
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple N-heterocyclic carbene for the catalytic up-conversion of aldehydes into stoichiometric super electron donors†
Nadhrata
Assani
 a,
Ludivine
Delfau
b,
Preslav
Smits
b,
Sébastien
Redon
a,
Youssef
Kabri
a,
Eder
Tomás-Mendivil
b,
Patrice
Vanelle
a,
David
Martin
*b and
Julie
Broggi
*a
a,
Ludivine
Delfau
b,
Preslav
Smits
b,
Sébastien
Redon
a,
Youssef
Kabri
a,
Eder
Tomás-Mendivil
b,
Patrice
Vanelle
a,
David
Martin
*b and
Julie
Broggi
*a
aAix Marseille Univ, CNRS, ICR, Institut de Chimie Radicalaire, Faculté de Pharmacie, 13005, Marseille, France. E-mail: julie.broggi@univ-amu.fr
bUniv. Grenoble-Alpes, UMR CNRS-UGA 5250, CS 40700, 38058 Grenoble, France. E-mail: david.martin@univ-grenoble-alpes.fr
First published on 16th August 2024
Abstract
Catalytic amounts of 1,3-di(methyl)imidazole-2-ylidene, one of the simplest and most prototypical N-heterocyclic carbenes, can up-convert aldehydes into powerful stoichiometric sources of electrons (Super Electron Donors) for reductive transformations of iodoaryls (Ered < −2 V). In particular, the hydroarylation of 1,1′-diarylethylenes, which may require high temperatures and inherently generate stoichiometric amounts of oxidized waste, was performed at room temperature, with the concomitant formation of esters as oxidized co-products.
Introduction
The development of Organic Electron Donors (OEDs) has allowed for a broad range of selective metal-free reductive transformations.1 The design of compounds with increasingly reducing power, termed Super Electron Donors (SEDs), has culminated so far in the report of tetra(iminophosphorano)-substituted bispyridinylidene A, which can undergo reversible oxidation at E1/2(A/A2+) = −1.7 V versus SCE and perform the challenging reduction of iodoarenes (Fig. 1a).2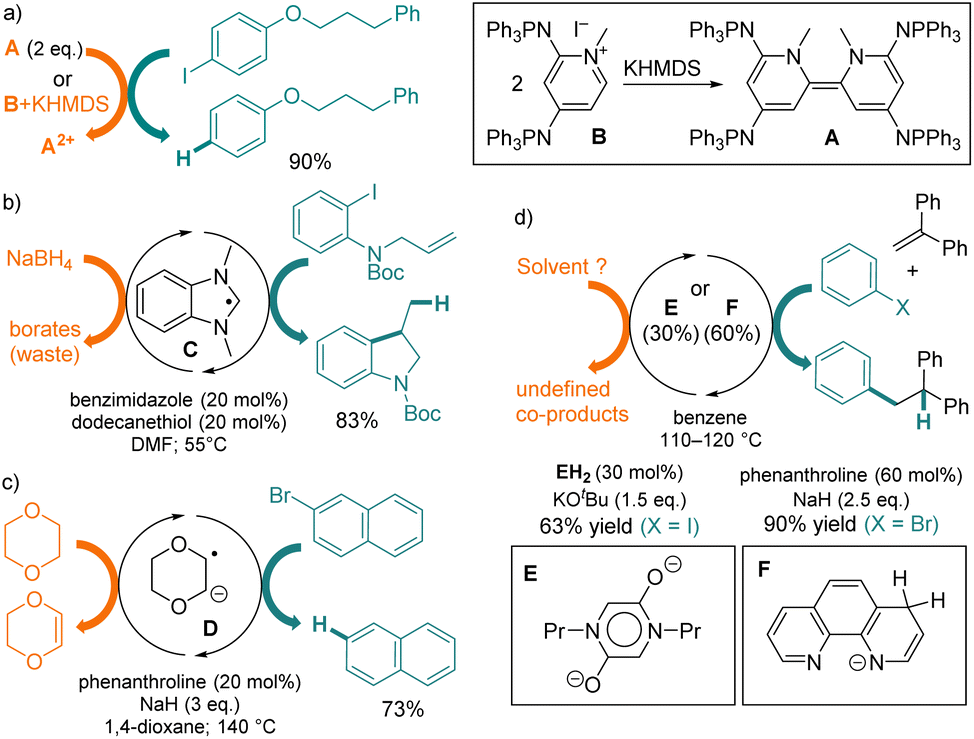 | ||
| Fig. 1 (a) Reduction of aryl halides with a stoichiometric organic Super Electron Donors (SED, 2 equiv.) A or (b–d) with catalytically up-converted SEDs C–F. | ||
Note that, similarly to other OEDs with dimeric patterns,3A can be generated in situ by the deprotonation of precursor B, which is air-stable and easier to handle. More generally, a variety of reductant up-conversions4,5 of weak reagents into various SEDs have been considered, ranging from in situ stoichiometric chemical reactions3 to photoactivation.6 Several groups described reactions involving catalytic amounts of chemically upconverted SEDs, such as benzimidazole radical C7 (Eoxp = −2.1 V),8 dioxane-derived radical anion D,9 diketopiperazine bis-enolate E,10 or phenanthroline-based anion F,11 which promote the reductive transformations of halogenoaryls at high temperatures (Fig. 1b–d). In these processes, the stoichiometric sacrificial electron donors are solvents or metallic hydrides. They are required for feeding electrons into the catalytic cycle, but yield wasteful oxidized co-products and can cause side reactions.
On the other hand, aldehydes are ubiquitous synthons in organic synthesis, some of which are available from biomass resources, and they represent a valuable source of electrons. They are up-converted by N-heterocyclic carbenes (NHCs) into Breslow intermediates G·H (Fig. 2a).12,13 The oxidation of the latter into acyliums G+ yields numerous valuable products upon reaction with nucleophiles.14 Yet, very few studies have considered the fate of the oxidative partner. In 1980, Inoue and Higashiuba showed that phenazine (Ered = −0.6 V)15 and azobenzene (Ered = −1.3 V)16 are reduced in the presence of aldehydes under NHC catalysis, yielding dihydrophenazine and benzidine, respectively.17 The group of Chi also designed a few NHC-catalyzed reductive functionalization reactions of activated nitrobenzyl bromides (Ered < −0.8 V) or nitroalkenes (Ered < −0.6 V)18 in the presence of methanol, with the concomitant formation of a methyl ester.19 More recently, NHC-catalysed radical transformations of aldehydes20 have advanced to include the activation of more challenging substrates, such as iodoaryls (Ered < −2 V),21via single electron transfer (SET) from Breslow-type enolates G22 stemming from thiazol-2-ylidenes (Ohmiya group),23 1,3,4-triazol-2-ylidenes (Chi group)24 or meso-ionic 1,4,5-triazol-2-ylidenes (Bertrand and Yan groups).25 However, so far, these radical reactions have remained redox-neutral overall, because the transient radicals are intercepted by persistent Breslow-type radicals G˙, affording ketone derivatives (Fig. 2b).
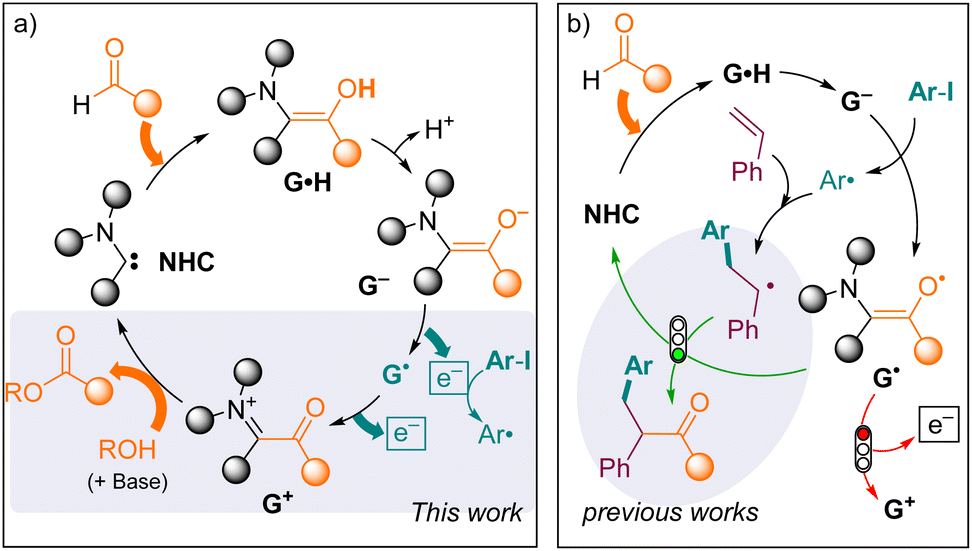 | ||
| Fig. 2 (a) Reductant up-conversion of aldehydes under NHC catalysis. (b) Interception of G˙ by transient radicals in the redox-neutral NHC-catalysed acylarylation of styrenes. | ||
Herein, we show that catalytic amounts of 1,3-di(methyl) imidazole-2-ylidene, one of the simplest and most prototypical N-heterocyclic carbenes (NHCs), can up-convert aldehydes into strong SED reagents for the light-free reductive transformations of iodoaryls (Ered < −2 V) at room temperature, with the concomitant formation of esters as oxidized co-products.
Results and discussion
We were intrigued by electrochemical26 and computational27 studies, indicating that imidazole-2-ylidene NHCs should provide some of the strongest electron-donor enolates G− with oxidation potentials (ca. −2.1 V) lower than those of enolates stemming from thiazolylidenes (−1.4 V) or mesoionic carbenes (−1.8 V), which are known to perform the reductive radical activation of iodoaryls. Thus, although imidazolylidenes have been disregarded as mediocre catalysts for the radical transformations of aldehydes,19b,28 we attempted the reductive addition of iodobenzene 2a to 1,1′-diphenylethylene 3a, in the presence of benzaldehyde 1a, potassium tert-butoxide and a catalytic amount of the imidazolium precursor of NHC C2, featuring bulky 1,3-di(isopropyl)phenyl N-substituents (Fig. 3a). After one day at room temperature, NMR monitoring of the crude mixture indicated the presence of 1,1,2-triphenylethane 5aa with a low, but encouraging, 11% conversion, and the concomitant formation of tert-butyl benzoate ester 4a.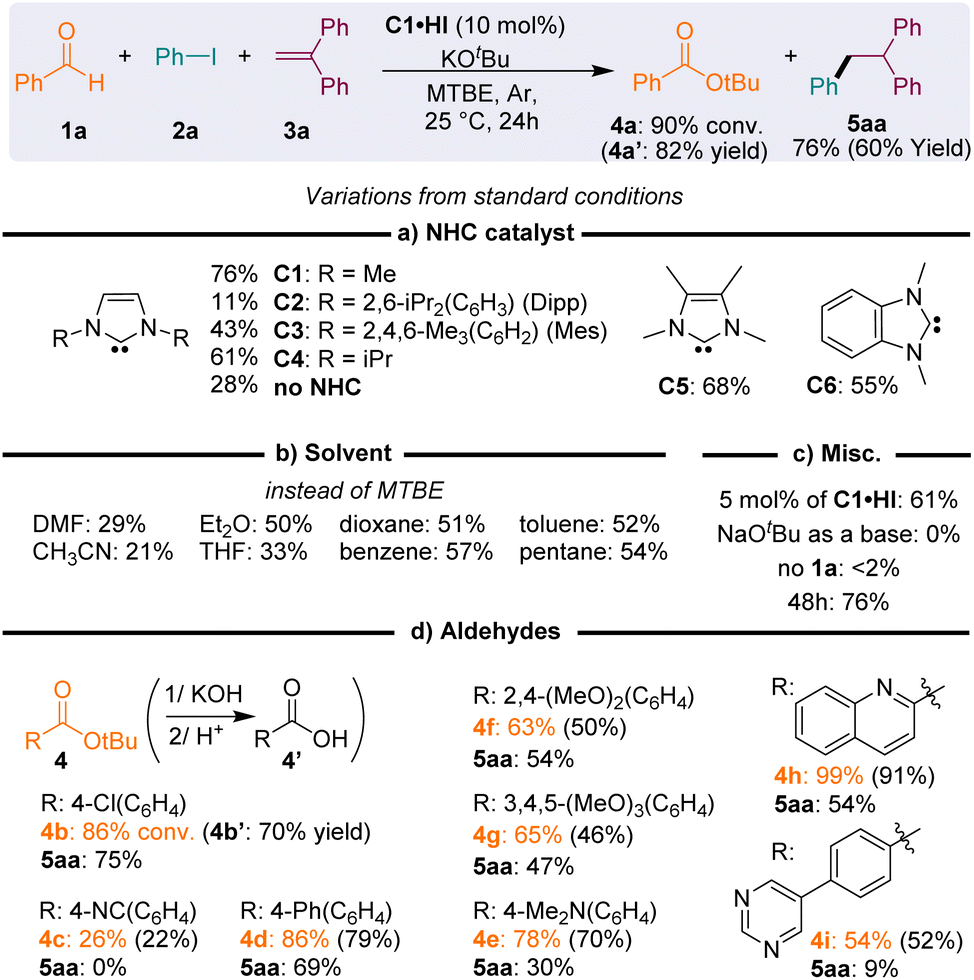 | ||
| Fig. 3 Optimization of NHC-catalyzed reductive anti-Markovnikov hydroarylation of 1,2-diphenylethylene with phenyl iodide; standard reaction conditions were: 1a (0.3 mmol), 2a (0.36 mmol), 3a (0.20 mmol), C1·HI (10 mol%), and KOtBu (0.3 mmol) in dry MTBE (0.6 mL) under an argon atmosphere for 24 h at 25 °C. Percentages are for the conversion of 3a into product 5aa, unless otherwise noted. Values in brackets are yields. | ||
The use of NHCs C1–6 with decreasing steric bulk increased the conversion of 5aa, up to 76% with 1,3-di(methyl)imidazolylidene C1. The reaction also worked with N-methyl substituted NHCs C5 and C6 (with 3,4-dimethylimidazole and benzimidazole backbones, respectively), though with slightly lower conversions. The clear reactivity difference of NHCs C1–6 could not be attributed to the similar reducing power of their Breslow derivative (Ep ≈ −1.1, −1.8 V vs. SCE, see the ESI†). The deleterious effect of steric hindrance at the N-substituents of the NHC catalyst was interpreted as resulting from a significant decrease of the nucleophilicity of the NHC, therefore precluding the fast formation of the Breslow intermediate G·H. The steric hindrance of G·H itself could also impede the formation of a tight charge transfer complex (CTC) with the substrate, leading to slow electron transfers. Interestingly, 28% conversion was observed in the absence of the NHC, indicating the existence of some background reactions, likely promoted by stoichiometric potassium tert-butoxide.29 In the absence of aldehyde 1a, starting iodoarene 2a was recovered (<2% 5aa).
Further optimization established tert-butyl methyl ether (MTBE) as the solvent and the reaction performed better at room temperature (25 °C) with a 10 mol% catalyst loading (Fig. 3b and c). The best conversion rates were obtained with non-polar solvents. Note that the conversion rate was not influenced by their C–H bond dissociation energy (BDEC–H). Solvents with high BDEC–H (benzene: 113 kcal mol−1 and pentane: 100 kcal mol−1) exhibited the same reactivity as solvents with lower BDEC–H (diethyl ether: 93 kcal mol−1 and toluene: 89 kcal mol−1). No conversion was observed with sodium tert-butoxide as a base, likely due to its poor solubility in MTBE.
Other aryl aldehyde partners were tolerated, provided that they were neither too electron-rich nor too electron-poor (Fig. 3d). This balance is in line with the expected necessity for the aldehyde to be both electrophilic enough for fast addition of the NHC and electron-rich enough to yield a highly reducing Breslow enolate. Satisfyingly, the ester co-product was easily removed from the crude reaction and recovered in good yields by saponification, followed by acidic precipitation of the corresponding carboxylic acid 4′ from the aqueous solution.
Next, we considered various substrates under these optimized conditions (Fig. 4). Aryl iodides bearing electron-donating or electron-withdrawing groups reacted well, affording the corresponding arylation products 5aa–5ma in moderate to good yields. Sterically hindered aryl iodides (2f, 2h, and 2j–l) were also tolerated. Substrate 2m, featuring both iodoaryl and alkene moieties, underwent intramolecular cyclisation to afford dihydro-benzofuran 5ma, which was isolated in a modest 30% yield.
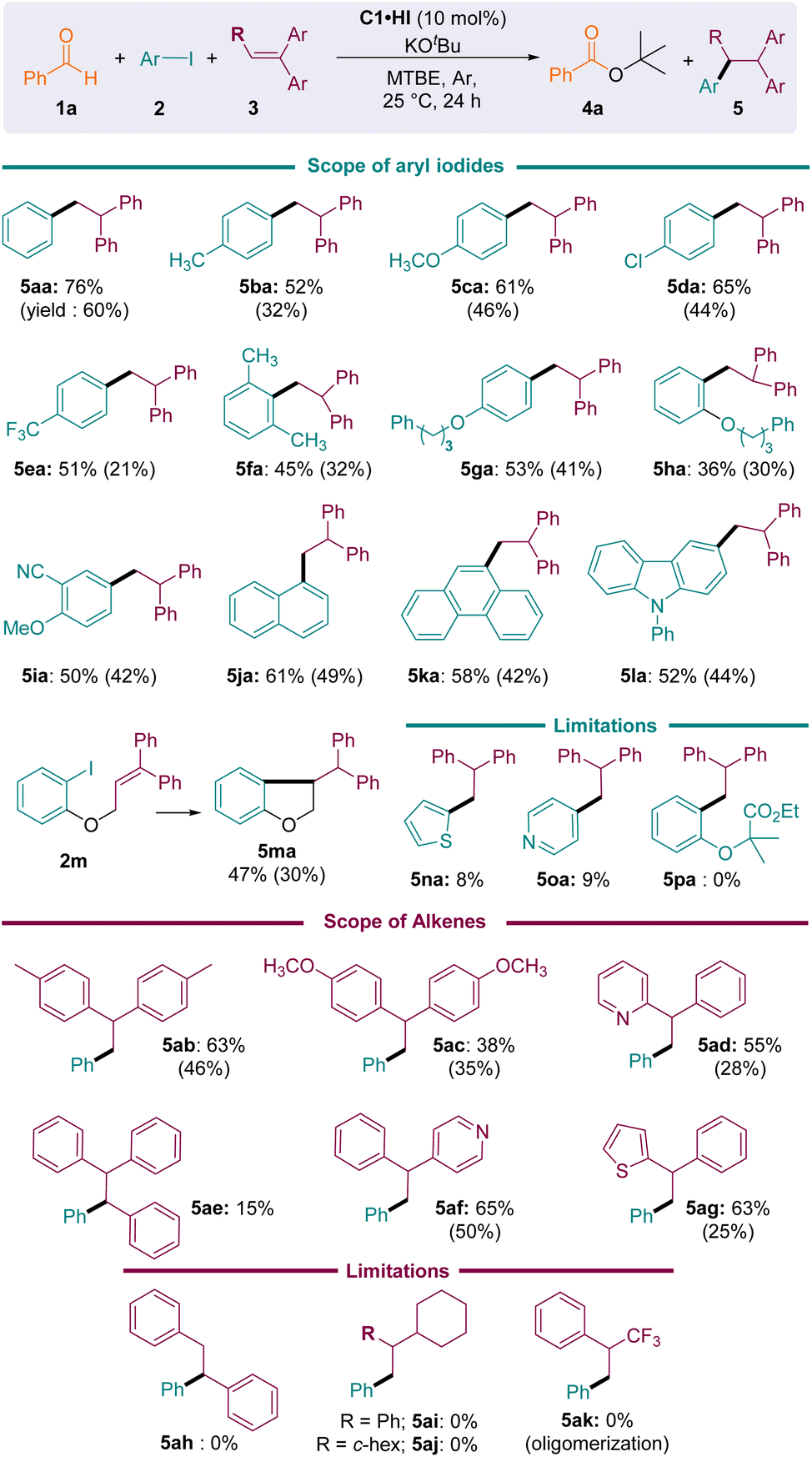 | ||
| Fig. 4 Substrate scope of the NHC-catalysed hydroarylation of 1,1′-diarylethylenes with aryl iodides under pre-optimized conditions. Conversions were evaluated from the 1H NMR spectra of the crude reaction. Values in brackets are yields. | ||
A limitation was reached with heterocycles: 2-iodo thiophene 2n and 4-iodopyridine 2o were reduced, but less than 10% of the desired addition products 5na and 5oa was observed. Indeed, the 1,1′-diphenylethylene 3a was not sufficiently electron-rich to trap these electrophilic pyridyl and thienyl radicals under our mild room-temperature conditions.30 On the other hand, the reduction of the more challenging iodoester 2p failed and the starting materials were mainly recovered.
The reductive coupling of iodobenzene also performed well with geminal diaryl- and heteroaryl-ethylenes 3a–g. Addition to trisubstituted ethylene 3e was also possible, albeit sluggish (5ae conv. 15%). However, neither trans-stilbene 3h nor aliphatic counterparts 3i–k could trap the phenyl radical, as the formation of the corresponding products 5ah–5ak was not observed. Note that in the case of the electron poor α-(trifluoromethyl)styrene 3k, broad band shapes in the 1H NMR spectra of the crude mixture suggested the formation of oligomers.
Importantly, when styrene was introduced as a radical trap, aryl-acylation products 6 were obtained, indicating the interception of the persistent Breslow-type radical G˙ (Fig. 5a). Although the conditions have not been optimized for this specific reaction, compounds 6 were isolated in 26–45% yields, which compared well with the results of the previously reported methodology of Ohmiya et al., employing thiazolylidene NHC catalysts in DMSO at 80 °C (6aa: 34%; 6ja: 52%; 6ab: 34%).23
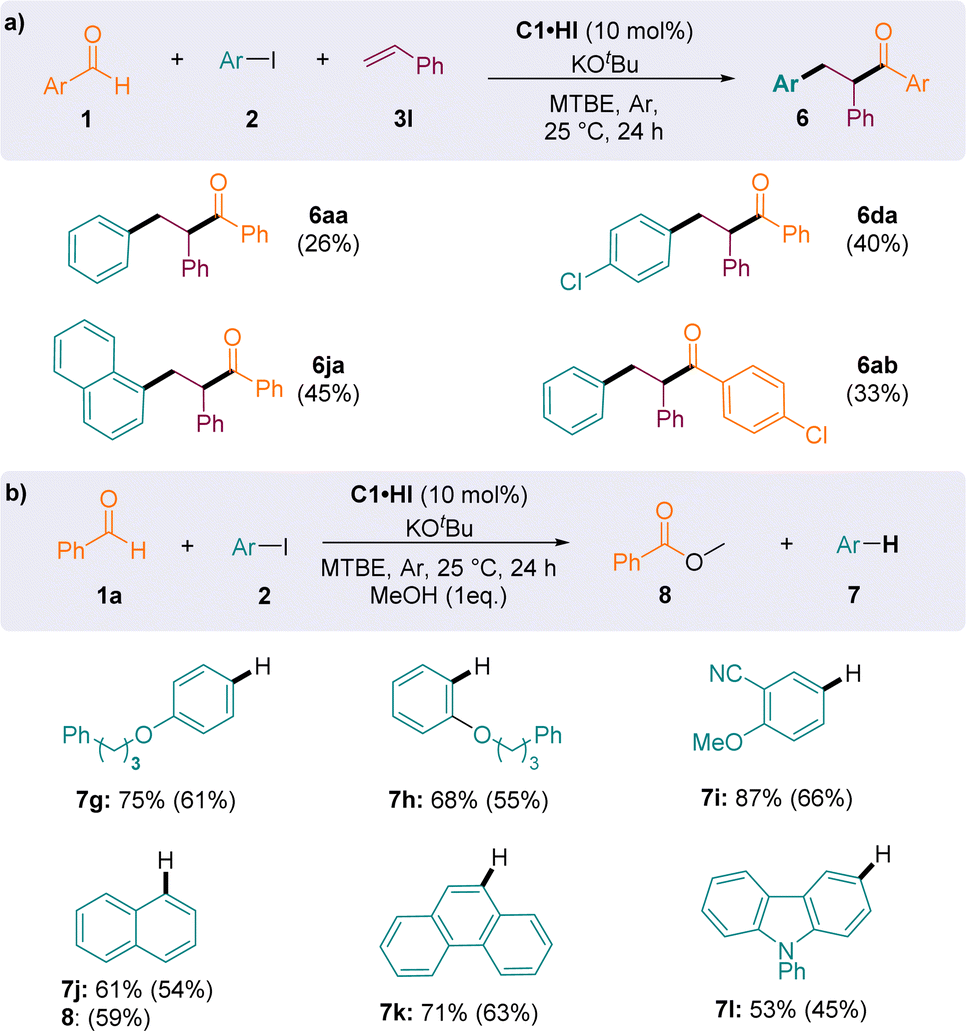 | ||
| Fig. 5 (a) NHC-catalysed (redox-neutral) aryl-acylation of styrene. (b) NHC-catalysed reduction of iodoaryls to arenes with benzaldehyde as a source of electron. Values in brackets are yields. | ||
When the reaction was performed in the absence of alkene and in methanol, two main products could be observed in the 1H NMR spectra of crude reaction mixtures: the arene 7 resulting from the reduction of iodoaryl 2 and the oxidized ester 8 (Fig. 5b). Using one equivalent of methanol as a nucleophile to release the NHC catalyst C1 from the oxidized acyliums G+ maintains the catalytic cycle and leads to the formation of the methyl benzoate 8 (59% yield consecutively to the formation of 54% of 7j). Both oxidative and reductive cycles were concomitant and interdependent, allowing the formation of two distinct redox products in a similar ratio. Arenes 7g–l were isolated in good yields, after purification by column chromatography.
We performed isotopic labeling experiments in order to get further mechanistic insights. The reduction of iodonaphthalene 2j with deuterated aldehyde d-1a in the presence of one equivalent of deuterated methanol CD3OD yielded naphthalene with no incorporation of deuterium (Fig. 6a). These protons are likely transferred by the conjugate acid of the base within the catalytic cycle. Therefore, this suggested that aryl radicals Ar˙ stemming from the reduction of iodoaryls were not further reduced to aryl anions Ar−, which would incorporate acidic protons, but most likely abstracted H˙ from solvent or other molecules.
 | ||
| Fig. 6 (a) Experiments with deuterated substrates and additives. (b) Ensuing mechanistic proposition. (c) Cyclic voltammograms of chloride salts of G1+ and G2+ in 0.1 M nBu4NPF6 electrolyte at 100 mV s−1 rates in acetonitrile. | ||
In contrast, the hydroarylation of 1,1′-diphenylethylene 3a with iodobenzene 2a and d-1a yielded 5aa with 20% deuterium incorporation6d at the C1 position. The incorporation increased to 39% when the reaction was performed with an additional equivalent of CD3OD. This indicated that radical Y˙, stemming from the trapping of Ar˙ by 3a, could be further reduced by Breslow-type enolate G˙ to yield anion Y− and acylium G+ (Fig. 6b). Conversely, in the case of styrene, the reduction of Y˙ is slower than its radical coupling with G˙, leading to aryl-acylation products 6.31
To test this mechanistic proposition further, we synthesized chloride salts of Breslow-type acyliums G1+ and G2+, stemming from benzoyl chloride and NHC catalysts C1 and C2, respectively. Cyclic voltammograms in acetonitrile indicated a first reversible reduction to radical G˙ at −1.1 and −0.9 V, respectively. These values indicate that Breslow-type radicals G˙ could, indeed, reduce radicals Y˙ stemming from 1,1′-diarylethylenes (for diphenylmethyl radicals: E1/2 (Y−/Y˙) = −1.3–−0.7 V),32 whereas SET to radicals Y˙ stemming from styrene would be disfavoured (for benzyl and cumyl radicals: E1/2 (Y−/Y˙) = −1.8–−1.4 V).32
Cyclic voltammograms feature a second reduction wave, which confirmed the strong reducing power of enolates G− stemming from imidazole-2-ylidenes, although the −1.8 V values for Ep(G1−/G1˙) and E1/2 (G2−/G2˙) in acetonitrile are slightly less negative than that reported in the literature (about −2.1 V in dichloromethane).26 Of course, the reducing properties of enolates G− are expected to depend on the nature of the solvent because polarity stabilizes charged molecules. As a matter of fact, a computational study by Bertrand et al. showed that the oxidation potentials of enolates G− increase with polarity. These compounds are predicted to be especially reducing in low polarity solvents, including MTBE.27 Indeed, compared to MTBE, we observed a drastic drop in the reactivity in polar solvents, which are known to favor electron-transfer reactions (DMF, CH3CN or THF, Fig. 3b). Note that SET from G− to aryl iodides is not necessarily favored in non-polar solvents, since the reduction potentials of aryl iodides also increase with polarity. Importantly, despite the necessary caution in interpreting electrochemical data, they once again allowed for rationalizing the main mechanistic outlines of the NHC-catalysed redox processes and must provide useful guidelines for further improvements.
Conclusions
We showed that catalytic amounts of 1,3-di(methyl)imidazole-2-ylidene up-convert aldehydes into powerful stoichiometric sources of electrons, allowing light-free reductive transformations of iodoaryls at room temperature and leading to isolable organic oxidized co-products instead of inorganic wastes. This preliminary study shows the efficiency of this NHC in the reduction of iodoaryls to the corresponding arenes 7, as well as the hydroarylation of 1,1′-diarylethylenes 3. In the latter case, electrochemical data and isotopic labeling suggest that the outcome of the reaction is determined by the ability of Breslow-type persistent radicals G˙ to further reduce radicals Y˙. In particular, this mechanistic proposition accounts for the formation of aryl-acylation products 6 when styrene is the co-reactant, with Y˙ being then a more difficult-to-reduce benzylic radical.Prior to our investigations, imidazole-2-ylidenes had been unsuccessfully tested and considered unfit for the promotion of radical transformations of aldehydes. In contrast, this work reveals the unforeseen potential of these NHCs for the generation of strong SED enolates. We are currently reinvestigating the use and scope of this family of organic catalysts for the reductive activation of challenging substrates.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. B. and D. M. conceived the project and acquired funding. J. B., D. M., E. T.-M. and P. V. provided resources and supervised the project. N. A., L. D., E. T.-M. and P. S. conducted the investigation and formal analysis of the methodology. All authors contributed to the reviewing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the French National Agency for Research (ANR-20-CE07-0010). P. Smits acknowledges Labex Arcane and CBHEUR-GS (ANR-17-EURE-0003) for funding. We thank the ICMG analytic platform of Grenoble (FR 2607) and the Spectropole of the Fédération des Sciences Chimiques de Marseille for spectroscopic analyses.Notes and references
- For reviews, see: (a) J. Broggi, T. Terme and P. Vanelle, Angew. Chem., Int. Ed., 2014, 53, 384–413 CrossRef CAS; (b) J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS; (c) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073–6087 RSC.
- S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236–11239 CrossRef CAS.
- For the in situ formation of organic electron donors from air-stable precursors, see: (a) G. Tintori, P. Nabokoff, R. Buhaibeh, D. Bergé-Lefranc, S. Redon, J. Broggi and P. Vanelle, Angew. Chem., Int. Ed., 2018, 57, 3148–3153 CrossRef CAS; (b) G. Tintori, A. Fall, N. Assani, Y. Zhao, D. Bergé-Lefranc, S. Redon, P. Vanelle and J. Broggi, Org. Chem. Front., 2021, 8, 1197–1205 RSC.
- “Reductant up-conversion” was originally introduced in a broader context by analogy with photon up-conversion: (a) M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532–5550 CrossRef CAS; (b) P. Eckhardt, Q. Elliot, I. V. Alabugin and T. Opatz, Chem.–Eur. J., 2022, 28, e202201637 CrossRef CAS PubMed; (c) V. A. Balycheva, B. K. Chabuka, L. R. Kuhn, P. G. Shangin, A. Y. Akyeva, I. V. Krylova, V. A. Korolev, A. V. Lalov, M. P. Egorov, I. V. Alabugin and M. A. Syroeshkin, J. Phys. Chem. C, 2024, 128, 4581–4599 CrossRef CAS.
- See also: (a) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765–773 CrossRef CAS PubMed; (b) O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC; (c) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed; (c) I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS; (d) H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS PubMed; (e) X. Mao, M.-M. Li, P. Wang, Q. Cao, W. Zhou and W. Ding, Org. Lett., 2024, 26, 1265–1270 CrossRef CAS.
- S. Rohrbach, R. S. Shah, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2019, 58, 11454–11458 CrossRef CAS.
- K. A. Ogawa and A. J. Boydston, Chem. Lett., 2014, 43, 907–909 CrossRef CAS.
- T. Hokamp, A. Dewanji, M. Lübbesmeyer, C. Mück-Lichtenfeld, E.-U. Würthwein and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 13275–13278 CrossRef CAS.
- (a) E. Doni, S. Zhou and J. A. Murphy, Molecules, 2015, 20, 1755–1774 CrossRef PubMed; (b) S. Zhou, E. Doni, G. M. Anderson, R. G. Kane, S. W. MacDougall, V. Ironmonger, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2014, 136, 17818–17826 CrossRef CAS PubMed.
- K. Nozawa-Kumada, S. Onuma, K. Ono, T. Kumagai, Y. Iwakawa, K. Sato, M. Shigeno and Y. Kondo, Chem.–Eur. J., 2023, 29, e202203143 CrossRef CAS.
- (a) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS; (b) R. Breslow and C. Schmuck, Tetrahedron Lett., 1996, 37, 8241–8242 CrossRef CAS; (c) M. Pareek and Y. Reddi, Chem. Sci., 2021, 12, 7973–7992 RSC.
- (a) A. Berkessel, V. R. Yatham, S. Elfert and J.-M. Neudörfl, Angew. Chem., Int. Ed., 2013, 52, 11158–11162 CrossRef CAS PubMed; (b) M. Paul, P. Sudkaow, A. Wessels, N. E. Schlörer, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2018, 57, 8310–8315 CrossRef CAS PubMed; (c) M. Paul, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2019, 58, 10596–10600 CrossRef CAS PubMed; (d) M. Paul, K. Peckelsen, T. Thomulka, J. Martens, G. Berden, J. Oomens, J.-M. Neudörfl, M. Breugst, A. J. H. M. Meijer, M. Schäfer and A. Berkessel, Chem.–Eur. J., 2021, 27, 2662–2669 CrossRef CAS PubMed.
- Selected reviews on NHC catalysis: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (c) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem.–Eur. J., 2013, 19, 4664–4678 CrossRef CAS; (d) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (e) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS; (f) M. H. Wang and K. A. Scheidt, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed.
- A. Hollas, X. Wei, V. Murugesan, Z. Nie, B. Li, D. Reed, J. Liu, V. Sprenkle and W. Wang, Nat. Energy, 2018, 3, 508–514 CrossRef CAS.
- J. L. Sadler and A. J. Bard, J. Am. Chem. Soc., 1968, 90, 1979–1989 CrossRef CAS.
- H. Inoue and K. Higashiuba, J. Chem. Soc., Chem. Commun., 1980, 12, 549–550 RSC.
- J. A. Squella, J. C. Sturm, B. Weiss-lopez, M. Bonta and L. J. Nunez-Vergara, J. Electroanal. Chem., 1999, 466, 90–98 CrossRef CAS.
- (a) Y. Du, Y. Wang, X. Li, Y. Shao, G. Li, R. D. Webster and Y. R. Chi, Org. Lett., 2014, 16, 5678–5681 CrossRef CAS; (b) S. Li, Y. Wang, R. S. J. Proctor, Y. Zhang, R. D. Webster, S. Yang, B. Song and Y. R. Chi, Nat. Commun., 2016, 7, 12933 CrossRef PubMed; (c) Y. Wang, Y. Du, X. Huang, X. Wu, Y. Zhang, S. Yang and Y. R. Chi, Org. Lett., 2017, 19, 632–635 CrossRef CAS PubMed; (d) Y. Wang, X. Wu and Y. R. Chi, Chem. Commun., 2017, 53, 11952–11955 RSC.
- For reviews, see: (a) A. V. Bay and K. A. Scheidt, Trends Chem., 2022, 4, 277–290 CrossRef CAS PubMed; (b) K. Liu, M. Schwenzer and A. Studer, ACS Catal., 2022, 12, 11984–11999 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- (a) J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS; (b) L. Delfau, S. Nichilo, F. Molton, J. Broggi, E. Tomás-Mendivil and D. Martin, Angew. Chem., Int. Ed., 2021, 60, 26783–26789 CrossRef CAS.
- Y. Matsuki, N. Ohnishi, Y. Kakeno, S. Takemoto, T. Ishii, K. Nagao and H. Ohmiya, Nat. Commun., 2021, 12, 3848 CrossRef CAS PubMed.
- F. Su, J. Zou, X. Lv, F. Lu, Y. Long, K. Tang, B. Li, H. Chai, X. Wu and Y. R. Chi, Angew. Chem., Int. Ed., 2023, 62, e202303388 CrossRef CAS.
- (a) C. Liu, W. Liu, A. Vianna, Z. Zhang, S. Huang, L. Huang, M. Melaimi, G. Bertrand and X. Yan, Chem Catal., 2021, 1, 196–206 CrossRef; (b) Z. Zhang, L.-L. Zhao, G. Bertrand and X. Yan, Angew. Chem., Int. Ed., 2023, 62, e202303478 CrossRef; (c) F. Su, F. Lu, K. Tang, X. Lv, Z. Luo, F. Che, H. Long, X. Wu and Y. R. Chi, Angew. Chem., Int. Ed., 2023, 62, e202310072 CrossRef CAS.
- C. L. Deardorff, E. R. Sikma, C. P. Rhodes and T. W. Hudnall, Chem. Commun., 2016, 52, 9024–9027 RSC.
- F. F. Mulks, M. Melaimi, X. Yan, M.-H. Baik and G. Bertrand, J. Org. Chem., 2023, 88, 2535–2542 CrossRef CAS PubMed.
- For examples, see: (a) W.-D. Liu, W. Lee, H. Shu, C. Xiao, H. Xu, X. Chen, K. N. Houk and J. Zhao, J. Am. Chem. Soc., 2022, 144, 22767–22777 CrossRef CAS PubMed; (b) Y. Q. Liu, Q.-Z. Li, X.-X. Kou, R. Zeng, T. Qi, X. Zhang, C. Peng, B. Han and J.-L. Li, J. Org. Chem., 2022, 87, 5229–5241 CrossRef CAS PubMed; (c) K. She, F. Liang, S. Tian, H. Wang, G. C. Tsui and Q. Wang, Org. Lett., 2022, 24, 4840–4844 CrossRef CAS PubMed; (d) Z. Zhang, X. Zou, Z. Li, Y. Gao, Y. Qu, Y. Quan, Y. Zhou, J. Li, J. Sun and K. Guo, Org. Chem. Front., 2021, 8, 6074–6079 RSC; (e) K. Ota, K. Nagao and H. Ohmiya, Org. Lett., 2020, 22, 3922–3925 CrossRef CAS; (f) T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed.
- Stoichiometric OED species can be in situ formed upon the reaction of KOtBu with electrophiles: (a) S. Zhou, G. M. Anderson, B. Mondal, E. Doni, V. Ironmonger, M. Kranz, T. Tuttle and J. A. Murphy, Chem. Sci., 2014, 5, 476–482 RSC; (b) J. Cuthbertson, V. J. Gray and J. D. Wilden, Chem. Commun., 2014, 50, 2575–2578 RSC; (c) G. Nocera, A. Young, F. Palumbo, K. J. Emery, G. Coulthard, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2018, 140, 9751–9757 CrossRef CAS PubMed.
- In the literature, the addition of these electrophilic radicals to electron-rich alkenes is at high temperatures or under photoirradiation, see: D. Mirizzi, S. T. Hilton and K. Jones, Adv. Heterocycl. Chem., 2010, 100, 101–143 CrossRef CAS.
- A more detailed mechanism scheme for the formation of ketone 6 is given in the supporting information.†.
- B. A. Sim, P. H. Milne, D. Griller and D. D. M. Wayner, J. Am. Chem. Soc., 1990, 112, 6635–6638 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04011b |
| This journal is © The Royal Society of Chemistry 2024 |