
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselectivity switches between anthraquinone precursor fissions involved in bioactive xanthone biosynthesis†‡
Xiao Jing
Lv§
a,
Chun Zhi
Ai§
b,
Li Rong
Zhang
a,
Xiu Xiu
Ma
c,
Juan Juan
Zhang
d,
Jia Peng
Zhu
c and
Ren Xiang
Tan
 *ad
*ad
aState Key Laboratory Cultivation Base for TCM Quality and Efficacy, School of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China. E-mail: rxtan@nju.edu.cn
bState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (Ministry of Education of China), Collaborative Innovation Center for Guangxi Ethnic Medicine, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin 541004, China
cSchool of Medicine and Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China
dState Key Laboratory of Pharmaceutical Biotechnology, Institute of Functional Biomolecules, School of Life Sciences, Nanjing University, Nanjing 210023, China
First published on 5th November 2024
Abstract
Xanthone-based polyketides with complex molecular frameworks and potent bioactivities distribute and function in different biological kingdoms, yet their biosynthesis remains under-investigated. In particular, nothing is known regarding how to switch between the C4a–C10 (C4a-selective) and C10a–C10 bond (C10a-selective) cleavages of anthraquinone intermediates involved in biosynthesizing strikingly different frameworks of xanthones and their siblings. Enabled by our characterization of antiosteoporotic brunneoxanthones, a subfamily of polyketides from Aspergillus brunneoviolaceus FB-2, we present herein the brunneoxanthone biosynthetic gene cluster and the C10a-selective cleavage of anthraquinone (chrysophanol) hydroquinone leading ultimately to the bioactive brunneoxanthones under the catalysis of BruN (an undescribed atypical non-heme iron dioxygenase) in collaboration with BruM as a new oxidoreductase that reduces the anthraquinone into its hydroquinone using NADPH as a cofactor. The insights into the driving force that determines whether the C10a- or C4a-selective cleavages of anthraquinone hydroquinones take place were achieved by a combination of multiprotein sequence alignment, directed protein evolution, theoretical simulation, chemical capture of hydroquinone tautomer, 18O chasing, and X-ray crystal structure of the BruNN441M mutant, eventually allowing for the protocol establishment for the on-demand switch between the two ways of anthraquinone openings. Collectively, the work paves the way for the synthetic biology-based regeneration of uniquely structured high-value xanthones present in low abundance in complex mixtures, and helps to deepen the understanding on why and how such xanthones and their congeners are biosynthesized by different (micro)organisms in nature.
Introduction
Xanthone-based compounds form and function in different biological kingdoms, and constitute a unique family of natural products characterized by diverse molecular frameworks and various biological activities.1–3 On one hand, the xanthone tricyclic scaffold facilitates the interaction of these molecules with diverse biological targets to display an array of important effects (e.g., anti-cancer, neuroprotective, antimicrobial, anti-inflammatory, antiosteoarthritic, antimalarial, and anti-cardiovascular disease).4,5 On the other hand, the type and position of substituent(s) anchoring on the tricyclic nucleus substantially or decisively influence their ultimate efficacy.6,7 The natural xanthones are biosynthesized roughly in a life domain-dependent manner,1–3 but the substitution patterns of the tricyclic system are largely inherited from precursors such as anthraquinones in fungi.3 However, the mechanism underlying the downstream reaction steps of such precursors remains neglected or overlooked.Distinct from plants1 and bacteria,2 fungi seem to be a richer source of xanthones with intriguing structures and potent bioactivities, as exemplified by brunneoxanthone E,8 shamixanthone,9,10 blennolide A,11 secalonic acid A,12 neosartorin,13 beticolin 1,14 geodin,15,16 and trypacidin17 (Fig. 1). As showcased in Fig. 1, most of the fungal xanthones and their oligomers are biosynthetically mediated by the ring fission of anthraquinone intermediates such as emodin (1) and chrysophanol (2).3 This has been repetitiously substantiated by the fast-evolving sequencing technologies and genome assembling approaches, which collectively enabled the identification of some unexpected biosynthetic gene clusters encoding anthraquinones and anthraquinone-derived xanthones.3 Interestingly, the C4a–C10 (C4a-selective) and C10a–C10 bond (C10a-selective) cleavages of anthraquinone intermediates lead to distinct xanthone molecules with different bioactivities (Fig. 1). Accordingly, the C4a- or C10a-regioselectivities in the anthraquinone precursor cleavages as well as their switchability are of paramount importance for the de novo regeneration of substitution-oriented xanthones desirable for the drug discovery pipeline.
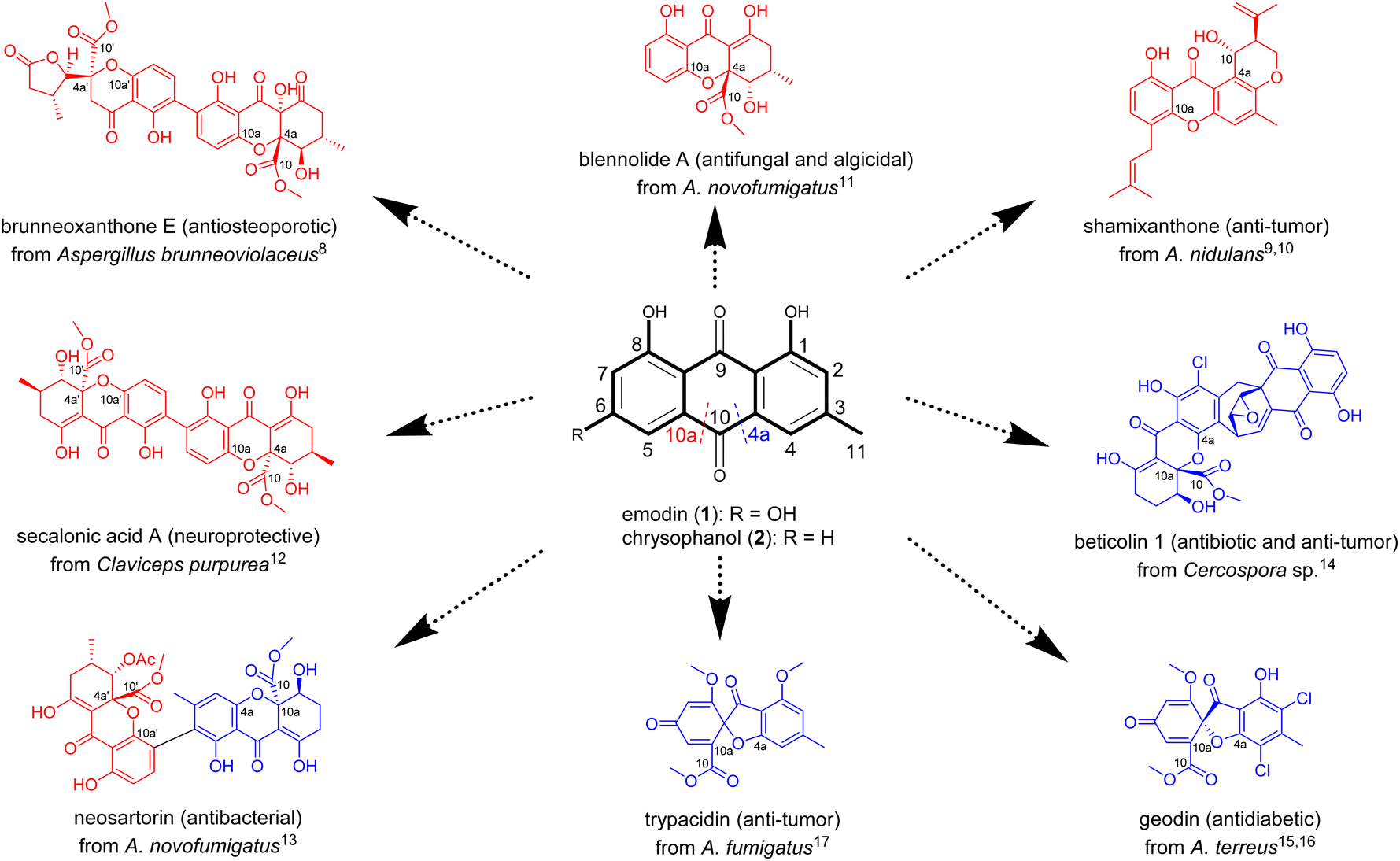 | ||
| Fig. 1 A selection of bioactive fungal xanthones with the carbon skeleton formation depending biosynthetically on the C4a–C10 (C4a-selective, in blue) or C10a–C10 (C10a-selective, in red) bond cleavages of anthraquinone intermediates such as emodin (1) and chrysophanol (2). | ||
The C4a- and C10a-selective fissions of anthraquinones for the xanthone-related polyketide biosynthesis were believed to be catalyzed by Baeyer–Villiger monooxygenases (BVMO).3 However, this was disproved by recent insights into the ring openings of anthraquinone precursors. In 2021, Lu et al. unraveled the two-enzyme (GedF and GedK) catalyzed C4a-selective anthraquinone (questin) fission; or more specifically, in the presence of NADPH, GedF promotes the reduction of questin into its hydroquinone, which is subsequently accepted as substrate by GedK (an atypical cofactor-free dioxygenase) and eventually oxidized into desmethylsulochrin.18 A year later, Rao and his co-workers reported the C4a-selective cleavage of 2 in the chrysophanol hydroquinone (3) form by another enzyme pair consisting of a non-heme iron dioxygenase (BTG13) and a reductase (BTG7, also using NADPH as an electron donor).14 In both cases, no oxidation happened upon the direct exposure of anthraquinones to GedK or BTG13, which became catalytically active after addition of sodium dithionite (Na2S2O4) into the reaction media. The findings reinforced that GedK and BTG13 do not interact “effectively” with the anthraquinone molecules, but rather recognize as substrates the corresponding hydroquinones forming from the two-electron reduction of anthraquinones.14,18,19 Another oxygenase (NsrF) was evidenced to catalyze the unselective cleavage of 2 to form diversely modified xanthones as a result of simultaneous fissions of C4a–C10 and C10a–C10 bonds of the same anthraquinone precursor.13 We envisioned, and wished to experimentally prove, the interchangeability between the C4a- and C10a-selective fissions of anthraquinones; and if proven true, we were more curious about how to steer the trajectory of such cleavage reactions hopefully in an on-demand manner.
The reports describing the C4a-selective14,19 and unselective13 anthraquinone fissions tempted us to search for an enzyme counterpart that catalyzes the C10a-selective cleavage of such quinone precursors. Catching our eyes was Aspergillus brunneoviolaceus FB-2 (A. brunneoviolaceus) which produces various xanthone dimers including brunneoxanthone E with an antiosteoporotic activity (Fig. 1).8 In particular, brunneoxanthone E and its siblings (brunneoxanthones A–D and penibishexahydroxanthone A) belong to the 2,2′-linked dimers with all tailored xanthone monomers derived from monodictyphenone (4a) rationalized to form from the C10a-selective cleavage of 2.8 With our confidence in the fungal (A. brunneoviolaceus) enzyme that catalyzes the C10a-selective cleavage, we performed the present investigation to identify the brunneoxanthone biosynthetic gene (shortened as “bru”) cluster through a combination of genome sequencing, multiprotein sequence alignment, gene inactivation, and heterologous expression. From the bru cluster, BruMN were evidenced to play catalytic roles in the C10a-selective fission of 2 into 4a. Starting from the enzyme, we established the first protocol for freely operational switches between the C4a- and C10a-selective cleavages of 2, the common anthraquinone precursor of diverse xanthones and their oligomer in nature.1–3,8 Taken together, the work offers a versatile foundation for the synthetic biology-based access to high-value xanthones and their congeners existing as (very) minor components in complex mixtures, and helps to understand why and how xanthones and their variants are, or must be, produced in different (micro)organisms.
Results and discussion
BruMN mediate the C10a-selective cleavage of chrysophanol
The brunneoxanthone structures8 suggested that the C10a-selective fission of chrysophanol (2) was most likely a key step for constructing these highly tailored xanthone dimers in A. brunneoviolaceus. This motivated us to identify the gene cluster for biosynthesizing brunneoxanthone E from the fungal genome which was sequenced and subsequently analyzed using antiSMASH (antibiotics and secondary metabolite analysis shell) (fungal version).20 The attempt identified a total of 26 polyketide synthase (PKS)-containing biosynthetic gene clusters (Fig. S1‡), of which six were predicted to express non-reducing PKSs (NR-PKSs). Typical of containing an NR-PKS gene, the bru cluster was proposed to encode likely the brunneoxanthone E biosynthesis (Fig. 2A and Table S1‡) from our sequence alignment with the nsr, agn, sec and dmx clusters, which are responsible for the biosynthesis of neosartorin in A. novofumigatus,13 agnestins in Paecilomyces variotii,21 secalonic acids in Claviceps purpurea,11,12 and cryptosporioptides in Cryptosporiopsis sp,22 respectively (Table S2 and Fig. S2‡). Next, we sought out to confirm the proposal through the gene inactivation strategy as described.23 The individual gene function in the bru cluster (Fig. 2A and Table S2‡) was annotated to be similar to those in the nsr cluster from A. novofumigatus.13 This agreed with the structural comparability between the xanthone monomers produced by the two Aspergillus species.8,13 In particular, the bruN gene in the bru cluster was annotated to encode an oxygenase with a 47% sequence identity to NsrF assumed to be an oxygenase involved in sculpting 2 into the xanthone monomers leading to neosartorin.13 We therefore deleted the bruN gene from the bru cluster to obtain the ΔbruN mutant. As indicated by the liquid chromatography-mass spectrometry (LC-MS) analysis, 2 was abundant in the ΔbruN culture but undetectable in that of the wild-type (WT) strain (Fig. 2C and S3‡).8 Owing to the deprival of the BruN-catalyzed oxidative cleavage, the ΔbruN strain did not produce brunneoxanthone E, which remained detectable in the WT culture (Fig. 2C and S3‡). The observation pinpointed that the bru cluster governed the brunneoxanthone biosynthesis with BruN catalyzing the C10a-selective cleavage of 2 (Fig. 2B). The assumption agreed with the predicted function of the 2-expressing six-gene set (bruHIJKMO), encoding respectively a short chain dehydrogenase (BruH), a thioesterase (BruI), an NR-PKS (BruJ), a dehydratase (BruK), an oxidoreductase (BruM), and a decarboxylase (BruO). Particularly noteworthy was that these enzymes displayed a 52%∼89% amino acid (AA) sequence identity to the counterparts that sculpted 2 into diverse xanthone derivatives (Table S2‡), such as neosartorin,13 agnestins,21 secalonic acids,12 and cryptosporioptides.22 | ||
Fig. 2 Identification of BruMN cleaving C10a-selectively chrysophanol (2) from the brunneoxanthone E biosynthetic gene (bru) cluster in the A. brunneoviolaceus genome. (A) Predicted functions of enzymes in the bru cluster. (B) The brunneoxanthone E biosynthesis is mediated by the BruN catalyzed C10a-selective cleavage of chrysophanol hydroquinone (3) into monodictyphenone (4a). The BruNN441F/Q437A/V347L mutant found herein catalyzed the C4a-selective cleavage of 3 into cephalanone F (4b) with a 93% C4a-selectivity. (C) The bruN deletion led to the 2 boost. The EtOAc extracts from the cultures of the ΔbruN and wild-type (WT) strains of A. brunneoviolaceus were analyzed by LC-MS using an acetonitrile/water gradient (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :70 → 100:0, within 13 min). (D) BruN accepts 3, rather than 2, as substrate that was C10a-selectively cleaved into 4a. The bruMN, bruM, and bruN genes were expressed in A. oryzae (AO) followed by exposure to 2, indicating that the AO-bruMN co-transformant allowed for the 4a production. The LC-MS analysis was performed using the duration-dependent methanol/water (containing 0.1% formic acid) gradients (0 → 10 min, 5:95 → 35:65; 10 → 35 min, 35:65 → 100:0; 35 → 40 min, pure methanol). :70 → 100:0, within 13 min). (D) BruN accepts 3, rather than 2, as substrate that was C10a-selectively cleaved into 4a. The bruMN, bruM, and bruN genes were expressed in A. oryzae (AO) followed by exposure to 2, indicating that the AO-bruMN co-transformant allowed for the 4a production. The LC-MS analysis was performed using the duration-dependent methanol/water (containing 0.1% formic acid) gradients (0 → 10 min, 5:95 → 35:65; 10 → 35 min, 35:65 → 100:0; 35 → 40 min, pure methanol). | ||
As reported,14,18 the ring opening of 2 could be accomplished by an enzyme pair consisting of a reductase and a dioxygenase. Our attention was therefore focused on BruM, the only oxidoreductase in the bru cluster. BruM was shown homologous both to NsrR (58%) that catalyzes the emodin (1) reduction to mediate the neosartorin (xanthone dimer) biosynthesis,13 and to BTG7 (51%) involved in the 2 reduction to facilitate the construction of beticolin 1, a xanthone–anthraquinone hybrid.14 To confirm their partnership, we generated the AO-bruM and AO-bruN transformants as well as the AO-bruMN co-transformant using A. oryzae NSAR1 strain (shortened as “AO” hereafter), a quadruple auxotrophic mutant strain (niaD−, sC−, ΔargB, adeA−) applicable for the biosynthetic studies on fungal natural products.23,24 With that, 2 was supplemented separately in the cultures of the three transformants. The results showed that the AO-bruMN co-transformant produced a major product identified as 4a (Fig. 2D(iv))25 by its MS and 1H and 13C NMR data (Table S5 and Fig. S24–S26‡). However, the AO-bruN transformant generated as well a lower abundance of 4a (Fig. 2D(iii)) presumably owing to an unknown AO-expressed reductase similar to our earlier observation.26 Furthermore, AO-bruM transformant and AO yielded a common byproduct identified as emodinic acid (Fig. 2D(i) and (ii)) by comparing its MS and 1H NMR data with those reported (Table S9, Fig. S34 and S35‡).23 Emodinic acid forms from emodin (1) via the C9–C9a cleavage catalysed by an AO-produced BVMO.23 Our characterization of emodinic acid from the 2-exposed culture signified the presence of another AO oxidase that catalyzed the 6-hydroxylation of 2 (ahead of C9–C9a cleavage) or 6-deshydroxy emodinic acid (after the C9–C9a cleavage of 2). Such lines of evidences underpinned that the BruMN enzyme pair plays a decisive role in the C10a-selective fission of 2 to facilitate the brunneoxanthone biosynthesis.
Regioselectivity and substrate promiscuity of BruN
To get more insights into the catalytic property of BruN, the codon-optimized N-terminally His-tagged BruN was cloned into the pET28a vector and overexpressed in E. coli BL21 (DE3) with the obtained yellow enzyme protein purified by an Ni-NTA affinity chromatography (Fig. S4‡). As predicted, no activity was appreciable when 2 was incubated with purified BruN in the (co-)presence of cofactors including NADPH, NADH, FAD, and FMN (Fig. S5A‡). However, upon its co-exposure to BruN and Na2S2O4, 2 was oxidized into 4a, but not cephalanone F (4b) as a C4a-selectively cleaved product of 2 under the BTG7/BTG13 catalysis (Fig. S5A‡).14 This experimentation confirmed that BruN did not directly accept 2 as substrate although bioinformatically predicted to be a BVMO that might catalyze the C4a- or C10a-selective cleavage of anthraquinones. To reinforce its catalyst partnership with BruN, BruM was heterologously expressed and purified as was done for the pure BruN protein (Fig. S4‡). In the presence of NADPH (2 mM), 2 (250 μM) was incubated with an equimolar (15 μM) mixture of BruM and BruN, leading ultimately to the production of 4a as indicated by our LC-MS analysis of the obtained reaction mixture (Fig. S5A‡). Despite its lower abundance, 3 was detected in the extracted ion chromatogram of the enzymatic reaction mixture resulting from the first 2 minutes exposure of 2 to BruM and NADPH, but became undetectable after reacted for 10 minutes (Fig. S5B‡), thus highlighting that 3 is fairly labile and tends to be re-oxidized into 2 in air.14 The attempt failed to identify the formation of 4b, thereby establishing the BruN's C10a-selectivity which is independent of BruM (Fig. 2B and S5‡). To test its substrate promiscuity, the equimolar mixture of BruM and BruN was exposed to each of chrysophanol analogs (available in our laboratory) in the presence of NADPH, followed by the LC-MS analysis of resultant products (Fig. S6‡). Unexpectedly, aloe emodin (5) was shown to be the most favorable substrate (Fig. S7‡). Also cleaved by the enzyme pair were 2, rhein (7), emodin (1), and physcion (6) with the transformation rates around 58%, 32%, 5%, and 2%, respectively (Fig. S6 and S7‡). However, no reaction was discerned after the two enzymes were co-exposed to emodin 8-O-β-D-glucopyranoside (8) (Fig. S7‡). To ascertain the regioselectivity in the ring opening, we scaled up the BruMN catalyzed reaction of 1 and 5–7, leading ultimately to the identification of 11-hydroxylated monodictyphenone (5a), a C10a-selective fission product from 5 (Table S7, Fig. S29, and S30‡). Although detected by the LC-MS analysis (Fig. S6‡), the cleaved products from other test compounds failed to be purified in sufficient quantities for the NMR measurement, presumably because of their (much) lower reaction rate in exposure to the enzyme pair (vide supra). Collectively, this set of experimentations reinforced the C10a-selectivity and a broader substrate scope of BruN in cleaving 2 and its analogs.Regioselectivity reversal of BruN and BTG13 by directed protein evolution
From a biochemical viewpoint,27 the enzyme catalysis is usually accomplished through (i) self-adjusting conformation to accept substrate, (ii) distorting or directing the substrate to follow a specific mechanism, and (iii) re-positioning its key AA residues to create a particular microenvironment to allow the reaction to proceed efficiently. In most or all cases, the reaction selectivity depends largely on several residues in the enzyme molecule, which are spatially close enough to the catalytic center. This dogma gave us an impetus to reverse the C10a-selectivity of BruN in opening anthraquinone ring of 2 through the directed protein evolution, which was proved effective in changing the catalytic trajectory of ChaP, a bacterial dioxygenase.28 To validate or generalize the strategy, BTG13 was chosen and processed in parallel owing to its oppositely selective (C4a–C10 bond) fission of 2 and crystal structure availability.14To minimize randomness, we aligned the sequences of all enzymes reported to catalyze the C10a- and C4a-selective cleavages of anthraquinones (Fig. S8‡). Thanks to the AlphaFold3 program,29 such multi-protein sequence alignments facilitated our prediction of the three-dimensional (3D) BruN structure, which was comparable to the crystal structures of its single-site mutant (BruNN441M) and BTG13 (Fig. S9‡).14 But our mutagenesis efforts were further frustrated by too many “selectivity-related” AA residues of the C10a- or C4a-cleaving enzymes that nested in different phylogenetic clades (Fig. 3A and S8‡). Docking between BruN and chrysophanol hydroquinone (3) was therefore conducted to suggest the selectivity-sensitive (closer than 5 Å from 3) AA residues (Fig. 3B), such as F55, R58, I91, A164, F243, G244, N245, K246, M247, V347, T435, D436, Q437, V440, N441, F442, and R452 (Fig. 3C). These “sensitive residues” contributing (most) likely to the C10a-selectivity of BruN were mutated individually to the AA residues appearing (more) frequently in the C4a-selective enzyme counterparts. All BruN variants obtained were heterologously expressed and purified as N-terminally His-tagged proteins as done for BruN (Fig. S10‡), followed by the catalysis assessment by being incubated at 30 °C for 2 h in the co-presence of BruM (15 μM), 2 (250 μM), and NADPH (2 mM). Particular attention was paid to the product detection by the LC-MS analysis to roughly quantify the amounts of 4a and 4b resulting from, and thus adopted as indicators of, the C10a- and C4a-selective fissions of 2, respectively. Notably, two BruN variants (BruNN441M and BruNN441W) exhibited substantially reversed regioselectivity as reflected by their catalysis for the conversion of 2 into 4b as a product with the 4a/4b ratio around 77:23 (BruNN441M) and 70:30 (BruNN441W), respectively. Other variants (e.g., BruNF243G, BruNG244P, BruNN245V, BruND436P, BruNV440I, and BruNF442G) were also generated but their 4a/4b ratios were found higher than 90:10 (Fig. 4 and S11‡). Unfortunately, such single-site mutations of BruN failed to afford any mutant capable of catalyzing the purely C4a-selective fission of 2 as does BTG13.14 Such a partially negative consequence was counteracted by our identification of two BTG13 mutants, BTG13M427N and BTG13M427V, both catalyzing a 100% conversion of 2 into 4a in a regioselective manner entirely different from intact BTG13 that allowed the 4b formation only.14 In other words, the findings strengthened our confidence in reversing BruN's regioselectivity via the directed protein evolution, although other single-site BTG13 mutants (e.g., BTG13M427F and BTG13M427P) were shown catalytically identical to BTG13 (Fig. 4, S12, and S13‡). As signified by the BruNN441M and BruNN441W catalysis (vide supra), the N441 mutation for BruN was most likely substantial or essential for its reversal to the C4a-selectivity, but not necessarily mutated to Met and Trp despite their frequent presence in the C4a-selectively cleaving enzymes (Fig. 3C). This encouraged us to perform the saturation mutagenesis28,30,31 by mutating N441 to each of the rest 17 protein AAs (Fig. S10‡). As a step forward, the BruNN441F mutant displayed an improved regioselectivity reversal (Fig. 4 and S14‡).
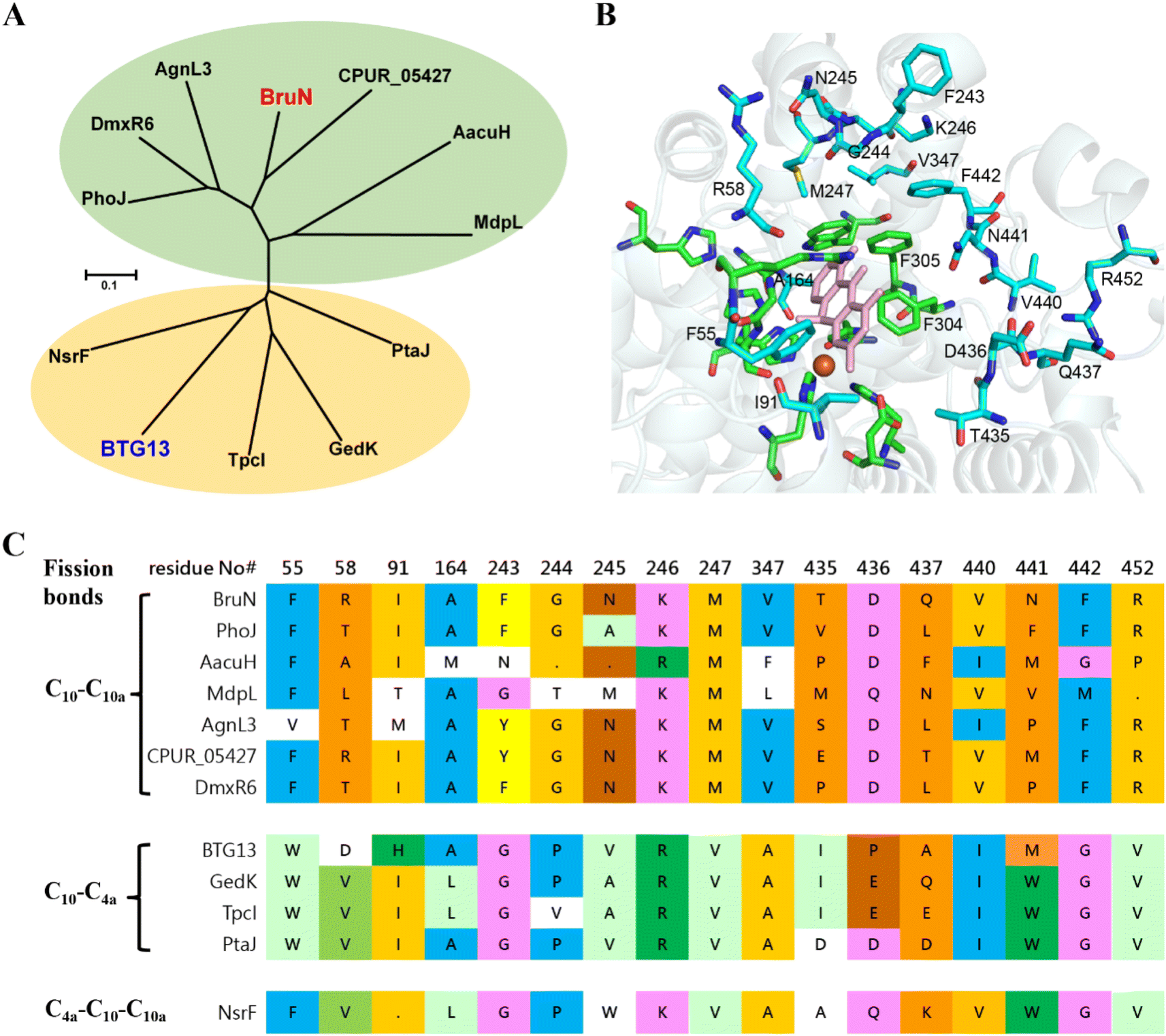 | ||
| Fig. 3 Sequence alignment of BruN with reported homologues that cleave the C4a–C10 and C10a–C10 bonds of anthraquinones. (A) Nesting in strikingly different clades were BruN (this work) and BTG13,14 which catalyze respectively C10a- and C4a-selective fissions of chrysophanol (2). (B) The docking of BruN (Alphafold3 modeled)29 to chrysophanol hydroquinone (3) highlighted the conserved (green) and varied residues (cyan) that are ≤5 Å distant from the 3 molecule in collaboration with multi-protein sequence alignments. (C) Multi-protein sequence alignments suggested a total of 17 amino acid (AA) residues associated possibly with the regioselective fission of C4a–C10 and C10a–C10 bonds of anthraquinones. | ||
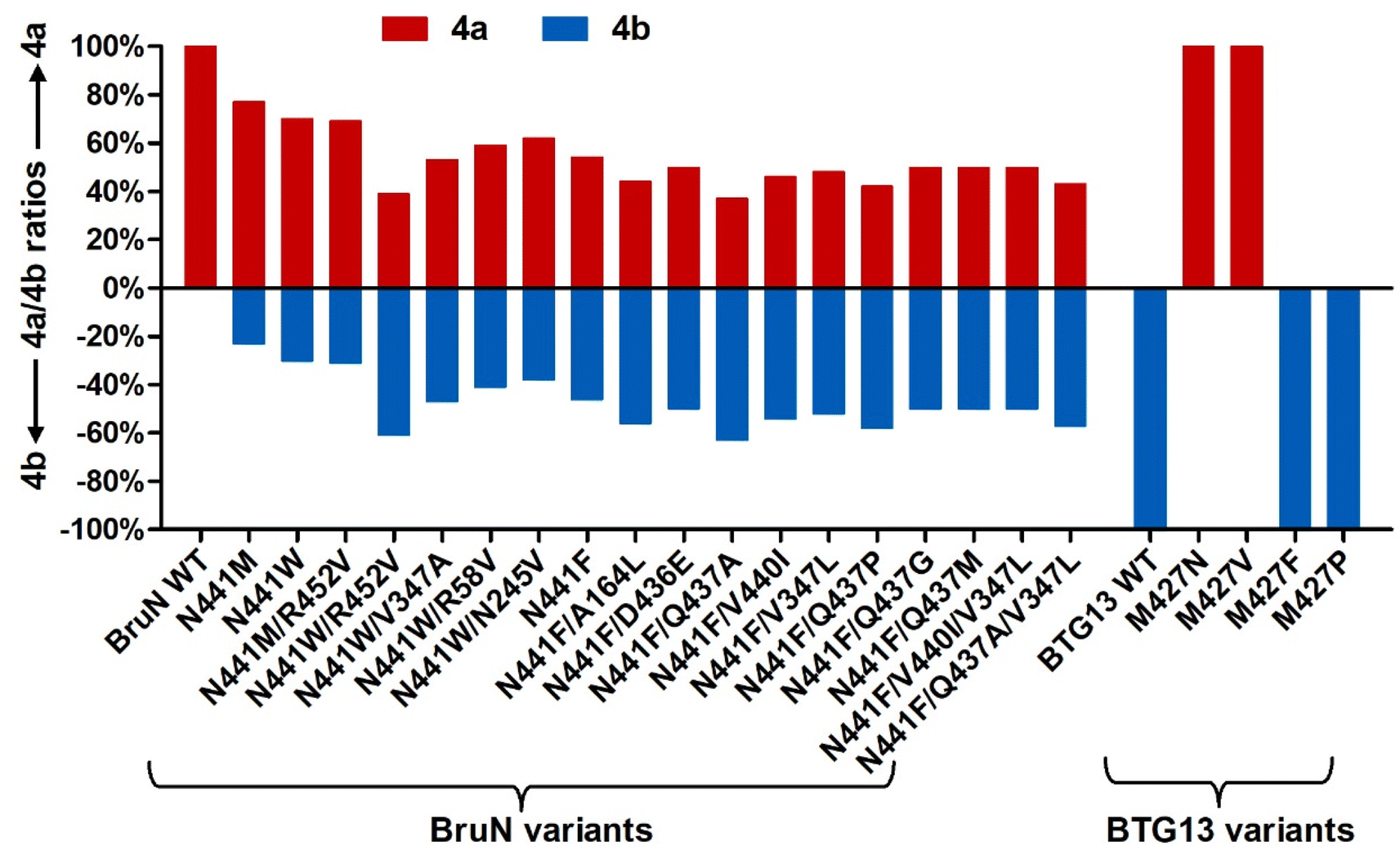 | ||
| Fig. 4 Yield comparison between monodictyphenone (4a) and cephalanone F (4b) formed as the C10a- and C4a-selective fission products, respectively, from chrysophanol (2) under the catalysis of BruN variants (NVs) and BTG13 mutants in the presence of equimolar amount BruM (15 μM), substrate 2 (250 μM), and NADPH (2 mM, as a cofactor). The regioselectivity reversal assay was performed at 30 °C for 2 h. Depicted were the NVs' mutants displaying appreciable selectivity reversals signified by the 4b formation (see Fig. 2B). The M427N and M427V mutants of BTG13, a C4a-selective catalyst,14 were demonstrated by the assay to catalyze the conversion of 2 into 4a with a 100% C10a-selectivity. | ||
In pursuing the highly regioselective BruN variant(s), the relatively effective single-site mutants, BruNN441F, or BruNN441M, or BruNN441W were subjected to our double mutation efforts. Thus, the rest 16 AA residues that predicted to be “selectivity-sensitive” (Fig. 3C) were individually changed into the counterparts of C4a-selective enzymes (Fig. 3C) and others if perceived necessary (Fig. S10‡). Interestingly, the abundance of 4b was found escalated in the reaction solution of 2 under the BruNN441W/R452V catalysis with the 4a/4b ratio approaching 39:61, although poor regioselectivity reversal was discerned with other double-site mutants, such as BruNN441M/R452V, BruNN441W/R58V, BruNN441W/N245V, and BruNN441W/V347A (Fig. 4 and S15‡). Moreover, starting from the N441-to-Phe mutant, we obtained several double-site variants like BruNN441F/Q437A, BruNN441F/V440I, BruNN441F/V347L, BruNN441F/Q437P, BruNN441F/A164L, BruNN441F/Q437G, and BruNN441F/Q437M, all exhibiting improved yields of 4b in the same catalysis assay (Fig. 4 and S16‡).
To obtain more efficient mutants, we were motivated to generate multi-site BruN mutants from the promising variants such as BruNN441W/R452V, BruNN441F/Q437A, and BruNN441F/V440I. The subsequent catalysis assay showed that BruNN441F/V440I/V347L and BruNN441F/Q437A/V347L possessed the substantially improved regioselectivity reversals as reflected by the 4a/4b ratios around 50:50 and 43:57 (Fig. 4, S16, and S17‡). Furthermore, during such repeated mutation attempts, we observed and thus envisioned that enzymatic reaction condition (RC) might play roles in reversing the regioselectivity. Thus, we re-evaluated the 4a/4b ratio values after 2 was treated with the promising mutants (vide supra) at varied concentrations within differentiated reaction durations (Fig. 5A). Surprisingly, the higher (>90%) C4a-selectivity was discerned with BruNN441F/Q437A, BruNN441F/V347L, BruNN441F/Q437P, BruNN441F/V440I/V347L, and BruNN441F/Q437A/V347L variants with the 4a/4b ratios around 10:90, 9:91, 9:91, 8:92, and 7:93, respectively (Fig. 2B, 5B and S18‡). Overall, the aforementioned findings realized the switch between the C4a- and C10a-selective fissions of chrysophanol (2) under the catalysis of BruN and BTG13.
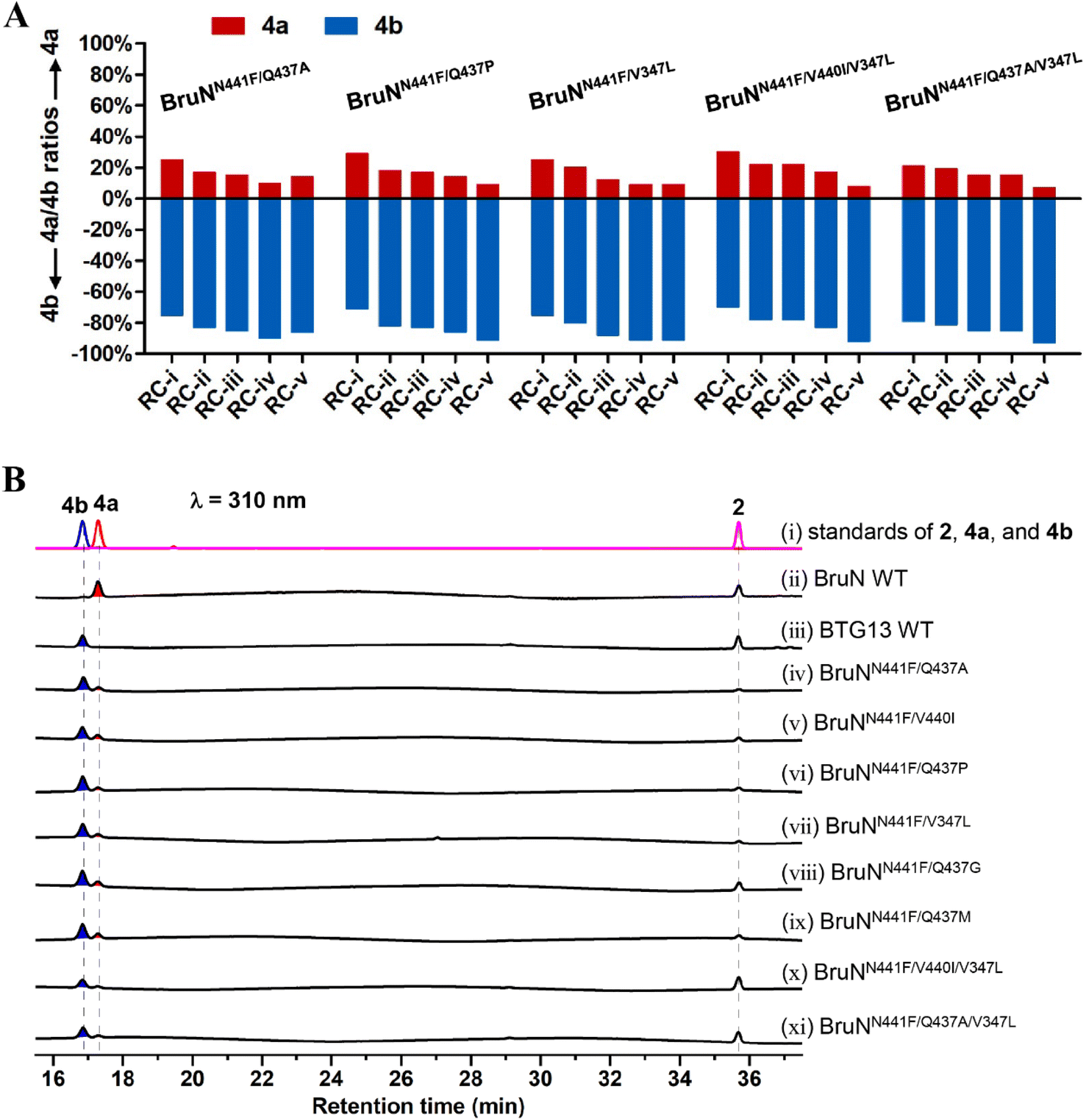 | ||
| Fig. 5 Reaction condition (RC) optimization for multiply muted BruN variants (NVs). (A) Reaction duration and protein concentration were optimized at 30 °C for the NVs capable of catalyzing the cleavage of chrysophanol (2) into cephalanone F (4b) with a substantial (>70%) C4a-selectivity in the presence of BruM and NADPH as specified below: (RC-(i)) 60 μM NVs, 25 μM BruM, 4 mM NADPH, 1 hour; (RC-(ii)) 80 μM NVs, 40 μM BruM, 4 mM NADPH, 30 min; (RC-(iii)) 100 μM NVs, 100 μM BruM, 5 mM NADPH, 15 min; (RC-(iv) and RC-(v)) 120 μM NVs, 120 μM BruM, 6 mM NADPH, 10 min. Substrate 2 was either 250 (from RC-(i) through RC-(iv)) or 125 μM (RC-(v)). (B) HPLC profiling for monodictyphenone (4a) and (4b) generating from 2 under the NVs' catalysis. The triple mutant (BruNN441F/Q437A/V347L) exhibited the highest (93%) C4a-selectivity. The C10a- and C4a-selective fission products, 4a and 4b, were quantified by the LC-MS analysis (Fig. S18‡). | ||
Insights into the regioselectivity and interchangeability of BruN and BTG13 catalyses
To deepen our understanding on the regioselectivity in the enzymatic catalysis, we were motivated to obtain the BruN crystal by consulting the BTG13 crystallization protocol.14 Unfortunately, it was unsuccessful. We therefore tried to get the crystal structure of BruN mutants generated for investigating the regioselectivity (vide supra). Fortunately, the BruNN441M crystal was afforded and determined at 3.6 Å resolution to resemble that of BTG13 (Table S10 and Fig. S9‡). As discerned with BTG13,14 an iron ion at the crystal center coordinated with four histidines (H63, H166, H308, and H386), a carboxylated-lysine (kcx389), and a water molecule. The iron ion was ascertained by the divalent cation replacement32 and the inductively coupled plasma mass spectrometry (ICP-MS) (Fig. S19‡). The AA residues around the iron center are conserved among the BruN homologs (Fig. S8‡) and were indispensable for BruN's catalytic activity as verified by our site-directed mutations (Fig. S20 and S21‡). Despite the same iron coordination mode, BruNN441M and BTG13 crystals showed different binding pockets above the iron cofactor (Fig. S9‡). This observation, along with the directed protein evolution experiments, prompted us to hypothesize that the shared substrate, chrysophanol hydroquinone (3), may exist in several or diverse tautomers (e.g., 3a and 3b) in the BruN- or BTG13-created microenvironments; but only particularly structured tautomers could effectively interact with enzymes to form the regioselectively cleaved products from 3. The assumption agreed with the keto–enol tautomerism of the anthraquinone hydroquinone intermediate involved in the aflatoxin B1 biosynthesis.33 While re-oxidizable into 2 in exposure to air,14,18,34 hydroquinone 3 tautomerizes rapidly, and the tautomers are too interchangeable to be captured individually. We were tempted to chemically confirm the tautomerism. Thus, chrysophanol (2) was methylated with Me2SO4 to obtain its methylated product, which was further reduced by Na2S2O4 followed by immediate methylation with Me2SO4 once again to give mainly 12, the permethylated derivative of 3 (Fig. S22‡), whose structure was proven identical to that defined earlier.35 Such efforts verified the tautomerism of 3 since both 3a and 3b can be permethylated into 12. Moreover, the isotope labeling experiment was performed using 18O2, thereby confirming the dioxy catalytic mechanism of dioxygenase BruN rather than Baeyer–Villiger oxidation (Fig. S24‡). Accordingly, as illustrated in Scheme 1, the BruM/BTG7-catalyzed (two-electron) reduction of chrysophanol (2) gave chrysophanol hydroquinone (3) that tends to tautomerize into 3a and 3b, and probably others. In the presence of dioxygen, BruN interacts fruitfully with the tautomer 3a with 10,10a-double bond involved in the catalytic complex I, but BTG13 prefers to act towards 3b with 10,4a-double bond bound to the enzyme to form complex I′. Next complexes I and I′ undergo the double bond cleavages to afford 4a and 4b presumably via intermediates II–V and II′–V′, respectively, as if carotenoids are converted into ketone and/or aldehyde products under the catalysis of carotenoid cleavage oxygenases with the iron cofactor coordinated with four His residues, too.36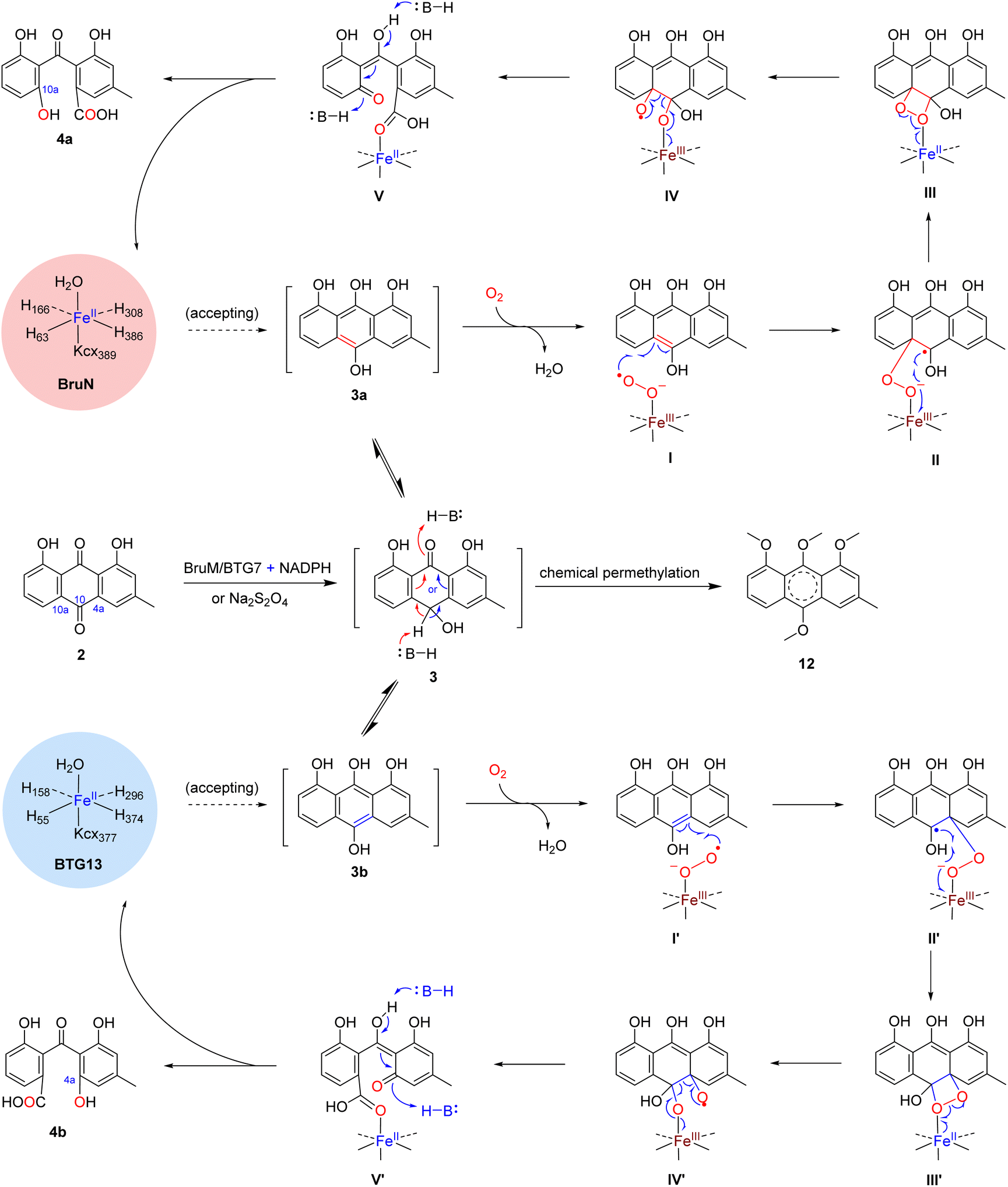 | ||
| Scheme 1 Proposed catalytic mechanism underlying the regioselectivity difference between BruN and BTG13. After its reduction by Na2S2O4, or by BruM (or BTG7) in the presence of NADPH, chrysophanol (2) was transformed into chrysophanol hydroquinone (3) which was cleaved by BruN and BTG13 into monodictyphenone (4a) and cephalanone F (4b) in C10a- and C4a-selective manners, respectively. | ||
The above proposal agreed with our theoretical simulations. The geometric optimization and relative energy were calculated at B3LYP level to suggest four lower-energy conformations (LECs) for each of 3a and 3b (see 3a1–3a4 and 3b1–3b4 in Fig. S23‡). All the eight LECs were individually docked to BruN, BruNN441M, and BTG13. Interestingly, the average binding free energies (ΔGbinding) of BruN to 3a (−25.32 ∼ −23.42 kJ mol−1) are lower than that to 3b (−23.21 ∼ −22.28 kJ mol−1). In contrast, the ΔGbinding value of BTG13 to 3a (−24.86 ∼ −23.37 kJ mol−1) were higher than that to 3b (−27.34 ∼ −23.74 kJ mol−1) (Table S11‡). More interestingly, molecular dioxygen was shown to be able to access the 10,10a-double bond of 3a3 interacted with BruN (Fig. 6A), and the 10,4a-double bond of 3b1 docked with BTG13 (Fig. 6B). In compliance with the mutation experiments (Fig. S21‡), we did detect the π–π stacking interactions of 3a and 3b with F304 (in BruN) and F292 (in BTG13), respectively, thereby confirming the previously reported important role of this phenylalanine residue in the enzymatic catalysis.14
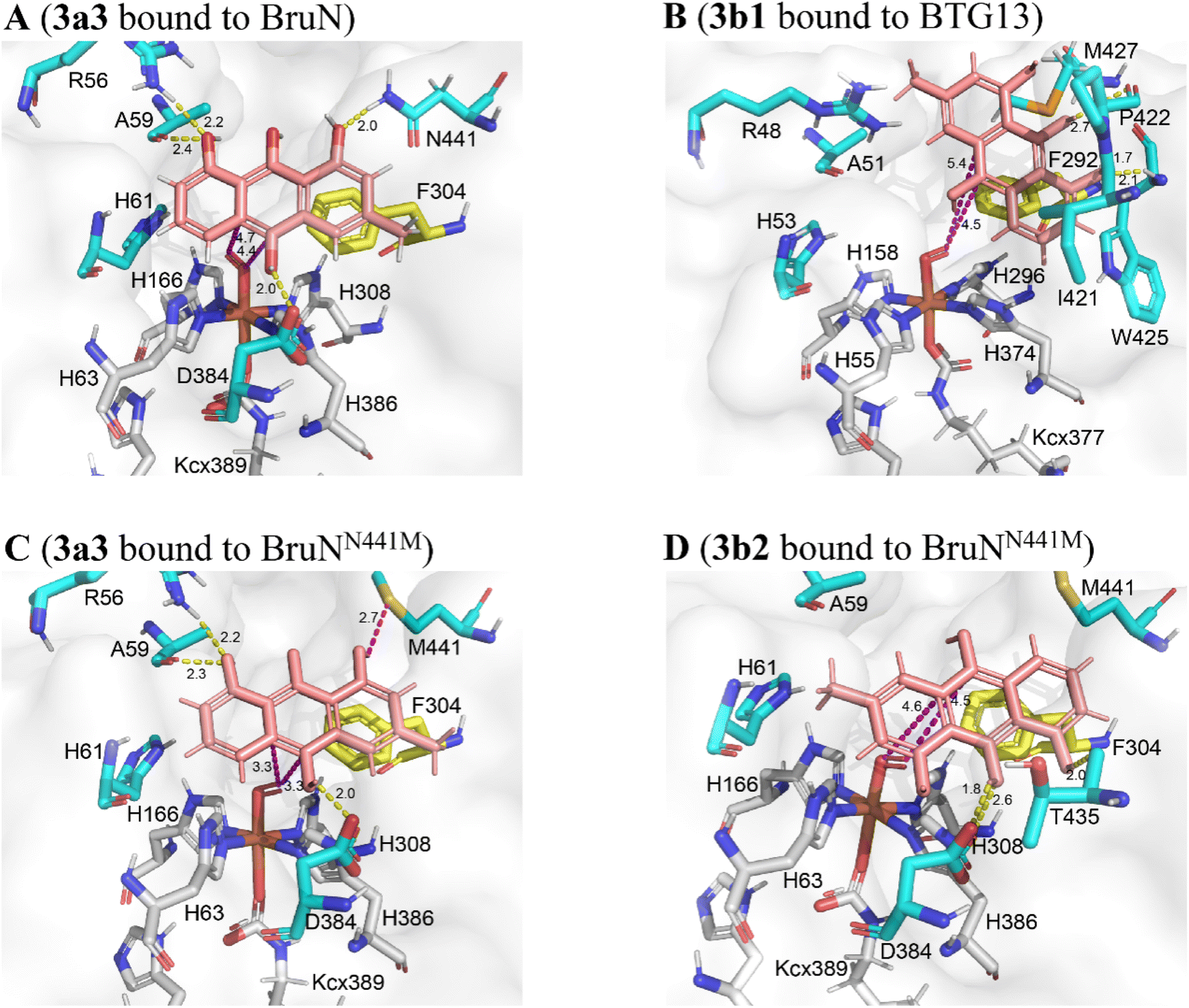 | ||
| Fig. 6 Theoretical simulations of chrysophanol hydroquinone (3) tautomers (3a and 3b) with BruN, BruNN441M (PDB: 8YXS), and BTG13 (PDB: 7Y3W). (A) BruN preferred to bind 3a3, a conformation of 3a. (B) BTG13 interacted more effectively with 3b1, a conformation of 3b. (C and D) BruNN441M was more interactive with 3a3 than with 3b2, another conformation of 3b. The magentas and yellow dashes indicated the electrostatic and hydrogen-bonding interactions, respectively. As marked in yellow, the F304 (in BruN and BruNN441M) and F292 (in BTG13) residues formed the π–π stacking interactions with the 3a and 3b conformations. Using the AlphaFold3 program,29 BruNN441F/Q437A/V347L was shown to resemble BruNN441M (Fig. S9‡). | ||
Also important is BruN's N441 residue that hydrogen-bond with 3a to drive the 10,10a-double bond closer to the catalytic site (Fig. 6A). However, such hydrogen-bonding interaction between 3a and the N441 residue in BruN was deprived in the BruNN441M mutant (Fig. 6C and D), which was thus rendered “liberated” to form other optional binding conformations with both 3a3 (Fig. 6C) and 3b2 (Fig. 6D). Agreeing with the experimental observation (Fig. 4), dioxygen was accessible when BruNN441M interacted with 3a3 (Fig. 6C) and 3b2 (Fig. 6D) to drive respectively the 10,10a- and 10,4a-double bonds closer to the catalytic center. This observation explained at least in part why both 4a and 4b were produced from the BruNN441M catalyzed fission of 3 that was cleaved in its tautomer forms 3a and 3b, respectively. Moreover, the binding energies of BruNN441M with 3a was lower than that with 3b (Table S11‡) agreed roughly with the 4a/4b ratio around 77:23. Collectively, our experimentation and computation corroborated the C10a- and C4a-selectivity and interchangeability of BruN and BTG13 in catalyzing the ring-openings of chrysophanol (2) after the two-electron reduction into chrysophanol hydroquinone (3).
The C4a- and C10-selective cleavage of anthraquinones is widely involved in the fungal biosynthesis of structurally diverse bioactive xanthones as enumerated in Fig. 1. But the bond fission mechanism was misunderstood to be results of the Baeyer–Villiger oxidation reaction before Lu's and Rao's groups recognized the two-electron reduction of anthraquinone substrates into the corresponding hydroquinone forms as a prerequisite for such ring openings.14,18 As additional insights into the topic, this work unravels the tunability between the C10a- and C4a-selectivities in cleaving 2 after its reduction into 3 by deepening the understanding on the catalytic features of BruN and BTG13 through directed protein evolution, chemical capture of chrysophanol hydroquinone tautomers (e.g., 3a and 3b), 18O chasing experiment, theoretical simulations starting from the structures of BruN (AlphaFold3 modeled),29 BruNN441M, and BTG13.14 According to our experimental results, the N441 site in BruN corresponding to M427 site in BTG13 is essential for the reversal between C10a- and C4a-selectivity. For BruN, polar AA (N441) was mutated to non-polar aromatic amino acid phenylalanine (BruNN441F) to facilitate the C10a-to-C4a selectivity switch, which could be further improved by simultaneous double or multiple mutation of its neighboring AAs (e.g., V347L, T435–F442), as evidenced from the superior catalytic performance of BruNN441F/Q437P, BruNN441F/V440I/V347L, and BruNN441F/Q437A/V347L. The observation could be rationalized by the mutation-reshaped binding cavity that favored the binding of 3b tautomer with the mutants such as BruNN441M (Fig. 6D). For BTG13, the mutation of non-polar AA (M427) to a polar AA (asparagine) facilitated the C4a-to-C10a selectivity switch. Interestingly, if M427 was replaced by other non-polar AAs such as phenylalanine and proline, these BTG13 variants remained to be C4a-selective. Next, we generated BTG13M427V by substituting M427 with valine, but unexpectedly, this mutant shared the same regioselectivity with BTG13M427N (vide supra). This could be due to the similarity in chain length between the valine and asparagine residues despite the difference in polarity, suggesting that the AA size also played a substantial role in re-shaping the binding pocket for their bindings with the 3a tautomer. Generally speaking, the smaller the binding cavity is, the fewer AA residues need to be muted for re-shaping the cavity capable of recognizing and accepting one particular tautomer of 3. We therefore compared the crystal structures to pinpoint that the substrate binding pocket of BTG13 is substantially smaller than that of BruNN441M (Fig. S9‡). Such a difference could be among the key reason why the single-site mutation of BTG13 (M427N and M427V) could completely switch the regioselectivity, whereas BruN could only be muted to achieve a maximal (93%) conversion rate as discerned with BruNN441F/Q437A/V347L (Fig. 5).
Many fungal xanthones are biosynthetically mediated by the anthraquinone-forming step,14,18 and the carbon skeletons of end products depend crucially on the xanthone monomers and their oligomerizations.3 To our observation, such a big family of polyketides consists largely of the homodimers of tailored xanthone monomers derived from C10a- or C4a-selective fissions of anthraquinone intermediates whereas the heterodimeric counterparts are much rarer (Fig. 1).3,13 Here defined is the tunability between the C10a- and C4a-selectivities, which can give “non-native” cleaved anthraquinone intermediates for generating otherwise unobtainable xanthones. In this sense, the work lays the foundation for mining so far undescribed xanthones that are structurally too complex to be synthesized chemically at a reasonable or acceptable cost. Taken together, our findings are of fundamental significance in (i) facilitating more profound investigation of these families of structurally complex bioactive xanthones, and (ii) enabling the synthetic biology-based regeneration of high-value xanthones, many of which exist for unknown reasons as minor components in nature.
Conclusions
The broad involvement of regiospecific anthraquinone cleavages in the xanthone biosynthesis directed our attention to A. brunneoviolaceus characterized by its generation of the brunneoxanthone structures signifying the homodimerization of modified xanthone monomers forming from the C10a–C10 bond (C10a-selective) fission of anthraquinone chrysophanol (2).8 Following the whole-genome sequencing of the fungus, our deletion and heterologous expression of genes led to the attribution of the C10a-selectivity to the BruN (an atypical non-heme iron dioxygenase)-catalyzed fission of the 10,10a-double bond of tautomer (3a) of chrysophanol hydroquinone (3) forming from 2 under the BruM catalysis in the presence of NADPH. In correlation to our own (vide supra) and others' insights into the C4a–C10 bond (C4a-selective) cleavage of 2,14,18 we were able to disclose, and subsequently decipher the mechanism of, the tunability between the C10- and C4a-selectivities by a combination of directed protein evolution, theoretical simulation, chemical capture of the hydroquinone tautomer, 18O chasing, and X-ray crystal structure of the BruNN441M mutant. In aggregation, the findings shed light on how to reverse or tune the regioselectively differed ring openings of anthraquinones as biosynthetic intermediates, and are thus of fundamental value for the deeper mining and/or unnatural (e.g., synthetic biology-enabled) regeneration of useful polyketides via biotechnologies.Data availability
The data supporting this article have been reported as part of the ESI.‡Author contributions
X. J. L. and R. X. T. designed research and wrote paper; X. J. L., L. R. Z., and J. J. Z. performed experiments; X. J. L., X. X. M., J. P. Z., and R. X. T. analyzed data; C. Z. A. performed the computational work.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was cofinanced by the grants from NSFC (81991524 and 22207057), the Natural Science Foundation of Jiangsu Province of China (BK20220480), and the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (22KJB350002).Notes and references
- C. Badiali, V. Petruccelli, E. Brasili and G. Pasqua, Plants, 2023, 12, 694 CrossRef PubMed.
- L. X. Kong, Z. X. Deng and D. L. You, Nat. Prod. Rep., 2022, 39, 2057–2095 RSC.
- K. M. J. de Mattos-Shipley and T. J. Simpson, Nat. Prod. Rep., 2023, 40, 174–201 RSC.
- H. R. El-Seedi, H. M. S. Ibrahim, N. Yosri, M. A. A. Ibrahim, M. E. F. Hegazy, W. N. Setzer, Z. M. Guo, X. B. Zou, M. S. Refaey, S. E. Salem, S. G. Musharraf, A. Saeed, S. E. Salem, B. J. Xu, C. Zhao and S. A. M. Khalifa, Curr. Med. Chem., 2024, 31, 62–101 CrossRef.
- M. Alam, S. Rashid, K. Fatima, M. Adnan, A. Shafie, M. S. Akhtar, A. H. Ganie, S. M. Eldin, A. Islam, I. Khan and I. Hassan, Biomed. Pharmacother., 2023, 163, 114710 CrossRef.
- V. V. Vanessa and S. H. Mah, Mini-Rev. Med. Chem., 2021, 21, 2507–2529 CrossRef.
- M. Gallorini, S. Carradori, D. I. S. P. Resende, L. Saso, A. Ricci, A. Palmeira, A. Cataldi, M. Pinto and E. Sousa, Int. J. Mol. Sci., 2022, 23, 13319 CrossRef PubMed.
- X. J. Lv, F. Ding, Y. J. Wei and R. X. Tan, Chin. J. Chem., 2021, 39, 1580–1586 CrossRef.
- J. F. Sanchez, R. Entwistle, J. H. Hung, J. Yaegashi, S. Jain, Y. M. Chiang, C. C. C. Wang and B. R. Oakley, J. Am. Chem. Soc., 2011, 133, 4010–4017 CrossRef CAS PubMed.
- S. Pornpakakul, J. Liangsakul, N. Ngamrojanavanich, S. Roengsumran, P. Sihanonth, J. Piapukiew, E. Sangvichien, S. Puthong and A. Petsom, Arch. Pharmacal Res., 2006, 29, 140–144 CrossRef CAS.
- X. X. Wei and Y. Matsuda, Org. Lett., 2020, 22, 1919–1923 CrossRef CAS.
- L. Neubauer, J. Dopstadt, H. U. Humpf and P. Tudzynski, Fungal Biol. Biotechnol., 2016, 3, 2 CrossRef PubMed.
- Y. Matsuda, C. H. Gotfredsen and T. O. Larsen, Org. Lett., 2018, 20, 7197–7200 CrossRef CAS PubMed.
- X. D. Hou, H. B. Xu, Z. W. Deng, Y. J. Yan, Z. B. Yuan, X. Z. Liu, Z. P. Su, S. Yang, Y. Zhang and Y. J. Rao, Angew. Chem., Int. Ed., 2022, 61, e202208772 CrossRef CAS.
- M. T. Nielsen, J. B. Nielsen, D. C. Anyaogu, D. K. Holm, K. F. Nielsen, T. O. Larsen and U. H. Mortensen, PLoS One, 2013, 8, e72871 CrossRef CAS PubMed.
- S. Sato, N. Okusa, A. Ogawa, T. Ikenoue, T. Seki and T. Tsuji, J. Antibiot., 2005, 58, 583–589 CrossRef CAS PubMed.
- K. Throckmorton, F. Y. Lim, D. P. Kontoyiannis, W. F. Zheng and N. P. Keller, Environ. Microbiol., 2016, 18, 246–259 CrossRef CAS PubMed.
- F. F. Qi, W. Zhang, Y. Y. Xue, C. Geng, X. N. Huang, J. Sun and X. F. Lu, J. Am. Chem. Soc., 2021, 143, 16326–16331 CrossRef CAS.
- Z. W. Deng, H. Su, X. D. Hou, H. B. Xu, Z. B. Yuan, X. Sheng and Y. J. Rao, ACS Catal., 2024, 14, 797–811 CrossRef CAS.
- K. Blin, S. Shaw, K. Steinke, R. Villebro, N. Ziemert, S. Y. Lee, M. H. Medema and T. Weber, Nucleic Acids Res., 2019, 47, W81–W87 CrossRef CAS PubMed.
- A. J. Szwalbe, K. Williams, Z. S. Song, K. de Mattos-Shipley, J. L. Vincent, A. M. Bailey, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 233–238 RSC.
- C. Greco, K. de Mattos-Shipley, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Sci., 2019, 10, 2930–2939 RSC.
- Y. B. Han, W. Bai, C. X. Ding, J. Liang, S. H. Wu and R. X. Tan, J. Am. Chem. Soc., 2021, 143, 14218–14226 CrossRef CAS PubMed.
- G. Z. Dai, W. B. Han, Y. N. Mei, K. Xu, R. H. Jiao, H. M. Ge and R. X. Tan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1174–1180 CrossRef CAS.
- A. Krick, S. Kehraus, C. Gerhäuser, K. Klimo, M. Nieger, A. Maier, H. H. Fiebig, I. Atodiresei, G. Raabe, J. Fleischhauer and G. M. König, J. Nat. Prod., 2007, 70, 353–360 CrossRef CAS.
- Z. Z. Zhou, H. J. Zhu, L. P. Lin, X. Zhang, H. M. Ge, R. H. Jiao and R. X. Tan, Chem. Sci., 2019, 20, 73–82 RSC.
- D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS.
- Y. S. Wang, W. Zheng, N. Jiang, Y. X. Jin, Z. K. Meng, M. X. Sun, Y. L. Zong, T. Xu, J. P. Zhu and R. X. Tan, Angew. Chem., Int. Ed., 2022, 61, e202201321 CrossRef PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C. C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Zemgulyte, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Zídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. X. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef.
- R. M. Phelan and C. A. Townsend, J. Am. Chem. Soc., 2013, 135, 7496–7502 CrossRef.
- Y. S. Wang, B. Zhang, J. P. Zhu, C. L. Yang, Y. Guo, C. L. Liu, F. Liu, H. Q. Huang, S. W. Zhao, Y. Liang, R. H. Jiao, R. X. Tan and H. M. Ge, J. Am. Chem. Soc., 2018, 140, 10909–10914 CrossRef.
- D. Conradt, M. A. Schatzle, J. Haas, C. A. Townsend and M. Müller, J. Am. Chem. Soc., 2015, 137, 10867–10869 CrossRef PubMed.
- M. A. Schätzle, S. M. Husain, S. Ferlaino and M. Müller, J. Am. Chem. Soc., 2012, 134, 14742–14745 CrossRef.
- H. Miyabe, M. Torieda, K. Inoue, K. Tajiri, T. Kiguchi and T. Naito, J. Org. Chem., 1998, 63, 4397–4407 CrossRef.
- A. Daruwalla and P. D. Kiser, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2020, 1865, 158590 CrossRef PubMed.
Footnotes |
| † The crystal structure of BruNN441M has been deposited in the Protein Data Bank (PDB), https://www.rcsb.org, with accession number 8YXS. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06369d |
| § Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |