
R
2
|
|
Eqn (2)
|
1 |
ΔG |
— |
45 |
(MBO(N–H+) ammonium) |
[−681.7] |
[626.0] |
0.527 |
| Pyr > 0.450 |
29 |
(MBO(N–H+) ammonium) |
[−1032.2] |
[935.2] |
0.715 |
| 2 |
— |
45 |
(E(LUMO) amine) |
[−204.6] |
[39.3] |
0.731 |
| (Molecular area) |
[−0.3] |
| Pyr > 0.450 |
29 |
(E(LUMO) amine) |
[−304.7] |
[45.6] |
0.898 |
| (Molecular area) |
[−0.4] |
| 3 |
— |
45 |
(E(LUMO) amine) |
[−154.5] |
[290.4] |
0.785 |
| (Molecular area) |
[−0.2] |
| (MBO(N–H+) ammonium) |
[−287.1] |
| Pyr > 0.450 |
29 |
(E(LUMO) amine) |
[−249.4] |
[274.9] |
0.914 |
| (Molecular area) |
[−0.3] |
| (MBO(N–H+) ammonium) |
[−264.5] |
| 1 |
ΔG‡ |
— |
39 |
(Low %VBur amine) |
[0.3] |
[−11.6] |
0.502 |
| Pyr > 0.450 |
23 |
(High %VBur ammonium) |
[0.2] |
[−8.5] |
0.651 |
| 2 |
— |
39 |
(pKa amine) |
[−1.6] |
[−24.8] |
0.869 |
| (%VBur amine) |
[0.3] |
| Pyr > 0.450 |
23 |
(pKa amine) |
[−1.6] |
[−24.3] |
0.867 |
| (%VBur amine) |
[0.3] |
| 3 |
— |
39 |
(pKa amine) |
[−1.4] |
[−18.9] |
0.884 |
| (Pyramidalization) |
[−6.2] |
| (%VBur ammonium) |
[0.3] |
| Pyr > 0.450 |
23 |
(pKa amine) |
[−1.6] |
[493.0] |
0.903 |
| (%VBur amine) |
[0.2] |
| (d(N–H+) ammonium) |
[−504.8] |
|
Eqn (3)
|
1 |
ΔG |
— |
39 |
(d(Au–N)) |
[117.4] |
[−231.2] |
0.679 |
| Pyr > 0.450 |
26 |
(E(LUMO) amine) |
[−154.1] |
[26.4] |
0.688 |
| 2 |
— |
39 |
(d(Au–N)) |
[119.6] |
[−241.2] |
0.838 |
| (pKa amine) |
[−0.7] |
| Pyr > 0.450 |
26 |
(MBO(Au–N)) |
[−59.9] |
[−186.5] |
0.865 |
| (d(Au–N)) |
[112.1] |
| 3 |
— |
39 |
(d(Au–N)) |
[81.8] |
[−168.4] |
0.867 |
| (pKa amine) |
[−0.9] |
| (Low %VBur amine) |
[0.2] |
| Pyr > 0.450 |
26 |
(MBO(Au–N)) |
[−98.7] |
[85.1] |
0.904 |
| (Pyramidalization) |
[−23.8] |
| (Low %VBur ammonium) |
[0.3] |
|
Eqn (4)
|
1 |
ΔG |
— |
36 |
(Pyramidalization Pd) |
[−27.3] |
[33.0] |
0.603 |
| Pyr > 0.450 |
25 |
(MBO(Pd–N)) |
[−31.0] |
[24.2] |
0.580 |
| 2 |
— |
36 |
(MBO(Pd–N)) |
[−25.9] |
[30.8] |
0.766 |
| (Pyramidalization) |
[−13.2] |
| Pyr > 0.450 |
25 |
(MBO(Pd–N)) |
[−29.1] |
[20.4] |
0.767 |
| (Low %VBur ammonium) |
[0.1] |
| 3 |
— |
36 |
(MBO(Pd–N)) |
[−29.5] |
[35.9] |
0.803 |
| (Molecular area) |
[−0.1] |
| (Pyramidalization) |
[−15.2] |
| Pyr > 0.450 |
25 |
(MBO(Pd–N)) |
[18.4] |
[406.8] |
0.791 |
| (pKa amine) |
[−0.4] |
| (d(N–H+) ammonium) |
[−431.0] |
|
Eqn (5)
|
1 |
ΔG |
— |
17 |
(Pyramidalization) |
[−55.1] |
[32.7] |
0.808 |
| Pyr > 0.450 |
14 |
(%VBur amine) |
[0.5] |
[−23.0] |
0.767 |
| 2 |
— |
17 |
(MBO(Pd–N)) |
[−91.5] |
[11.0] |
0.875 |
| (Low %VBur amine) |
[0.5] |
| Pyr > 0.450 |
14 |
(MBO(Pd–N)) |
[−92.4] |
[13.3] |
0.883 |
| (Low %VBur amine) |
[0.5] |
| 3 |
— |
17 |
(MBO(Pd–N)) |
[−140.5] |
[−480.7] |
0.922 |
| (Low %VBur ammonium) |
[0.4] |
| (MBO(N–H+) ammonium) |
[582.9] |
| Pyr > 0.450 |
14 |
(Low %VBur amine) |
[0.4] |
[44.6] |
0.936 |
| (η amine) |
[−7.4] |
| (Longest C–N bond) |
[−9.9] |
In fact, for the kinetics, it is necessary to emphasize again the correlations, including the 39 amines. A great deal is achieved with two or three variables, with coefficients of 0.869 and 0.884, respectively. Despite the complexity of plotting all the systems in Fig. 3, we can perceive a series of facts that are remarkable in our opinion. All the substituents on the nitrogen are essentially electron-donating, perhaps unlike 10, which has electron-withdrawing ester groups that, in principle, hinder proton acceptance because the nitrogen loses electron density to stabilize more positive charge on it. It should be noted, however, that all these groups are not directly attached to N in the case of ester groups.51 This discussion of electrons is vaguely clear with just one system. In the case of the structural contribution, the differentiation is completely clear; thus, if the nitrogen is not substituted, i.e., bearing hydrogen substituents, as in the case of amines 2, 3, 22, and 23, the protonation is in equilibrium. However, increasing the steric component of the nitrogen substituents has the opposite effect, but it is not really palpable if there are not at least two substituents that are highly sterically hindered, for example, with two adamantyl groups as in systems 38 and 39, and even more so if they are tert-butyls as in amines 33 and 36. Thus, for the latter systems, protonation holds significant value, kinetically speaking.
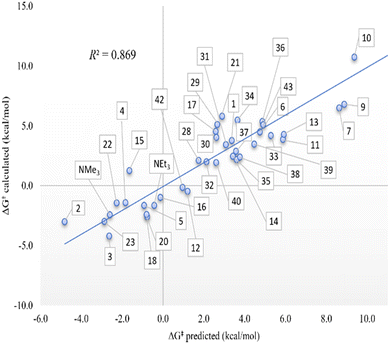 |
| | Fig. 3 Computed theoretical Gibbs energies (ΔG‡) in kcal mol−1 in front of the modeled ones according to the multilinear adjustment with two variables (pKa amine and pyramidalization) according to eqn (2). | |
For the robustness of the results, we proceeded to perform two measurements, i.e., convert the negative transition state energies to 0 and eliminate these systems. In the first case, the best correlations slightly worsened in terms of fit, with an R2 of 0.454, 0.821, and 0.842 for one, two, and three variables, respectively. On the other hand, the same variables involved in the best correlation reported in Table 2 still lead to the best correlation with two variables, whereas by using one variable, the amine LUMO energy is the descriptor that best fits the data rather than the amine Low %VBur descriptor in the previous case reported. The addition of NPA charge on nitrogen as the third variable only slightly improves the fitting up to 0.842. In the second case, the fit results better with the amine pKa (R2 = 0.559), but yet with two variables, the addition of amine %VBur leads to a worse correlation than the previous one (R2 = 0.750 vs. R2 = 0.869). Finally, since pyramidalization on nitrogen is linearly dependent on amines %VBur for a pyramidalization index greater than 0.450, we looked at what occurs when removing the amines with values below 0.450. For thermodynamics, the fit improves up to 0.715, 0.898, and 0.914, respectively, for one, two, and three variables, even though we had to say the number of systems involved decreases to 29. Similar to thermodynamics, there is an improvement in fit up to 0.651, 0.867, and 0.904, respectively, for one, two, and three variables in the case of kinetics.
Metal complexation
The exchange in eqn (3) of the phosphine by the amine in Au(I) complexes, using chlorine as an anionic ligand, led us to correlations that did not manage to exceed 0.679 with one variable with the Au–N distance. By increasing the number of variables, the correlations improve up to 0.838 with two variables and up to 0.867 with three variables, showing the influence of amine pKa and Low %VBur amine on phosphine substitution. Furthermore, since the Au–N bond distance seems to influence more the thermodynamics of the phosphine-amine substitution, we looked for which descriptor may affect that distance more. Scanning along the previously used descriptors, the pyramidalization on the nitrogen atom (see Fig. 4) turns out to be the descriptor with the most impact in determining the Au–N distance, and as we show previously, it is indirectly linked in some way with amine %VBur. Thus, to improve the thermodynamics of eqn (3), we can promote the shortening of the Au–N bond distance by acting on the pyramidalization and on %VBur.
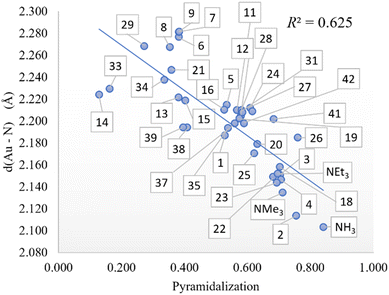 |
| | Fig. 4 Pyramidalization vs. Au–N distance (in Å) correlation. | |
Removing the amines with a pyramidalization index below 0.450, the fit improves up to 0.688, 0.865, and 0.904, respectively, for one, two, and three variables, with the amine LUMO energy as the best descriptor with one variable, while MBO of the Au–N bond becomes more important with two and three variables, and pKa is no longer involved in the correlation. The exchange of trimethylphosphine for the amines in eqn (4) represented our next approach. With one variable, the thermodynamics of eqn (4) led to the best correlation when nitrogen pyramidalization is involved when bonded to Pd (Pd pyramidalization in Table 2), reaching an R2 of 0.603. Using two and three variables, the pyramidalization still contributes, but the MBO of the Pd–N bond and the amine molecular area increase the fit up to 0.766 (with two variables) and up to 0.803 (with three variables). In contrast, applying the same filter as previously, pyramidalization loses importance and does not appear among the variables involved in the correlation, while MBO of the Pd–N bond still does. Unexpectedly, the best correlations have a worse overall fit. On the other hand, if we take into account the coordination of the amine not on Pd(0) as it was in eqn (4), but in Pd(II) with two chlorides as in eqn (5), the pyramidalization on nitrogen still leads to the best correlation with one variable (R2 = 0.808). It is worth mentioning that most amines with a pyramidalization index less than 0.450 do not bond to Pd(II). By completely excluding those amines from the correlations, the thermodynamics of eqn (5) correlate better with amine %VBur as it would be possible to imagine since %VBur and pyramidalization index are somehow dependent. However, in the case of two variables, the best correlation (R2 = 0.875) no longer involves the pyramidalization index, but the process seems to depend on steric (Low %VBur amine) and structural (MBO(Pd–N)) factors, while with three variables, the fit improves only slightly (R2 = 0.922). Last, applying the filter pyramidalization >0.450 with three variables, the fitting improves up to 0.936, but it is worth mentioning that MBO of Pd–N is not one of the variables, while the process depends on a steric factor (Low %VBur amine), an electronic factor (η amine), and the longest C–N bonds of amine.
4. Conclusions
In conclusion, steric effects surrounding the nitrogen atom of amines are of fundamental importance to the geometry and reactivity of this prominent functional group. We reported a blueprint for correlating the geometry of extremely hindered alkylamines with their key thermodynamic and kinetic properties. This is modest compared to machine learning,52 but also a real example of predictive catalysis.38,53 In particular, the optimization and evaluation of sterically hindered amines in coordination with gold and palladium highlights the focus of the study, based on the understanding of steric effects around the nitrogen atom and the ability of such amines to trap protons. The %VBur steric parameter of Cavallo and coworkers, directly or indirectly through the pyramidalization index, emerged as the key factor permitting the assessment of the geometrical effects of sterically hindered amines. The role of steric hindrance, represented by %VBur values, is highlighted, as is the importance of selective and robust parameters in understanding the reactivity of sterically hindered amines. The analysis involved various equations and statistical measures to establish correlations. For gold, the bond energies were influenced mainly by the Au–N distance, which correlates with the pyramidalization and indirectly with the %VBur. For Pd(0), pyramidalization contributes more in combination with MBO of the Pd–N bond, while for Pd(II), the relationship is similar to %VBur rather than pyramidalization. For proton thermodynamics, the LUMO is the most important parameter in combination with molecular area and N–H MBO, whereas kinetics is mainly described by %VBur parameters and amine pKa. Therefore, the steric contribution of sterically hindered amines can be summarized by the %VBur or the pyramidalization, while in some cases, additional weight should be given to the electronic part through the energy of the frontier orbitals or the structural part like distances or MBOs.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†
Author contributions
M. T.: data curation, formal analysis, investigation, methodology, visualization, writing – original draft. M. V.: data curation, formal analysis, investigation, methodology, visualization, writing – original draft. L. C: data curation, formal analysis, investigation, validation, supervision, funding acquisition, review & editing. M. S.: conceptualization, data curation, formal analysis, investigation, funding acquisition, methodology, project administration, supervision, validation, visualization, review & editing. A. P.: conceptualization, data curation, funding acquisition, methodology, project administration, supervision, visualization, writing – original draft, review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. S. thanks the NIH (1R35GM133326), the NSF (CAREER CHE-1650766), and Rutgers University for generous financial support. Supplement funding for this project was provided by the Rutgers University – Newark Chancellor's Research Office. A. P. is a Serra Húnter Fellow. A. P. received ICREA Academia Prize 2019. We thank the Spanish Ministerio de Ciencia e Innovación for project PID2021-127423NB-I00 and the Generalitat de Catalunya for project 2021SGR623. We gratefully acknowledge financial support from CIRCC, Interuniversity Consortium Chemical Reactivity and Catalysis. We thank the support from BSC supercomputing facilities.
Notes and references
-
Science of Synthesis, ed. D. Enders, Thieme, Stuttgart, 2008, vol. 40a Search PubMed.
-
(a)
S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 1st edn, 2004 Search PubMed;
(b)
A. Ricci, Amino Group Chemistry. From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 1st edn, 2008 Search PubMed.
-
(a)
T. C. Nugent, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, 1st edn, 2010 CrossRef;
(b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed;
(c) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Privileged Scaffolds for Library Design and Drug Discovery, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS;
(d) L. E. Vine and J. M. Schomaker, Nat. Chem., 2022, 14, 1093–1094 CrossRef CAS PubMed.
-
(a) R. P. Rucker, A. M. Whittaker, H. Dang and G. Lalic, Angew. Chem., Int. Ed., 2012, 51, 3953–3956 CrossRef CAS PubMed;
(b) M. Mailig, R. P. Rucker and G. Lalic, Chem. Commun., 2015, 51, 11048–11051 RSC;
(c) K. Sakai, K.-i. Yamada, T. Yamasaki, Y. Kinoshita, F. Mito and H. Utsumi, Tetrahedron, 2010, 66, 2311–2315 CrossRef CAS.
- N. Mandal, A. K. Pal, P. Gain, A. Zohaib and A. Datta, J. Am. Chem. Soc., 2002, 142, 5331–5337 CrossRef PubMed.
-
(a) A. Greenberg and C. A. Venanzi, J. Am. Chem. Soc., 1993, 115, 6951–6957 CrossRef CAS;
(b) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS;
(c) J. Aubé, A New Twist on Amide Solvolysis, Angew. Chem., Int. Ed., 2012, 51, 3063–3065 CrossRef PubMed;
(d) S. Adachi, N. Kumagai and M. Shibasaki, Chem. Sci., 2017, 8, 85–90 RSC.
-
(a)
The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, ed. A. Greenberg, C. M. Breneman and J. F. Liebman, Wiley, New York, 2000 Search PubMed;
(b) G. Li, S. Ma and M. Szostak, Trends Chem., 2020, 2, 914–928 CrossRef CAS;
(c) G. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS PubMed;
(d)
Amide Bond Activation: Concepts and Reactions, ed. M. Szostak, Wiley-VCH, Weinheim, 2022 Search PubMed.
- S. Hünig and M. Kiessel, Chem. Ber., 1958, 91, 380–392 CrossRef.
-
(a) D. Kampmann, G. Stuhlmüller, R. Simon, F. Cottet, F. Leroux and M. Schlosser, Synthesis, 2005, 2005, 1028–1029 CrossRef;
(b) M. Dagonneau, E. S. Kagan, V. I. Mikhailov, E. G. Rozantsev and V. D. Sholle, Synthesis, 1984, 1984, 895–916 CrossRef.
- S. Barriga, Synlett, 2001, 2001, 0563 CrossRef.
-
(a) T. J. Stone, T. Buckman, P. L. Nordio and H. M. McConnell, Proc. Natl. Acad. Sci. U. S. A., 1965, 54, 1010–1017 CrossRef CAS PubMed;
(b) J. F. W. Keana, Chem. Rev., 1978, 78, 37–64 CrossRef CAS.
-
(a) E. N. Step, N. J. Turro, M. E. Gande and P. P. Klemchuk, Macromolecules, 1994, 27, 2529–2539 CrossRef CAS;
(b) M. Dagonneau, V. B. Ivanov, E. G. Rozantsev, V. D. Sholle and E. S. Kagan, J. Macromol. Sci., Polym. Rev., 1982, 22, 169–202 CrossRef;
(c) T. Kurumada, H. Ohsawa, T. Fujita, T. Toda, T. Yoshioka and O. Oda, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 1477–1491 CrossRef CAS.
-
(a) G. Sartori and D. W. Savage, Ind. Eng. Chem. Fundam., 1983, 22, 239–249 CrossRef CAS;
(b) R. J. Hook, Ind. Eng. Chem. Res., 1997, 36, 1779–1790 CrossRef CAS.
-
(a) R. Hiltmann, H. Wollweber, W. Wirth and R. Gösswald, Angew. Chem., 1960, 72, 1001 CrossRef CAS;
(b) L. Kürti, Science, 2015, 348, 863–864 CrossRef.
- S. I. Druzhinin, S. R. Dubbaka, P. Knochel, S. A. Kovalenko, P. Mayer, T. Senyushkina and K. A. Zachariasse, J. Phys. Chem. A, 2008, 112, 2749–2761 CrossRef CAS PubMed.
- M. Yang, T. Albrecht-Schmitt, V. Cammarata, P. Livant, D. S. Makhanu, R. Sykora and W. Zhu, J. Org. Chem., 2009, 74, 2671–2678 CrossRef CAS PubMed.
- N. Z. Yagafarov, P. N. Kolesnikov, D. L. Usanov, V. V. Novikov, Y. V. Nelyubina and D. Chusov, Chem. Commun., 2016, 52, 1397–1400 RSC.
- V. Chaplinski and A. de Meijere, Angew. Chem., Int. Ed. Engl., 1996, 35, 413–414 CrossRef CAS.
- H. Bock, I. Göbel, Z. Havlas, S. Liedle and H. Oberhammer, Angew. Chem., Int. Ed. Engl., 1991, 30, 187–190 CrossRef.
-
(a) T. C. Wong, L. R. Collazo and F. S. Guziec Jr, Tetrahedron, 1995, 51, 649–656 CrossRef CAS;
(b) J. E. Anderson, D. Casarini and L. Lunazzi, J. Org. Chem., 1996, 61, 1290–1296 CrossRef CAS.
- R. Boese, D. Bläser, M. Y. Antipin, V. Chaplinski, A. de Meijere, R. Boese and M. Y. Antipin, Chem. Commun., 1998, 781–782 RSC.
-
(a)
J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 1968, p. 123 Search PubMed;
(b)
M. B. Smith, March's Advanced Organic Chemistry, Wiley, Hoboken, NJ, 7th edn, 2013, p. 206 Search PubMed.
-
(a) P. D. Bartlett and A. Schneider, J. Am. Chem. Soc., 1945, 67, 141–144 CrossRef CAS;
(b) P. D. Bartlett and E. B. Lefferts, J. Am. Chem. Soc., 1955, 77, 2804–2805 CrossRef CAS;
(c) E. Hemmer, C. Cavelius, V. Huch and S. Mathur, Inorg. Chem., 2015, 54, 6267–6280 CrossRef CAS PubMed;
(d) E. Hemmer, V. Huch, M. Adlung, C. Wickleder and S. Mathur, Eur. J. Inorg. Chem., 2011, 2011, 2148–2157 CrossRef;
(e) I. Majerz and I. Natkaniec, J. Mol. Struct., 2006, 788, 93–101 CrossRef CAS.
-
A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, Germany, 102nd edn, 2007, p. 2006 Search PubMed.
- S. Hellmann, H.-D. Beckhaus and C. Rüchardt, Chem. Ber., 1983, 116, 2219–2237 CrossRef CAS.
- K. Banert, M. Heck, A. Ihle, J. Kronawitt, T. Pester and T. Shoker, J. Org. Chem., 2018, 83, 5138–5148 CrossRef CAS.
-
(a) A. K. Hoffmann, A. M. Feldman, E. Gelblum and A. Henderson, Org. Synth., 1973, 5, 355–358 Search PubMed;
(b) K. S. Chan, X. Z. Li and S. Y. Lee, Organometallics, 2010, 29, 2850–2856 CrossRef CAS;
(c) H. Sarker, M. L. Greer and S. C. Blackstock, J. Org. Chem., 1996, 61, 3177–3182 CrossRef CAS PubMed;
(d) V. Dhayalan and P. Knochel, Synthesis, 2015, 47, 3246–3256 CrossRef CAS.
- J. S. Amato, J. Y. L. Chung, R. J. Cvetovich, X. Gong, M. McLaughlin and R. A. Reamer, J. Org. Chem., 2005, 70, 1930–1933 CrossRef CAS PubMed.
-
B. U. Schlottmann, PhD dissertation, Universität Marburg, Germany, 1972.
-
(a) G. Wieland and G. Simchen, Liebigs Ann. Chem., 1985, 1985, 2178–2193 CrossRef;
(b) T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968–5969 CrossRef CAS PubMed;
(c) G. H. Coleman, J. Am. Chem. Soc., 1933, 55, 3001–3005 CrossRef CAS.
- T. Hatakeyama, Y. Yoshimoto, S. K. Ghorai and M. Nakamura, Org. Lett., 2010, 12, 1516–1519 CrossRef CAS PubMed.
-
(a) B. D. Sherry and A. Fürstner, Chem. Commun., 2009, 46, 7116–7118 RSC;
(b) V. Dimitrov, Bulg. Chem. Commun., 2014, 46, 16–20 Search PubMed.
- F. Kuffner and W. Koechlin, Monatsh. Chem., 1962, 93, 476–482 CrossRef CAS.
-
(a) L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed;
(b) K. Takeuchi, T. Moriyama, T. Kinoshita, H. Tachino and K. Okamoto, Chem. Lett., 1980, 9, 1395–1398 CrossRef.
-
(a) C. H. Bushweller, W. G. Anderson, P. E. Stevenson, D. L. Burkey and J. W. O'Neil, Tetrahedron, 1983, 39, 305–308 CrossRef;
(b) J. Reny, C. Y. Wang, C. H. Bushweller and W. G. Anderson, Tetrahedron Lett., 1975, 16, 503–506 CrossRef.
- T. G. Back and D. H. R. Barton, J. Chem. Soc., Perkin Trans. 1, 1977, 924–927 RSC.
- R. Monreal-Corona, À. Díaz-Jiménez, A. Roglans, A. Poater and A. Pla-Quintana, Adv. Synth. Catal., 2023, 365, 760–766 CrossRef CAS.
-
(a) S. Escayola, N. Bahri-Laleh and A. Poater, Chem. Soc. Rev., 2024, 53, 853–882 RSC;
(b) R. Monreal-Corona, A. Pla-Quintana and A. Poater, Trends Chem., 2023, 5, 935–946 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed;
(c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
-
(a) W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef;
(b) T. Leininger, A. Nicklass, H. Stoll, M. Dolg and P. Schwerdtfeger, J. Chem. Phys., 1996, 105, 1052–1059 CrossRef CAS.
-
(a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS;
(b) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
-
(a) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS;
(b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- The pyramidalization was calculated using the Python package Morfeus (https://github.com/digital-chemistry-laboratory/morfeus/tree/main/morfeus); T. P. Radhakrishnan and I. Agranat, Struct. Chem., 1991, 2, 107–115 CrossRef CAS.
-
(a) G. J. Tawa, I. A. Topol, S. K. Burt, R. A. Caldwell and A. A. Rashin, J. Chem. Phys., 1998, 109, 4852–4963 CrossRef CAS;
(b) J. Wappel, R. C. Fischer, L. Cavallo, C. Slugovc and A. Poater, Beilstein J. Org. Chem., 2016, 12, 154–165 CrossRef CAS PubMed;
(c) F. Acuña-Parés, Z. Codolà, M. Costas, J. M. Luis and J. Lloret-Fillol, Chem.–Eur. J., 2014, 20, 5696–5707 CrossRef PubMed.
-
(a) J. Sala, L. Capdevila, C. Berga, A. de Aquino, L. Rodríguez, S. Simon and X. Ribas, Chem.–Eur. J., 2024, 30, e202303200 CrossRef CAS PubMed;
(b) J. Wang, C. Shen, G. Zhang, F. Gan, Y. Ding and H. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202115979 CrossRef CAS PubMed.
- Examples of metal(0) species:
(a) E. S. Isbrandt, A. Nasim, K. Zhao and S. G. Newman, J. Am. Chem. Soc., 2021, 143, 14646–14656 CrossRef CAS PubMed;
(b) J. Heitkämper, S. Posada-Pérez, S. Escayola, M. Solà, J. Kästner and A. Poater, Chem.–Eur. J., 2023, 29, e202300193 CrossRef PubMed;
(c) G. M. Meconi, S. V. C. Vummaleti, J. A. Luque-Urrutia, P. Belanzoni, S. P. Nolan, H. Jacobsen, L. Cavallo, M. Solà and A. Poater, Organometallics, 2017, 36, 2088–2095 CrossRef CAS;
(d) G. Li, P. Lei, M. Szostak, E. Casals-Cruañas, A. Poater, L. Cavallo and S. P. Nolan, ChemCatChem, 2018, 10, 3096–3106 CrossRef CAS;
(e) G. Li, T. Zhou, A. Poater, L. Cavallo, S. P. Nolan and M. Szostak, Catal. Sci. Technol., 2020, 10, 710–716 RSC;
(f) T. Zhou, S. Ma, F. Nahra, A. M. C. Obled, A. Poater, L. Cavallo, C. S. J. Cazin, S. P. Nolan and M. Szostak, iScience, 2020, 23, 101377 CrossRef CAS PubMed;
(g) S. Yang, X. Yu, A. Poater, L. Cavallo, S. C. J. Cazin, S. P. Nolan and M. Szostak, Org. Lett., 2022, 24, 9210–9215 CrossRef CAS PubMed.
-
(a) G. Ciancaleoni, N. Scafuri, G. A. Bistoni, A. Macchioni, F. Tarantelli, D. Zuccaccia and L. Belpassi, Inorg. Chem., 2004, 53, 9907–9916 CrossRef PubMed;
(b) D. Setiawan, R. Kalescky, E. Kraka and D. Cremer, Inorg. Chem., 2016, 55, 2332–2344 CrossRef CAS PubMed;
(c) R. Dorta, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
-
(a) L. Falivene and L. Cavallo, Coord. Chem. Rev., 2017, 344, 101–114 CrossRef CAS;
(b) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS;
(c) S. Diez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef CAS PubMed;
(d) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- Examples of electron-withdrawing substituents on the catalysis:
(a) N. Joly, M. Gimferrer, S. Escayola, M. Cendra, S. Coufourier, J.-F. Lohier, Q. Gaignard Gaillard, S. Gaillard, M. Solà, J.-L. Renaud and A. Poater, Organometallics, 2023, 42, 1784–1792 CrossRef CAS;
(b) M. Gimferrer, N. Joly, S. Escayola, E. Viñas, S. Gaillard, M. Solà, J.-L. Renaud, P. Salvador and A. Poater, Organometallics, 2022, 41, 1204–1215 CrossRef CAS.
-
(a) R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed;
(b) P. Friederich, G. Dos Passos Gomes, R. De Bin, A. Aspuru-Guzik and D. Balcells, Chem. Sci., 2020, 11, 4584–4601 RSC;
(c) B. Sánchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef PubMed;
(d) M. Foscato, G. Occhipinti, S. H. Hopen Eliasson and V. R. Jensen, J. Chem. Inf. Model., 2024, 64, 412–424 CrossRef CAS PubMed;
(e) J. V. Alegre-Requena, S. Sowndarya S. V., R. Pérez-Soto, T. M. Alturaifi and R. S. Paton, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13, e1663 CAS;
(f) R. C. Cammarota, W. Liu, J. Bacsa, H. M. L. Davies and M. S. Sigman, J. Am. Chem. Soc., 2022, 144, 1881–1898 CrossRef CAS PubMed.
- M. P. Maloney, B. A. Stenfors, P. Helquist, P.-O. Norrby and O. Wiest, ACS Catal., 2023, 13, 14285–14299 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Maria
Voccia‡
ab,
Lucia
Caporaso
b,
Michal
Szostak
*c and
Albert
Poater
ab,
Maria
Voccia‡
ab,
Lucia
Caporaso
b,
Michal
Szostak
*c and
Albert
Poater