
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclic peptides targeting the SARS-CoV-2 programmed ribosomal frameshifting RNA from a multiplexed phage display library†
Jacob A.
Iannuzzelli‡§
a,
Rachel
Bonn¶§
bc,
Andrew S.
Hong§
 a,
Abhijith Saseendran
Anitha
ad,
Jermaine L.
Jenkins
bc,
Joseph E.
Wedekind
*bc and
Rudi
Fasan
*ad
a,
Abhijith Saseendran
Anitha
ad,
Jermaine L.
Jenkins
bc,
Joseph E.
Wedekind
*bc and
Rudi
Fasan
*ad
aDepartment of Chemistry, University of Rochester, Rochester, NY 14627, USA
bDepartment of Biochemistry and Biophysics, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642, USA. E-mail: joseph.wedekind@rochester.edu
cCenter for RNA Biology, University of Rochester School of Medicine and Dentistry, Rochester, NY 14642, USA
dDepartment of Chemistry & Biochemistry, The University of Texas at Dallas, Richardson, TX 75080, USA. E-mail: Rudi.Fasan@UTDallas.edu
First published on 17th October 2024
Abstract
RNA provides the genetic blueprint for many pathogenic viruses, including SARS-CoV-2. The propensity of RNA to fold into specific tertiary structures enables the biomolecular recognition of cavities and crevices suited for the binding of drug-like molecules. Despite increasing interest in RNA as a target for chemical biology and therapeutic applications, the development of molecules that recognize RNA with high affinity and specificity represents a significant challenge. Here, we report a strategy for the discovery and selection of RNA-targeted macrocyclic peptides derived from combinatorial libraries of peptide macrocycles displayed by bacteriophages. Specifically, a platform for phage display of macrocyclic organo-peptide hybrids (MOrPH-PhD) was combined with a diverse set of non-canonical amino acid-based cyclization modules to produce large libraries of 107 structurally diverse, genetically encoded peptide macrocycles. These libraries were panned against the −1 programmed ribosomal frameshifting stimulatory sequence (FSS) RNA pseudoknot of SARS-CoV-2, which revealed specific macrocyclic peptide sequences that bind this essential motif with high affinity and selectivity. Peptide binding localizes to the FSS dimerization loop based on chemical modification analysis and binding assays and the cyclic peptides show specificity toward the target RNA over unrelated RNA pseudoknots. This work introduces a novel system for the generation and high-throughput screening of topologically diverse cyclopeptide scaffolds (multiplexed MOrPH-PhD), and it provides a blueprint for the exploration and evolution of genetically encoded macrocyclic peptides that target specific RNAs.
Introduction
RNA has emerged as a promising therapeutic target because of its key regulatory roles in numerous biological processes.1–4 Although traditionally viewed as a passive carrier of genetic information, whole-genome sequencing indicates that many transcripts comprise noncoding RNAs (ncRNAs)5 that regulate transcription, RNA processing, translation, and innate immunity.6–9 RNA also plays a pivotal role in controlling viral replication and bacterial homeostasis, providing new targets for antimicrobial development.10–13 The ability of RNA to adopt tertiary structures with deep grooves and concave surfaces as well as motifs for protein binding renders them susceptible to recognition by drug-like molecules.14–20Macrocyclic peptides represent an attractive class of bioactive agents capable of inhibiting biomolecular interactions.21–27 Their modest size (800–3000 Da) and conformational rigidity impart advantages over their linear counterparts, including increased target affinity,28,29 enhanced proteolytic stability30,31 and cell permeability.32–36 These beneficial characteristics make them ideal candidates for targeting intracellular complexes such as RNA-protein interactions. For example, representative work from our groups and others achieved disruption of the essential HIV Tat–TAR binding interaction through use of a focused group of cyclic peptides that bind the viral TAR RNA major-groove bulge with affinity and specificity.37,38 Albeit, cyclic peptides that target specific RNA molecules have been identified thus far via low throughput, structure-guided-design approaches, which are laborious and limited in terms of the sequence space that can be explored.39,40 Accordingly, high-throughput platforms capable of probing structurally and functionally diverse libraries of macrocyclic peptides against specific RNA targets are highly desirable.
Recently, some of us introduced an innovative platform to discover bioactive cyclic peptides that combines the production of genetically encoded macrocyclic organo-peptide hybrids (MOrPHs) using non-canonical amino acids with M13 bacteriophage display (i.e., MOrPH-PhD).41 This system allows for the exploration of large libraries up to 109 macrocyclic peptides against various target proteins and protein–protein interactions (PPIs). In this system, cyclization of a ribosomally derived peptide is achieved via a spontaneous, post-translational cross-linking reaction between a genetically encoded electrophilic unnatural amino acid (eUAA) and a proximal cysteine residue, leading to a side-chain-to-side-chain linked macrocyclic peptide.42,43 The eUAA cyclization module is incorporated into the precursor polypeptide via amber stop codon (UAG) suppression using an engineered aminoacyl tRNA synthetase/tRNACUA pair derived from Methanococcus jannaschii. In the MOrPH-PhD system, the cyclic peptide is fused to the N-terminus of the pIII phage coat protein. Via helper phage-assisted assembly, mature M13 phage particles display the MOrPH library on their surface, establishing a physical linkage between genotype and phenotype.41
Building upon this work, we were interested in exploring the utility of this platform for the discovery of cyclic peptides capable of targeting a RNA molecule, using the −1 programmed ribosomal frameshifting (−1 PRF) RNA of SARS-CoV-2 as a model target (Fig. 1). This RNA element plays a key role in the viral replication of SARS-CoV-2,44 which folds as a conserved three-stem H-type pseudoknot (PK) that functions as part of a ‘frameshift-stimulating sequence’ (FSS).45 We chose to target the FSS PK because it interacts with the host ribosome, leading to a −1 frameshift46 and the translation of two distinct viral polyproteins (encoded by ORF1a and ORF1ab) essential for the SARS-CoV-2 lifecycle.47 The −1 PRF FSS system is structurally and functionally conserved within the betacoronavirus genus, and mutations to this sequence are detrimental to viral propagation.10 Therefore, the FSS is an attractive target for antiviral therapy, as evident in recent studies showing that chemical targeting of this motif can interfere with the FSS-mediated frameshifting mechanism48,49 and reduce SARS-CoV-2 replication in cells.11,48 Despite progress in the development of platforms for high-throughput screening of genetically-encoded41,50–55 or phage-encoded56–60 macrocyclic peptide libraries, there are no reports of their successful application to target RNA to our knowledge. While the use of a phage-encoded library led to the discovery of a bicyclic peptide with sub-micromolar affinity for a DNA G-quadruplex,61 the application of a phage-display library of cyclic peptides constrained by a disulfide linkage against BIV TAR RNA resulted in compounds with no specificity against the target RNA.62
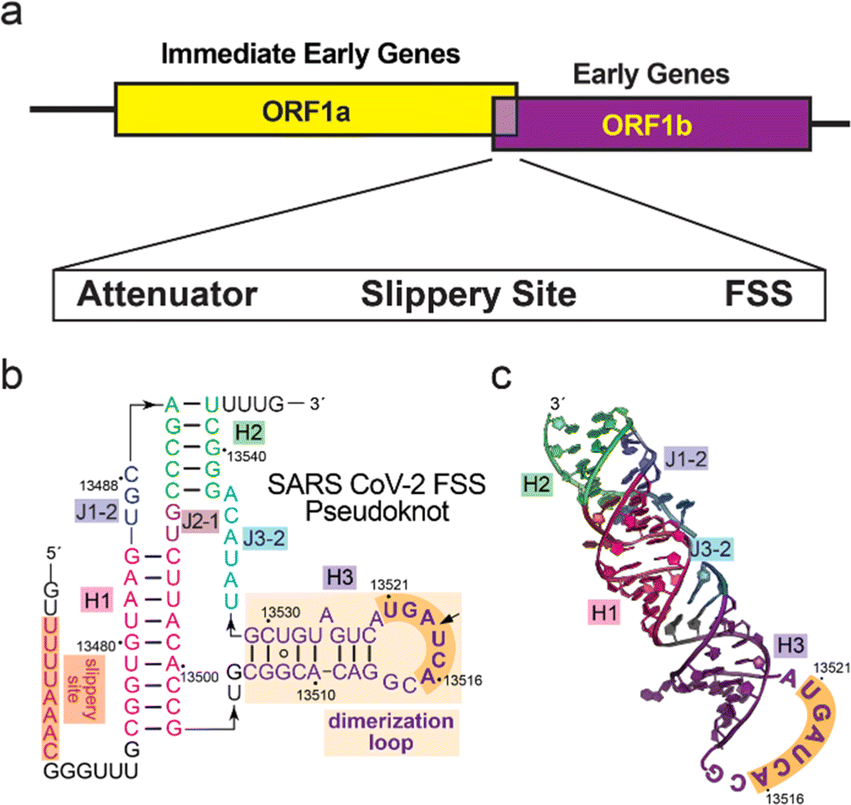 | ||
| Fig. 1 Overview of the −1 programmed ribosomal frameshifting stimulatory sequence (FSS) of SARS-CoV-2. (a) Genomic organization of the SARS-CoV-2 open reading frames (ORFs) whose expression is controlled by the FSS element. (b) Secondary structure of the FSS pseudoknot (PK) of SARS-CoV-2 with the dimerization loop hairpin (DL) boxed (orange). The PK sequence used for this study began at C13476 and ended at U13543. (c) Crystal structure of the FSS PK variant (PDB 7mky). The GNRA tetra loop in the original structure was replaced with the actual dimerization loop sequence. | ||
Here, we report the discovery and characterization of RNA targeting cyclic peptides using a “multiplexed MOrPH-PhD” system, in which phage-displayed MOrPH libraries are diversified by means of different eUAA cyclization modules (Fig. 2). This method led to the selection of multiple macrocyclic peptides that specifically and selectively target the −1 PRF RNA of SARS-CoV-2 (Fig. 2). Details of our multiplexed MOrPH-PhD approach are presented along with binding analysis to the FSS PK and localization using chemical mapping. These findings have broader implications for the identification and development of new antiviral and antibacterial molecules.
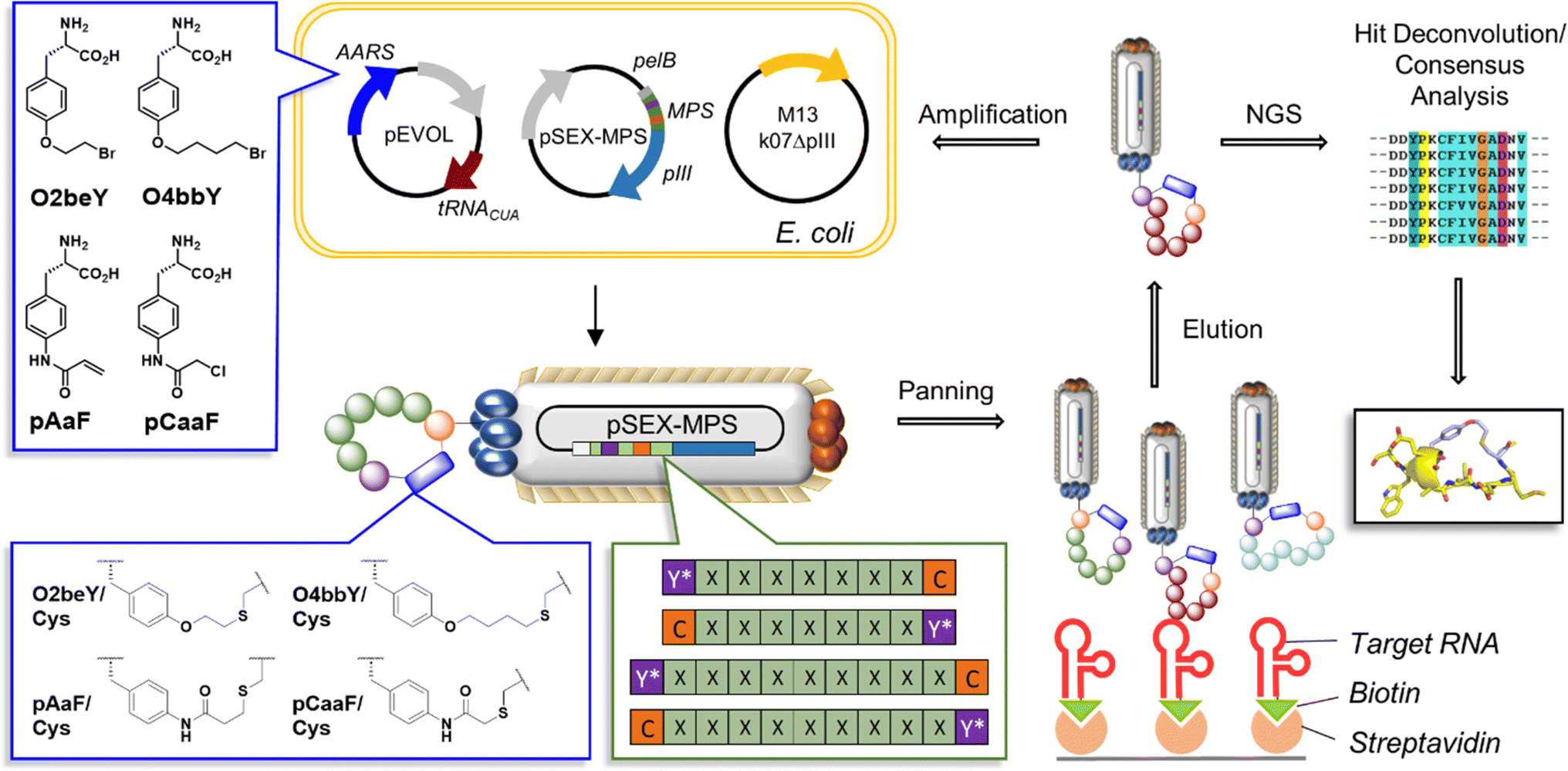 | ||
| Fig. 2 Multiplexed MOrPH-PhD system for the selection of SARS-CoV-2 FSS-targeting cyclic peptides. MOrPH-PhD libraries were diversified through cyclization of 11-mer (i/i ± 10) and 9-mer (i/i ± 8) peptide sequences using four different eUAAs and two different orientations for the eUAA/Cys linkage (X = randomized amino acid position, C = cysteine residue, Y* = eUAA). See Fig. S1† for further information about the library design. | ||
Results
Multiplexing of the MOrPH-PhD library enhances diversity yielding a library of >50 million unique peptides
We first sought to design a set of MOrPH-PhD libraries capable of targeting the FSS pseudoknot (PK) of SARS-CoV-2. To this end, for randomization of the peptide sequence we chose to use restricted amino acid alphabets that comprise amino acid residues found to be statistically more prevalent at protein-RNA interfaces (i.e., Lys, Arg, His, Phe, Tyr, Leu, Asn, Gln).63–65 Accordingly, the macrocycle peptide sequences were randomized using different patterns of the following degenerate codons: (MRW = R, N, Q, K, H, S), (YWT = H, L, F, Y) and (HWS = N, Q, H, I, L, K, M, F, Y) (Fig. S1†). Peptide libraries were further diversified by varying the ring size (9-mer vs. 11-mer) and orientation of the eUAA/Cys linkage (e.g., i/i + 8 vs. i/i − 8, where the ith position indicates the eUAA relative to the invariant cysteine nucleophile). Previous studies demonstrated that the orientation of the thioether linkage can drastically impact the target binding properties of these peptides.43,66 Because of these structural variations, the overall library comprises ∼13.6 million unique peptide sequences (Fig. 2).While O-2-bromoethyl tyrosine (O2beY) was the initial eUAA for cyclization, this choice was extended subsequently to a broader suite of eUAAs based on recent work demonstrating additional methodologies to establish thioether crosslinks.43 Variation of the thioether crosslink was shown to impact the functional properties of bioactive cyclic peptides. Accordingly, an additional element of structural diversification was incorporated into our libraries through the multiplexed integration of our MOrPH-PhD libraries with different eUAAs as cyclization modules, namely O2beY, O-4-bromobutyl tyrosine (O4bbY), p-acrylamido phenylalanine (pAaF) and p-chloroacetamido phenylalanine (pCaaF). Altogether, the resulting library comprises 54.5 million unique macrocyclic peptides.
Panning against the target RNA reveals consensus peptides
Our initial experiments determined that phage particles — produced in bacterial cell cultures — contain significant amounts of RNase, which led to rapid degradation of the target RNA (Fig. S2†). This phenomenon highlights a fundamental challenge when applying high-throughput techniques to RNA targets. We found the addition of a protein-based RNase inhibitor resulted in significant reduction in RNA degradation with the majority of the full-length FSS PK remaining intact after overnight incubation with the phage solution (Fig. S2†). Based on these results, we included RNase inhibitor in the phage solution prior to panning against the target RNA. Furthermore, we also envisioned that the full-length FSS PK, which comprises 68 nucleotides, may be susceptible to chemical degradation, particularly within less structured joining regions. In this context, the dimerization loop (DL) hairpin, which comprises 26 conserved nucleotides that play a key role in the −1 PRF mechanism of action, is a shorter structural motif contained within the FSS and would be less susceptible to degradation than the full-length FSS PK (Fig. 2).67 Accordingly, in addition to the full-length FSS PK, MOrPH-PhD libraries were screened in parallel against a more compact motif corresponding to the DL hairpin of the FSS.Based on these considerations, selection experiments were carried out by panning the four MOrPH-PhD libraries, each constructed using a different eUAA, against both the DL and the full-length FSS biotinylated RNA immobilized on streptavidin-coated magnetic beads. All libraries were subjected to three rounds of affinity-based selection and amplification (Fig. 3A and B). Notably, the MOrPH-PhD libraries show differential levels of post-selection recovery depending on the nature of the eUAA module (Fig. 3A and B), likely reflecting the differential ability of these cyclization modules to generate cyclic peptides capable of interacting with the target RNA. In particular, MOrPH libraries cyclized via O4bbY or pCaaF showed substantially higher levels of phage recovery during the second and third rounds of affinity selection, relative to the MOrPH libraries cyclized via O2beY or pAaF (Fig. 3A and B). Based on these results, the library members recovered after the second and third round of selection from O4bbY- and pCaaF-based libraries against the DL and FSS PK targets were analyzed via next-generation sequencing (NGS). In addition, libraries recovered from the second round and third round of selection against the DL for pAaF- and O2beY-containing libraries were similarly subjected to NGS.
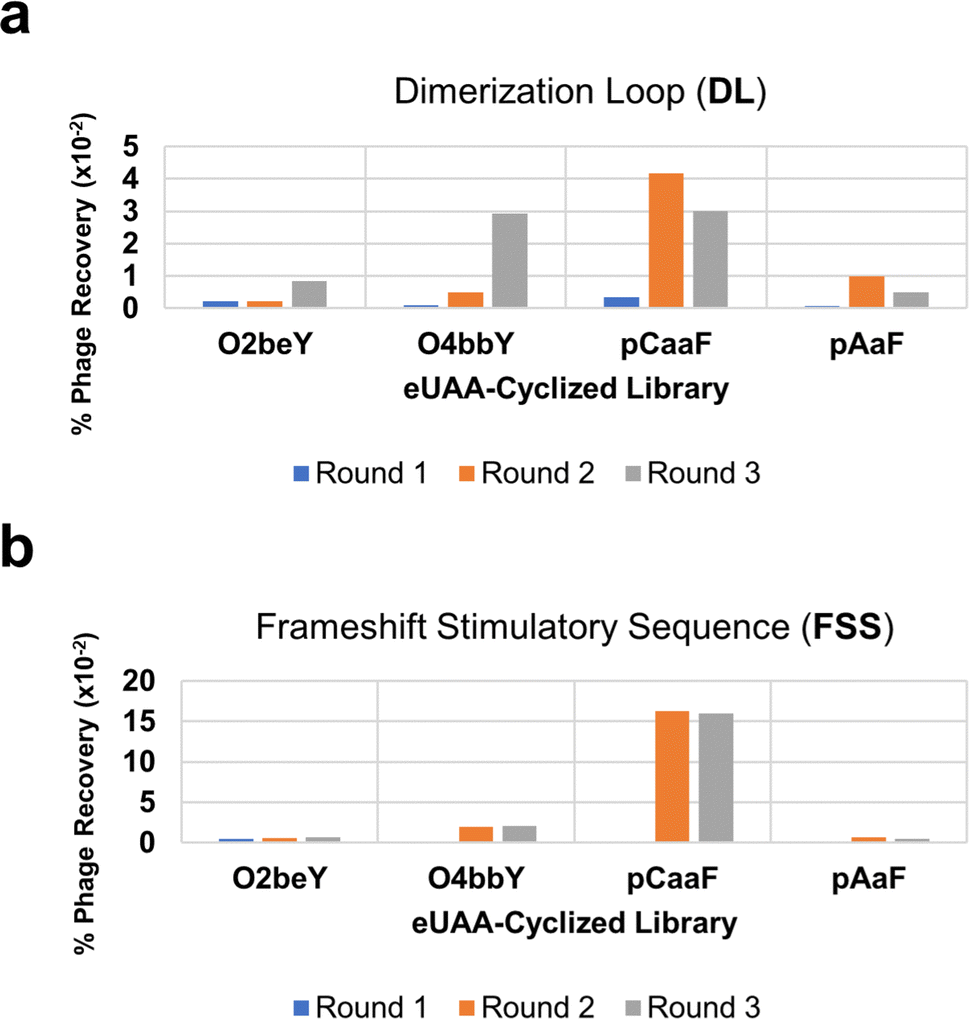 | ||
| Fig. 3 Phage recovery after each round of panning. Percent phage recovery of individual eUAA-containing libraries through three rounds of affinity selection and amplification against the (a) DL and (b) FSS. | ||
Consistent with the trend observed from phage recovery analyses, NGS showed enrichment of peptide sequences primarily in the O4bbY- and pCaaF-based libraries. By contrast, sequencing results from O2beY- and pAaF-containing libraries failed to yield peptide sequences with significant enrichment. Accordingly, macrocyclic peptides selected for further characterization were chosen solely from the O4bbY- and pCaaF-based cyclic peptide libraries. Interestingly, enriched macrocyclic peptides (‘hits’) were obtained from all four O4bbY-based libraries (i.e., for both the 9-mer and 11-mer libraries and for both linkage orientations) with distinct consensus sequences associated with each library (Fig. 4A). By contrast, sequence ‘hits’ derived from the pCaaF-containing libraries were found solely in the Cys/pCaaF orientation (i.e., i/i − 8 or i/i − 10), indicating a clear advantage of the Cys/pCaaF connectivity over the pCaaF/Cys connectivity for binding to the target RNAs (Fig. 4A). Overall, these results evidence the impact of different cyclization modules and linkage orientations on phage recovery and enrichment, highlighting the distinct advantage of the present multiplexed MOrPH system for exploring a larger macrocycle space than possible using a single eUAA module and thus increasing chances of success in hit identification against a desired target.
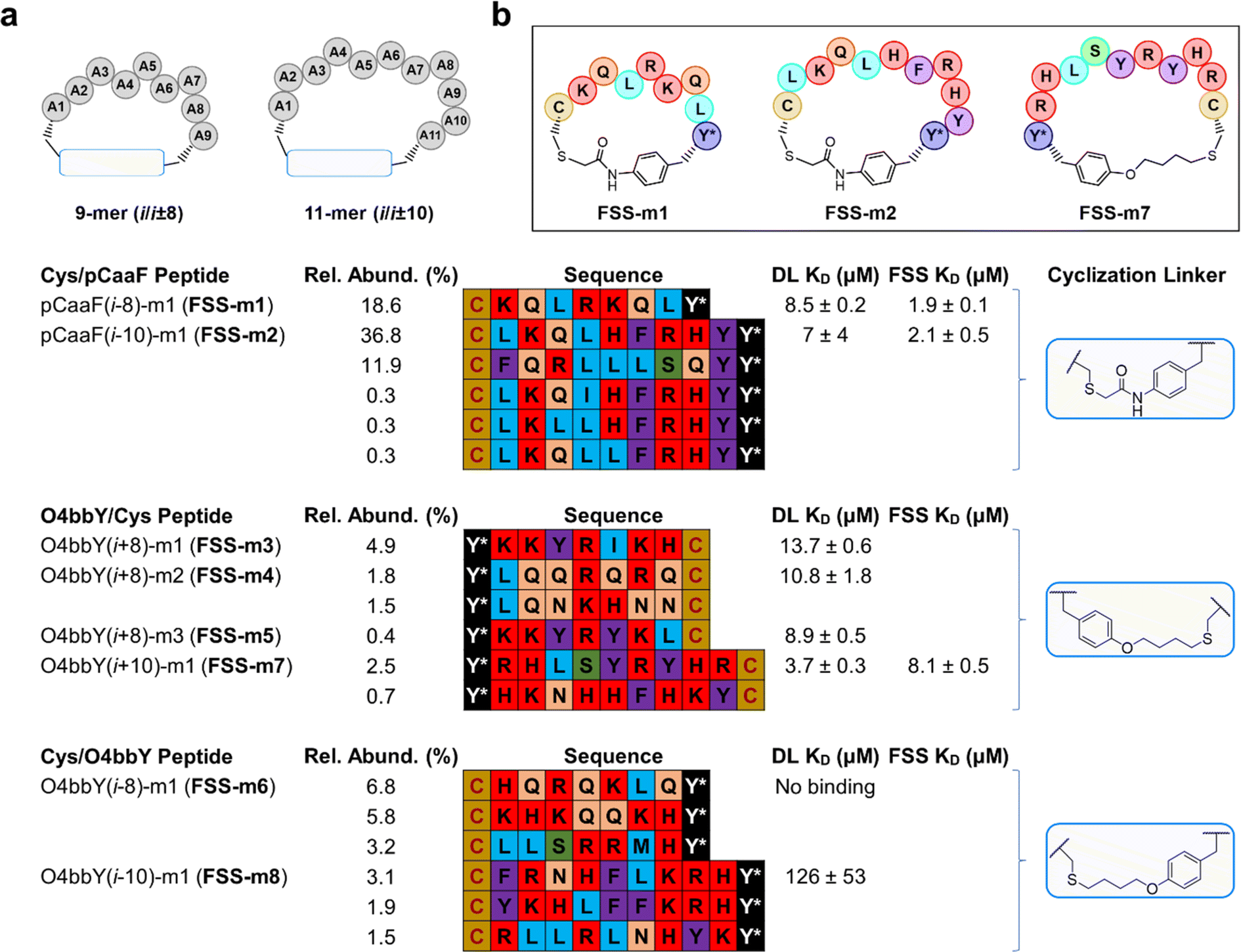 | ||
| Fig. 4 Macrocyclic peptides identified from deconvoluted MOrPH-PhD libraries. (a) Top enriched cyclic peptides were identified from i/i ± 8 and i/i ± 10 libraries. Cyclic peptides identified from sequenced libraries panned against the FSS comprise the pCaaF i/i − 10 11-mer library. All other peptides were identified from NGS deconvoluted libraries panned against the DL RNA from SARS-CoV-2. (b) Cyclic peptides with the highest binding affinity for the DL RNA. | ||
Analysis of the sequences selected from the 9-mer (i/i − 8) pCaaF-based library showed a single, highly enriched peptide (i.e., pCaaF(i − 8)-m1, called FSS-m1) (Fig. 4A) with markedly higher abundance over the other top-scoring sequences (19% vs. <0.6%). On the other hand, the 11-mer (i/i − 10) pCaaF-based library yielded two highly enriched peptides, one of which represented as much as 37% of the sequence reads (i.e., pCaaF(i − 10)-m1, called FSS-m2). In addition, several other sequences among the top-scoring ones show a high level of homology to FSS-m2 (Fig. 4A). Notably, FSS-m1 and FSS-m2, the top hits from the 9-mer and 11-mer libraries, respectively, share nearly identical motifs (–KQL(R/H)–).
Sequencing results from the 9-mer (i/i + 8) O4bbY-linked library (Fig. 4A) revealed four peptides with significant abundance within the library (0.4–4.9%). Interestingly, the top sequences identified from this library show a high level of homology, displaying two distinct consensus groups of cyclic peptides. The top enriched peptide (i.e., O4bbY(i + 8)-m1, called FSS-m3) shares a –KKYR– motif with the less abundant (0.4%) O4bbY(i + 8)-m3 peptide (called FSS-m5). The second and third top enriched peptides share a similar –LQ(Q/N)– motif (Fig. 4A). Furthermore, a consensus was observed among the sequences identified from the 11-mer (i/i − 10) O4bbY-based library with the top sequence (i.e., O4bbY(i − 10)-m1, called FSS-m8) sharing a similar –KRH– motif with the second most highly enriched peptide. In addition, a preference for aromatic amino acid residues (Phe/Tyr) is observed at the i + 1 position relative to the preinstalled cysteine at the ith position, with the exception of the third top enriched peptide, which contains an amphipathic Arg residue at the i + 1 position (Fig. 4A).
In general, panning experiments against the DL resulted in more substantial enrichment and stronger consensus sequences among hits compared to those targeting the FSS PK. For most of the libraries, sequence hits were identified from affinity selection experiments against the DL, although the same sequences were also found among those selected from panning experiments against the FSS PK. As an exception, the opposite result (i.e., stronger enrichment and consensus for hits selected against the FSS PK vs. DL) was observed for the pCaaF-based libraries. Notably, sequence hits identified from the FSS PK binding experiments included FSS-m2, which showed the largest degree of enrichment (and strongest consensus) among hits from all libraries (Fig. 4A). The FSS PK panning experiments notably yielded strong hits from only one sub library (i.e., that comprising 11-mer cyclic peptides constrained by a pCaaF-based thioether linkage in the Cys/pCaaF orientation). These results indicate that — among the 16 different macrocyclic peptide scaffolds tested — this cyclopeptide topology is optimal and nearly uniquely suited for interaction with the FSS PK — a result consistent with the distinctly higher phage recovery observed for the pCaaF-based libraries during panning against this target (Fig. 3B).
Solid-phase synthesis of selected cyclic peptides
Based on the sequencing results and consensus motifs derived above, we selected eight representative cyclic peptides for synthesis and further characterization derived from the O4bbY- and pCaaF-based MOrPH libraries. To this end, solid-phase peptide synthesis (SPPS) protocols were developed to prepare peptides cyclized via Cys/pCaaF, O4bbY/Cys and Cys/O4bbY linkages (Scheme 1A–C). Briefly, the Cys/pCaaF-cyclized peptides were synthesized via the use of N′-allyloxycarbonyl (Alloc)-protected p-aminophenylalanine (pAmF) and Acm-protected cysteine. After incorporation of the protected pAmF residue and assembly of the remainder of the peptide, the side-chain Alloc group was removed using a Pd(PPh3)4 catalyst and PhSiH3 followed by acylation with chloroacetyl chloride. After peptide assembly, the cysteine residue was deprotected with a PdCl2 catalyst followed by on-resin cyclization under basic conditions (Scheme 1A). The O4bbY/Cys-linked peptides were prepared via a different strategy using an acetamidomethyl (Acm)-protected cysteine and 9-fluorenylmethyloxycarbonyl (Fmoc)-protected O4bbY. Upon the completion of peptide assembly, cysteine deprotection and on-resin cyclization were performed in a similar manner to that described above (Scheme 1B). Synthesis of Cys/O4bbY cyclized peptides involved the use of an allyl-protected dipeptide encompassing the Cys/O4bbY linkage (Scheme S1†). Upon incorporation of the dipeptide and assembly of the remainder of the peptide, the side-chain allyl group was removed using a Pd(PPh3)4 catalyst and PhSiH3 followed by cyclization via amide bond formation (Scheme 1C). Further details regarding methods for solid-phase synthesis and characterization of the cyclic peptides are described in the ESI.†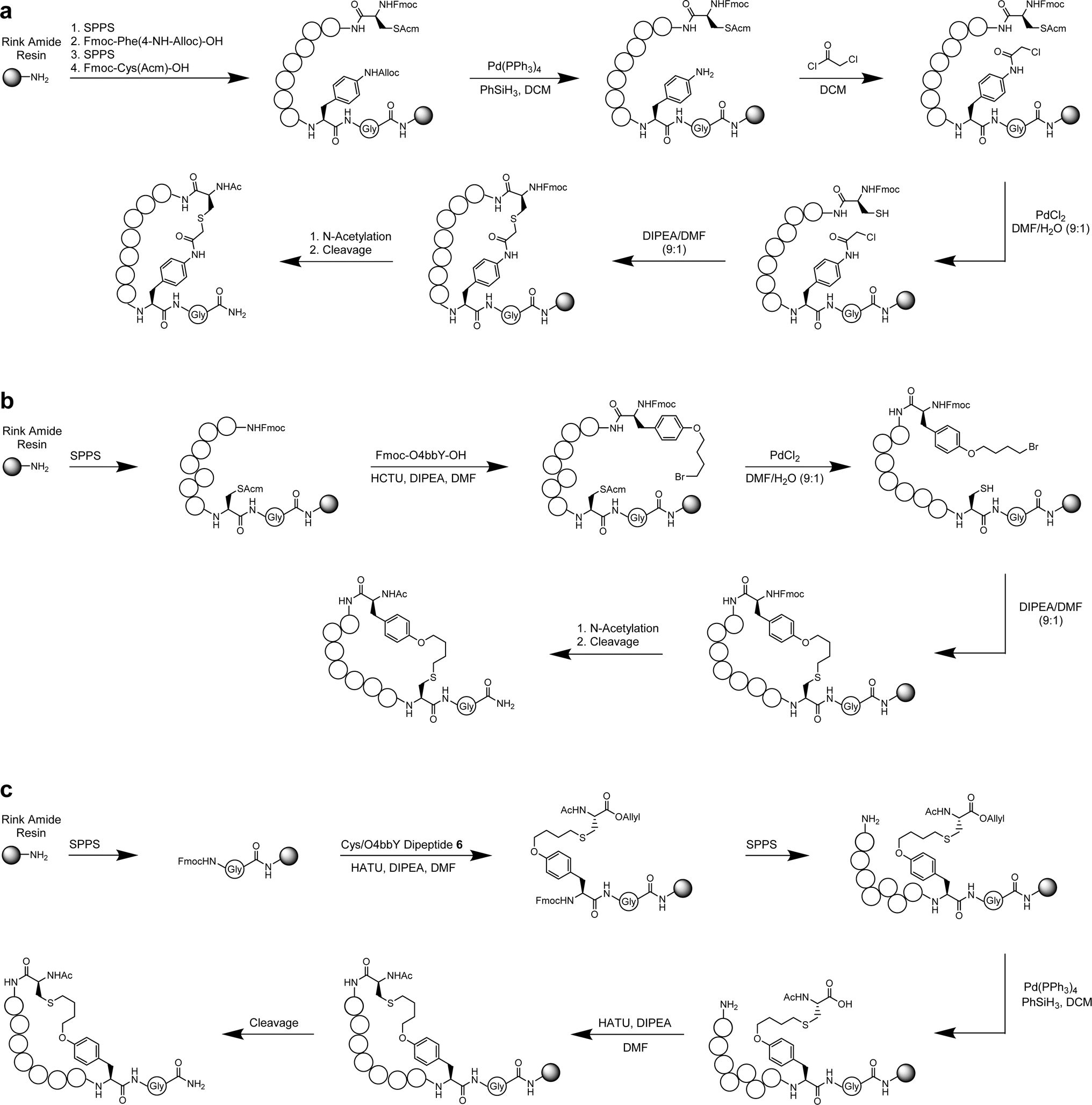 | ||
| Scheme 1 SPPS methods for the generation of cyclic peptides analyzed in this study. (a) SPPS of peptides comprised by a Cys/pCaaF linkage. (b) SPPS of peptides comprised by an O4bbY/Cys linkage. (c) SPPS of peptides comprised by a Cys/O4bbY linkage. | ||
Cyclic peptides bind SARS-CoV-2 RNA targets
After synthesis, cyclic peptides FSS-m1 through FSS-m8 were characterized for DL binding affinity (apparent KD) and binding kinetics (kon and koff) using surface plasmon resonance (SPR) (Table 1 for averaged binding parameters and Fig. S4† for representative binding sensorgrams and curve fits). These experiments showed that peptides FSS-m1, FSS-m2, and FSS-m7 bind the dimerization loop with micromolar affinity (KD = 3.6–4.6 μM). To test target specificity, the SPR experiments were repeated in the presence of a 100-fold molar excess of yeast tRNA in the SPR buffer relative to the immobilized DL RNA target. Importantly, each cyclic peptide maintained the same or only slightly weakened (2–3 fold) affinity for binding (KD = 3.7–8.5 μM), demonstrating the specificity of each peptide for the target RNA. Peptides FSS-m1 and FSS-m2, which comprise 9-mer and 11-mer cyclic peptides, share a common KQL motif, and display similar DL binding affinities (KD values of 3.6 ± 0.2 μM and 4.6 ± 0.2 μM). In addition, each showed only a slight increase in KD in the presence of tRNA (1.6–2.4-fold), suggesting each peptide forms a specific interaction with the DL structure. Peptide FSS-m7, which comprises an 11-mer cyclized via an O4bbY/Cys linkage, maintained full affinity in the presence of tRNA, as indicated by no discernible difference in the KD under conditions with and without tRNA (i.e., KD values of 3.7 ± 0.3 μM vs. 3.7 ± 1.6 μM, Table 1).| Peptide | k on × 102 (M−1 s−1) | S.E. × 102 (M−1 s−1) | k off × 10−2 (s−1) | S.E. × 10−2 (s−1) | K D (μM) | S.E. (μM) | X 2 (RU2) | S.E. |
|---|---|---|---|---|---|---|---|---|
| a Peptides tested in SPR buffer lacking 100-fold molar excess yeast tRNA. b Equilibrium dissociation constants determined via steady-state analysis. c S.E. indicates standard error. d n.a. indicates not assessed. e n.b. indicates no detectable binding. | ||||||||
| FSS-m1 | 11.3 (7.9a) | 0.05 (0.6a) | 0.96 (0.28a) | 0.01 (0.01a) | 8.5 (3.6a) | 0.2 (0.2a) | 1.9 (2.3a) | 0.05 (0.1a) |
| FSS-m2 | n.a. | n.a. | n.a. | n.a. | 7.4b (4.6a,b) | 3.5b (0.2a,b) | 0.5b (12.8a,b) | 0.2b (2.1a,b) |
| FSS-m3 | 17.4 | 0.8 | 2.38 | 0.004 | 13.7 | 0.6 | 222 | 13.2 |
| FSS-m4 | 32.8 | 3.0 | 3.59 | 0.84 | 10.8 | 1.8 | 4.2 | 0.4 |
| FSS-m5 | 31.2 | 0.9 | 2.76 | 0.09 | 8.9 | 0.5 | 100 | 1.3 |
| FSS-m6 | n.b. | n.b. | n.b. | n.b. | n.b. | n.b. | n.b. | n.b. |
| FSS-m7 | 44.9 (21.4a) | 3.8 (1.3a) | 1.65 (1.06a) | 0.01 (0.02a) | 3.7 (3.7a) | 0.3 (1.6a) | 10.0 (96.5a) | 0.7 (6.9a) |
| FSS-m8 | n.a | n.a | n.a. | n.a | 126.3 | 52.8 | 2.7 | 1.0 |
Encouraged by these results, we investigated other peptides for DL binding in the presence of tRNA. Peptides FSS-m3, FSS-m4, and FSS-m5 (Fig. 4A) — identified from the 9-mer library containing an i/i + 8 O4bbY/Cys linkage — yielded micromolar affinity to DL (KD values from 9–14 μM; Table 1). On the other hand, peptide FSS-m6, which comprises an i/i − 8 Cys/O4bbY linkage in the reverse orientation, did not exhibit binding. By contrast, peptide FSS-m8, which comprises an i/i − 10 Cys/O4bbY linkage, exhibited high micromolar affinity to the DL (KD >120 μM; Table 1). This stark contrast highlights a clear advantage of the O4bbY/Cys cyclization topology over the Cys/O4bbY topology for binding to the DL.
The highest affinity cyclic peptides identified in the DL binding experiments (i.e., peptides FSS-m1, FSS-m2, and FSS-m7) were tested next for binding to the FSS PK in the presence of tRNA (Fig. 4B). Notably, pCaaF-based cyclic peptides FSS-m1 and FSS-m2, which share the –KQL–consensus motif, displayed three- to four-fold increases in binding affinity for the FSS PK compared to DL (8.5 ± 0.2 μM vs. 1.9 ± 0.01 μM and 7.4 ± 3.5 μM vs. 2.1 ± 0.5 μM; Tables 1 and 2 for averaged binding parameters and Fig. S3F and G† for representative binding sensorgrams and curve fits). It is worth noting that peptide FSS-m2 (pCaaF(i − 10)-m1) was identified from the libraries panned against the FSS PK. By contrast, 11-mer cyclic peptide FSS-m7 cyclized via an O4bbY/Cys linkage, produced a poorer KD of 8.1 μM for binding to the FSS PK (Table 2 for averaged binding parameters and Fig. S4H† for a representative binding sensorgram and curve fit). This value is 2-fold worse than binding to the shorter DL, which yielded a KD of 3.7 ± 0.3 μM (Table 1). The molecular basis for this difference in affinity is unknown at present.
| Peptide | k on × 102 (M−1 s−1) | S.E. × 102 (M−1 s−1) | k off × 10−2 (s−1) | S.E. × 10−2 (s−1) | K D (μM) | S.E. (μM) | X 2 (RU2) | S.E |
|---|---|---|---|---|---|---|---|---|
| FSS-m1 | 16.3 | 4.8 | 0.31 | 0.10 | 1.9 | 0.01 | 0.03 | 0.01 |
| FSS-m2 | 65.8 | 13.0 | 1.24 | 0.03 | 2.1 | 0.47 | 0.14 | 0.01 |
| FSS-m7 | 10.6 | 0.6 | 0.85 | 0.01 | 8.1 | 0.52 | 92.9 | 0.10 |
To evaluate whether the cyclic peptides exhibited off-target RNA interactions with pseudoknots, we tested FSS-m1 and FSS-m2 for binding to riboswitch RNAs that adopt different pseudoknot folds. Specifically, we chose the type II preQ1-I riboswitch, which adopts an H-type pseudoknot that binds a single preQ1 (7-aminomethyl-7-deazaguanine) molecule.68 We also tested a type I preQ1-I riboswitch that folds as an H-type pseudoknot to cooperatively bind two preQ1 equivalents in a single binding pocket.69 The third riboswitch adopts an HHH pseudoknot fold that recognizes a single equivalent of guanine.70 These experiments revealed that neither peptide showed evidence of binding to off-target pseudoknots at concentrations that elicit a binding response in the presence of FSS PK (Fig. S5†vs.Table 2 and Fig. S4†).
Chemical modification localizes peptide binding to the FSS dimerization loop
We next sought to identify the binding location for each of the three highest affinity cyclic peptides. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) followed by next-generation sequencing was used to probe peptide binding to the RNA target.71 The 26-nucleotide dimerization stem loop of the SARS-CoV-2 FSS PK was embedded in a folding cassette comprising flanking 5′ and 3′ hairpins and a downstream primer binding site (Fig. 5A);72 two additional G–C pairs were added to the stem for stability. As an internal control to monitor non-specific binding, the 5′ hairpin was composed of the HIV-1 FSS hairpin loop. The RNA cassette was folded and bound to either FSS-m1, FSS-m2 or FSS-m7 and subsequently acylated by 2-methylnicotinic acid imidazolide (NAI).73 Differential reactivity was quantified by deep-sequence reads measured for the bound and unbound RNA. Each peptide revealed a significant decrease in reactivity at base U13518 (Fig. 5B–D). In addition, flanking nucleotides C13517 and A13521 showed minor reductions in acylation. Whereas the 9-mer pCaaF(i − 8)-m1 (FSS-m1) showed almost no change at A13516, 11-mer cyclic peptides pCaaF(i − 10)-m1 (FSS-m2) and O4bbY(i + 10)-m1 (FSS-m7) revealed a slight decrease in reactivity. Reductions in acylation suggest a loss in flexibility in the presence of ligand,74 possibly through a direct interaction or through allosteric changes due to cyclic peptide binding (Fig. 5B–D). Although the stem loop nucleotides are predicted to reside in an unpaired region, A13516 through U13521 engage in palindromic intermolecular Watson–Crick pairing via a kissing loop that is key for effective frameshifting and viral RNA synthesis.67 Most notably, U13518 shows the greatest reduction in acylation in the presence of each cyclic peptide and is predicted to pair intermolecularly with A13519′.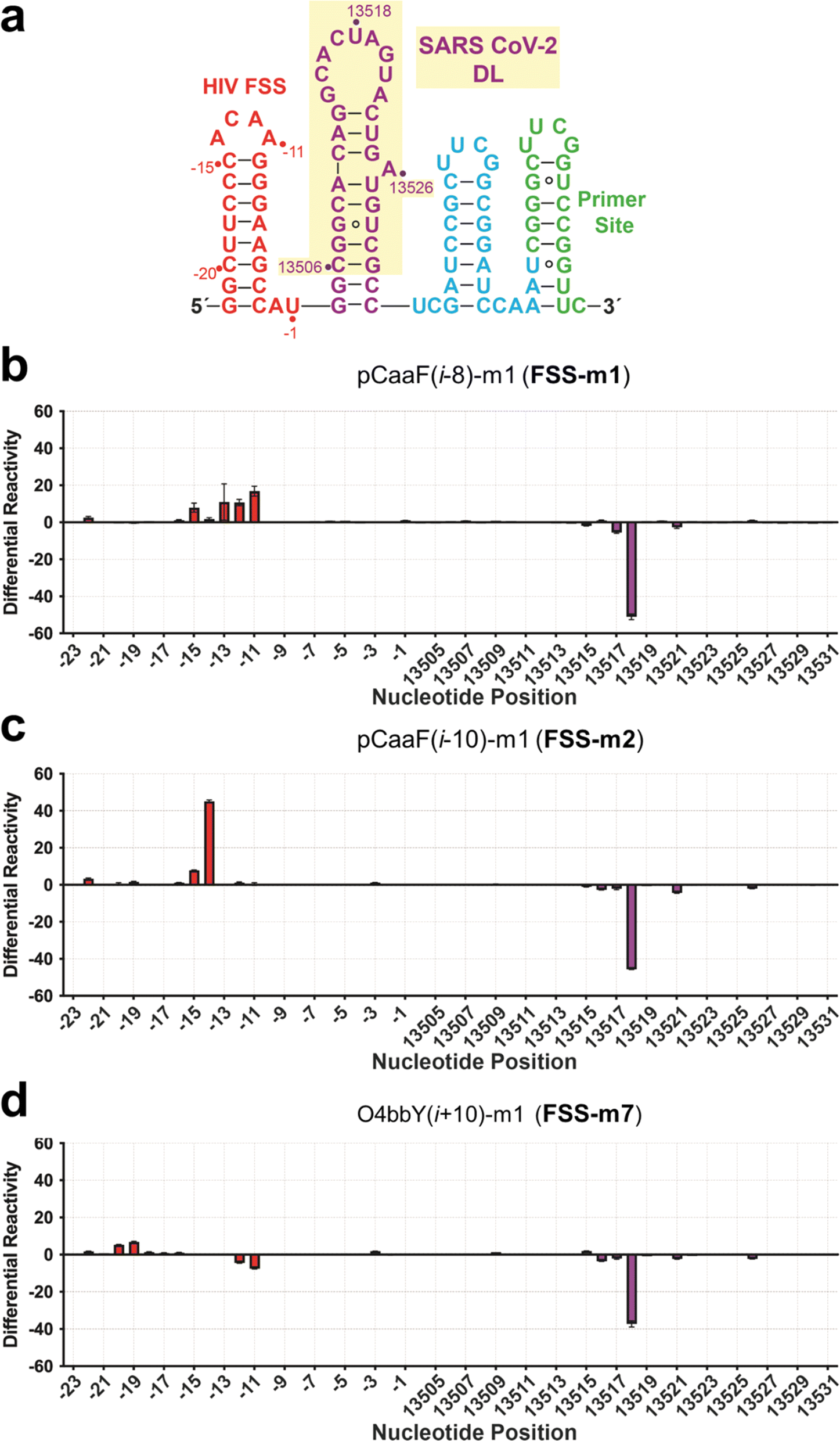 | ||
| Fig. 5 SHAPE-Seq data localizes peptide binding to U13518 within the dimerization loop. (a) Secondary structure of the RNA cassette containing the stable HIV-1 FSS hairpin at the 5′ end, followed by the SARS-CoV-2 FSS dimerization loop (DL) upon its stem, a strong 3′ linker hairpin, and the reverse transcriptase primer binding site. SARS-CoV-2 numbering corresponds to reference genome NC_045512.2. (b) Differential SHAPE reactivity (Δρ) profiles of the SARS-CoV-2 DL showing average differential acylation in the presence and absence of peptide (i.e., ρ+(pCaaF(i−8)-m1) − ρ−(pCaaF(i−8)-m1)) versus sequence position. (c) Differential SHAPE reactivity (Δρ) profiles of the SARS-CoV-2 DL showing average differential acylation in the presence and absence of peptide (i.e., ρ+(pCaaF(i−10)-m1) − ρ−(pCaaF(i−10)-m1)) versus sequence position. (d) Differential SHAPE reactivity (Δρ) profiles of the SARS-CoV-2 DL showing average differential acylation in the presence and absence of peptide (i.e., ρ+(O4bbY(i+10)-m1) − ρ−(O4bbY(i+10)-m1)) versus sequence position. Each bar represents the average of two replicates with standard deviations shown. | ||
In terms of off-target binding, our workflow allowed parallel monitoring of an upstream 5′ HIV-1 FSS element (Fig. 5). Specifically, each cyclic peptide produced acylation changes in the stem loop. Cyclic peptides FSS-m1 and FSS-m2 produced increases in flexibility (Fig. 5B and C), whereas FSS-m7 produced minor decreases in flexibility (Fig. 5D). Such differences may reflect the different sequences of these peptides since FSS-m1 and FSS-m2 share a –KQL(R/H)– consensus motif, whereas FSS-m7 is rich in positive charge but devoid of glutamine (Fig. 4). Notably, the latter peptide prompts fewer off-target changes while eliciting a strong acylation decrease on the SARS CoV-2 FSS stem loop at U13518.
Scanning mutagenesis of RNA-targeting MOrPHs reveals key amino acids
We next asked whether individual amino acids could be identified in a representative cyclic peptide that alter SARS CoV-2 dimerization loop recognition. One key advantage of the MOrPH system is that peptides can be expressed recombinantly in E. coli, where they undergo spontaneous and chemoselective cyclization to form the desired macrocyclic peptide.41,43,66 Accordingly, constructs for recombinant production of cyclic peptide FSS-m1 and alanine-scanning mutants at each position of the peptide were designed to comprise the macrocyclic peptide precursor with N-terminal Met–Gly sequence fused to an polyhistidine-tagged Mxe GyrA intein (Table S3†). Tyr was inserted at the junction between the macrocyclic peptide precursor and the intein to promote thiophenol-mediated intein cleavage after expression and isolation via Ni-affinity chromatography, resulting in macrocyclic peptide MG-FSS-m1 and its alanine variants containing an additional Gly and Tyr at the N- and C-terminus, respectively (Table S6†).After purification following intein cleavage, SPR analysis of the parent recombinant peptide MG-FSS-m1 and its alanine variants was performed to elucidate key residues for DL binding (Fig. S7†). Responses for mutants K1A, R4A and K5A were comparable to the response for MG-FSS-m1, suggesting the side chains are not critical for target binding. By contrast, the response for L7A was lower than the parent peptide. Notably, the response for Q6A was substantially higher than the response for MG-FSS-m1, while the responses for Q2A and L3A were substantially lower, suggesting these side chains contribute to RNA affinity or cyclic peptide conformation. Interestingly, Q2 and L3 are each contained within the consensus motif (–KQL(R/H)–) shared by FSS-m1 (pCaaF(i − 8)-m1) and FSS-m2 (pCaaF(i − 10)-m1), suggesting this region of the motif may be important for targeting the SARS CoV-2 FSS dimerization loop. By contrast, mutation of R4A was approximately neutral, which is unexpected if the guanidinium group participates in RNA recognition.37,75 Q6A was the only position that enhanced binding affinity relative to the parent peptide, consistent with the lack of conservation at this position in cyclic peptide FSS-m2 (Fig. 4).
Discussion
Due to the emerging appreciation of RNA as a promising target,4,14,49,76,77 we investigated here the potential of genetically encoded macrocyclic peptide libraries for targeting RNA, using the −1 programmed ribosomal frameshifting stimulatory sequence (FSS) RNA pseudoknot of SARS-CoV-2 as a model of a therapeutically relevant RNA molecule.46,78 In particular, we have introduced and applied here a multiplexed version of the MOrPH-PhD platform,41 in which libraries of phage displayed macrocyclic peptides are diversified through the use of multiple eUAAs (i.e., O2beY, O4bbY, pAaF, and pCaaF) and thus produce different types of intramolecular thioether crosslinks. By exploiting the modularity of the MOrPH architecture, these cyclopeptide libraries were further diversified through variation of the ring size (9-mer vs. 11-mer) and orientation of the eUAA/Cys linkage (e.g., i/i + 8 vs. i/i − 8, where i is the position of the eUAA with respect to the invariant cysteine residue). Finally, to favor RNA targeting, the amino acid sequences of these peptide libraries were randomized using amino acid residues that are found to be statistically more prevalent at protein-RNA interfaces, including Lys, Arg, His, Phe, Tyr, Leu, Asn, and Gln.63–65 Altogether, the resulting RNA-targeting library contains ∼55 million unique macrocyclic peptides.Additional elements were implemented to adapt the present phage display system for RNA targeting. As anticipated, our experiments were hindered initially by RNase degradation during library panning. This setback was overcome by use of a protein-based RNase inhibitor that allows broad application of our phage display approach against RNA targets. We also found that RNase degradation could be minimized by applying our method to the shorter 26-nucleotide dimerization loop hairpin (DL), which is substantially less complex in terms of its fold and the number of non-helical joining regions (Fig. 1).
Of the ∼55 million peptide sequences subjected to the panning procedure, we found enrichment primarily of cyclopeptide members from the O4bbY- and pCaaF-based libraries. Sequencing results from O2beY- and pAaF-containing libraries failed to yield peptide sequences with significant levels of enrichment, suggesting such cyclic peptides cannot adopt conformations suited for molecular recognition of the DL or full-length FSS PK. Moreover, panning of the same libraries against the smaller DL and the full-length FSS PK produced a larger number of hits for the former target RNA molecule. On the one hand, these results highlight the value of the present multiplexed MOrPH-PhD system toward enabling the exploration of different cyclopeptide topologies against a target of interest. On the other hand, the results suggest that the pursuit of small, well-defined motifs — such as the dimerization loop — is more tractable than more complex targets, such as the full-length FSS. This observation may be related to the inherently dynamic structural properties of the full-length FSS pseudoknot.46,79–82 Our findings have broader implications for target choices using high-throughput ligand screening platforms.
Based on the sequencing results and consensus analysis, we chose eight representative cyclic peptides derived from the O4bbY- and pCaaF-based MOrPH libraries for synthesis and characterization. Binding was assessed by SPR, which resulted in the identification of three high affinity peptides for the target RNA, namely FSS-m1, FSS-m2 and FSS-m7. Binding was established using both the DL and the full-length FSS PK. Only a small change in affinity between the two RNAs was observed, suggesting that the cyclic peptides bind structural features shared by the DL and FSS PK RNAs.
SHAPE-seq analysis revealed that the peptides localize to the DL sequence, which is expected to show non-canonical base-pairing properties.78 The importance of maintaining a precise level of frameshifting is underscored by the fact that a single nucleotide mutation in the slippery sequence of the viral RNA abolishes replication.83 Similarly, point mutants in the DL that ablate dimerization have a deleterious effect on −1 PRF. While the exact mode of cyclic peptide binding remains unknown, our present data indicated that it is localized to the apical loop region (Fig. 5). Binding of cyclic peptides within the DL may be analogous to recognition of the HIV-1 TAR UCU bulge wherein regions of non-canonical base pairing allow arginine- and glutamine-mediated readout of the major groove.37,84,85 Similar comparisons can be made to the HCV IRES and FMN riboswitch, which recognize small molecules within a helical bulge and multi-helix junction.17 Importantly, as demonstrated by our in vitro binding experiments in the presence of tRNA (Fig. S4†) and our control experiments with unrelated RNA pseudoknots (Fig. S5†), the cyclic peptides isolated through the present strategy show high specificity toward the target RNA molecule.
It is also instructive to compare these FSS targeting cyclic peptides with other compounds previously developed against this RNA target. In previous studies, the best hit isolated from the screening of ∼4000 small molecule drugs was reported to exhibit an IC50 of ∼20 μM in a frameshifting reporter assay,11 while an optimized analog from an initial screen of ∼40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 small molecules had a KD of 60 μM against the frameshifting element RNA in an SPR assay.48 In this context, the (low micromolar) cyclopeptides reported here constitute alternative and promising starting points for the development of cyclopeptide agents directed against this RNA target, e.g., through affinity maturation via site-saturation and/or combinatorial mutagenesis as done previously by our groups for other bioactive cyclic peptides.37,84 Finally, while further studies will be required to assess the activity of these compounds in cellular assays,86 it is promising that no significant cytotoxicity was observed against mammalian cells (HEK293T) even after incubation of FSS-m1 at 50 μM for 24 hours (Fig. S6†).
000 small molecules had a KD of 60 μM against the frameshifting element RNA in an SPR assay.48 In this context, the (low micromolar) cyclopeptides reported here constitute alternative and promising starting points for the development of cyclopeptide agents directed against this RNA target, e.g., through affinity maturation via site-saturation and/or combinatorial mutagenesis as done previously by our groups for other bioactive cyclic peptides.37,84 Finally, while further studies will be required to assess the activity of these compounds in cellular assays,86 it is promising that no significant cytotoxicity was observed against mammalian cells (HEK293T) even after incubation of FSS-m1 at 50 μM for 24 hours (Fig. S6†).
Conclusion
The emergence of RNA as a key regulator of cellular functions and human disease heightens the importance of developing strategies that can accelerate the discovery of molecules that target RNA with high affinity and specificity. While libraries of genetically encoded macrocyclic peptides have provided a valuable source of chemical agents for modulating protein–protein interactions,41,50–60 their application toward developing RNA-targeting molecules has remained underexplored. This work demonstrates the implementation and validation of a multiplexed MOrPH phage display platform for the discovery of macrocyclic peptides that target SARS-CoV-2 RNA with specificity and low micromolar affinity. Given the importance of the SARS-CoV-2 −1 PRF FSS PK in the viral life cycle and its conservation within the betacoronavirus genus, these results represent a first step toward the development of first-in-class cyclopeptide inhibitors of the −1 PRF pathway. We further envision these compounds can be useful for the development of systems for RNA degraders.49,77,87 More broadly, these results pave the way to the application of multiplexed MOrPH-PhD for the discovery of chemical entities directed against RNAs and other biomolecular targets.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
R. F. and J. E. W. conceived the project and supervised the work. J. A. I., R. B., A. S. H., A. S. A., and J. L. J. performed the experimental work. J. A. I., R. B., A. S. H., J. E. W., and R. F. wrote the manuscript, with input from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NIH grant R01GM134076 to R. F. and in part by NIH grants R01AI150463 & R01GM063162 to J. E. W. R. F. acknowledges endowed chair support from the Robert A. Welch Foundation (Chair, AT-0051). We thank members of the Wedekind and Fasan labs for technical support and discussions about this work. We thank Dr E. Pritchett and C. Baker (U. Rochester) for assistance with SHAPE-seq and Shelby Phelps in the Dodani Lab at UT Dallas for materials for the cytotoxicity experiments. R. B. recognizes support from NIH training grant T32 GM135134 and an E. Huntington Hooker graduate fellowship. A. S. H. is a trainee in the Medical Scientist Training Program funded by NIH grant T32 GM07356 and T32 GM152318.References
- K. V. Morris and J. S. Mattick, The rise of regulatory RNA, Nat. Rev. Genet., 2014, 15(6), 423–437, DOI:10.1038/nrg3722.
- G. St Laurent, C. Wahlestedt and P. Kapranov, The Landscape of long noncoding RNA classification, Trends Genet., 2015, 31(5), 239–251, DOI:10.1016/j.tig.2015.03.007.
- K. K. Ebbesen, J. Kjems and T. B. Hansen, Circular RNAs: Identification, biogenesis and function, Biochim. Biophys. Acta, 2016, 1859(1), 163–168, DOI:10.1016/j.bbagrm.2015.07.007.
- M. D. Disney, B. G. Dwyer and J. L. Childs-Disney, Drugging the RNA World, Cold Spring Harbor Perspect. Biol., 2018, 10(11), a034769, DOI:10.1101/cshperspect.a034769.
- T. R. Cech and J. A. Steitz, The noncoding RNA revolution-trashing old rules to forge new ones, Cell, 2014, 157(1), 77–94, DOI:10.1016/j.cell.2014.03.008.
- P. A. Latos, F. M. Pauler, M. V. Koerner, H. B. Senergin, Q. J. Hudson, R. R. Stocsits, W. Allhoff, S. H. Stricker, R. M. Klement and K. E. Warczok, et al., Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing, Science, 2012, 338(6113), 1469–1472, DOI:10.1126/science.1228110.
- C. L. Peebles, P. S. Perlman, K. L. Mecklenburg, M. L. Petrillo, J. H. Tabor, K. A. Jarrell and H. L. Cheng, A self-splicing RNA excises an intron lariat, Cell, 1986, 44(2), 213–223, DOI:10.1016/0092-8674(86)90755-5.
- R. Barrangou, C. Fremaux, H. Deveau, M. Richards, P. Boyaval, S. Moineau, D. A. Romero and P. Horvath, CRISPR provides acquired resistance against viruses in prokaryotes, Science, 2007, 315(5819), 1709–1712, DOI:10.1126/science.1138140.
- T. M. Schmeing, K. S. Huang, S. A. Strobel and T. A. Steitz, An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA, Nature, 2005, 438(7067), 520–524, DOI:10.1038/nature04152.
- J. A. Kelly, M. T. Woodside and J. D. Dinman, Programmed -1 Ribosomal Frameshifting in coronaviruses: A therapeutic target, Virology, 2021, 554, 75–82, DOI:10.1016/j.virol.2020.12.010.
- Y. Sun, L. Abriola, R. O. Niederer, S. F. Pedersen, M. M. Alfajaro, V. Silva Monteiro, C. B. Wilen, Y. C. Ho, W. V. Gilbert and Y. V. Surovtseva, et al., Restriction of SARS-CoV-2 replication by targeting programmed -1 ribosomal frameshifting, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(26), e2023051118, DOI:10.1073/pnas.2023051118.
- N. N. Patwardhan, L. R. Ganser, G. J. Kapral, C. S. Eubanks, J. Lee, B. Sathyamoorthy, H. M. Al-Hashimi and A. E. Hargrove, Amiloride as a new RNA-binding scaffold with activity against HIV-1 TAR, Medchemcomm, 2017, 8(5), 1022–1036, 10.1039/c6md00729e.
- T. A. Hilimire, J. M. Chamberlain, V. Anokhina, R. P. Bennett, O. Swart, J. R. Myers, J. M. Ashton, R. A. Stewart, A. L. Featherston and K. Gates, et al., HIV-1 Frameshift RNA-Targeted Triazoles Inhibit Propagation of Replication-Competent and Multi-Drug-Resistant HIV in Human Cells, ACS Chem. Biol., 2017, 12(6), 1674–1682, DOI:10.1021/acschembio.7b00052.
- J. L. Childs-Disney, X. Yang, Q. M. R. Gibaut, Y. Tong, R. T. Batey and M. D. Disney, Targeting RNA structures with small molecules, Nat. Rev. Drug Discovery, 2022, 21(10), 736–762, DOI:10.1038/s41573-022-00521-4.
- C. M. Connelly, M. H. Moon and J. S. Schneekloth Jr, The Emerging Role of RNA as a Therapeutic Target for Small Molecules, Cell Chem. Biol., 2016, 23(9), 1077–1090, DOI:10.1016/j.chembiol.2016.05.021.
- J. P. Falese, A. Donlic and A. E. Hargrove, Targeting RNA with small molecules: from fundamental principles towards the clinic, Chem. Soc. Rev., 2021, 50(4), 2224–2243, 10.1039/D0CS01261K.
- S. S. Chavali, R. Bonn-Breach and J. E. Wedekind, Face-time with TAR: Portraits of an HIV-1 RNA with diverse modes of effector recognition relevant for drug discovery, J. Biol. Chem., 2019, 294(24), 9326–9341, DOI:10.1074/jbc.REV119.006860.
- J. Davila-Calderon, N. N. Patwardhan, L. Y. Chiu, A. Sugarman, Z. Cai, S. R. Penutmutchu, M. L. Li, G. Brewer, A. E. Hargrove and B. S. Tolbert, IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex, Nat. Commun., 2020, 11(1), 4775, DOI:10.1038/s41467-020-18594-3.
- R. J. Marcheschi, M. Tonelli, A. Kumar and S. E. Butcher, Structure of the HIV-1 frameshift site RNA bound to a small molecule inhibitor of viral replication, ACS Chem. Biol., 2011, 6(8), 857–864, DOI:10.1021/cb200082d.
- M. Zafferani and A. E. Hargrove, Small molecule targeting of biologically relevant RNA tertiary and quaternary structures, Cell Chem. Biol., 2021, 28(5), 594–609, DOI:10.1016/j.chembiol.2021.03.003.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Constraining cyclic peptides to mimic protein structure motifs, Angew Chem. Int. Ed. Engl., 2014, 53(48), 13020–13041, DOI:10.1002/anie.201401058.
- E. Valeur, S. M. Gueret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, New Modalities for Challenging Targets in Drug Discovery, Angew Chem. Int. Ed. Engl., 2017, 56(35), 10294–10323, DOI:10.1002/anie.201611914.
- T. A. F. Cardote and A. Ciulli, Cyclic and Macrocyclic Peptides as Chemical Tools To Recognise Protein Surfaces and Probe Protein-Protein Interactions, Chemmedchem, 2016, 11(8), 787–794, DOI:10.1002/cmdc.201500450.
- M. Buyanova and D. Pei, Targeting intracellular protein-protein interactions with macrocyclic peptides, Trends Pharmacol. Sci., 2022, 43(3), 234–248, DOI:10.1016/j.tips.2021.11.008.
- S. Taguchi and H. Suga, Targeting of extracellular protein-protein interactions with macrocyclic peptides, Curr. Opin. Chem. Biol., 2021, 62, 82–89, DOI:10.1016/j.cbpa.2021.02.013.
- N. S. Robertson and D. R. Spring, Using Peptidomimetics and Constrained Peptides as Valuable Tools for Inhibiting Protein–Protein Interactions, Molecules, 2018, 23(4), 959, DOI:10.3390/molecules23040959.
- D. M. Kruger, A. Glas, D. Bier, N. Pospiech, K. Wallraven, L. Dietrich, C. Ottmann, O. Koch, S. Hennig and T. N. Grossmann, Structure-Based Design of Non-natural Macrocyclic Peptides That Inhibit Protein-Protein Interactions, J. Med. Chem., 2017, 60(21), 8982–8988, DOI:10.1021/acs.jmedchem.7b01221.
- R. M. Cardoso, F. M. Brunel, S. Ferguson, M. Zwick, D. R. Burton, P. E. Dawson and I. A. Wilson, Structural basis of enhanced binding of extended and helically constrained peptide epitopes of the broadly neutralizing HIV-1 antibody 4E10, J. Mol. Biol., 2007, 365(5), 1533–1544, DOI:10.1016/j.jmb.2006.10.088.
- C. Ngambenjawong, J. M. B. Pineda and S. H. Pun, Engineering an Affinity-Enhanced Peptide through Optimization of Cyclization Chemistry, Bioconjugate Chem., 2016, 27(12), 2854–2862, DOI:10.1021/acs.bioconjchem.6b00502.
- T. L. Aboye, H. Ha, S. Majumder, F. Christ, Z. Debyser, A. Shekhtman, N. Neamati and J. A. Camarero, Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity, J. Med. Chem., 2012, 55(23), 10729–10734, DOI:10.1021/jm301468k.
- J. M. Smith, J. R. Frost and R. Fasan, Designer macrocyclic organo-peptide hybrids inhibit the interaction between p53 and HDM2/X by accommodating a functional α-helix, Chem. Commun., 2014, 50(39), 5027–5030, 10.1039/c4cc01199f.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix, Science, 2004, 305(5689), 1466–1470, DOI:10.1126/science.1099191.
- W. M. Hewitt, S. S. F. Leung, C. R. Pye, A. R. Ponkey, M. Bednarek, M. P. Jacobson and R. S. Lokey, Cell-Permeable Cyclic Peptides from Synthetic Libraries Inspired by Natural Products, J. Am. Chem. Soc., 2015, 137(2), 715–721, DOI:10.1021/ja508766b.
- L. Peraro, Z. Zou, K. M. Makwana, A. E. Cummings, H. L. Ball, H. Yu, Y.-S. Lin, B. Levine and J. A. Kritzer, Diversity-Oriented Stapling Yields Intrinsically Cell-Penetrant Inducers of Autophagy, J. Am. Chem. Soc., 2017, 139(23), 7792–7802, DOI:10.1021/jacs.7b01698.
- A. F. L. Schneider, M. Kithil, M. C. Cardoso, M. Lehmann and C. P. R. Hackenberger, Cellular uptake of large biomolecules enabled by cell-surface-reactive cell-penetrating peptide additives, Nat. Chem., 2021, 13(6), 530–539, DOI:10.1038/s41557-021-00661-x.
- F. Milletti, Cell-penetrating peptides: classes, origin, and current landscape, Drug Discovery Today, 2012, 17(15), 850–860, DOI:10.1016/j.drudis.2012.03.002.
- S. S. Chavali, S. M. Mali, R. Bonn, A. Saseendran Anitha, R. P. Bennett, H. C. Smith, R. Fasan and J. E. Wedekind, Cyclic peptides with a distinct arginine-fork motif recognize the HIV trans-activation response RNA in vitro and in cells, J. Biol. Chem., 2021, 297(6), 101390, DOI:10.1016/j.jbc.2021.101390.
- M. D. Shortridge, P. T. Wille, A. N. Jones, A. Davidson, J. Bogdanovic, E. Arts, J. Karn, J. A. Robinson and G. Varani, An ultra-high affinity ligand of HIV-1 TAR reveals the RNA structure recognized by P-TEFb, Nucleic Acids Res., 2019, 47(3), 1523–1531, DOI:10.1093/nar/gky1197.
- M. D. Shortridge, M. J. Walker, T. Pavelitz, Y. Chen, W. Yang and G. Varani, A Macrocyclic Peptide Ligand Binds the Oncogenic MicroRNA-21 Precursor and Suppresses Dicer Processing, ACS Chem. Biol., 2017, 12(6), 1611–1620, DOI:10.1021/acschembio.7b00180.
- A. K. Manna, A. Kumar, U. Ray, S. Das, G. Basu and S. Roy, A cyclic peptide mimic of an RNA recognition motif of human La protein is a potent inhibitor of hepatitis C virus, Antiviral Res., 2013, 97(3), 223–226, DOI:10.1016/j.antiviral.2012.12.026.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, MOrPH-PhD: An Integrated Phage Display Platform for the Discovery of Functional Genetically Encoded Peptide Macrocycles, ACS Cent. Sci., 2020, 6(3), 368–381, DOI:10.1021/acscentsci.9b00927.
- N. Bionda, A. L. Cryan and R. Fasan, Bioinspired strategy for the ribosomal synthesis of thioether-bridged macrocyclic peptides in bacteria, ACS Chem. Biol., 2014, 9(9), 2008–2013, DOI:10.1021/cb500311k.
- J. A. Iannuzzelli and R. Fasan, Expanded toolbox for directing the biosynthesis of macrocyclic peptides in bacterial cells, Chem. Sci., 2020, 11(24), 6202–6208, 10.1039/d0sc01699c.
- P. V'Kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Coronavirus biology and replication: implications for SARS-CoV-2, Nat. Rev. Microbiol., 2021, 19(3), 155–170, DOI:10.1038/s41579-020-00468-6.
- J. A. Kelly, A. N. Olson, K. Neupane, S. Munshi, J. San Emeterio, L. Pollack, M. T. Woodside and J. D. Dinman, Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2), J. Biol. Chem., 2020, 295(31), 10741–10748, DOI:10.1074/jbc.AC120.013449.
- P. R. Bhatt, A. Scaiola, G. Loughran, M. Leibundgut, A. Kratzel, R. Meurs, R. Dreos, K. M. O'Connor, A. McMillan and J. W. Bode, et al., Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome, Science, 2021, 372(6548), 1306–1313, DOI:10.1126/science.abf3546.
- Y. H. Wang, M. Grunewald and S. Perlman, Coronaviruses: An Updated Overview of Their Replication and Pathogenesis, Methods Mol. Biol., 2020, 2203, 1–29, DOI:10.1007/978-1-0716-0900-2_1.
- M. Yang, F. P. Olatunji, C. Rhodes, S. Balaratnam, K. Dunne-Dombrink, S. Seshadri, X. Liang, C. P. Jones, S. F. J. Le Grice and A. R. Ferre-D'Amare, et al., Discovery of Small Molecules Targeting the Frameshifting Element RNA in SARS-CoV-2 Viral Genome, ACS Med. Chem. Lett., 2023, 14(6), 757–765, DOI:10.1021/acsmedchemlett.3c00051.
- H. S. Haniff, Y. Tong, X. Liu, J. L. Chen, B. M. Suresh, R. J. Andrews, J. M. Peterson, C. A. O'Leary, R. I. Benhamou and W. N. Moss, et al., Targeting the SARS-CoV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RIBOTAC) Degraders, ACS Cent. Sci., 2020, 6(10), 1713–1721, DOI:10.1021/acscentsci.0c00984.
- A. Tavassoli, SICLOPPS cyclic peptide libraries in drug discovery, Curr. Opin. Chem. Biol., 2017, 38, 30–35, DOI:10.1016/j.cbpa.2017.02.016.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, Development and Application of Yeast and Phage Display of Diverse Lanthipeptides, ACS Cent. Sci., 2018, 4(4), 458–467, DOI:10.1021/acscentsci.7b00581.
- M. Zheng, F.-J. Chen, K. Li, R. M. Reja, F. Haeffner and J. Gao, Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display, J. Am. Chem. Soc., 2022, 144(34), 15885–15893, DOI:10.1021/jacs.2c07375.
- A. R. Horswill, S. N. Savinov and S. J. Benkovic, A systematic method for identifying small-molecule modulators of protein-protein interactions, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(44), 15591–15596, DOI:10.1073/pnas.0406999101.
- S. M. Howell, S. V. Fiacco, T. T. Takahashi, F. Jalali-Yazdi, S. W. Millward, B. Hu, P. Wang and R. W. Roberts, Serum stable natural peptides designed by mRNA display, Sci. Rep., 2014, 4(1), 6008, DOI:10.1038/srep06008.
- C. J. Hipolito and H. Suga, Ribosomal production and in vitro selection of natural product-like peptidomimetics: the FIT and RaPID systems, Curr. Opin. Chem. Biol., 2012, 16(1–2), 196–203, DOI:10.1016/j.cbpa.2012.02.014.
- P. Diderich and C. Heinis, Phage selection of bicyclic peptides binding Her2, Tetrahedron, 2014, 70(42), 7733–7739, DOI:10.1016/j.tet.2014.05.106.
- K. Deyle, X.-D. Kong and C. Heinis, Phage Selection of Cyclic Peptides for Application in Research and Drug Development, Acc. Chem. Res., 2017, 50(8), 1866–1874, DOI:10.1021/acs.accounts.7b00184.
- S. S. Kale, C. Villequey, X.-D. Kong, A. Zorzi, K. Deyle and C. Heinis, Cyclization of peptides with two chemical bridges affords large scaffold diversities, Nat. Chem., 2018, 10(7), 715–723, DOI:10.1038/s41557-018-0042-7.
- S. Ng, M. R. Jafari and R. Derda, Bacteriophages and Viruses as a Support for Organic Synthesis and Combinatorial Chemistry, ACS Chem. Biol., 2012, 7(1), 123–138, DOI:10.1021/cb200342h.
- T. R. Oppewal, I. D. Jansen, J. Hekelaar and C. Mayer, A Strategy to Select Macrocyclic Peptides Featuring Asymmetric Molecular Scaffolds as Cyclization Units by Phage Display, J. Am. Chem. Soc., 2022, 144(8), 3644–3652, DOI:10.1021/jacs.1c12822.
- K. C. Liu, K. Roder, C. Mayer, S. Adhikari, D. J. Wales and S. Balasubramanian, Affinity-Selected Bicyclic Peptide G-Quadruplex Ligands Mimic a Protein-like Binding Mechanism, J. Am. Chem. Soc., 2020, 142(18), 8367–8373, DOI:10.1021/jacs.0c01879.
- V. A. Burns, B. G. Bobay, A. Basso, J. Cavanagh and C. Melander, Targeting RNA with cysteine-constrained peptides, Bioorg. Med. Chem. Lett., 2008, 18(2), 565–567, DOI:10.1016/j.bmcl.2007.11.096.
- D. M. Kruger, S. Neubacher and T. N. Grossmann, Protein-RNA interactions: structural characteristics and hotspot amino acids, RNA, 2018, 24(11), 1457–1465, DOI:10.1261/rna.066464.118.
- S. Jones, D. T. Daley, N. M. Luscombe, H. M. Berman and J. M. Thornton, Protein-RNA interactions: a structural analysis, Nucleic Acids Res., 2001, 29(4), 943–954, DOI:10.1093/nar/29.4.943.
- Y. Yi, Y. Zhao, Y. Huang and D. Wang, A Brief Review of RNA-Protein Interaction Database Resources, Noncoding RNAs, 2017, 3(1), 6, DOI:10.3390/ncrna3010006.
- Y. Gu, J. A. Iannuzzelli and R. Fasan, MOrPH-PhD: A Phage Display System for the Functional Selection of Genetically Encoded Macrocyclic Peptides, Methods Mol. Biol., 2022, 2371, 261–286, DOI:10.1007/978-1-0716-1689-5_14.
- D. Ishimaru, E. P. Plant, A. C. Sims, B. L. Yount Jr, B. M. Roth, N. V. Eldho, G. C. Perez-Alvarado, D. W. Armbruster, R. S. Baric and J. D. Dinman, et al., RNA dimerization plays a role in ribosomal frameshifting of the SARS coronavirus, Nucleic Acids Res., 2013, 41(4), 2594–2608, DOI:10.1093/nar/gks1361.
- J. L. Jenkins, J. Krucinska, R. M. McCarty, V. Bandarian and J. E. Wedekind, Comparison of a preQ1 riboswitch aptamer in metabolite-bound and free states with implications for gene regulation, J. Biol. Chem., 2011, 286(28), 24626–24637, DOI:10.1074/jbc.M111.230375.
- G. M. Schroeder, C. E. Cavender, M. E. Blau, J. L. Jenkins, D. H. Mathews and J. E. Wedekind, A small RNA that cooperatively senses two stacked metabolites in one pocket for gene control, Nat. Commun., 2022, 13(1), 199, DOI:10.1038/s41467-021-27790-8.
- S. Hamal Dhakal, K. Kavita, S. S. S. Panchapakesan, A. Roth and R. R. Breaker, 8-oxoguanine riboswitches in bacteria detect and respond to oxidative DNA damage, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(40), e2307854120, DOI:10.1073/pnas.2307854120.
- D. Loughrey, K. E. Watters, A. H. Settle and J. B. Lucks, SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing, Nucleic Acids Res., 2014, 42(21), e165, DOI:10.1093/nar/gku909.
- K. A. Wilkinson, E. J. Merino and K. M. Weeks, Selective 2'-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution, Nat. Protoc., 2006, 1(3), 1610–1616, DOI:10.1038/nprot.2006.249.
- R. C. Spitale, P. Crisalli, R. A. Flynn, E. A. Torre, E. T. Kool and H. Y. Chang, RNA SHAPE analysis in living cells, Nat. Chem. Biol., 2013, 9(1), 18–20, DOI:10.1038/nchembio.1131.
- J. L. McGinnis, J. A. Dunkle, J. H. Cate and K. M. Weeks, The mechanisms of RNA SHAPE chemistry, J. Am. Chem. Soc., 2012, 134(15), 6617–6624, DOI:10.1021/ja2104075.
- S. S. Chavali, C. E. Cavender, D. H. Mathews and J. E. Wedekind, Arginine Forks Are a Widespread Motif to Recognize Phosphate Backbones and Guanine Nucleobases in the RNA Major Groove, J. Am. Chem. Soc., 2020, 142(47), 19835–19839, DOI:10.1021/jacs.0c09689.
- K. D. Warner, C. E. Hajdin and K. M. Weeks, Principles for targeting RNA with drug-like small molecules, Nat. Rev. Drug Discovery, 2018, 17(8), 547–558, DOI:10.1038/nrd.2018.93.
- Y. Tong, Y. Lee, X. Liu, J. L. Childs-Disney, B. M. Suresh, R. I. Benhamou, C. Yang, W. Li, M. G. Costales and H. S. Haniff, et al., Programming inactive RNA-binding small molecules into bioactive degraders, Nature, 2023, 618(7963), 169–179, DOI:10.1038/s41586-023-06091-8.
- K. Zhang, I. N. Zheludev, R. J. Hagey, R. Haslecker, Y. J. Hou, R. Kretsch, G. D. Pintilie, R. Rangan, W. Kladwang and S. Li, et al., Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome, Nat. Struct. Mol. Biol., 2021, 28(9), 747–754, DOI:10.1038/s41594-021-00653-y.
- N. C. Huston, H. Wan, M. S. Strine, R. de Cesaris Araujo Tavares, C. B. Wilen and A. M. Pyle, Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms, Mol. Cell, 2021, 81(3), 584–598, DOI:10.1016/j.molcel.2020.12.041.
- C. P. Jones and A. R. Ferre-D'Amare, Crystal structure of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) frameshifting pseudoknot, RNA, 2022, 28(2), 239–249, DOI:10.1261/rna.078825.121.
- C. Roman, A. Lewicka, D. Koirala, N. S. Li and J. A. Piccirilli, The SARS-CoV-2 Programmed -1 Ribosomal Frameshifting Element Crystal Structure Solved to 2.09 A Using Chaperone-Assisted RNA Crystallography, ACS Chem. Biol., 2021, 16(8), 1469–1481, DOI:10.1021/acschembio.1c00324.
- T. C. T. Lan, M. F. Allan, L. E. Malsick, J. Z. Woo, C. Zhu, F. Zhang, S. Khandwala, S. S. Y. Nyeo, Y. Sun and J. U. Guo, et al., Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells, Nat. Commun., 2022, 13(1), 1128, DOI:10.1038/s41467-022-28603-2.
- E. P. Plant, R. Rakauskaite, D. R. Taylor and J. D. Dinman, Achieving a golden mean: mechanisms by which coronaviruses ensure synthesis of the correct stoichiometric ratios of viral proteins, J. Virol., 2010, 84(9), 4330–4340, DOI:10.1128/JVI.02480-09.
- S. S. Chavali, S. M. Mali, J. L. Jenkins, R. Fasan and J. E. Wedekind, Co-crystal structures of HIV TAR RNA bound to lab-evolved proteins show key roles for arginine relevant to the design of cyclic peptide TAR inhibitors, J. Biol. Chem., 2020, 295(49), 16470–16486, DOI:10.1074/jbc.RA120.015444.
- I. A. Belashov, D. W. Crawford, C. E. Cavender, P. Dai, P. C. Beardslee, D. H. Mathews, B. L. Pentelute, B. R. McNaughton and J. E. Wedekind, Structure of HIV TAR in complex with a Lab-Evolved RRM provides insight into duplex RNA recognition and synthesis of a constrained peptide that impairs transcription, Nucleic Acids Res., 2018, 46(13), 6401–6415, DOI:10.1093/nar/gky529.
- M. J. Waring, Lipophilicity in drug discovery, Expert Opin. Drug Discovery, 2010, 5(3), 235–248, DOI:10.1517/17460441003605098.
- M. G. Costales, Y. Matsumoto, S. P. Velagapudi and M. D. Disney, Small Molecule Targeted Recruitment of a Nuclease to RNA, J. Am. Chem. Soc., 2018, 140(22), 6741–6744, DOI:10.1021/jacs.8b01233.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04026k |
| ‡ Present address: Merck & Co., Inc., Rahway, NJ 07065, USA. |
| § These authors contributed equally to this work. |
| ¶ Present address: Renaissance School of Medicine, Stony Brook University, Stony Brook, NY 11794, USA. |
| This journal is © The Royal Society of Chemistry 2024 |