
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral aggregates of rod-coil molecules inside nanopores as efficient nanoreactors for asymmetric synthesis†
Hui-Yu
Zhao
,
Qing
Xu
,
Gui-Lang
Liu
,
Yi-Rong
Pei
* and
Long Yi
Jin
 *
*
Department of Chemistry, National Demonstration Centre for Experimental Chemistry Education, Yanbian University, Yanji 133002, People's Republic of China. E-mail: pyr@ybu.edu.cn; lyjin@ybu.edu.cn
First published on 12th August 2024
Abstract
An important subject of porous organic materials is their capacity to access enantioselectivity due to their high surface area, controllable pore size, and ease of functionalization. However, recyclability of enantio-separation is a challenge, mainly due to the complex procedures of recovery and refreshing from enantiomers. For the first time, we combined nanochannel technology and supramolecular chiral assembly to achieve efficient enantioselectivity. Fine-designed amphiphilic chiral rod-coil molecules 1–3 were immobilized to SBA-15 pore walls to form SA-M1–3 (abbreviation for amino-functionalized SBA-15 connected to molecules 1–3), which commenced chiral aggregation inside the channels. The experimental results indicated that the strong π–π stacking interaction between the rigid terphenyl groups, as well as hydrophilic–hydrophobic interaction of the amphiphiles, assisted in chiral arrangement in aqueous solution, and was accompanied by amplification of chirality. As a result, porous chiral channels exhibiting enhanced efficiency in asymmetric synthesis were manufactured, where enantioselectivity can be controlled by the initial structural design of amphiphiles that induce chiral aggregation behaviors. The chiral centers of SA-M1 and SA-M2 are located on hydrophobic and hydrophilic coils, respectively, while SA-M3 possesses both chiral coils. The SA-M materials proceeded with chiral aggregation and behaved efficiently for enantioselectivity. SA-M3, which contained the most chiral centers, showed the most optimal enantioselectivity with an enantiomeric excess (ee) value up to 71.75%, which occurred because of the strongly driven chiral aggregation of the hydrophobic and hydrophilic chiral coils. The covalent hybrid structures of the SA-M materials can be easily refreshed simply through washing, and exhibited excellent recyclability with negligible loss of efficiency. Therefore, the SA-M materials have the ability to provide sustainable and reliable application value for enantiomer separation.
Introduction
Enantioselectivity is of great significance for the life sciences, pharmaceutical chemistry, and human health.1–4 In the current era where food and drug safety is of great concern, research related to enantioselectivity is very important. Recently, chiral materials have rapidly been developed with outstanding application prospects in the fields of enantio-separation5–9 and asymmetric synthesis.10–16 Normally, enantioselective sites are modified with chiral receptors, such as amino acids17,18 and cyclodextrins,19,20 and their asymmetric synthesis is costly.21–23 Therefore, the preparation of functional chiral nanomaterials from self-assembly has become a burgeoning topic for improving enantioselectivity, because well-designed chiral molecules can form ordered aggregates with advanced features such as chiral transfer, amplification, and recognition.24–27Amphiphilic molecules are known to effectively self-assemble into various nanostructures in specific solvents. In our previous work, we confirmed that implanting chiral centers into amphiphilic rod-coil block molecules induces steric hindrance to the molecular interactions, giving rise to various aggregation morphologies.28–33 It was also found that chiral groups on different sites of molecules lead to great changes in aggregation behaviors.34,35 In detail, molecules with chiral groups on a rigid site prefer twisting stacking mode to weaken the steric hindrance of π–π packing interaction from overlap stacking mode.
The twisted assembly can effectively amplify chirality in an orderly manner. Chiral groups on hydrophilic coil sites also result in a relatively tight assembly mainly with twists, according to hydrophilic–hydrophobic interaction. These chiral amphiphiles self-assemble in aqueous solution, giving rise to an enantioselective environment. Thus, the obtained chiral aggregates exhibit obvious enantioselectivity in the process of enantioseparation and asymmetric synthesis of small molecules. However, the current issue is that the enantiomeric excess (ee) value of asymmetric synthesis reactions still needs to be improved. In addition, the advantage of aggregates as a nanoreactor is not significant, as the refresh process is indeed cumbersome.35 Therefore, developing a simple, efficient, and recyclable asymmetric synthesis system to enhance the catalytic activity and structural stability of nanoreactors remains a challenge.
As a well-known molecular sieve, SBA-15 possesses wide application prospects in catalysis, separation, biology, and nanomaterials due to its large specific surface area, uniform pore diameter, and structural heat-resistant stability.36–43 For chiral materials based on SBA-15, Liu et al. reported the modification of SBA-15 pore walls with chiral molecules for enantio-separation,44 and simultaneously emphasized the importance of pore size in improving the separation performance. However, in their work, the ee value only reached 2.9% because the separation process was mainly based on weak molecular interaction. Obviously, it is difficult to achieve high chiral selectivity only through the interaction between small molecules. Based on these current studies, we formulated an idea that chiral aggregation in a confined space could improve enantioselectivity, and for the first time, we constructed an efficient asymmetric synthesis nanoreactor by introducing an SBA-15 channel as the assembly space.
Thus, in this study, we designed a series of coil-rod-coil molecules with shorter hydrophilic chains and lateral methyl groups in either hydrophilic chains or hydrophobic chains, or both hydrophilic and hydrophobic chains, which were connected to the SBA-15 pore walls to form SA-M composites. Structural characterization of the composites precisely proceeded, as well as analysis of chiral aggregation behaviors. The structure of SA-M possesses a high pore volume with a rich S-environment constructed from chiral assembly, providing a nano chiral reactor. As expected, the obtained SA-M series materials showed superior asymmetric synthesis in nucleophilic substitution synthesis of small molecules. These materials also exhibited good recyclability with almost negligible errors. In this work, we fully demonstrate the potential applications of SA-M series materials, which provide a platform for asymmetric synthesis and catalysis.
Results and discussion
Material design and synthesis
Amphiphiles with rigid blocks and flexible coils self-assemble into regularly packed aggregates through cooperation of π–π stacking interactions and hydrophilic–hydrophobic interactions. Furthermore, chiral amphiphiles carry on tilted stacking to overcome steric hindrance from additional chiral groups, which has been proven in previous studies.33,34 The chiral aggregation generated amplified chirality and exhibited significant enantioselectivity in asymmetric synthesis. To achieve a highly efficient enantioselective system with enhanced ee value and recyclability, we conceived chiral assembly in the confined pore space of SBA-15. In addition, the hydrophobic environment inside the pore will be conducive to the reactivity, compared to the aqueous environment outside the hole. To verify this hypothesis, as shown in Fig. 1, rod-coil amphiphilic molecules 1–3 were grafted onto modified mesoporous silica SBA-APT (the abbreviation for SBA-15 modified with APTES (3-aminopropyl triethoxysilane)) by covalent bond, resulting in materials SA-M1, SA-M2, and SA-M3.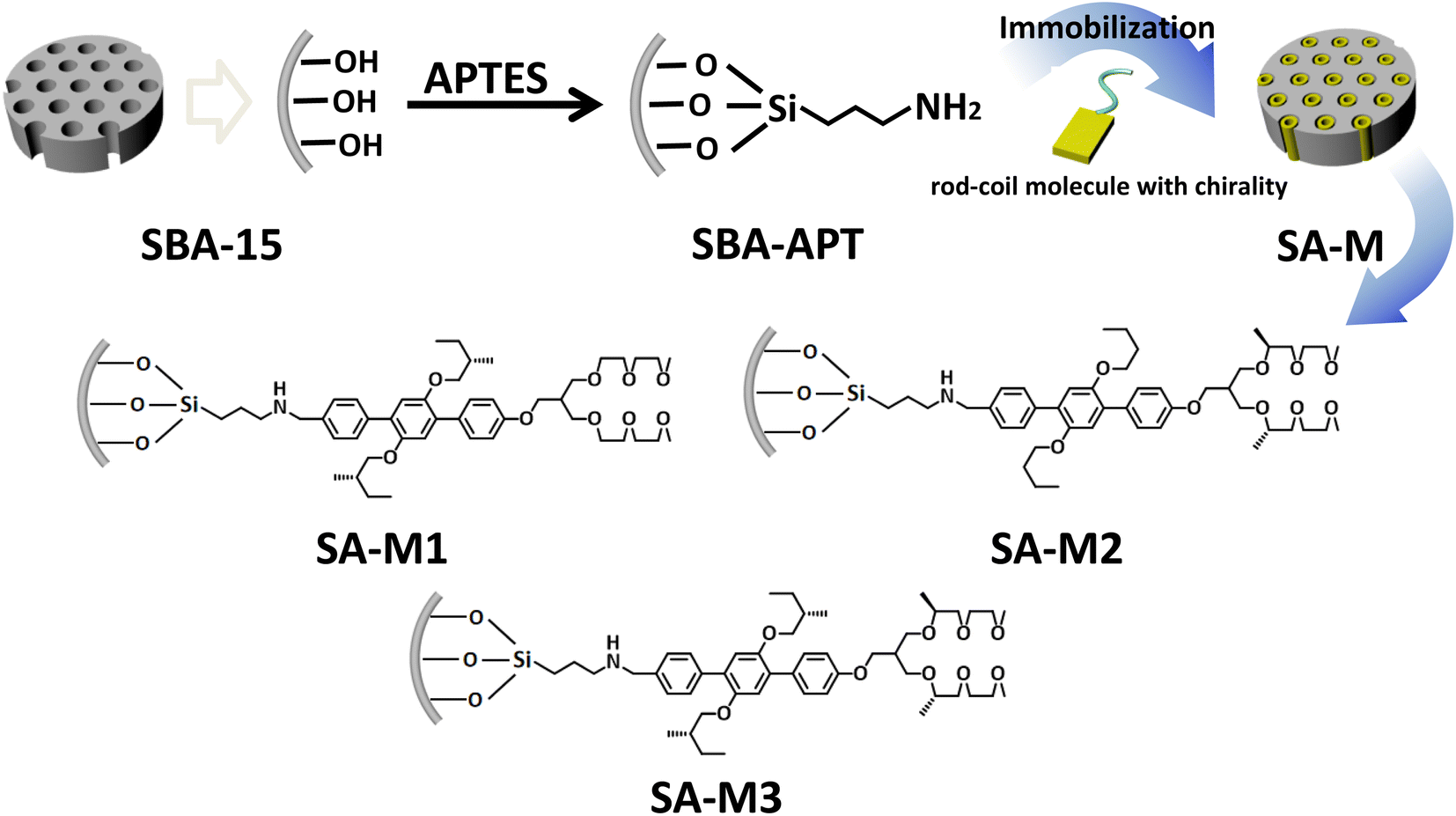 | ||
| Fig. 1 Schematic illustration of preparation routes for SA-M materials, and detailed structures of amphiphilic rod-coil molecules 1–3 covalently bonded to the pore walls. | ||
Rod-coil amphiphilic molecules 1–3 were designed and synthesized with a triphenyl group as a rigid segment connecting hydrophilic/hydrophobic chiral coils, as shown in Scheme S1.† The molecular structure was confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy and matter-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS), as shown in Fig. S1–S6.†
Structural characterization
For characterizing structural changes of nanochannels during SA-M synthesis, we initially carried out N2 adsorption–desorption experiments for all the materials at 77 K, as shown in Fig. 2. The N2 adsorption capacity was gradually decreased upon surface modification of SBA-15 to SBA-APT. Further, molecules 1–3 were connected to SBA-APT and generated SA-M1, SA-M2, and SA-M3, respectively, for which the adsorption tendency showed a more significant downward trend (see Fig. 2a and c). In addition, typical type I and type IV gas adsorption isotherms were observed, revealing microporous and mesoporous characteristics according to the IUPAC classification.45 It can be inferred that the channel type porous structure of SBA-15 is maintained after hybridizing with molecules.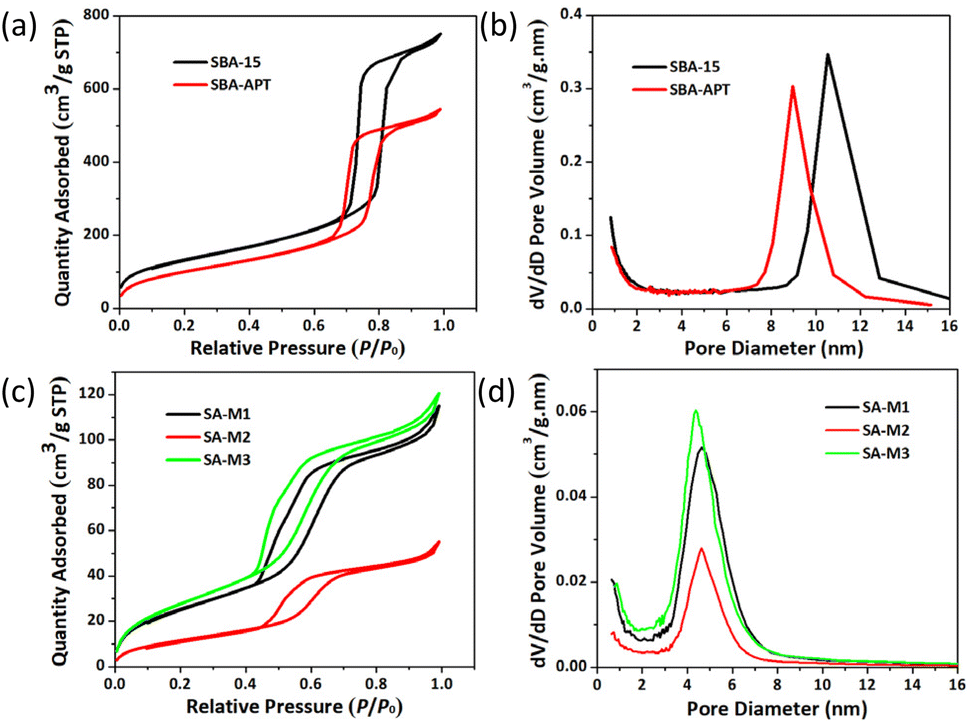 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms and (b) pore size distribution of SBA-15 and SBA-APT. (c) N2 adsorption–desorption isotherms and (d) pore size distribution of SA-M materials. | ||
The desorption curve lags behind the adsorption curve due to the capillary condensation effect of the pore structure that originated from the SBA-15 matrix structure. The BJH pore size distribution shown in Fig. 2b and d shows that the pore size also gradually decreased, but remained a uniform narrow distribution. All the results fully explain that molecules 1–3 were successfully loaded inside the pores. In detail, the pore size values of SA-M1, SA-M2, and SA-M3 were confirmed as 4.62, 4.62, and 4.40 nm from the Barret–Joyner–Halenda (BJH) porosity analysis, and the mesopore volume values were 0.11, 0.06, and 0.12 cm3 g−1, respectively, which are 19.2, 9.9, and 20.0% relative to that of SBA-APT (see Table S1†).
To analyze the exact content of molecules 1–3 loaded inside pores, thermogravimetric analysis (TGA) experiments were conducted for the above materials. The TGA curves in Fig. 3a clearly show weight loss and the corresponding temperatures. The initial SBA-15 showed a total weight loss of 1.6%, which should be attributed to the evaporation of gases (air) and water molecules adsorbed onto the surface, as well as the dehydration of –OH groups on the surface of silica within the full temperature range of room temperature to 873 K.44,46 The total weight of SBA-APT was decreased to 90.3%, possibly due to the thermal decomposition of APTES in the pores. In the case of SA-M1, SA-M2, and SA-M3, there was a considerable decrease in the final weight values compared to the intermediate SBA-APT, which was attributed to the grafted block molecule 1–3. The molecular content was calculated to be 12.7%, 16%, and 14.5%, for SA-M1, SA-M2, and SA-M3, respectively.
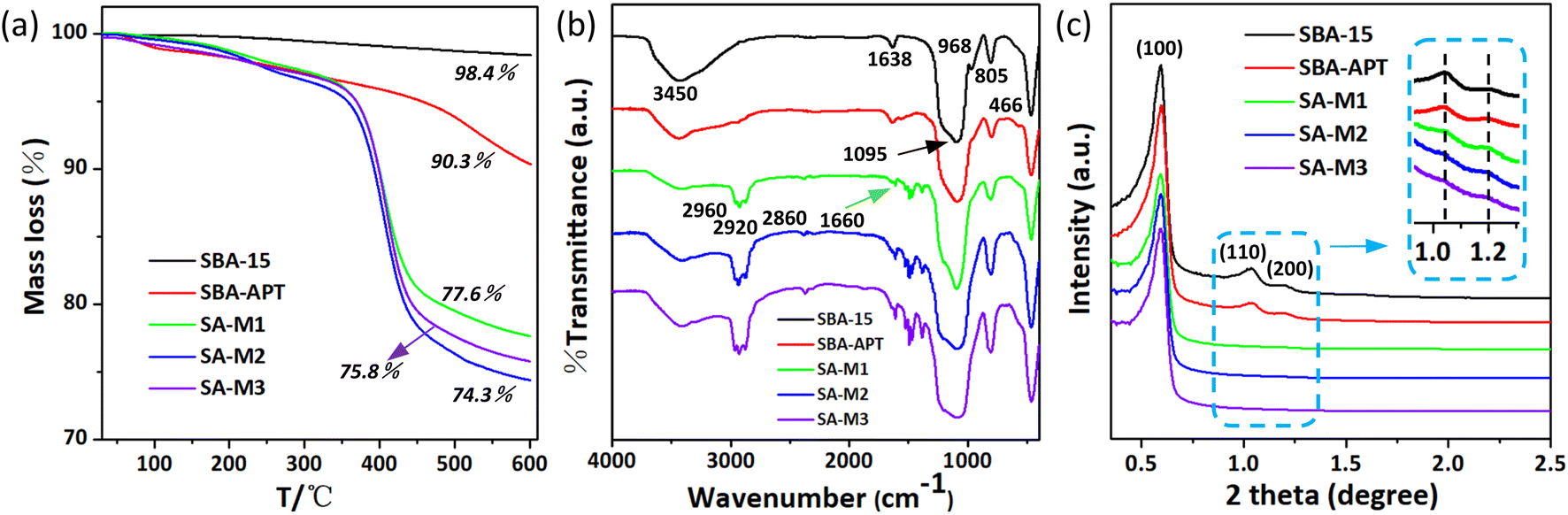 | ||
| Fig. 3 (a) TGA graph, (b) FT-IR spectra, and (c) SAXS patterns for SBA-15, SBA-APT, and SA-M materials. | ||
SA-M2 showed the maximum weight loss, which can be explained from molecular aggregation behaviors inside pores. As shown in Fig. 1, there was no methyl side group on the hydrophobic chain for molecule 2, and a relatively small cross-sectional area was exhibited among the three. Thus, there was low steric hindrance during the aggregation process for molecule 2, resulting in a packed arrangement inside pores to form SA-M2. Therefore, SA-M2 accommodated more loading. This phenomenon was in agreement with the N2 adsorption results in which SA-M2 accommodated the least porosity, indicating that the pores were greatly occupied.
According to TGA verification, significant weight loss of the SA-M materials occurred above 623 K (350 °C), suggesting that molecules 1–3 were stably bonded to the matrix. To further demonstrate the successful synthesis of the SA-M materials, their detailed chemical structures were characterized by infrared spectroscopy (see the FT-IR spectra in Fig. 3b). For the initial SBA-15 matrix, we observed a strong and broad absorption band at 1095 cm−1 that was attributed to the Si–O–Si anti-symmetric stretching vibration, with the peak at 968 cm−1 as the Si–OH bending vibration, and the peaks at 805 cm−1 and 466 cm−1 as the Si–O bond symmetric stretching vibration. The broad peak at 3450 cm−1 and the peak near 1638 cm−1 were attributed to the OH anti-symmetric stretching vibration of SA-M and the H–O–H bending vibration, respectively.
After APTES grafting, we observed several significant changes: the vibration peak intensity of silanol at 968 cm−1 weakened, confirming the consumption of Si–OH through APTES silanization, which indicated successful grafting of APTES. The new peaks at 2860–2960 cm−1 and 1380–1430 cm−1 were attributed to the stretching vibration of the organic molecular units methyl and methylene and the deformation vibration of methyl, respectively.44 The above analysis indicates that the SA-M materials contain SBA-APT and organic molecular structural units. In addition, the N–H stretching vibration and N–H bending vibration of secondary amine were observed at 3450 and 1660 cm−1, respectively, and originated from reaction between –NH2 of SBA-APT and –CHO of molecules 1–3. The above analysis indicates that SBA-APT materials are connected to organic block molecules 1–3 through C–N bonds, and the SA-M materials were successfully obtained.
Fig. 3c shows the small angle X-ray scattering (SAXS) pattern, and for all the materials, three significant scattering peaks were observed, which were indexed as (100), (110), and (200) reflections of two-dimensional symmetry (p6mm). These results indicate that all samples possess a clear hexagonal mesoporous structure.47 More importantly, these hexagonal ordered pore structures remained upon modification and further reaction, illustrating that molecules 1–3 were regularly bonded inside the pores, which was consistent with N2 adsorption and the FT-IR results mentioned above. Moreover the gradual decrease in scattering intensity of these three peaks was attributed to the difference in scattering contrast between pores and walls, as well as the influence of organic groups on the surface of nanochannels.
Microstructures of SBA-15, SBA-APT, and SA-M materials were further identified through transmission electron microscopy (TEM) experiments, as shown in Fig. 4 and S7.† Parallel channels and hexagonally arranged pores can be clearly observed for all the materials, and were attributed to the (110) and (100) directions of the p6mm phase, respectively, which are consistent with the results of the SAXS study. These arrays are distributed along the same parallel direction and are assembled perpendicular to their hexagonal pore arrangement. In addition, we marked lattice fringes and measured intervals, as shown in Fig. 4, which were in agreement with the N2 adsorption results, displaying approximately 10 nm intervals on the (100) plane.
 | ||
| Fig. 4 Negative-stained TEM images of (a) SBA-APT, (b) SA-M1, (c) SA-M2, and (d) SA-M3 with lattice fringes. | ||
The morphology was also observed by scanning electron microscopy (SEM), as shown in Fig. S8.† Mesochannels on the surface are clearly displayed for SBA-15, and the pore arrays of longitudinal nanochannels are arranged at equal intervals for all the samples. In the case of the SA-M materials, the channels were partially occupied and exhibited relatively blurry stripes. These phenomena can be attributed to the high molecular content of small molecules up to 16%, as characterized by TGA.
Co-assembly behaviors with achiral molecules
Similar rod-coil molecules with chiral side groups, which were studied in our previous work, can self-assemble into nanofibers, nanotubes, and nanosheets via slight changes of the molecular structures for nanoreactors. The experimental results showed that the assemblies exhibited significant circular dichroism (CD) adsorption due to the chiral aggregation.34,35 Based on the structural characterization of the SA-M series materials, we focused on the chiral assembly behavior of molecules 1–3 inside the confined pores. As shown in Fig. 5, weak CD signals were exhibited at approximately 350 nm for SA-M in aqueous solution, which is in agreement with the UV absorption wavelength shown in Fig. S9,† and suggests the presence of the chiral assembly of the organic molecules inside the channel.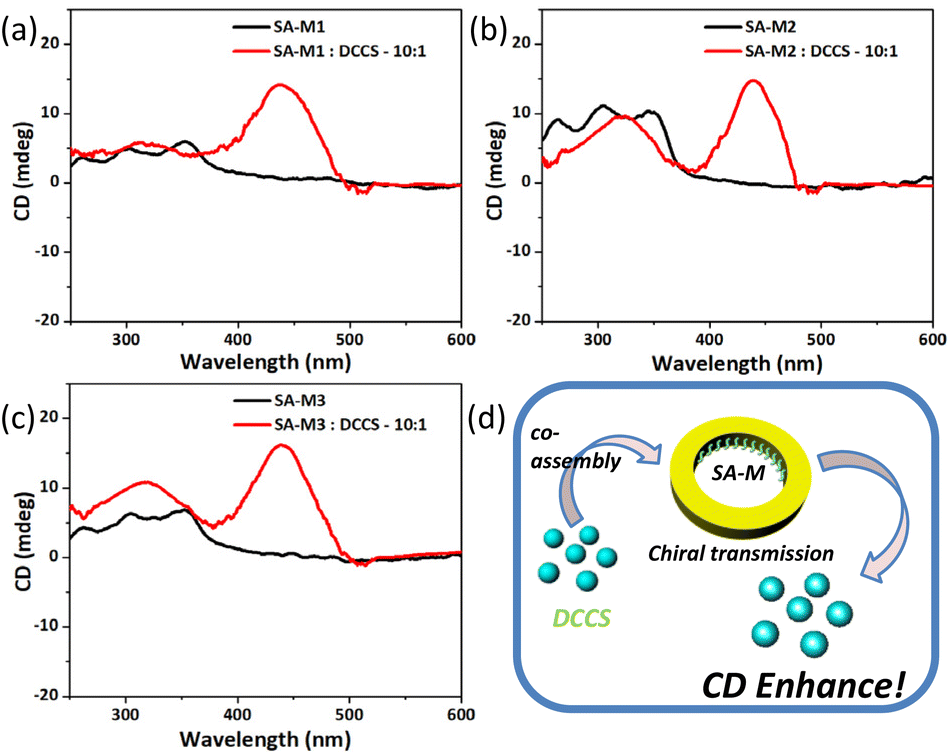 | ||
| Fig. 5 (a–c) CD spectra of SA-M materials before and after co-assembly with DCCS in H2O/THF (v/v = 9/1). (d) Schematic representation of chiral transfer through co-assemblies. | ||
To characterize the chiral aggregation inside the pores, we introduced 7-(diethylamino)coumarin-3-carboxylic acid N-succinimidyl ester (DCCS), an achiral molecule, into the system as an intermediate for signal conversion to analyze the co-aggregation behavior with SA-M. Upon the addition of DCCS, a new strong absorption peak was generated at 445 nm, which was consistent with the UV absorption peak position of DCCS (see Fig. S10†). Because DCCS is an achiral molecule, the corresponding CD signal resulted from chiral transfer from chiral aggregation inside the pores, as shown in Fig. 5d.
To further verify this, we replaced the aqueous solvent with pure THF under the same concentration, for which no significant absorption was found, as shown in Fig. S11.† On the subject of chirality transfer, it required very close distance and an adapted arrangement between the subject and object.48 An effective chirality transfer can only occur under assembly conditions for the SA-M molecular system. In organic solvents, amphiphilic molecules are dissolved into a relatively free state compared with aqueous environments, which would only weakly enable chiral transfer. Based on the above results, we confirmed the chiral self-assembly behavior in the pores of SA-M, which is consistent with our expected results.
Nanoreactors with enantioselectivity in nucleophilic substitution
Based on the research results above, we attempted to apply SA-M materials as enantioselective nanoreactors in the asymmetric synthesis of small molecules. SA-M materials were composed of SBA-APT pores and chiral amphiphilic rod-coil molecules connected by covalent bonds.Chiral assemblies inside the confined pores would promote selective capillary adsorption effects and provide an S- or R-environment for asymmetric synthesis. To verify this hypothesis, we chose the nucleophilic substitution reaction between p-methylbenzenesulfonyl chloride (TsCl) and 1-methoxypropane-2-ol (PM) as the model reaction to estimate the capacity of enantioselectivity in nucleophilic substitution (see Fig. 6). The two reactants were added to a THF/H2O (v/v = 1/9) solution containing SA-M, to which pyridine was added as a base catalyst. The system was then stirred overnight at room temperature to synthesize asymmetric 1-methoxypropan-2-yl-4-methylbenzene sulfonate (PM-Ts).
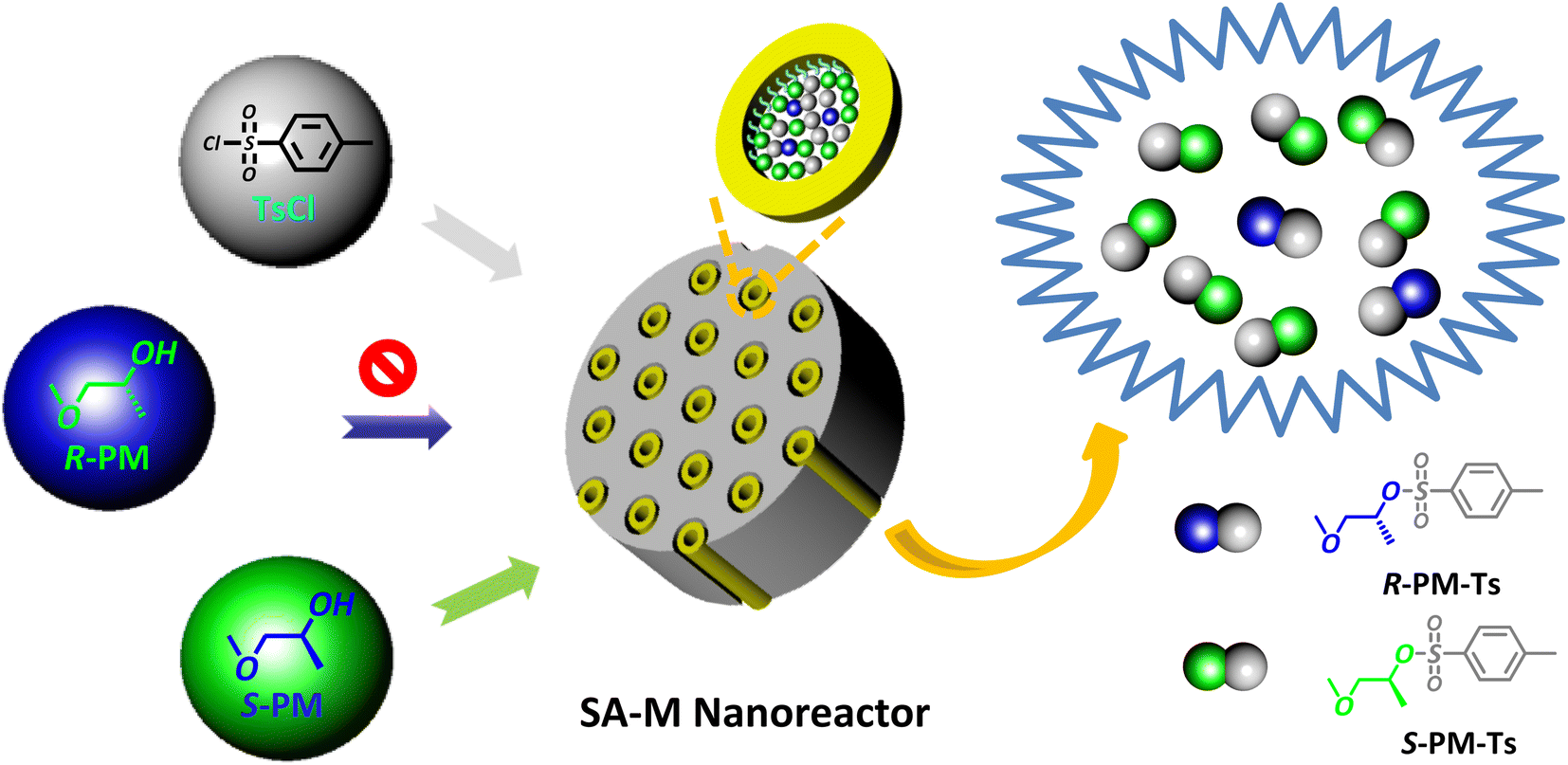 | ||
| Fig. 6 Schematic illustration of the asymmetric nucleophilic substitution reaction between PM and TsCl in the presence of SA-M materials as nanoreactors. | ||
The molecular weight of the obtained product PM-Ts was confirmed by HPLC-MS analysis, as shown in Fig. S12.† Subsequently, to determine and evaluate the enantioselectivity of SA-M materials, chiral HPLC determination was carried out to analyze the enantiomeric purity according to the experimental conditions and analytical methods previously reported.34,35 HPLC analysis for the target products in Fig. 7a indicated that all the SA-M materials exhibited S-preferred synthesis with ee values of 37.82%, 62.24%, and 71.75% for SA-M1, SA-M2, and SA-M3, respectively. SA-M provided a hydrophobic environment inside the pores compared to the external aqueous solution, which was favorable for the hydrophobic molecule TsCl and pyridine to enter inside the pore. Additionally, S-PM would take priority over R-PM in entering the pores, due to the S-type chiral coils of rod-coil molecules, which is conducive to adsorption of S-PM.35 Thus, it tended to produce S-preferred synthesis, with ee values up to 72%.
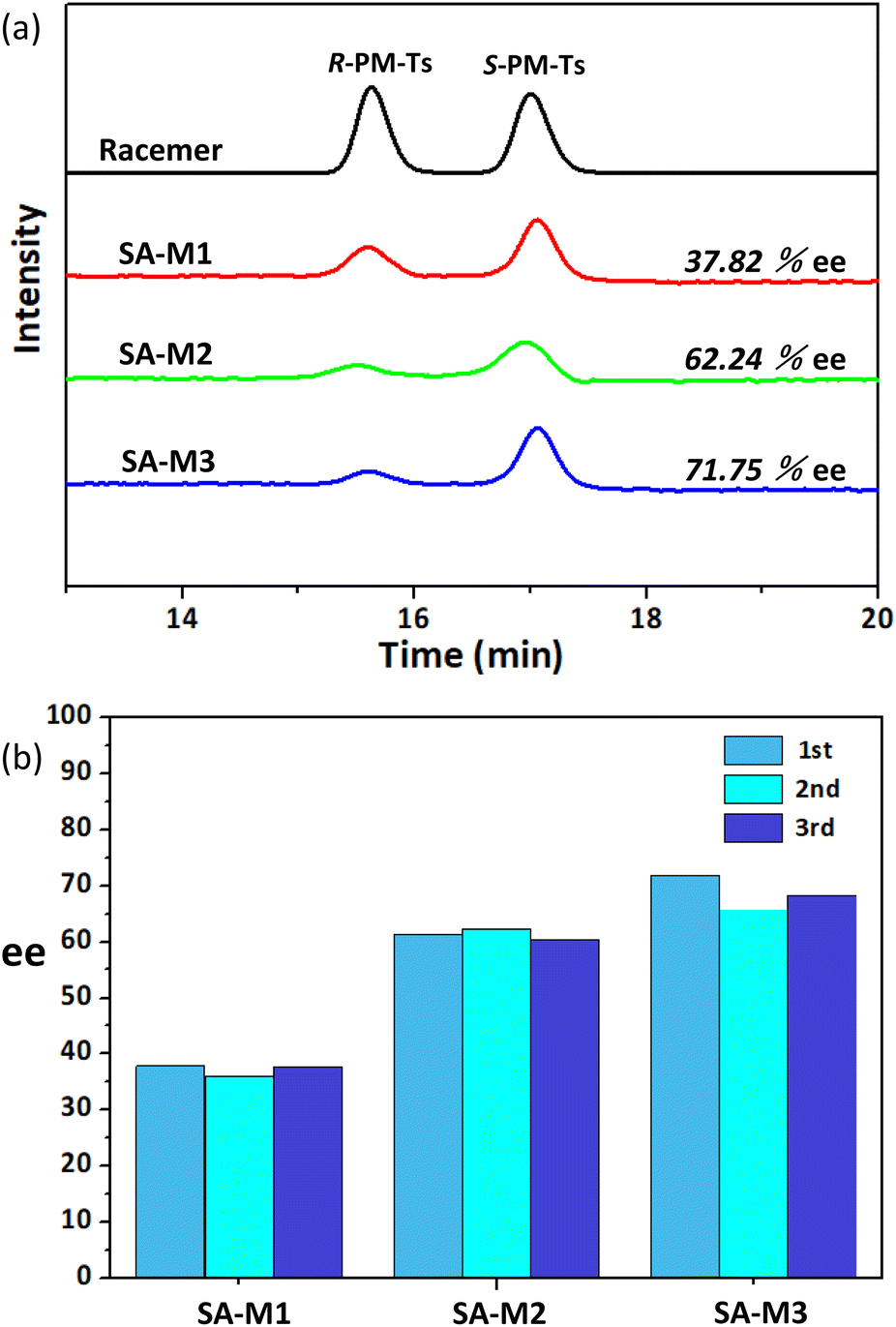 | ||
| Fig. 7 (a) Chiral HPLC chromatograms of the nucleophilic substitution reaction between PM and TsCl: results from racemic PM in the presence and absence of SA-M materials; (b) cycles of asymmetric synthesis. | ||
The ee value can be determined by the degree of chiral aggregation. SA-M3, which contains six chiral centers per molecular portion, proceeded to conduct maximum chiral assembly, and efficient enantioselectivity resulted among the three. There was a certain impact of the loading amount of chiral molecules on the results for SA-M2. For SA-M1, hydrophobic chiral groups bulked the cross-sectional area of rods, which hindered the π–π stacking of terphenyl groups, while SA-M2 and SA-M3 containing hydrophilic chiral coils on the end position behaved with magnified chirality as well as higher enantioselectivity.
Consequently, SA-M3 contained the most chiral centers, and showed stronger enantioselectivity through the synergistic effects of hydrophobic–hydrophilic and π–π stacking interactions. It is worth noting that the grafted porous materials exist in a precipitated form in solution and can be reused after simple filtration. To verify the recyclability of SA-M materials, we refreshed the precipitates (SA-M materials) through washing and filtering to conduct repeated experiments. Taking SA-M1 as an example, the ee values obtained from the three repeated experiments were 37.82%, 35.98%, and 37.78%, with a maximum error less than 2%, which demonstrated the possibility of effective practical application (see Fig. 7b and S13†).
Compared with the previously reported catalysts for asymmetric nucleophilic substitution reactions of small molecules, the SA-M materials possessed advanced properties, as summarized in Table S2.† The catalytic activity of the SA-M material was significantly improved, with an ee value of 71.75%. Because the chiral aggregation occurred within nanochannels, the confined chiral environment led to superior catalytic activity: capillary condensation as well as the hydrophobicity inside pores prompted small molecules to enter the nanoreactor. Additionally, the S-PM prioritized due to the enantioselectivity of the S-aggregates. Compared with pure organic aggregates, covalently bonded SA-M materials can maintain a stable porous chiral structure in various solvents and at a certain higher temperature with an efficient, environmentally friendly recycling performance through a simple washing process.
Conclusions
In summary, we successfully synthesized SA-M materials containing amphiphilic chiral rod-coil molecules covalently bonded to modified SBA-15 pore walls, and demonstrated chiral assembly behavior inside pores. Due to the excellent selectivity of the SA-M series materials towards enantiomeric compounds, these materials were applied as enantioselective nanoreactors for asymmetric synthesis of small molecules. There was a maximum ee value of 71.75% for the nucleophilic substitution reaction between p-methylbenzenesulfonyl chloride and 1-methoxypropane-2-ol. These materials also exhibited good durability and recyclability, with a maximum error of less than 2% after three cycles of reuse, which is important in practical applications. Therefore, our research results can provide an important porous materials platform for chiral asymmetric synthesis.Experimental
Synthetic method for materials SA-M1–3
Amphiphilic block organic molecules 1, 2, and 3 were synthesized by the well-known Suzuki coupling reaction (see Scheme S1†). SBA-15 was surface functionalized with 3-amino-propyl triethoxysilane to produce SBA-APT with sufficient amino groups on the pore walls. Taking the synthesis of SA-M1 as an example, molecule 1 (0.36 g) was dissolved in 50 mL of ethanol solvent and placed in a 100 mL one-neck round-bottom flask together with SBA-APT (0.127 g). Then, it was refluxed for 24 h, followed by concentrating and re-dissolving in 50 mL mixed solvent of methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane = 1:1 for the net reduction reaction. In an ice bath, 0.075 g NaBH4 was added, and the solution was stirred for 12 h. The residue was washed with ethanol 3 times, and subsequently freeze-dried for 12 h, producing the SA-M1 final material. The synthetic methods for SA-M2 and SA-M3 are similar to that of SA-M1.
:dichloromethane = 1:1 for the net reduction reaction. In an ice bath, 0.075 g NaBH4 was added, and the solution was stirred for 12 h. The residue was washed with ethanol 3 times, and subsequently freeze-dried for 12 h, producing the SA-M1 final material. The synthetic methods for SA-M2 and SA-M3 are similar to that of SA-M1.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Hui-Yu Zhao: methodology, validation, formal analysis, and writing – original draft. Qing Xu and Gui-Lang Liu: assisted in preparation of the molecules and analyzing the experimental results. Yi-Rong Pei and Long Yi Jin: conceptualization, methodology, investigation, resources, supervision, project administration, funding acquisition, and writing – review and editing. All authors read and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jilin Province (YDZJ202301ZYTS297), the National Natural Science Foundation of China (grant numbers 21961041 and 21562043), and the Higher Education Discipline Innovation Project (D18012).Notes and references
- X. Zhao, S. Zang and X. Chen, Chem. Soc. Rev., 2020, 49, 2481 RSC.
- B. Kasprzyk-Hordern, Chem. Soc. Rev., 2010, 39, 4466 RSC.
- H. Y. Xu and X. D. Zou, Science, 2019, 364, 632 CrossRef PubMed.
- P. Peluso and B. Chankvetadze, Chem. Rev., 2022, 122, 13235 CrossRef.
- Q. Cheng, Q. Ma, H. Pei and Z. Mo, Sep. Purif. Technol., 2022, 292, 121034 CrossRef CAS.
- S. M. Xie, X. X. Chen, J. H. Zhang and L. M. Yuan, TrAC, Trends Anal. Chem., 2020, 124, 115808 CrossRef CAS.
- Y. Chen, S. Huang, L. Xia, Y. Hu and G. Li, Anal. Chem., 2024, 96, 1380 CrossRef CAS.
- Y. Chen, L. Xia and G. Li, J. Chromatogr. A, 2022, 1677, 463341 CrossRef CAS.
- T. Liu, Z. Li, J. Wang, J. Chen, M. Guan and H. Qiu, Chem. Eng. J., 2021, 410, 128247 CrossRef CAS.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825 CrossRef CAS.
- P. Pecchini, M. Fochi, F. Bartoccini, G. Piersanti and L. Bernardi, Chem. Sci., 2024, 15, 5832 RSC.
- J. Sui, N. Wang, J. Wang, X. Huang, T. Wang, L. Zhou and H. Hao, Chem. Sci., 2023, 14, 11955 RSC.
- J. Y. C. Lim, I. Marques, V. Felix and P. D. Beer, Angew. Chem. Int. Ed., 2018, 57, 584 CrossRef CAS PubMed.
- S. Li, M. Veksler, Z. Xu, L. Xu, C. Xu and N. A. Kotov, ACS Energy Lett., 2021, 6, 1405 CrossRef CAS.
- H. Guan, C. H. Tung and L. Liu, J. Am. Chem. Soc., 2022, 144, 5976 CrossRef CAS.
- L. Chen, Y. Chen, Y. Zhang and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 7654 CrossRef CAS.
- L. Huang, Y. X. Li, Q. Lin, B. Y. Lou and Y. T. Chen, Amino Acids, 2018, 50, 1549 CrossRef CAS PubMed.
- S. Y. Zhang, M. Cheng, M. K. Dhinakaran, Y. Sun and H. B. Li, ACS Nano, 2021, 15, 13148 CrossRef CAS.
- S. Y. Zhang, X. Chen, L. D. Sun and H. B. Li, ACS Appl. Nano Mater., 2020, 3, 4351 CrossRef CAS.
- Q. Huang, L. Jiang, W. Lian, J. Gui, D. Xu, W. Wu, Y. Nakai, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Org. Chem., 2016, 81, 3430 CrossRef CAS PubMed.
- D. N. Dybtsev and K. P. Bryliakov, Coord. Chem. Rev., 2021, 437, 213845 CrossRef CAS.
- H. Zhang, L. Lou, K. Yu and S. Liu, Small, 2021, 17, 2005686 CrossRef CAS PubMed.
- J. Shen and Y. Okamoto, Chem. Rev., 2016, 116, 1094 CrossRef PubMed.
- Y. Sang and M. Liu, Chem. Sci., 2022, 13, 633 RSC.
- C. Liu, J.-C. Yang, J. W. Y. Lam, H.-T. Feng and B. Z. Tang, Chem. Sci., 2022, 13, 611 RSC.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef.
- Q. Ye, F. Zheng, E. Zhang, H. K. Bisoyi, S. Zheng, D. Zhu, Q. Lu, H. Zhang and Q. Li, Chem. Sci., 2020, 11, 9989 RSC.
- N. Ye, Y. R. Pei, Q. Han, M. Lee and L. Y. Jin, Soft Matter, 2021, 17, 6661 RSC.
- N. Ye, Y. R. Pei, Q. Han and L. Y. Jin, Soft Matter, 2023, 19, 1540 RSC.
- Y. Yang, F. Chen, X. Tian, T. Chen, L. Wu and L. Y. Jin, Soft Matter, 2019, 15, 6718 RSC.
- S. Yu, R. Sun, T. Chen and L. Y. Jin, Soft Matter, 2018, 14, 6822 RSC.
- S. Yu, Y. Yang, T. Chen, J. Xu and L. Y. Jin, Nanoscale, 2017, 9, 17975 RSC.
- X. Gou, J. Lu, H. Y. Zhao, Y. R. Pei and L. Y. Jin, Soft Matter, 2023, 19, 6683 RSC.
- H. Y. Zhao, X. Gou, Y. R. Pei and L. Y. Jin, Langmuir, 2023, 39, 8824 CrossRef CAS.
- H. Y. Zhao, G. L. Liu, Q. Xu, Y. R. Pei and L. Y. Jin, Soft Matter, 2024, 20, 1884 RSC.
- H. Moon, S. Han and S. L. Scott, Chem. Sci., 2020, 11, 3702 RSC.
- Y. Xie, Y. Lai, X. Wu, J. Qiu, X. Wang, J. Zuo, L. Ye and Y. Yuan, Appl. Surf. Sci., 2024, 654, 159399 CrossRef.
- Z. A. Akbar, S. V. Situmorang, I. Yati, R. T. Yunarti, S. N. Surip and M. Ridwan, Int. J. Hydrogen Energy, 2024, 57, 1506 CrossRef.
- X. Shang, X. Yang, G. Liu, T. Zhang and X. Su, Chem. Sci., 2024, 15, 4631 RSC.
- Y. Zhang, C. Zhang, G. Liu, L. Wang and Z. Pan, Appl. Surf. Sci., 2023, 616, 156515 CrossRef.
- R. H. Maricely, J. Norambuena, H. Hu, B. Thomas, C. Tang, J. M. Boyd and T. Asefa, ACS Appl. Mater. Interfaces, 2023, 15, 17459 CrossRef PubMed.
- E. N. Kusumawati, T. Sasaki and M. Shirai, ACS Appl. Nano Mater., 2023, 6, 17913 CrossRef CAS.
- P. Shanmugam, S. M. Smith, S. Boonyuen and A. Luengnaruemitchai, Environ. Res., 2023, 224, 115496 CrossRef CAS.
- X. Li, C. Meng, Y. Meng, L. Gu, Q. Chen and H. Liu, Colloids Surf., A, 2019, 581, 123789 CrossRef CAS.
- M. J. B. De souza, T. H. A. Silva, T. R. S. Ribeiro, A. O. S. Da Silva and A. M. G. Pedrosa, J. Therm. Anal. Calorim., 2020, 140, 167 CrossRef.
- U. Patil, A. Fihri, A.-H. Emwasa and V. Polshettiwar, Chem. Sci., 2012, 3, 2224 RSC.
- F. Vibert, S. R. A. Marque, E. Bloch, S. Queyroy, M. P. Bertrand, S. Gastaldi and E. Besson, Chem. Sci., 2014, 5, 4716 RSC.
- S. Pandey, S. Mandal, M. B. Danielsen, A. Brown, C. Hu, N. J. Christensen, A. V. Kulakova, S. Song, T. Brown, K. J. Jensen, J. Wengel, C. Lou and H. Mao, Nat. Commun., 2022, 13, 76 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization and analytical data. See DOI: https://doi.org/10.1039/d4sc03171g |
| This journal is © The Royal Society of Chemistry 2024 |