
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular synthesis of α-branched secondary alkylamines via visible-light-mediated carbonyl alkylative amination†
Milo A.
Smith
 ,
Ryan J. D.
Kang
,
Roopender
Kumar
,
Biswarup
Roy
and
Matthew J.
Gaunt
*
,
Ryan J. D.
Kang
,
Roopender
Kumar
,
Biswarup
Roy
and
Matthew J.
Gaunt
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: mjg32@cam.ac.uk
First published on 23rd August 2024
Abstract
The development of methods for the assembly of secondary α-alkyl amines remains a central challenge to chemical synthesis because of their critical importance in modulating the physical properties of biologically active molecules. Despite decades of intensive research, chemists still rely on selective N-alkylation and carbonyl reductive amination to make most amine products. Here we report the further evolution of a carbonyl alkylative amination process that, for the first time, brings together primary amines, aldehydes and alkyl iodides in a visible-light-mediated multicomponent coupling reaction for the synthesis of a wide range of α-branched secondary alkylamines. In addition to exploring the tolerance and limitations in each reaction component, we also report preliminary applications to the telescoped synthesis of α-branched N-heterocycles and an N-alkylation protocol that is selective for primary over cyclic secondary amines. Our data support a mechanism involving addition of an alkyl radical to an uncharged alkyl imine which, to the best of our knowledge, has not previously been described. We believe that this method will enable practitioners of synthetic chemistry in academic and industrial settings to approach the synthesis of these important molecules in a manner that is streamlined compared to established approaches.
Introduction
Secondary alkylamines are one of the most commonly occurring functional motifs in small molecules that perturb biological processes (Fig. 1A).1 Their capacity to function as hydrogen bond donors or acceptors with proteins, as modulators of solubility and bioavailability on protonation, and as scaffolds to support small molecule topology makes them privileged features in pharmaceuticals and agrochemicals. Accordingly, the development of new methods that lead to robust and modular preparations of secondary alkylamines remains a constant challenge to the synthetic organic chemistry community.2,3 While catalytic methods are emerging for their synthesis, most notably, alkene hydroamination,4 photoredox-mediated reactions,5 hydrogenation,6 biocatalytic transformations,7 C–H activation,8 and others,9 the majority of secondary alkylamines are still prepared by carbonyl reductive amination10 or N-alkylation.11,12 A possible reason for this preference is that methods such as reductive amination offer an operationally straightforward, robust and widely explored means by which to prepare secondary alkylamines via the union of two classes of extensive and diverse building blocks, namely primary amines and aldehydes or ketones. In reality, however, many of the established amine synthesis technologies often come up short in increasingly complex scenarios and so new robust synthetic methods for alkylamine synthesis are still needed.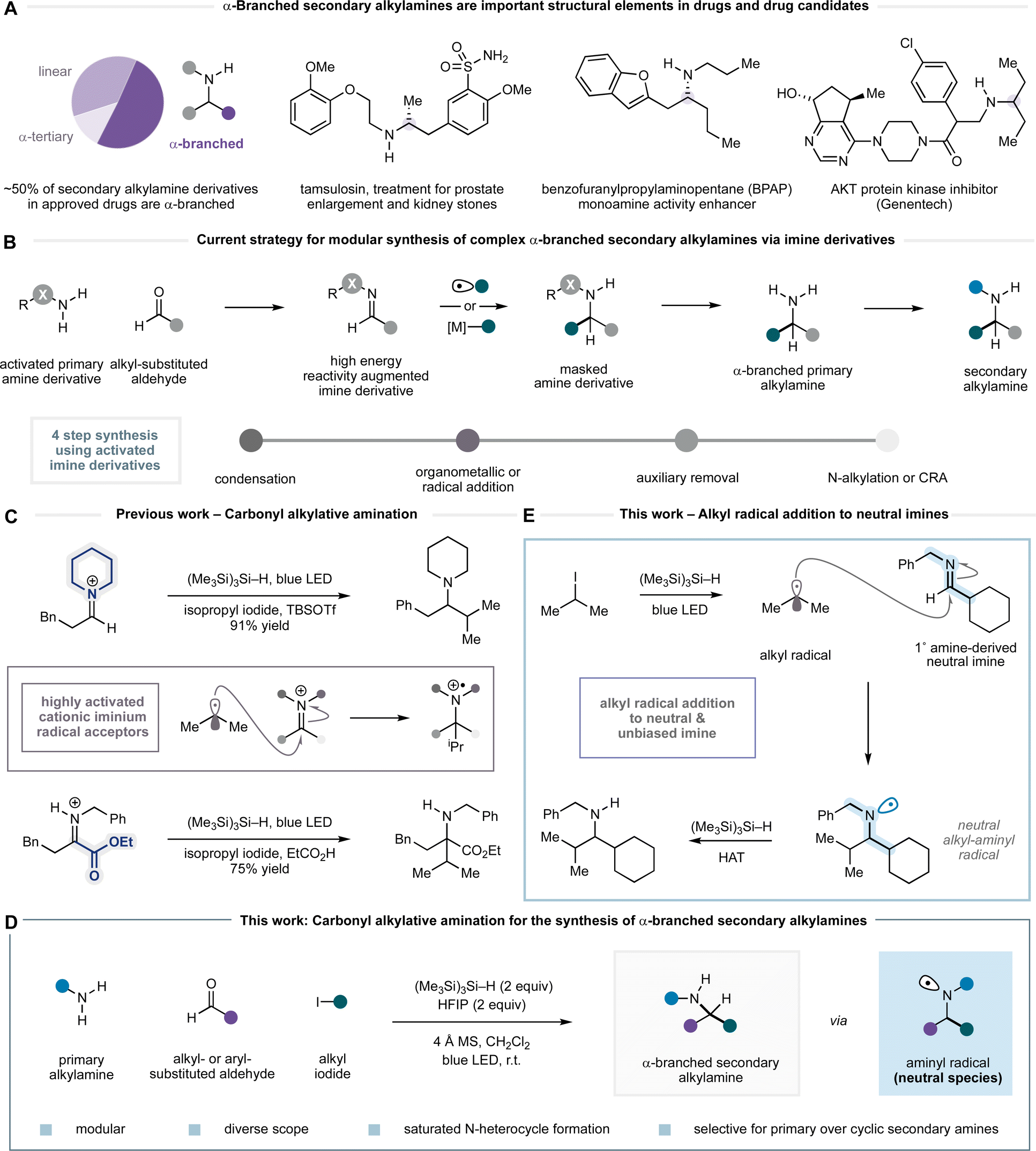 | ||
| Fig. 1 (A) The importance of secondary alkylamines in drug discovery and development. (B) Current strategies for the modular synthesis of α-branched alkylamines (auxiliary-based methods which require protection and deprotection). (C) Carbonyl alkylative amination (CAA) for the synthesis of α-branched tertiary alkylamines. (D) This work: CAA as a general method for the synthesis of complex α-branched secondary alkylamines. (E) This work: alkyl radical addition to neutral imines. | ||
The condensation of a primary alkylamine and a ketone adorned with alkyl substituents to form an alkyl imine is usually reliable, from which there are a plethora of reduction protocols to form the corresponding α-branched secondary alkylamine.10 In some cases, however, steric hindrance or adverse electronic effects on either the amine or ketone component can preclude alkyl imine formation, leading to failure of the reductive amination process. An alternative disconnection involves addition of a carbon nucleophile to an alkyl aldimine, thus partitioning the steric features of the product alkylamine between the two reacting components. Despite almost 100 years of effort towards a solution to this simple idea, a general method for the addition of organometallic nucleophiles to alkyl imines or iminium ions derived from primary alkylamines and alkyl-substituted aldehydes has largely eluded synthetic chemists.13 Only when reactivity-augmenting auxiliary groups are deployed on either the amine or carbonyl component does an effective and general transformation result (Fig. 1B). Organometallic additions to Ellman's tert-butanesulfinamide derivatives have become the benchmark for this type of reaction.14 While a distinct advantage of this auxiliary is the capacity to exert stereocontrol at the newly formed α-amino carbon centre, they nonetheless suffer from the well-established drawbacks of auxiliary use, namely the requirement for installation and removal steps, and poor overall atom economy. Several additional steps are also required to form secondary or tertiary amine products, with cyclic tertiary amine motifs presenting a notable challenge. The main reason that organometallic additions to non-activated alkyl imines fail lies with the low pKa of the C–H bonds adjacent to the carbon–nitrogen double bond, and the low electrophilicity of the carbon–nitrogen double bond. As a result, the treatment of alkyl imine/iminium species with organometallic reagents typically results in deprotonation of the acidic α-C–H as the major process, leading to formation of the corresponding enamine instead of the desired 1,2-addition product.14b
As an alternative, carbon-centred radicals have been shown to effectively add to a range of activated surrogates, such as hydrazones, oximes, carbamoyl and sulfonyl imines.15 However, although the charge-neutral and non-basic nature of alkyl radicals addresses the deprotonation problems associated with organometallic additions, tailored activating substituents on nitrogen are still required, in this case to promote radical 1,2-addition by stabilising the resulting nitrogen-centred radical.
Our laboratory has a longstanding interest in the development of methods for the direct synthesis of alkylamines that avoid the use of reactivity-enhancing auxiliaries. Recently, we introduced a process called carbonyl alkylative amination (CAA) for the synthesis of complex alkylamines from feedstock alkylamines, carbonyls and alkyl iodides (Fig. 1C).16 In this reaction, visible-light and a silane mediate the addition of an alkyl radical—generated from a non-activated alkyl iodide—to an in situ generated all-alkyl iminium ion to form a wide range of complex alkylamines. Central to the success of this process was the multifaceted role of the silane, which not only facilitated a unique alkyl-radical initiation step but also enabled a rapid hydrogen atom transfer (HAT) to an aminium radical cation intermediate to form the product as its ammonium salt. The addition of an alkyl radical to a highly reactive alkyl-substituted iminium ion represents a higher order variant of reductive amination and provides a particularly useful strategic bond forming reaction for the synthesis of alkylamines that would not be possible using the corresponding organometallic addition. The Qin laboratory recently reported a mechanistically distinct multicomponent platform for alkylamine synthesis using alkyl boronates as the alkyl radical source.17 While our CAA platform displayed excellent scope in the secondary amine, aldehyde and alkyl halide components, significantly lower yields of product were observed when using primary amines (unless the imine was derived from highly activated α-ketoesters) and precluded a general strategy for the synthesis of different types of alkylamine.16b
Herein, we report a reaction platform to accommodate the synthesis of α-branched secondary alkylamines through the modular coupling of primary amines with aldehydes and alkyl iodides (Fig. 1D).16d In contrast to our previous work on radical additions to iminium ions, and to the multitude of prior reports suggesting that reactivity-augmenting auxiliaries (or Lewis acid activation) are essential for effective intermolecular radical addition to imines,15 we present evidence that the reaction proceeds via visible-light and silane-mediated radical addition to an uncharged alkyl imine (Fig. 1E). In turn, this addition step forms a neutral aminyl radical, which undergoes HAT with the silane reagent. The reaction exhibits a good scope across the three components and offers a practical process for the synthesis of a wide range of α-branched secondary alkylamines. Furthermore, we demonstrate how the reaction can be adapted to generate saturated nitrogen heterocycles, to enable selective alkylation of primary over cyclic secondary amines, and to effect telescoped multicomponent processes to functionally and structurally diverse α-branched secondary alkylamine derivatives. This work substantially expands the scope of carbonyl alkylative amination towards its establishment as a general strategy for alkylamine synthesis.
Results and discussion
Beginning with the reaction conditions developed for CAA to form tertiary alkylamines,16a we investigated the synthesis of α-branched secondary alkylamines by irradiating a dichloromethane solution of benzylamine (1a, 1 equiv.), hydrocinnamaldehyde (2a, 1.2 equiv.) and 2-iodopropane 3a (3 equiv.) with a 40 W blue LED in the presence of (Me3Si)3Si–H (2 equiv.), TBSOTf (2 equiv.) and 4 Å MS. These conditions produced a complex mixture of degradation products, and none of the desired α-branched secondary amine 4a was detected by 1H NMR analysis of the crude reaction mixture (entry 1, Table 1). Surprisingly, when TBSOTf was omitted, 4a was formed in 11% assay yield (entry 2). Assuming formation of an iminium ion to be a requirement for effective product formation (see Fig. 1C) we subsequently conducted a screen of acid additives, the key results of which are presented in Table 1. The use of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) significantly improved the yield of 4a, as did the use of trimethylsilyl chloride (TMSCl) (entries 3 and 4).18,19 The combination of these two additives gave 4a with an assay yield of 52%, which proved to be optimal for this combination of amine and aldehyde.|
|
|||
|---|---|---|---|
| Entry | R | Additive | Assay yielda (%) |
| a Assay yield determined by 1H NMR using 1,1,2,2-tetrachloroethane as internal standard. Yield of isolated product shown in parentheses. | |||
| 1 | (CH2)2Ph | TBSOTf (2.0 equiv.) | 0 |
| 2 | (CH2)2Ph | None | 11 |
| 3 | (CH2)2Ph | HFIP (2.5 equiv.) | 29 |
| 4 | (CH2)2Ph | TMSCl (2.0 equiv.) | 37 |
| 5 | (CH2)2Ph | HFIP + TMSCl | 52 (41) |
| 6 | c-C6H11 | None | 88 |
| 7 | c-C6H11 | TBSOTf (2.0 equiv.) | 74 |
| 8 | c-C6H11 | HFIP (2.5 equiv.) | 91 |
| 9 | c-C6H11 | TMSCl (2.0 equiv.) | 78 |
| 10 | c-C6H11 | HFIP + TMSCl | 97 (86) |
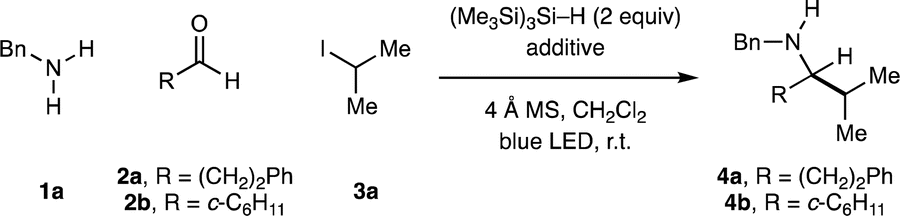
To our surprise, changing linear aldehyde 2a to the α-branched aldehyde cyclohexanecarboxaldehyde (2b) resulted in a greatly enhanced yield of the corresponding α-branched secondary amine 4b in the absence of any additive (entry 6), highlighting a significant difference in the reactivity of structurally distinct carbonyls. The use of TBSOTf, again, had a detrimental effect on the reaction and, in contrast to the case of 2a, so did the addition of TMSCl (entries 7 and 9). The addition of HFIP gave a small boost in the yield (entry 8). Interestingly, the combination of TMSCl and HFIP again proved optimal, providing 4b in 97% assay yield which could be isolated in 86% yield after reverse phase chromatography on C18. Given the efficacy of both HFIP alone and HFIP + TMSCl as additives in these studies, we evaluated the use of both for the majority of substrate combinations, and the most effective conditions are reported. Although we were unable to identify precise mechanistic rationale for the efficacy of these additives, 1H NMR analysis supports the formation of a hydrogen-bonded adduct with the intermediate imine when using HFIP alone, and the formation of a discrete N–H iminium ion when using HFIP + TMSCl. A detailed mechanistic discussion based on the data we have collected is presented in the ESI (Section 10, p. S80†).
With optimal reaction conditions in hand, the scope of the transformation with respect to the primary amine component was explored in combination with representative linear and α-branched aldehydes (2a–b) and isopropyl iodide (3a) (Fig. 2). Assay yields are reported alongside yields of isolated product for all amines to accurately represent the reaction efficiency. A range of functionalized primary alkylamines were found to be compatible with the CAA process and produced good yields of α-branched secondary alkylamines (4) after isolation. A series of linear primary alkylamines (4c–e), amines containing electron-deficient (4g–h), and electron-rich heteroaryl (4f, 4i) groups all worked well in the CAA reaction. Throughout the course of our studies, we found that substrates featuring basic heterocycles often benefitted from employing HFIP in co-solvent quantities (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2/HFIP): imidazole-containing product 4h was formed in quantitative yield under these conditions. The use of TMSCl was also of particular importance in this case. Omission of TMSCl, or use of the standard 2.5 equivalents of HFIP, resulted in low yields. Further observations included the accommodation of unprotected, potentially competitive two-electron nucleophiles (4f – furan; 4i – indole) in the primary alkylamine component. α-Branched primary amines were also good substrates and delivered the corresponding α,α′-branched products (4j–l). An aniline-derived amine was also a competent substrate (4m).
:1 CH2Cl2/HFIP): imidazole-containing product 4h was formed in quantitative yield under these conditions. The use of TMSCl was also of particular importance in this case. Omission of TMSCl, or use of the standard 2.5 equivalents of HFIP, resulted in low yields. Further observations included the accommodation of unprotected, potentially competitive two-electron nucleophiles (4f – furan; 4i – indole) in the primary alkylamine component. α-Branched primary amines were also good substrates and delivered the corresponding α,α′-branched products (4j–l). An aniline-derived amine was also a competent substrate (4m).
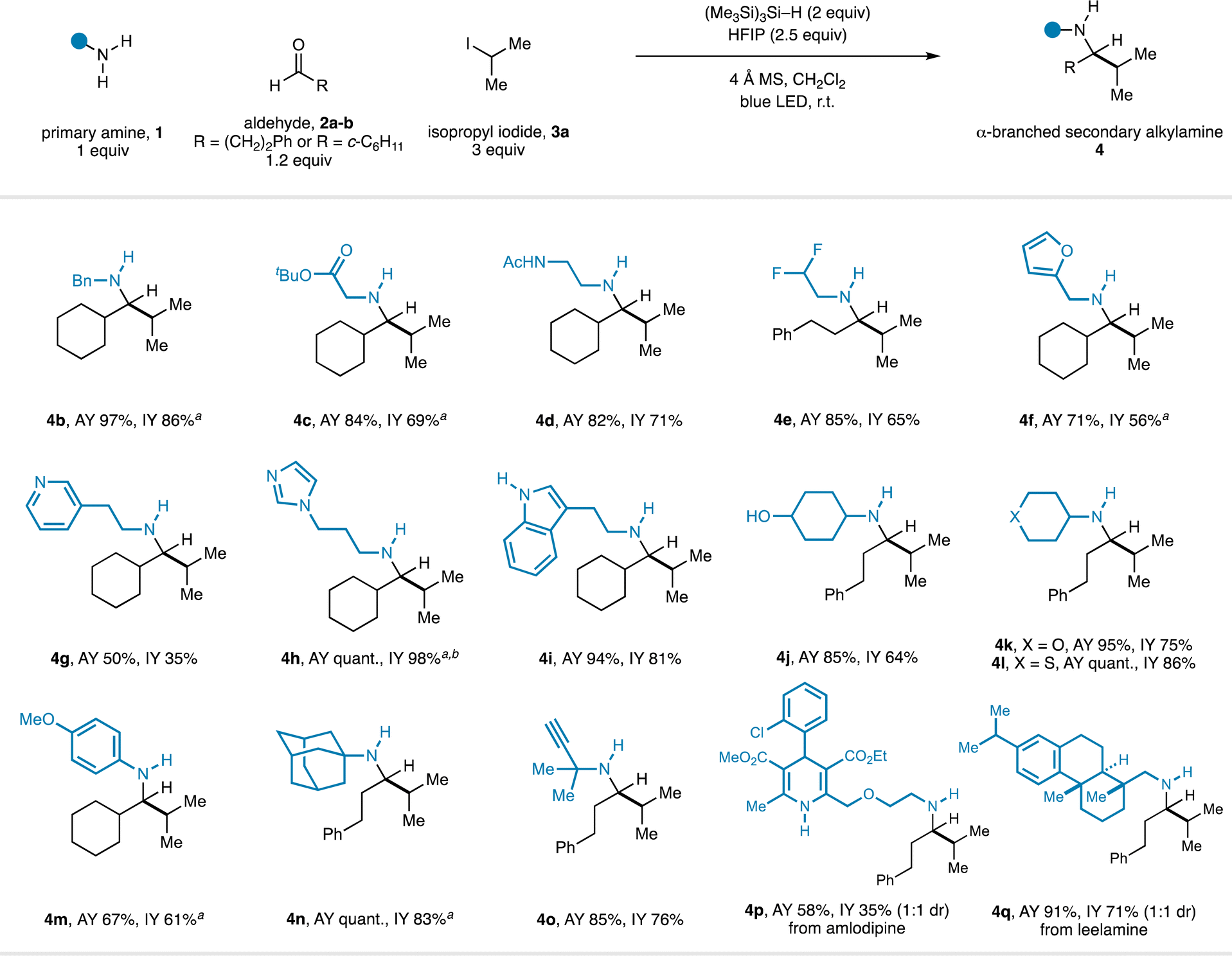 | ||
| Fig. 2 Scope of the primary amine component in CAA to α-branched secondary alkylamines. a TMSCl (2 equiv.) used as an additive. b 4:1 CH2Cl2/HFIP used as solvent. | ||
α-Tertiary amines are productive substrates in this CAA reaction, the use of which forges extremely hindered secondary alkylamines in good yields (4n–o). The synthesis of this type of product via classical carbonyl reductive amination would require forcing conditions for the imine formation step because of the steric hindrance in both α-tertiary primary amine and α-branched dialkyl ketone components, which may preclude the incorporation of more delicate functionality. Finally, we found that Hantzsch dihydropyridine and steroid-derived alkylamine-containing pharmaceuticals were effective substrates for the CAA reaction, delivering the corresponding products in good yield (4p–q). It is particularly notable that the CAA reaction tolerates the presence of unprotected protic functionality (4d, 4i, 4j, 4p) which would be incompatible with organometallic approaches to these structures.
Next, the scope of the reaction in the aldehyde component was examined using benzylamine (1a) in combination with isopropyl iodide (3a, Fig. 3). The CAA reaction accommodated the use of alkyl-substituted aldehydes featuring α-branching (4r–v) and could incorporate useful functionalities. The successful transformation of an aldehyde containing a cyclopropyl motif to the expected amine product (4v) proceeded without any products arising from β-scission of the strained ring, suggesting that the reaction is unlikely to proceed via an α-amino radical intermediate.20 A series of benzaldehydes also performed well in the CAA reaction (to 4w–y). Although aryl iodides can undergo halogen-atom transfer with the silyl radicals generated under our reaction conditions, reaction with alkyl iodides is generally faster;21 α-branched secondary amine product 4y derived from 3-iodobenzeldehyde could be obtained in good yield with the aryl iodide moiety intact. Products containing Lewis-basic heterocycles could be readily accessed in good yields through the use of heteroarene-derived aldehydes (4z–aa) with HFIP as co-solvent. Unbranched aldehydes were generally poor substrates when using benzylamine as the amine (entries 1–5, Table 1) and we currently do not have an explanation for this phenomenon. However, while evaluating the scope in the amine component, the performance of unbranched aldehydes was markedly improved when α-branching was present in the amine (e.g.4j–l, 4n–o), suggesting a subtle steric effect is at play. Accordingly, with 4-aminotetrahydropyran (1b) as the amine, product 4ab containing a remote alkene was formed from (±)-citronellal in excellent yield. The use of 4-pentenal as the aldehyde provided α-branched secondary amine 4ac, and no products derived from radical 5-exo-trig cyclisation were detected. This result contrasts to our previous study16b and suggests that, under the present conditions, the isopropyl radical adds to imine Int-I to form neutral aminyl radical Int-II (Fig. 4), rather than proceeding via cationic intermediates (Fig. 1C). Performing the same reaction in the presence of 2 equivalents of TMSCl led to the formation of pyrrolidine product 4ac′ in addition to 4ac. Pyrrolidine 4ac′ presumably arises from radical addition to iminium ion Int-III to form aminium radical cation Int-IV which, in contrast to Int-II, cyclises with a rate comparable to that of hydrogen atom transfer from the silane reductant.221H and 13C NMR studies suggest that iminium ion Int-III is formed quantitatively from imine Int-I in the presence of 2 equiv. TMSCl (see ESI†), lending further support to the involvement of two distinct mechanistic pathways. Moreover, HFIP is not required to promote radical addition to the neutral imine (entry 6, Table 1). To the best of our knowledge, the intermolecular addition of alkyl radicals to non-activated, uncharged, alkyl-substituted imines, has not previously been reported.15
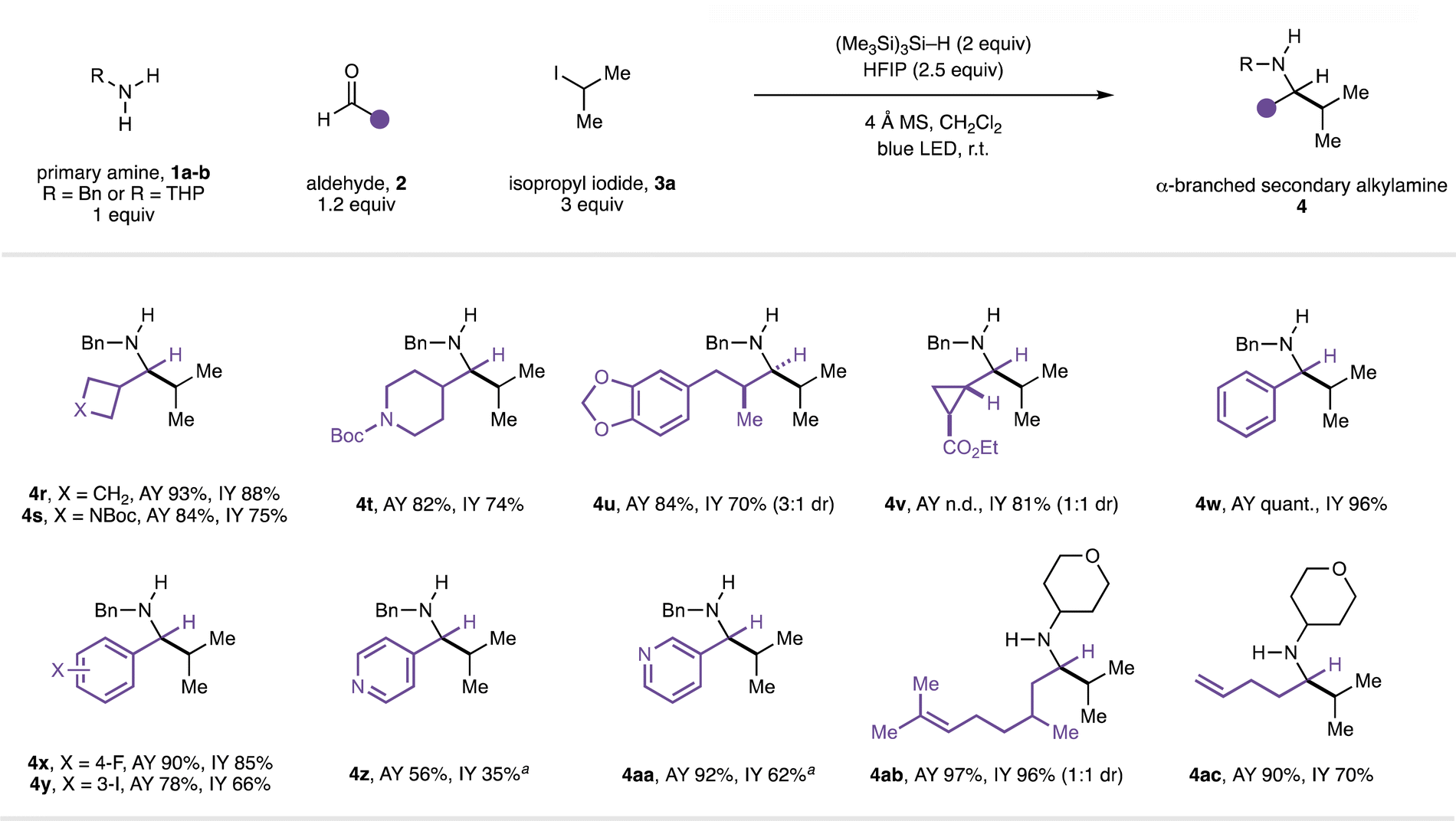 | ||
| Fig. 3 Scope of the aldehyde component in CAA to α-branched secondary alkylamines. a 4:1 CH2Cl2/HFIP used as solvent. n.d. = not determined. | ||
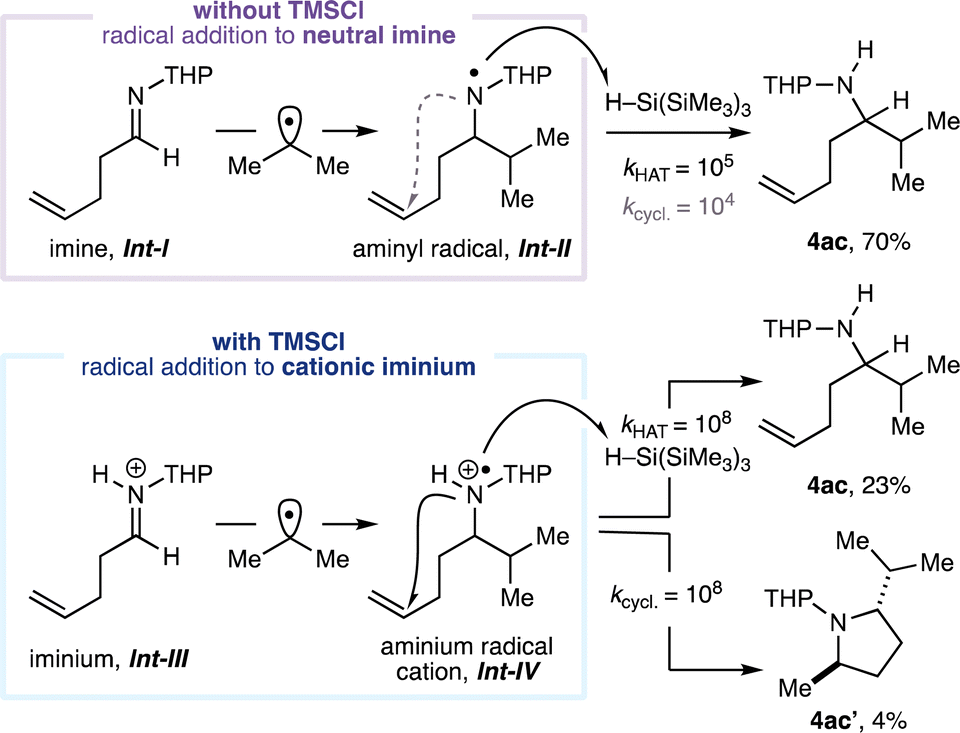 | ||
| Fig. 4 Evidence for distinct mechanistic pathways. Conditions as Fig. 3 with and without TMSCl. Isolated yields. Values for rate constants taken from ref. 22e and given as kHAT/M−1 s−1 and kcycl./s−1. THP = 4-tetrahydropyranyl. | ||
The scope in the alkyl iodide component was explored using benzylamine (1a) in combination with hydrocinnamaldehyde (2a) or cyclohexanecarboxaldehyde (2b) (Fig. 5). Linear alkyl fragments derived from primary alkyl iodides could be added to the in situ-generated imine intermediates to form a selection of α-branched secondary alkylamines displaying a variety of functionality in generally good yields (4ad–4ak). In these cases, we found the addition of two equivalents TMSCl to be beneficial—we speculate that this effect originates from the lower nucleophilicity of primary alkyl radicals, wherein full protonation (by in situ-generated HCl) of the intermediate aldimine to generate a more electrophilic species is advantageous.
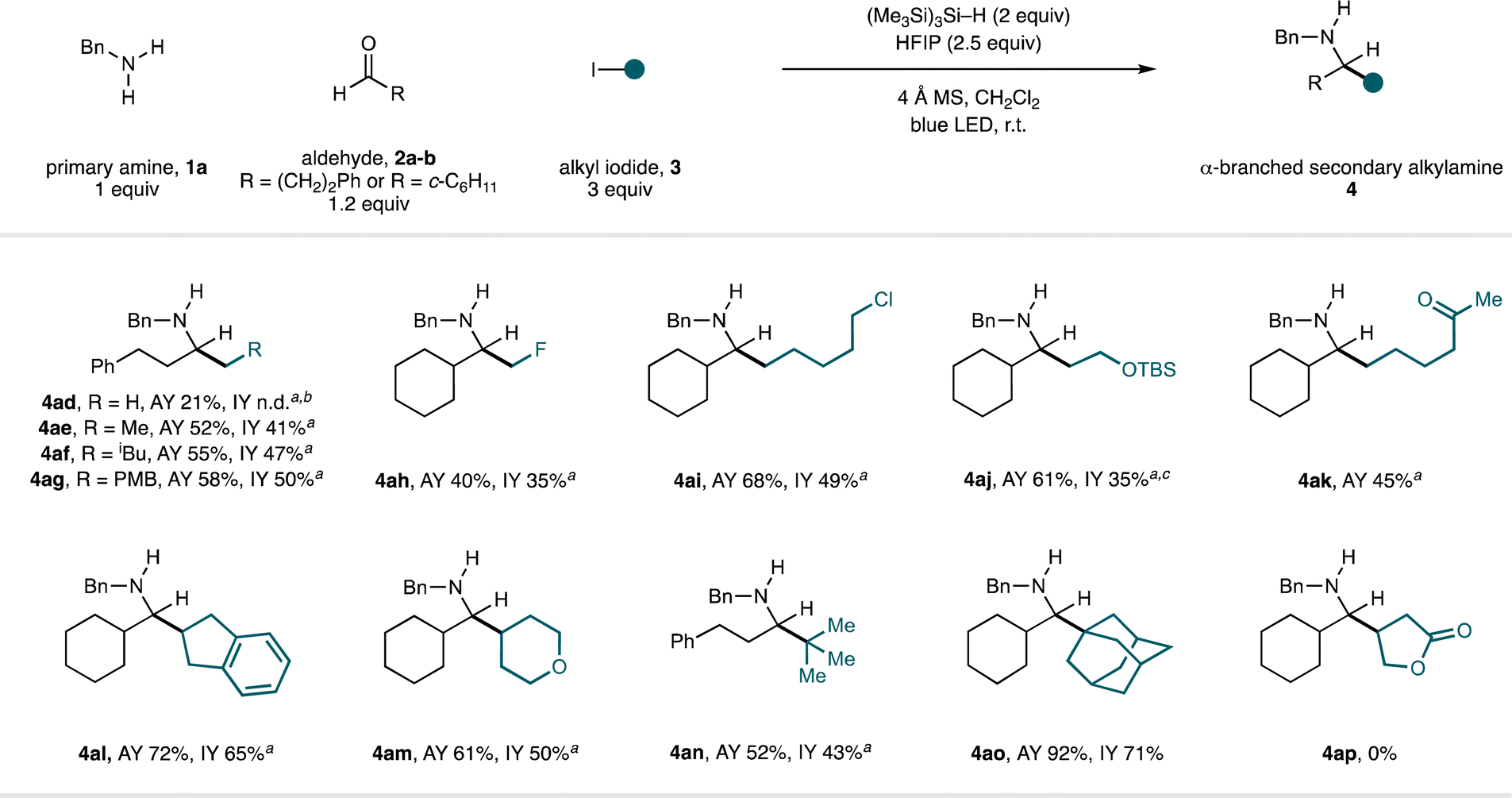 | ||
| Fig. 5 Scope of the alkyl iodide component in CAA to α-branched secondary alkylamines. a TMSCl (2 equiv.) used as an additive. b It was not possible to separate 4ad from the reductive amination side-product. c 13 mol% ethyl 2-methyl-2-iodopropionate used as additive. PMB = 4-methoxybenzyl. n.d. = not determined. | ||
Methyl iodide was productive in this process, but the yield of the amine product (4ad) was low and proved inseparable from a reductive amination by-product. By products of a formal reductive amination were commonly observed in low-yielding reactions; several possible mechanisms for their formation are provided in the ESI.† We were pleased to find that the fluoromethyl radical (derived from fluoroiodomethane) engaged in effective radical addition under these conditions, giving β-fluoroamine 4ah in synthetically useful yield.16c A C–Cl bond was retained through the reaction to deliver 4ai, which contains an electrophilic functional handle suitable for further elaboration, and use of an alkyl iodide derived from ethylene glycol provided protected γ-amino alcohol 4aj. Pleasingly, incorporation of a ketone into the iodide component, which could theoretically compete with the aldehyde in the condensation step, resulted in the formation of aminoketone product 4ak in good yield. Cyclic secondary alkyl iodides of varying ring sizes and substitution were also good substrates (forming 4al–am) and even tertiary alkyl iodides generated the hindered amine products (4an–ao) in good yield. As observed in the scope of the amine and aldehyde components, classical reductive amination methods to form 4an–ao would be challenging due to the difficulty in performing condensations at hindered ketones. Although a range of alkyl iodides are competent in this CAA reaction, our studies highlighted this as the component having the greatest impact on the success of the reaction. Specifically, the reaction is sensitive to the nucleophilicity of the carbon-centred radical; less nucleophilic radicals (such as primary radicals and/or those bearing proximal electron-withdrawing groups), lead to reductive amination being observed as a significant by-product.
Given the efficiency of this visible-light-mediated CAA process, we questioned whether deployment of an alkyl iodide containing a tethered leaving group might enable a subsequent cyclization to form α-substituted N-heterocyclic frameworks in a multicomponent fashion. Specifically, after the CAA process, cyclization via displacement of a pendent chloride would generate a cyclic amine bearing N-substitution controlled by choice of amine, α-substitution controlled by choice of aldehyde, and ring size controlled by choice of the chloroalkyl iodide reagent. The ability to obtain α-branched N-heterocycles directly from feedstock amines and aldehydes with a programmable ring size dictated by choice of a commercial bifunctional lynchpin would represent a valuable approach that is complementary to existing methods. Accordingly, the CAA reaction of p-anisidine, 4-fluorobenzaldehyde and chloroiodomethane (5a) successfully delivered β-chloro-amine 6a, and heating this compound in the presence of tBuOK for 30 min effected cyclisation to give the α-branched aziridine 7a in good yield (Fig. 6A). Chromatographic purification of the intermediate chloro-amines was not required, although we observed higher yields when removing excess silane, alkyl iodide and aldehyde by strong cation exchange (SCX) filtration before performing the cyclisation. The use of 1-chloro-3-iodopropane (5b) delivered pyrrolidines 7b and 7c directly after basic aqueous workup without the need for a separate cyclisation procedure, reflecting the facile 5-exo ring-closure of saturated 5-membered rings. Piperidine 7d was obtained by heating ε-chloro-amine CAA product 6d in the presence of base and sodium iodide in acetonitrile for 2 h. We note the high yields (≥70%) of the CAA reaction when using functionalised alkyl iodides 5a–c with amine 1c and benzaldehydes, demonstrating that consistently high efficiency can be achieved with challenging alkyl iodides under our conditions with favourable amine and aldehyde coupling partners.
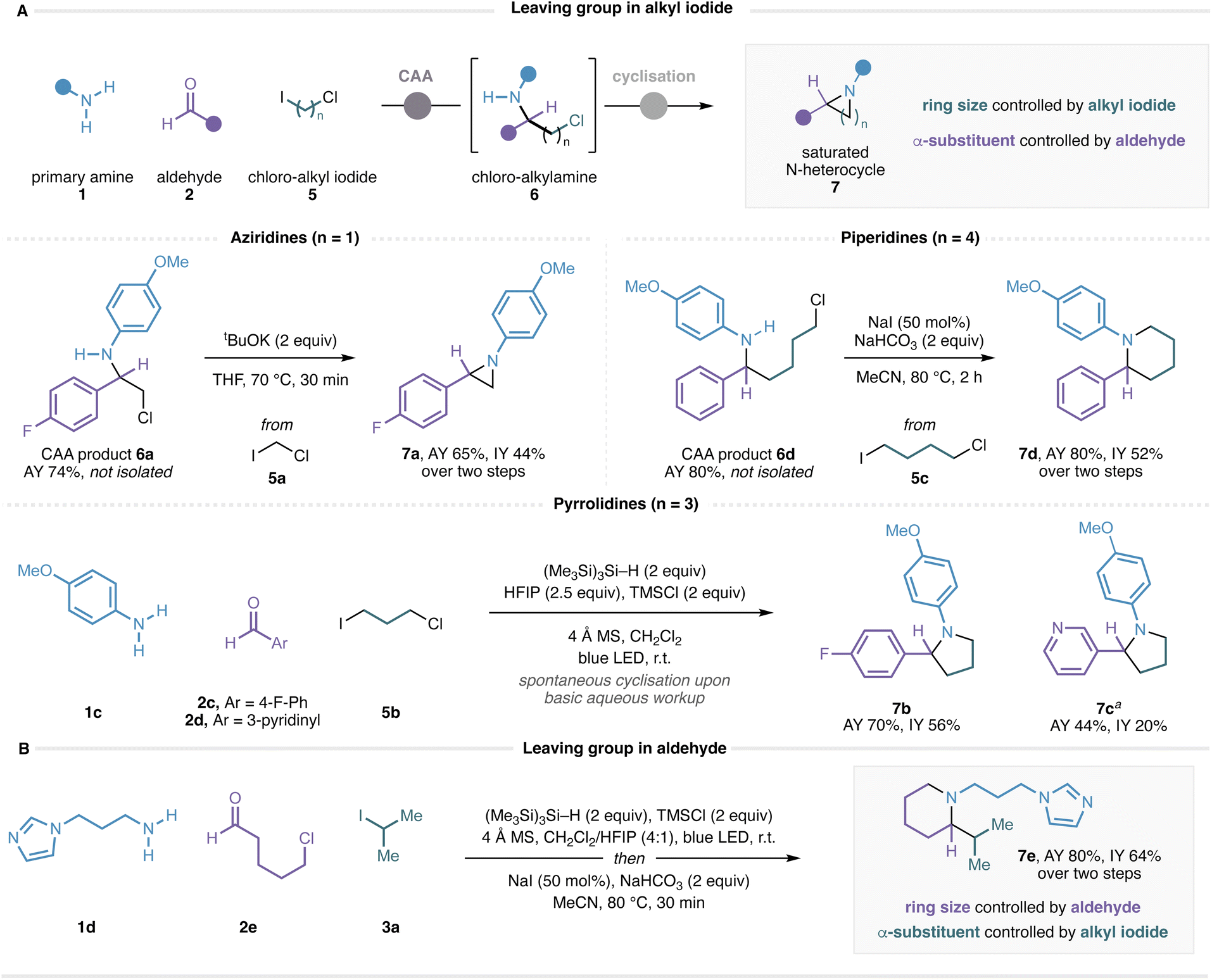 | ||
| Fig. 6 Preliminary scope of chloro-alkyl iodides in the application of carbonyl alkylative amination to α-branched saturated N-heterocycles. (A) Leaving group in alkyl iodide. CAA conditions given for the formation of 7b and 7c also apply to the formation of 6a and 6d. a 4:1 CH2Cl2/HFIP used as solvent. (B) Leaving group in aldehyde. | ||
We also recognized that the leaving group need not be confined to the alkyl iodide component, and that the same annulation strategy should be effective with the aldehyde component carrying the chloride leaving group and the alkyl iodide component carrying the eventual α-substituent. A reaction using primary alkylamine 1d, 5-chloropentanal (2e), and isopropyl iodide followed by cyclisation of the ε-chloro-amine CAA product provided α-branched piperidine 7e in 64% yield over two steps (Fig. 6B). These results suggest that a straightforward and general multicomponent approach to saturated nitrogen heterocycles may be possible with further optimization, and studies in this regard are ongoing and will be reported in due course.
A longstanding challenge in amine synthesis has been the development of alkylation processes that are selective between primary and secondary amines.23 With this in mind, we questioned how a molecule containing both a primary and a secondary amine would react under the new conditions. Accordingly, we submitted 4-aminomethylpiperidine (8a) to the HFIP-mediated CAA reaction with cyclohexanecarboxaldehyde (2b) and isopropyl iodide (3a) and were surprised to find that only trace quantities of product arising from the CAA reaction (at either amine) could be observed (Fig. 7A). Given the effectiveness of TMSCl as an additive in the main substrate scope, we also evaluated the reaction in the presence of two equivalents of TMSCl. Remarkably, 1H NMR analysis of the crude reaction mixture appeared to show the formation of a single product in 87% assay yield. While we were able to isolate diamine 9a in low yield (see ESI†) we were unable to find purification conditions that provided this compound in a yield commensurate with the assay. However, selective protection of the piperidine nitrogen as the Boc-carbamate gave the corresponding mono-Boc diamine (Boc-9a), which could be isolated in 66% yield via chromatography on neutral alumina (Fig. 7B). As such, all diamines generated through this reaction were isolated as their mono-Boc derivatives. Although analysis of the crude CAA reaction mixture by 1H NMR shows a remarkably clean reaction profile, analysis by LCMS elucidates that the reaction is not perfectly selective, and by-products derived from reductive amination and CAA reactivity can be observed (see ESI† for details). Nonetheless, the fact that products derived from a single CAA reaction at the primary amine can be isolated from the reaction mixture in yields typically exceeding 50% demonstrates a significant bias for the formation of these products. At the present stage of development, the origin of this unexpected selectivity remains unclear; several mechanistic possibilities are discussed in the ESI.†
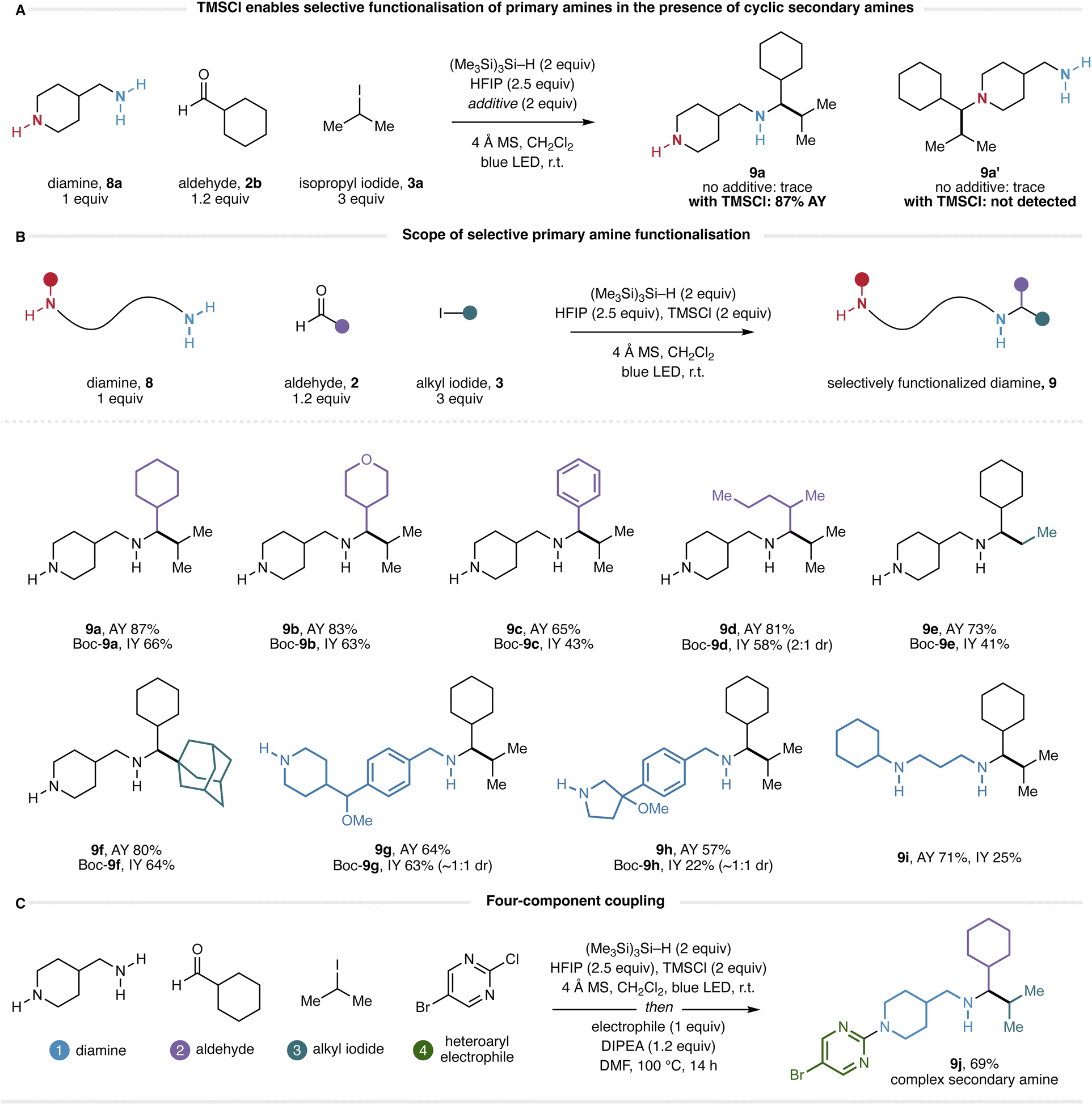 | ||
| Fig. 7 (A) TMSCl enables selective functionalization of primary amines in the presence of secondary amines. (B) Selective N-alkylation of diamines using CAA. (C) Selective functionalization of diamine 9a to effect a formal four-component coupling. | ||
The selective reactivity of the starting diamine was replicated over several combinations of aldehyde and alkyl iodide to form the desired products 9b–f with a scope reflecting that of the standard reaction: heterocyclic (9b), aromatic (9c) and acyclic (9d) aldehydes could be incorporated, as could primary (9e) and tertiary (9f) alkyl iodides (Fig. 7B). This selectivity could be extended to a diamine containing a benzylamine as the primary amine and a piperidine as the secondary amine, with the CAA reaction taking place selectively at the benzylamine nitrogen (9g). A diamine containing a benzylamine as the primary amine and a pyrrolidine as the secondary amine also reacted selectively at the benzylamine nitrogen (9h), however, a challenging purification led to a diminished isolated yield. A limitation of this method is the necessity of a cyclic secondary amine to impart selectivity: diamines containing acyclic secondary amines reacted unselectively (see ESI† for details). However, some selectivity was observed in a sterically biased system, and compound 9i was obtained in a good assay yield. Furthermore, we note that the use of α-branched aldehydes is required to impart this primary-over-cyclic-secondary amine reactivity; use of unbranched aldehydes resulted in complex mixtures. Finally, given our ability to selectively protect the cyclic secondary amine in the presence of the acyclic α-branched secondary amine derived from the CAA reaction, we questioned whether it might be possible to selectively functionalize the cyclic amine to achieve a formal four-component coupling to complex diamine-derived products. Accordingly, we heated 9a (obtained from SCX filtration of the crude CAA reaction mixture) to 100 °C in the presence of a heteroaryl electrophile (Fig. 7C). We were pleased to find that SNAr-type N-arylation took place exclusively at the piperidine nitrogen, providing 9j in 69% yield after chromatography. We believe that this four-component coupling demonstrates a powerful approach to the rapid assembly of complex nitrogen-containing scaffolds through the modular incorporation of commercial building blocks.
Conclusions
In summary, we have developed a visible-light and silane-mediated carbonyl alkylative amination method for the synthesis of α-branched secondary alkylamines from readily available primary alkylamines, aldehydes and alkyl iodides. This multicomponent protocol exhibits a good scope and tolerates a wide range of functional groups on all the three components. Although the key silane reagent is used in excess, it is relatively cheap and its by-products easy to remove, representing an advantage compared to the stoichiometric reductants (such as Hantzsch ester) that are commonly used in catalytic visible-light-mediated protocols. Further synthetic utility of the new method is demonstrated through a one-pot synthesis of saturated nitrogen-containing heterocycles, the synthesis of which would require multi-step routes using established methods. The new carbonyl alkylative amination is also chemoselective in the reaction of primary amines over cyclic secondary amines and provides a rare example of selective N-alkylation of diamines, which remains an important challenge in chemical synthesis. Our mechanistic data suggest that alkyl radicals undergo efficient intermolecular addition to uncharged alkyl imines when using HFIP as the sole additive—a process that is rare, and possibly unique. Overall, the operationally straightforward CAA reaction provides a strategically distinct transformation for alkylamine synthesis that is complementary to—and in many cases, provides products that would be inaccessible via—carbonyl reductive amination. We believe this transformation will be of interest to practitioners of biologically-relevant molecule synthesis in academic and industrial settings.Data availability
Data associated with this article, including experimental procedures and compound characterization, are available in the ESI.†Author contributions
R. K. and M. J. G. conceived the project. M. A. S. and R. J. D. K. and B. R. conducted the experiments. M. A. S., R. J. D. K., B. R. and M. J. G. analysed the data. M. A. S. and M. J. G. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding sources are the following: EPSRC Centre for Doctoral Training – SynTech EPS024220/1 to M. A. S.; Gates Cambridge Scholarship to R. J. D. K; Cambridge Trust International Scholarship and Trinity Henry Barlow Scholarship to B. R.; Swiss National Science Foundation Fellowship (P400P2_186730) to R. K.; and EPSRC (EP/S020292/1).Notes and references
- (a) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (b) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (d) J. Mayol-Llinàs, A. Nelson, W. Farnaby and A. Ayscough, Drug Discovery Today, 2017, 22, 965–969 CrossRef PubMed.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS.
- (a) M. Baumann, I. R. Baxendale, S. V. Ley and N. Nikbin, Beilstein J. Org. Chem., 2011, 7, 442–495 CrossRef CAS PubMed; (b) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed.
- (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (b) L. J. Gooßen, L. Huang, M. Arndt, K. Gooßen and H. Heydt, Chem. Rev., 2015, 115, 2596–2697 CrossRef PubMed; (c) S. Ma and J. F. Hartwig, Acc. Chem. Res., 2023, 56, 1565–1577 CrossRef CAS PubMed; (d) Y. Li and T. J. Marks, Organometallics, 1996, 15, 3770–3772 CrossRef CAS; (e) J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584–12605 CrossRef CAS PubMed; (f) H. F. Yuen and T. J. Marks, Organometallics, 2009, 28, 2423–2440 CrossRef CAS; (g) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984–8987 CrossRef CAS PubMed; (h) A. L. Reznichenko and K. C. Hultzsch, Organometallics, 2013, 32, 1394–1408 CrossRef CAS; (i) H. N. Nguyen, H. Lee, S. Audörsch, A. L. Reznichenko, A. J. Nawara-Hultzsch, B. Schmidt and K. C. Hultzsch, Organometallics, 2018, 37, 4358–4379 CrossRef CAS; (j) D. C. Miller, J. M. Ganley, A. J. Musacchio, T. C. Sherwood, W. R. Ewing and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 16590–16594 CrossRef CAS PubMed.
- (a) S. T. J. Cullen and G. K. Friestad, Synthesis, 2021, 53, 2319–2341 CrossRef CAS; (b) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986–16989 CrossRef CAS PubMed; (c) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed; (d) A. L. Fuentes de Arriba, F. Urbitsch and D. J. Dixon, Chem. Commun., 2016, 52, 14434–14437 RSC; (e) T. Rossolini, J. A. Leitch, R. Grainger and D. J. Dixon, Org. Lett., 2018, 20, 6794–6798 CrossRef CAS PubMed; (f) J. A. Leitch, T. Rossolini, T. Rogova, J. A. P. Maitland and D. J. Dixon, ACS Catal., 2020, 10, 2009–2025 CrossRef CAS.
- (a) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed; (b) M. Wang, S. Liu, H. Liu, Y. Wang, Y. Lan and Q. Liu, Nature, 2024, 631, 556–562 CrossRef CAS PubMed.
- (a) T. W. Thorpe, J. R. Marshall, V. Harawa, R. E. Ruscoe, A. Cuetos, J. D. Finnigan, A. Angelastro, R. S. Heath, F. Parmeggiani, S. J. Charnock, R. M. Howard, R. Kumar, D. S. B. Daniels, G. Grogan and N. J. Turner, Nature, 2022, 604, 86–91 CrossRef CAS PubMed; (b) A. K. Gilio, T. W. Thorpe, N. Turner and G. Grogan, Chem. Sci., 2022, 13, 4697–4713 RSC.
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CAS.
- (a) M. Hatano, S. Suzuki and K. Ishihara, J. Am. Chem. Soc., 2006, 128, 9998–9999 CrossRef CAS PubMed; (b) D. Qian, S. Bera and X. Hu, J. Am. Chem. Soc., 2021, 134, 1959–1967 CrossRef PubMed; (c) Y. He, H. Song, J. Chen and S. Zhu, Nat. Commun., 2021, 12, 638 CrossRef CAS PubMed; (d) J.-W. Wang, Y. Li, W. Nie, Z. Chang, Z.-A. Yu, Y.-F. Zhao, X. Lu and Y. Fu, Nat. Commun., 2021, 12, 1313 CrossRef CAS PubMed; (e) S. Wang, J.-X. Zhang, T.-Y. Zhang, H. Meng, B.-H. Chen and W. Shu, Nat. Commun., 2021, 12, 2771 CrossRef CAS PubMed; (f) T. Zhang, S. Jiang, M.-Y. Qian, Q.-L. Zhou and L.-J. Xiao, J. Am. Chem. Soc., 2024, 146, 3458–3470 CrossRef CAS PubMed.
- A. F. Abdel-Magid and S. J. Mehrman, Org. Process Res. Dev., 2006, 10, 971–1031 CrossRef CAS.
- J. Castillo, J. Orrego-Hernández and J. Portilla, Eur. J. Org Chem., 2016, 2016, 3824–3835 CrossRef CAS.
- (a) A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed; (b) X. Wu, J. Ren, Z. Shao, X. Yang and D. Qian, ACS Catal., 2021, 11, 6560–6577 CrossRef CAS.
- (a) R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS; (b) R. A. Volkmann in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 1, pp. 355–396 Search PubMed; (c) R. Bloch, Chem. Rev., 1998, 98, 1407–1438 CrossRef CAS PubMed.
- (a) G. Liu, D. A. Cogan and J. A. Ellman, J. Am. Chem. Soc., 1997, 119, 9913–9914 CrossRef CAS; (b) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984–995 CrossRef CAS PubMed; (c) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600–3740 CrossRef CAS PubMed.
- (a) H. Miyabe, R. Shibata, C. Ushiro and T. Naito, Tetrahedron Lett., 1998, 39, 631–634 CrossRef CAS; (b) G. K. Friestad, Tetrahedron, 2001, 57, 5461–5496 CrossRef CAS; (c) M. Ueda, H. Miyabe, O. Miyata and T. Naito, Tetrahedron, 2009, 65, 1321–1326 CrossRef CAS; (d) S. Lee and S. Kim, Tetrahedron Lett., 2009, 50, 3345–3348 CrossRef CAS; (e) H. Miyabe, E. Yoshioka and S. Kohtani, Curr. Org. Chem., 2010, 14, 1254–1264 CrossRef CAS; (f) G. K. Friestad, Top. Curr. Chem., 2012, 320, 61–91 CrossRef CAS PubMed; (g) K. Yamada, T. Konishi, M. Nakano, S. Fujii, R. Cadou, Y. Yamamoto and K. Tomioka, J. Org. Chem., 2012, 77, 1547–1553 CrossRef CAS PubMed; (h) J. A. Fernández-Salas, M. C. Maestro, M. M. Rodríguez-Fernández, J. L. García-Ruano and I. Alonso, Org. Lett., 2013, 15, 1658–1661 CrossRef PubMed; (i) J. Tauber, D. Imbri and T. Opatz, Molecules, 2014, 19, 16190–16222 CrossRef PubMed; (j) J. A. Fernández-Salas, M. M. Rodríguez-Fernández, M. C. Maestro and J. L. García-Ruano, Eur. J. Org Chem., 2014, 2014, 5265–5272 CrossRef; (k) S. Fujii, T. Konishi, Y. Matsumoto, Y. Yamaoka, K. Takasu and K. Yamada, J. Org. Chem., 2014, 79, 8128–8133 CrossRef CAS PubMed; (l) N. R. Patel, C. B. Kelly, A. P. Siegenfeld and G. A. Molander, ACS Catal., 2017, 7, 1766–1770 CrossRef CAS PubMed.
- (a) R. Kumar, N. J. Flodén, W. G. Whitehurst and M. J. Gaunt, Nature, 2020, 581, 415–420 CrossRef CAS PubMed; (b) J. H. Blackwell, R. Kumar and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 1598–1609 CrossRef CAS PubMed; (c) P. J. Deneny, R. Kumar and M. J. Gaunt, Chem. Sci., 2021, 12, 12812–12818 RSC; (d) Our laboratory recently reported a complementary solution to this problem involving the use of zinc (in place of silane) to activate the alkyl halide component: J. M. Phelps, R. Kumar, J. D. Robinson, J. C. K. Chu, N. J. Flodén, S. Beaton and M. J. Gaunt, J. Am. Chem. Soc., 2024, 146, 9045–9062 CrossRef CAS PubMed.
- C. Hu, J. Tsien, S.-J. Chen, M. Kong, R. R. Merchant, Y. Kanda and T. Qin, J. Am. Chem. Soc., 2024, 146(31), 21769–21777 CrossRef PubMed.
- M. Fleischmann, D. Drettwan, E. Sugiono, M. Rueping and R. M. Gschwind, Angew. Chem., Int. Ed., 2011, 50, 6364–6369 CrossRef CAS PubMed.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem, 2017, 1, 0088 CrossRef CAS.
- (a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS; (b) R. Hollis, L. Hughes, V. B. Bowry and K. U. Ingold, J. Org. Chem., 1992, 57, 4284–4287 CrossRef CAS; (c) D. C. Nonhebel, Chem. Soc. Rev., 1993, 22, 347–359 RSC.
- (a) C. Chatgilialoglu, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS; (b) D. P. Curran and A. I. Keller, J. Am. Chem. Soc., 2006, 128, 13706–13707 CrossRef CAS PubMed; (c) D. P. Curran, C. P. Jasperse and M. J. Totleben, J. Org. Chem., 1991, 56, 7169–7172 CrossRef CAS.
- (a) J. H. Horner, F. N. Martinez, O. M. Musa, M. Newcomb and H. E. Shahin, J. Am. Chem. Soc., 1995, 117, 11124–11133 CrossRef CAS; (b) M. Newcomb, N. Tanaka, A. Bouvier, C. Tronche, J. H. Horner, O. M. Musa and F. N. Martinez, J. Am. Chem. Soc., 1996, 118, 8505–8506 CrossRef CAS; (c) O. M. Musa, J. H. Horner, H. Shahin and M. Newcomb, J. Am. Chem. Soc., 1996, 118, 3862–3868 CrossRef CAS; (d) C. Ha, O. Musa, F. N. Martinez and M. Newcomb, J. Org. Chem., 1997, 62, 2704–2710 CrossRef CAS PubMed; (e) J. H. Horner, O. M. Musa, A. Bouvier and M. Newcomb, J. Am. Chem. Soc., 1998, 120, 7738–7748 CrossRef CAS.
- A. O. Gálvez, C. P. Schaack, H. Noda and J. W. Bode, J. Am. Chem. Soc., 2017, 139, 1826–1829 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03916e |
| This journal is © The Royal Society of Chemistry 2024 |