
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting photothermal conversion through array aggregation of metalloporphyrins in bismuth-based coordination frameworks†
Liang
He
 a,
Jing
He
a,
Er-Xia
Chen
*ac and
Qipu
Lin
*abd
a,
Jing
He
a,
Er-Xia
Chen
*ac and
Qipu
Lin
*abd
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: exchen@fjirsm.ac.cn; linqipu@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China, Fuzhou, Fujian 350108, China
dState Key Laboratory of Photocatalysis on Energy and Environment, Fuzhou University, Fuzhou, Fujian 350116, China
First published on 27th September 2024
Abstract
Materials capable of efficiently converting near-infrared (NIR) light into heat are highly sought after in biotechnology. In this study, two new three-dimensional (3D) porphyrin-based metal–organic frameworks (MOFs) with a sra-net, viz. CoTCPP-Bi/NiTCPP-Bi, were successfully synthesized. These MOFs feature bismuth carboxylate nodes interconnected by metalloporphyrinic spacers, forming one-dimensional (1D) arrays of closely spaced metalloporphyrins. Notably, the CoTCPP-Bi exhibits an approximate Co⋯C distance of 3 Å, leading to enhanced absorption of NIR light up to 1400 nm due to the presence of strong interlayer van der Waals forces. Furthermore, the spatial arrangement of the metalloporphyrins prevents axial coordination at the centers of porphyrin rings and stabilizes a CoII-based metalloradical. These characteristics promote NIR light absorption and non-radiative decay, thereby improving photothermal conversion efficiency. Consequently, CoTCPP-Bi can rapidly elevate the temperature from room temperature to 190 °C within 30 seconds under 0.7 W cm−2 energy power from 808 nm laser irradiation. Moreover, it enables solar-driven water evaporation with an efficiency of 98.5% and a rate of 1.43 kg m−2 h−1 under 1 sun irradiation. This research provides valuable insights into the strategic design of efficient photothermal materials for effective NIR light absorption, leveraging the principles of aggregation effect and metalloradical chemistry.
Introduction
Light-to-heat conversion materials have garnered significant attention for their potential applications in biomedicine, actuators, catalysis, and solar steam generation.1 Recent advancements have led to the emergence of a diverse array of photothermal materials, including carbon-based composites, metal-based inorganic materials, organic molecules and polymers, as well as inorganic–organic hybrids.2 The Jablonski diagram illustrates that improving the photothermal effect requires enhancements in the material's light absorption, especially in near-infrared (NIR) absorption, as well as the optimization of its non-radiative transition process, which involves energy dissipation and simultaneous heat generation.3 In addition, radical-based materials offer unique benefits for achieving efficient photothermal conversion owing to the existence of unpaired electrons and their spin effects.4 However, radicals are highly chemically reactive, making the actual preparation of photostable photothermal materials with a wide light absorption range challenging.The self-assembly of organic molecules through aggregation greatly facilitates the development of NIR photothermal materials by inducing a red-shifted spectral absorption.5 Porphyrin, a prototypical π-conjugated organic molecule, is increasingly recognized as a promising candidate for photothermal conversion due to its tunable optical properties and reactivity.6 While monomeric porphyrin has limitations in terms of NIR absorption and photothermal conversion efficiency, the broadening and red-shifting of the absorption peak can be achieved by forming suitable aggregates through interactions such as π–π stacking, electrostatic attraction, or hydrogen bonding.7 For instance, compared to the head-to-head approach of porphyrin H-aggregates, the head-to-tail configuration of J-aggregates results in stronger electronic coupling between molecules, leading to absorption redshift (Scheme 1).8 Additionally, metallized porphyrin endows advantages for NIR light absorption. The coordination of metal ions in porphyrin rings can significantly alter the orbital hybridization within the porphyrin chromophore.4b,9 This modification may alleviate the spin prohibition of porphyrin's Q bands, thereby enhancing light absorption and causing red shifting. Furthermore, the presence of metal ions in porphyrin rings can directly influence axial bonding and interactions between porphyrins in aggregates, potentially boosting light absorption and stabilizing metal radicals.
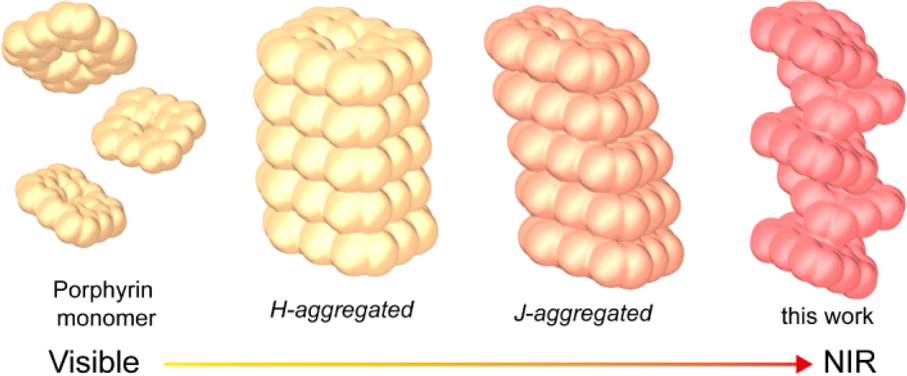 | ||
| Scheme 1 Enhanced light absorption caused by different ways of porphyrinic aggregation. | ||
A variety of spectroscopic techniques and theoretical models are employed to unravel the impact of metalloporphyrin aggregation on NIR light absorption enhancement. Nevertheless, the random arrangement and amorphous nature of most multi-metalloporphyrin assemblies present challenges in achieving a comprehensive understanding of the underlying principles using these methods.10 Therefore, there is considerable interest in transitioning metalloporphyrin-based disordered aggregates to crystalline phases, which offer well-defined and customizable structures that allow for the investigation of the role of metalloporphyrin aggregation mode in enhancing NIR light absorption at the molecular or atomic scale. Metalloporphyrin-based metal–organic frameworks (MOFs) have emerged as a class of crystalline materials with potential applications due to their precise spatial arrangement of components, tunable structures, and excellent light harvesting capabilities, rendering them an ideal platform for studying the role of metalloporphyrin aggregation in photothermal conversion.11 However, current design strategies for metalloporphyrin-based MOFs primarily concentrate on optimizing porosity to increase gas sorption or exposing active sites (e.g., metalloporphyrin centers) to enhance catalytic performance, often leading to the spatial separation of active centers and limiting the investigation of metalloporphyrin stacking patterns in MOFs.12
The efficient incorporation of densely packed metalloporphyrins into a MOF necessitates the selection of appropriate metal nodes and porphyrin ligands. The bismuth(III) oxidation state proves particularly advantageous due to its high coordination numbers and flexible coordination environments, enabling the creation of unique metal complexes with distorted conformations of inorganic building blocks (IBUs).13 Additionally, bismuth and its complexes are non-toxic, making them appealing for diverse applications such as sea water desalination, medicine and biotechnology.14 Herein, we have successfully fabricated two bismuth–metalloporphyrin-based MOFs, denoted as MTCPP-Bi, TCPP = tetra(4-carboxyphenyl)porphyrin, M = CoII and NiII, which consist of a one-dimensional (1D) rod-like aggregation of Co/NiTCPP (Fig. 1). The stacking pattern between porphyrins resembles the head-to-tail pattern typical of J-aggregates, albeit forming a helix. The strong Bi–O bonds and stacking force between adjacent metalloporphyrin moieties impart excellent chemical tolerance to MTCPP-Bi. Furthermore, the aggregation of metalloporphyrins and the abundance of CoII-based metalloradicals contribute to the broad light absorption of CoTCPP-Bi, extending into the NIR-II range. These attributes position CoTCPP-Bi as a promising candidate for photothermal conversion applications.
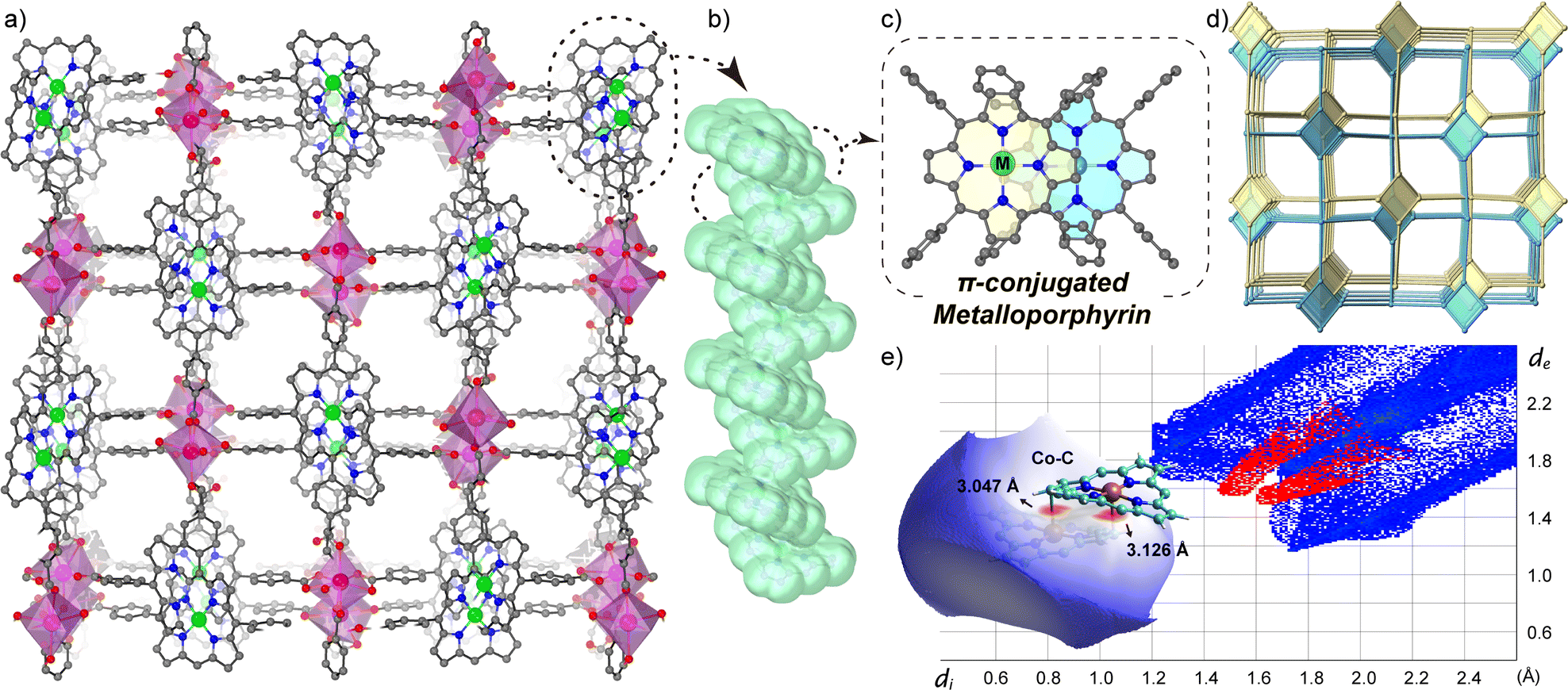 | ||
| Fig. 1 (a) Crystal structure of MTCPP-Bi viewed along the c axis. (b) Side view and (c) top view of 1D rod-like aggregation of MTCPP in MTCPP-Bi, illustrating the orbital overlap area between neighbouring porphyrin macrocycles. (d) Twofold interpenetrated network with sra topology. (e) Hirshfeld surface analysis and fingerprint plots for CoTCPP-Bi. M = Co/Ni in (a) and (c). | ||
Results and discussion
Synthetic procedures
All the samples can be formed in capped vials (see the ESI† for details). Their phase purities and formulas were confirmed by X-ray diffraction (XRD), elemental and thermogravimetric analyses, scanning electron microscopy (SEM), electron paramagnetic resonance (EPR), Fourier transformed-infrared (FT-IR) spectroscopy, ultraviolet-visible (UV-Vis) diffuse reflection spectroscopy, and fluorescence spectroscopy (ESI†).Structures and characterization of MTCPP-Bi
CoTCPP-Bi and NiTCPP-Bi were prepared using the solvothermal method, with a detailed protocol provided in the ESI (Section S1.2†). Single-crystal X-ray diffraction (XRD) analysis revealed that both CoTCPP-Bi and NiTCPP-Bi share an identical framework structure, possessing a three-dimensional (3D) structure with twofold interpenetration, aligned with the P21212 space group (Fig. 1a and Table S1†). During the solvothermal procedure, CoII or NiII metalated the porphyrinic macrocycles (TCPP) at their centers to be MTCPP that served as a four-connection square planar node (Fig. 1c), linking with four-connection [Bi(COO)4] IBUs to create a sra-net (Fig. 1d and S1†). The molar ratio of CoII/NiII-to-BiIII was determined as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 through site occupancy refinement, considering the distinct X-ray scattering factors and coordination environments of the metals. Metal content was validated by inductively coupled plasma-mass spectrometry (ICP-MS), element mapping images (Fig. S4†), energy dispersive spectroscopy (EDS) (Fig. S5 and S6†), and Fourier-transform infrared spectroscopy (FT-IR) (Fig. S7†). X-ray photoelectron spectroscopy (XPS) confirmed the local coordination environment of MII in MTCPP-Bi (Fig. S8 and S9†). The N1s XPS of CoTCPP-Bi showed identical binding energies (1087 eV) to CoTCPP, in contrast to the notable shifts in metal-free TCPP. Additionally, the Co2p binding energies for CoTCPP and CoTCPP-Bi remained consistent at 692 eV and 708 eV, respectively, with the Co2p binding energy peak absent in unmetallized TCPP. Similarly, the binding energies of N and Ni in both NiTCPP and NiTCPP-Bi samples are nearly identical, with Ni positioned at 855 eV and 872 eV, and N at 398 eV (Fig. S10†). These findings attest to the successful metallization of TCPP by CoII or NiII during the in situ synthesis of MTCPP-Bi. The unusual spatial arrangement of metalloporphyrins contributes to the specific twist angle and parallel orientation of IBUs within the interwoven networks, generating a continuous 1D rod-like aggregation facilitated by offset π–π interactions (Fig. 1b and c).
:1 through site occupancy refinement, considering the distinct X-ray scattering factors and coordination environments of the metals. Metal content was validated by inductively coupled plasma-mass spectrometry (ICP-MS), element mapping images (Fig. S4†), energy dispersive spectroscopy (EDS) (Fig. S5 and S6†), and Fourier-transform infrared spectroscopy (FT-IR) (Fig. S7†). X-ray photoelectron spectroscopy (XPS) confirmed the local coordination environment of MII in MTCPP-Bi (Fig. S8 and S9†). The N1s XPS of CoTCPP-Bi showed identical binding energies (1087 eV) to CoTCPP, in contrast to the notable shifts in metal-free TCPP. Additionally, the Co2p binding energies for CoTCPP and CoTCPP-Bi remained consistent at 692 eV and 708 eV, respectively, with the Co2p binding energy peak absent in unmetallized TCPP. Similarly, the binding energies of N and Ni in both NiTCPP and NiTCPP-Bi samples are nearly identical, with Ni positioned at 855 eV and 872 eV, and N at 398 eV (Fig. S10†). These findings attest to the successful metallization of TCPP by CoII or NiII during the in situ synthesis of MTCPP-Bi. The unusual spatial arrangement of metalloporphyrins contributes to the specific twist angle and parallel orientation of IBUs within the interwoven networks, generating a continuous 1D rod-like aggregation facilitated by offset π–π interactions (Fig. 1b and c).
The close proximity of adjacent metalloporphyrins enhances π–π interactions, resulting in a substantial increase in interlayer orbital overlap. Noteworthily, a strong interaction exists between the metal cobalt at the core of the porphyrin ring and the pyrrole β-carbon of the axially neighboring porphyrin layer, evidenced by a compressed metal–carbon (Co⋯C) distance of only 3 Å (Fig. S2†). This distance is significantly smaller than the Ni⋯C distance in the NiTCPP-Bi (minimum, 3.3 Å). In addition, the Co⋯C distance is shorter than the standard van der Waals contact observed in prior studies between graphite layers (3.4 Å) and adjacent porphyrins (>3.2 Å).15 As unveiled through investigations in the crystal database CCDC, the discovery of M⋯C contact within porphyrin aggregates marks a new finding. However, this distance is too long to denote coordination interaction, as it exceeds the length of robust M–C covalent bonds found in compounds like alkyl cobalamins (1.99–2.03 Å),16 as well as Co–C (C60, <2.7 Å)17 in the neutral complexes of fullerenes with metal-incorporating porphyrins. To gain deeper insights into the interlayer interactions in CoTCPP-Bi, density functional theory (DFT) calculations were utilized to examine the noncovalent interactions (NCIs) between adjacent metalloporphyrins in CoTCPP-Bi. Real-space visualization of both attractive and repulsive interactions in the structure, based on electron density, indicates that neighboring metalloporphyrins exhibit interlayer van der Waals interactions involving the porphyrin rings and peripheral benzene rings (Fig. S11†). Additionally, the analysis of density of states (DOS) displays a significant overlap between adjacent layers within the bonding region, indicating pronounced interlayer interactions (Fig. S12†). Fingerprint maps and further analysis of these interactions suggest that the stronger van der Waals forces originate from the metal-mediated interaction, specifically between cobalt and carbon (Fig. 1e). The electrostatic potential (ESP) analysis demonstrates that the presence of Co causes distortion in the electron density at the porphyrin core, and the aggregation of cobalt porphyrins facilitates a cohesive arrangement of 1D porphyrin arrays (Fig. S13†). In brief, the theoretical investigation underscores that the unique stacking pattern of metalloporphyrins engenders robust interlayer van der Waals interactions, indicative of a strong intermolecular interaction among neighboring metalloporphyrinic linkers and holding promise in elucidating the electronic and optical properties of the underlying porphyrin assemblies.
Metalloporphyrins typically aggregate in a 1D manner; however, the structures of MTCPP-Bi exhibit open square channels with aperture dimensions of 9.1 × 5.8 Å2. Pore characteristics were assessed using PLATON software, revealing small solvent-accessible voids with a volume of 1953 Å3, accounting for 21.1% of the unit cell volume. Despite pre-activation to remove lattice solvents, N2 adsorption–desorption isotherms of MTCPP-Bi at 77 K showed no detectable uptake, which could be attributed to blockage of porous channels by residual guest molecules, rendering the MOF inoperative for N2 sorption. Nonetheless, the permanent porosity of the representative CoTCPP-Bi was validated through CO2 capture measurements at 195 K, demonstrating a type-I isotherm indicative of microporous behavior (Fig. S14†). The Brunauer–Emmett–Teller (BET) surface area of CoTCPP-Bi was determined to be 464 m2 g−1. CoTCPP-Bi maintains structural integrity in aqueous solutions spanning a pH range of 3 to 12 (Fig. S15†), and remains stable in some common organic solvents such as methanol, N,N-dimethylformamide (DMF), acetonitrile, toluene, n-hexane, and acetone, as confirmed by powder XRD (PXRD) (Fig. S16†). Its thermal stability, evaluated using thermogravimetry (TG) and temperature-dependent PXRD, suggests that the framework of CoTCPP-Bi remains intact when exposed to air up to 300 °C (Fig. 2a and S17†).
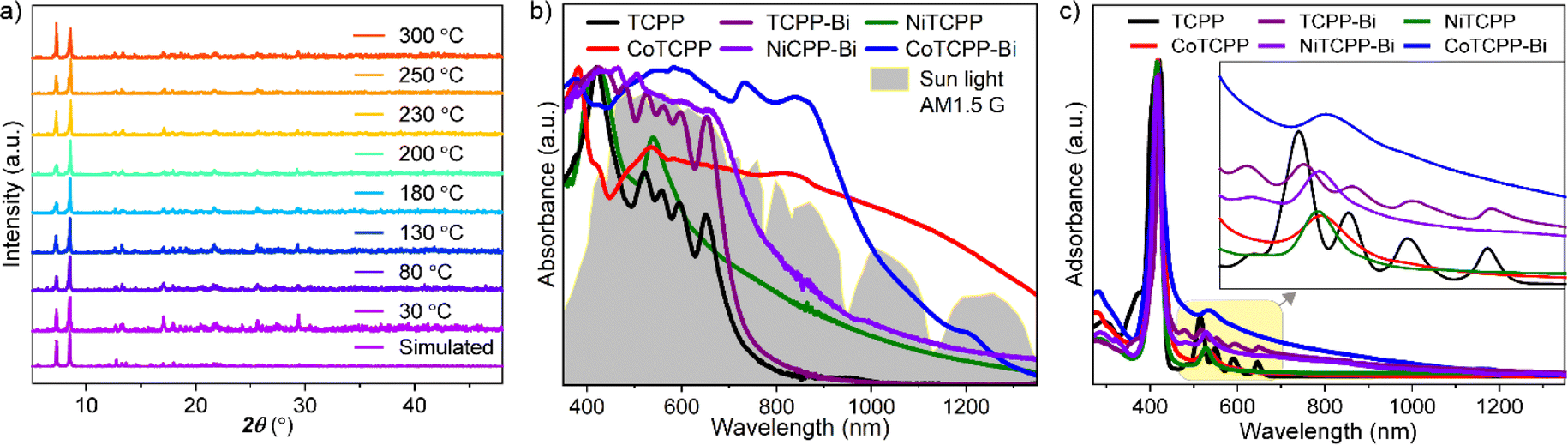 | ||
| Fig. 2 (a) Temperature-dependent PXRD patterns of CoTCPP-Bi under air atmosphere. (b) UV-Vis-NIR absorption spectra of powder samples of TCPP, TCPP-Bi, NiTCPP, CoTCPP, NiTCPP-Bi, CoTCPP-Bi, and the solar spectral irradiance (AM 1.5 G), and (c) their solutions in DMF. | ||
To explore the influence of metallization and aggregation modes of porphyrins on light absorption, we chose a 2D layered structure named Bi-TCPP for comparative testing (Fig. S3†).18 In Bi-TCPP, which is composed of TCPP organic ligands and Bi3+ inorganic nodes, only interlayer van der Waals interactions occur between the peripheral benzene ring and the central porphyrin rings due to the dislocated assignment of neighboring layers. The UV-Vis-NIR absorption spectra of powdered samples, including TCPP, TCPP-Bi, NiTCPP, CoTCPP, NiTCPP-Bi, and CoTCPP-Bi, are depicted in Fig. 2b. Compared to the monomeric TCPP, TCPP-Bi shows a noteworthy enhancement in light absorption up to 800 nm. Both the metalloporphyrin Co/NiTCPP and their assemblies, Co/NiTCPP-Bi, exhibit absorption peaks that extend into the NIR-II region (1400 nm). To further clarify the impact of metallization, Importantly, the absorption intensity of CoTCPP-Bi is greatly higher than that of the nickel-metalized samples. This expanded absorption range of CoTCPP-Bi closely matches the energy distribution of sunlight (AM1.5 G simulated sunlight), effectively covering a substantial portion of the solar spectrum (Fig. S18†). The presence of CoII and the short distance between the metalloporphyrin linkers may contribute to this outcome. Prior research indicates that the spin-allowed d–d transition of cobalt, characteristic of a CoII complex, enhances the NIR absorption of the sample.19 we conducted liquid mapping on light absorption and spectral absorption tests in a DMF solution (Fig. 2c). Compared with TCPP and TCPP-Bi, the changes in the position and quantity of the Q-band peaks of CoTCPP, NiTCPP, CoTCPP-Bi, and NiTCPP-Bi after metallization could be attributed to the asymmetry induced by the metal centers. The absorption spectra of TCPP-Bi, CoTCPP-Bi, and NiTCPP-Bi display a wider range of absorption compared to TCPP, CoTCPP, and NiTCPP. This broadening is partly attributed to the distinct molecular arrangements in solution: TCPP, CoTCPP, and NiTCPP are present as individual molecules in DMF, whereas TCPP-Bi, CoTCPP-Bi, and NiTCPP-Bi are dispersed as aggregates, which introduces scattering effects.
Density functional theory calculations, and EPR characterization
The role of metalloporphyrin aggregation in light absorption was further investigated through DFT calculations. The molecular geometry was optimized using the PBE0 functional. Fig. 3a clearly illustrates the profiles of the singly occupied molecular orbitals (SOMO) and singly unoccupied molecular orbitals (SUMO) for both the α and β spins of CoTCPP and CoTCPP-Bi. For the open-shell CoTCPP and CoTCPP-Bi, the majority of frontier orbitals are ligand-centered, and the calculated low energy bands primarily arise from π–π* transitions. Additionally, the contribution of cobalt to SUMO (β) suggests the involvement of d orbitals of the central metal in light absorption. This phenomenon is also noted in CoTCPP-Bi, where the aggregation of cobalt porphyrins promotes orbital overlap, reducing the band gap between SOMO and SUMO. The calculated band gap decreases from 2.89 eV for CoTCPP to 2.54 eV for CoTCPP-Bi, broadening the spectral region to sunlight. These results emphasize the significant influence of metalloporphyrin aggregation behavior on electronic spectra.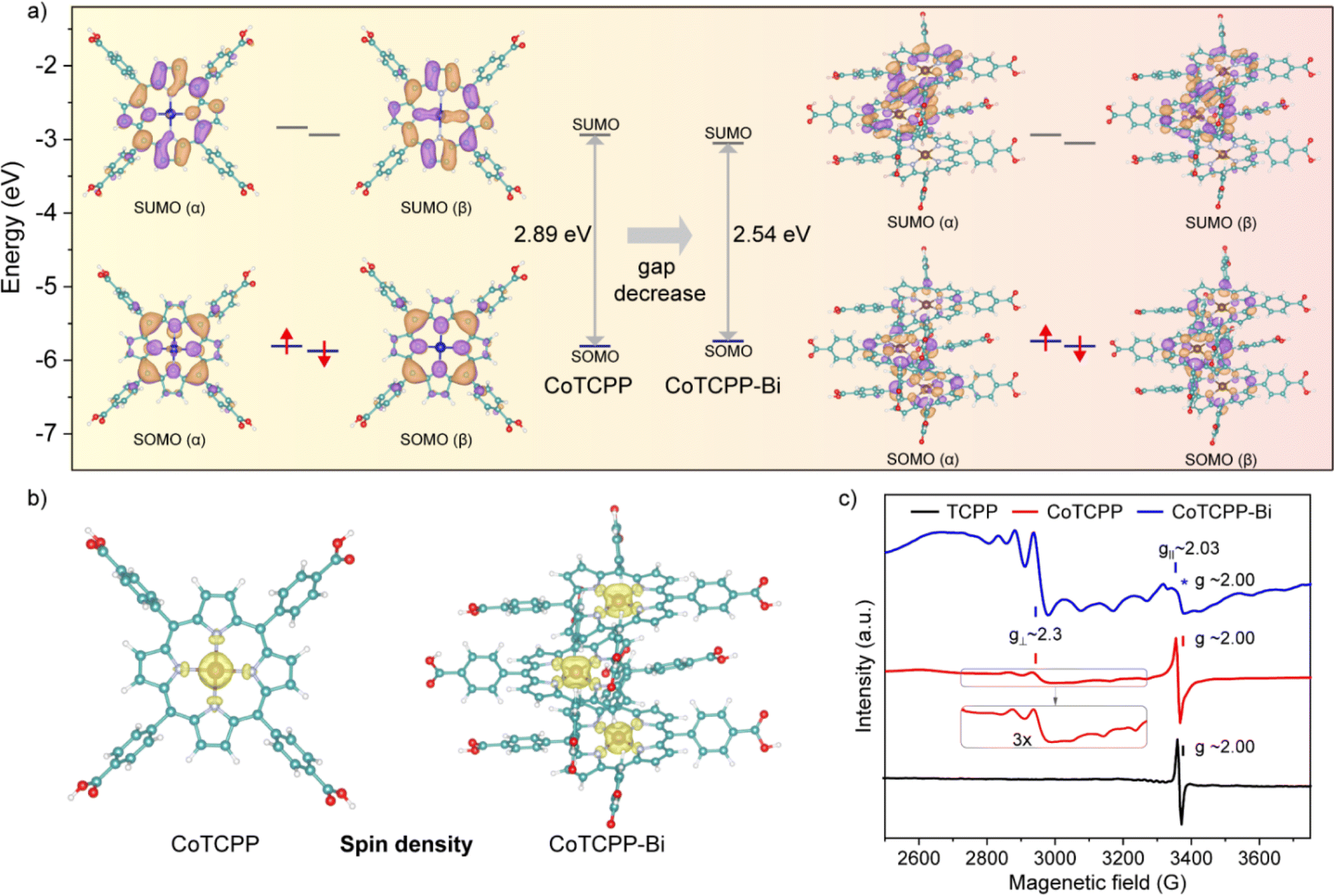 | ||
| Fig. 3 (a) Frontier molecular orbital diagrams of CoTCPP and CoTCPP-Bi. (b) Spin density maps of CoTCPP and CoTCPP-Bi. (c) EPR spectra of solid-state samples of TCPP, CoTCPP and CoTCPP-Bi collected at 100 K. | ||
To further examine the electronic structures of CoTCPP and CoTCPP-Bi, electron spin density was computed using the Multiwfn program (Fig. 3b). Analysis of spin distributions uncovered a predominance of single electrons on the central cobalt, followed by cobalt-coordinated nitrogen atoms, indicating the existence of a CoII-based metalloradical. Bulk samples were subjected to variable-temperature continuous-wave X-band EPR spectroscopy, showing a signal at approximately 3300 G with g = 2.00 at 100 K, as depicted in Fig. 3c. Similar signals have been observed in other porphyrins and associated with organic π-radicals based on carbon and nitrogen.20 Notably, cobaltous-porphyrin is a low-spin CoII-complex with a lone unpaired electron in the 3d orbital, making this unpaired electron highly sensitive to alterations in the axial coordination of the metal ion. External molecules like O2 induce a distortion of the coordination and adjust the oxidation state of Co, potentially forming Co–O2 adducts identified as CoIII superoxo (O2−˙) species.21 In addition, ligating molecules such as oxygen increase the electron density on Co, akin to the effect of an N base in cobaltous–porphyrin systems. Consequently, the solid-state sample based on cobaltous porphyrin exhibits remarkable instability; storage in air or some common solvents can result in the absence or alteration of EPR signaling. Prior studies have typically shielded cobaltous porphyrins from axial ligation by storing them in a vacuum or under an inert atmosphere, or by embedding them in solid-state diamagnetic matrices.4,22 In this work, the hyperfine coupling between unpaired electrons and the I = 7/2 59Co nucleus in CoTCPP-Bi produces a distinct eight-line splitting signal, characteristic of a well-known CoII-based metalloradical,23 whereas CoTCPP displays a weaker signal related to the CoII metalloradical. Comparison of experimental and theoretical data enabled the determination of spin-Hamiltonian parameters: S = 1/2, g⊥ = 2.3, g∥ = 2.03, consistent with values previously reported for the CoII center with a four-coordination environment, as indicated by XRD. Accordingly, the CoTCPP-Bi powder was intentionally exposed to air for an extended period, resulting in the observation of an intense radical signal, which could be ascribed to the closely spaced metalloporphyrinic arrays within the model. This is supported by the more prominent CoII-based metalloradical signal in CoTCPP-Bi compared to CoTCPP. Heating CoTCPP-Bi results in a singular broad resonance signal, while lower temperatures yield a more complex pattern with split peak formations (Fig. S19†). Conversely, the free-radical signal peaks with g = 2 consistently dominated in the samples TCPP and CoTCPP (Fig. S20 and S21†). CoTCPP-Bi serves as a model capable of stabilizing single-electron spins in the CoII center by preventing axial coordination.
As previously stated, the formation of robust and precisely arranged aggregations of the compound CoTCPP-Bi diminishes the energy gap between the S0 and S1 states, allowing for increased light absorption in the NIR region. Fig. S22† illustrates that the reduced bandgap of CoTCPP-Bi no longer matches the energy level of visible fluorescence, leading to fluorescence quenching, which promotes the conversion of NIR light into thermal energy. Moreover, the maintenance of air-stable functionality in organic conjugated radicals is vital for photothermal conversion, given their general susceptibility to oxygen. However, when these radicals are integrated into well-designed materials or systems, they can retain their functionality in air, which is essential for their practical application in photothermal devices. The effective aggregation of cobalt porphyrins produces plentiful and vigorous CoII-based metalloradicals that can attenuate radiative transitions, thus intensifying non-radiative processes.
Photothermal conversion performance
Leveraging its outstanding structural stability, UV-Vis-NIR absorption, and the abundance of CoII-based metalloradicals, CoTCPP-Bi appears to possess remarkable photothermal conversion properties. Fig. 4a shows the temperature change of CoTCPP-Bi powder when exposed to a 0.7 W cm−2 808 nm laser for 50 seconds, monitored by an IR thermal camera. Upon laser exposure, the temperature swiftly rose from room temperature (rt) to approximately 150 °C within just 10 seconds, peaking at 190 °C within 30 seconds. Subsequently, upon deactivation of the laser, the temperature rapidly dropped back to ambient levels. Under a comparable experimental setup, TCPP demonstrated a modest maximum temperature of 62 °C, while NiTCPP, TCPP-Bi, CoTCPP, and NiTCPP-Bi exhibited a notably higher maximum temperature of 81 °C, 94 °C, 130 °C, and 140 °C, respectively. These results suggest that the introduction of metallized porphyrins and denser organization of metalloporphyrins have led to an improved photothermal performance. Additionally, the temperatures of TCPP, CoTCPP, and CoTCPP-Bi consistently increased as the irradiation power of the 808 nm laser increased, spanning intensities of 0.1, 0.3, 0.5, and 0.7 W cm−2 (Fig. 4b and S23†). It is noteworthy that the temperature variations displayed a strong linear relationship with the laser's power density, demonstrating exceptional thermal regulation capabilities (Fig. S24†). These findings emphasize the remarkable photothermal conversion efficiency of CoTCPP-Bi, surpassing that of most recently reported MOFs and other types of materials with NIR photothermal transformation (Fig. 4c and Table S2†).2a,24 Following ten cycles of laser irradiation on CoTCPP-Bi, with the laser being on and off alternately after each cycle, no notable photobleaching or temperature loss was detected via photothermal assessment (Fig. S25†). Furthermore, PXRD analysis confirmed the retention of its crystalline structure in the sample, signifying the excellent photostability of CoTCPP-Bi (Fig. S26†).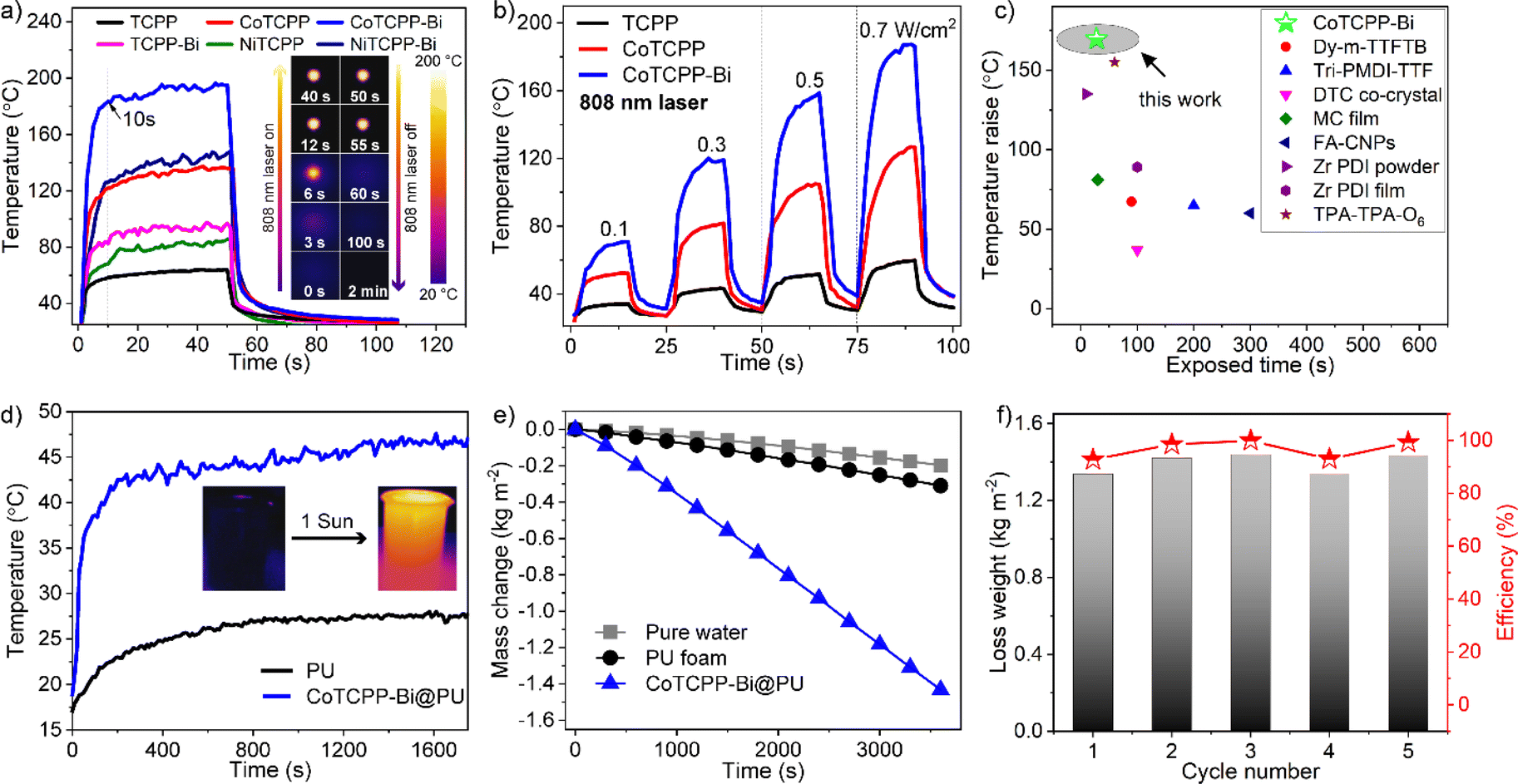 | ||
| Fig. 4 (a) Photothermal conversion curves of TCPP, CoTCPP, CoTCPP-Bi, TCPP-Bi, NiTCPP, and NiTCPP-Bi under irradiation of a 0.7 W cm−2 808 nm laser. Inset: IR-thermal images of CoTCPP-Bi under irradiation of a 0.7 W cm−2 808 nm laser. (b) Heating and cooling cycles for TCPP, CoTCPP and CoTCPP-Bi under different energy power 808 nm lasers. (c) Temperature variations for CoTCPP-Bi and some typical solid-state photothermal materials reported under an 808 nm laser of 0.7 W cm−2. (d) Photothermal conversion behavior of PU and CoTCP-Bi-loaded PU foams under 1 sun light irradiation (1 kW m−2). (e) Water evaporation curves with pure water, PU foam and CoTCPP-Bi-loaded PU under simulated sunlight with an intensity of 1 kW m−2 (1 sun). (f) Evaporation rate and efficiency for CoTCPP-Bi-loaded PU in five cycles. | ||
Interfacial solar vapor generation (ISVG) technology is a sustainable, environmentally friendly, and low-carbon alternative for seawater and wastewater treatment, offering a promising replacement for conventional desalination and purification techniques like reverse osmosis and ultrafiltration.25 Upon exposure to xenon lamp irradiation (1 kW m−2, filtered to match the AM1.5 G solar spectrum), the CoTCPP-Bi powder, with its enhanced NIR light absorption, registers a temperature rise from ambient to a peak of 60 °C. Conversely, the TCPP-Bi powder achieves a maximum temperature of 40 °C under the same experimental parameters (Fig. S27†). The impressive photothermal conversion capabilities of CoTCPP-Bi prompt us to assess its suitability for solar-driven water evaporation. Specifically, 30 mg of CoTCPP-Bi powder was loaded onto a commercially available white hydrophilic porous polyurethane (PU) to produce a brownish-black PU sponge. This sponge served as a foundation for establishing an efficient interfacial evaporation system, positioned atop the water to facilitate heat transfer (Fig. S28 and S29†). The solar–thermal conversion competence of CoTCPP-Bi@PU was recorded using an IR thermal camera under continuous irradiation of simulated sunlight (AM1.5 G, one sun with the light intensity of 1 kW m−2) for 30 minutes (Fig. S30†). The results revealed the effectiveness of CoTCPP-Bi@PU in promoting energy conversion at the water–air interface, as evidenced by the surface temperature of CoTCPP-Bi@PU reaching 45 °C while floating on the water, significantly higher than the pure PU temperature of 27 °C (Fig. 4d). After one hour, the water temperature rose and equilibrated at 32.2 °C (Fig. S31†). Mass change experiments were conducted to evaluate the efficacy of solar-driven water evaporation, involving time dependent water with PU foam and CoTCPP-Bi@PU (Fig. 4e and S32†). Under 1-sun exposure under ambient conditions, the evaporation rate of CoTCPP-Bi@PU reached 1.43 kg m−2 h−1 and 98.5% solar-driven water evaporation efficiency (η), significantly outperforming PU (0.2 kg m−2 h−1) and pure water (0.1 kg m−2 h−1), as well as other photothermal materials (Table S3†).24a,f,25,26 The average solar-driven water evaporation efficiency (η) for CoTCPP-Bi@PU was determined to be 94% over five cycles (Fig. 4f).
Conclusions
In conclusion, we synthesized a novel crystalline solid, through the combination of metalloporphyrin linkers with bismuth–carboxylate nodes. The stable CoII-based metalloradical species and the robust aggregation of metalloporphyrins in CoTCPP-Bi, caused by π–π stacking between neighboring metalloporphyrins, impart strong absorption capabilities to the material in the NIR region. Consequently, under an energy density of 0.7 W cm−2 using an 808 nm laser, CoTCPP-Bi demonstrates remarkable photothermal conversion efficiency, achieving a temperature rise from rt to 190 °C within 30 seconds, and efficient solar-driven water evaporation, with an efficiency of 94% and a rate of 1.43 kg m−2 h−1 under 1 sun irradiation. This work highlights the potential for developing materials with superior photothermal conversion and NIR light absorption abilities by enhancing the aggregation and stabilization of metalloradical moieties. Furthermore, the integration of functional photothermal materials opens up new possibilities for the consolidation of photothermal water evaporation with multifunctional applications, thereby improving the versatility and efficiency of photothermal technologies.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
L. He carried out the experimental sections and wrote the manuscript. E. Chen and Q. Lin supervised the study and reviewed the manuscript. J. He helped the characterization. All the authors discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (22201283), the National Key Research and Development Project (2022YFA1503900), the National Science Foundation of Fujian Province (2022J05090, 2022T3008), Fujian Science & Technology Innovation Laboratory for Optoelectronic Information of China (2021ZR138) and the Open Project Program of the State Key Laboratory of Photocatalysis on Energy and Environment (SKLPEE-KF202204), Fuzhou University.Notes and references
- (a) H. Hu, H. Wang, Y. Yang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200799 CrossRef CAS PubMed; (b) Y. Wang, J. Zhang, W. Liang, H. Yang, T. Guan, B. Zhao, Y. Sun, L. Chi and L. Jiang, CCS Chem., 2021, 4, 1153 CrossRef; (c) K. Li, X. Sun, Y. Wang, J. Wang, X. Dai, G. Li and H. Wang, Nano Energy, 2022, 93, 106789 CrossRef CAS; (d) Q. Huang, X. Ye, W. Chen, X. Song, Y.-t. Chen, X. Wen, M. Zhang, Y. Wang, S.-L. Chen, L. Dang and M.-D. Li, ACS Energy Lett., 2023, 8, 4179 CrossRef CAS; (e) H.-C. Ma, C.-C. Zhao, G.-J. Chen and Y.-B. Dong, Nat. Commun., 2019, 10, 3368 CrossRef PubMed; (f) D. Wang, Z. Zhang, L. Lin, F. Liu, Y. Wang, Z. Guo, Y. Li, H. Tian and X. Chen, Biomaterials, 2019, 223, 119459 CrossRef CAS PubMed; (g) K. Zhang, X. Meng, Y. Cao, Z. Yang, H. Dong, Y. Zhang, H. Lu, Z. Shi and X. Zhang, Adv. Funct. Mater., 2018, 28, 1804634 CrossRef; (h) J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356 CrossRef CAS PubMed.
- (a) Z. Wang, J. Zhou, Y. Zhang, W. Zhu and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202113653 CrossRef CAS PubMed; (b) Z. Wang, F. Alkan, C. M. Aikens, M. Kurmoo, Z.-Y. Zhang, K.-P. Song, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202206742 CrossRef CAS; (c) Y. Jiang, Y. Zhang, Y. Deng, S. Dong, B. Li, Y. Yi, Z. Zeng, H. Chen, H. Luo and Y. Geng, CCS Chem., 2022, 4, 3497 CrossRef CAS; (d) A. H. Proppe, Y. C. Li, A. Aspuru-Guzik, C. P. Berlinguette, C. J. Chang, R. Cogdell, A. G. Doyle, J. Flick, N. M. Gabor, R. van Grondelle, S. Hammes-Schiffer, S. A. Jaffer, S. O. Kelley, M. Leclerc, K. Leo, T. E. Mallouk, P. Narang, G. S. Schlau-Cohen, G. D. Scholes, A. Vojvodic, V. W.-W. Yam, J. Y. Yang and E. H. Sargent, Nat. Rev. Mater., 2020, 5, 828 CrossRef CAS; (e) G. Chen, J. Sun, Q. Peng, Q. Sun, G. Wang, Y. Cai, X. Gu, Z. Shuai and B. Z. Tang, Adv. Mater., 2020, 32, 1908537 CrossRef CAS PubMed; (f) B. Yang, Z. Zhang, P. Liu, X. Fu, J. Wang, Y. Cao, R. Tang, X. Du, W. Chen, S. Li, H. Yan, Z. Li, X. Zhao, G. Qin, X.-Q. Chen and L. Zuo, Nature, 2023, 622, 499 CrossRef CAS PubMed; (g) M. E. Matter, L. Čamdžić and E. E. Stache, Angew. Chem., Int. Ed., 2023, 62, e202308648 CrossRef CAS PubMed; (h) O. Shelonchik, N. Lemcoff, R. Shimoni, A. Biswas, E. Yehezkel, D. Yesodi, I. Hod and Y. Weizmann, Nat. Commun., 2024, 15, 1154 CrossRef CAS PubMed.
- M. Chen, X. Zhang, J. Liu, F. Liu, R. Zhang, P. Wei, H. Feng, M. Tu, A. Qin, J. W. Y. Lam, D. Ding and B. Z. Tang, ACS Nano, 2020, 14, 4265 CrossRef CAS PubMed.
- (a) X. Chen, H. Xie, E. R. Lorenzo, C. J. I. V. Zeman, Y. Qi, Z. H. Syed, A. E. B. S. Stone, Y. Wang, S. Goswami, P. Li, T. Islamoglu, E. A. Weiss, J. T. Hupp, G. C. Schatz, M. R. Wasielewski and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 2685 CrossRef CAS PubMed; (b) H. Gao, X. Zhi, F. Wu, Y. Zhao, F. Cai, P. Li and Z. Shen, Angew. Chem., Int. Ed., 2023, 62, e202309208 CrossRef CAS PubMed.
- L. Zhao, X. Ren and X. Yan, CCS Chem., 2021, 3, 678 CrossRef.
- C. Preston-Herrera, S. Dadashi-Silab, D. G. Oblinsky, G. D. Scholes and E. E. Stache, J. Am. Chem. Soc., 2024, 146, 8852 CrossRef CAS PubMed.
- (a) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed; (b) Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035 CrossRef CAS PubMed.
- Q. Zou, M. Abbas, L. Zhao, S. Li, G. Shen and X. Yan, J. Am. Chem. Soc., 2017, 139, 1921 CrossRef CAS PubMed.
- (a) T. Yamaguchi, T. Kimura, H. Matsuda and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 6350 CrossRef CAS PubMed; (b) B. Guo, G. Feng, P. N. Manghnani, X. Cai, J. Liu, W. Wu, S. Xu, X. Cheng, C. Teh and B. Liu, Small, 2016, 12, 6243 CrossRef CAS PubMed; (c) Q. Yin, E. V. Alexandrov, D.-H. Si, Q.-Q. Huang, Z.-B. Fang, Y. Zhang, A.-A. Zhang, W.-K. Qin, Y.-L. Li, T.-F. Liu and D. M. Proserpio, Angew. Chem., Int. Ed., 2022, 61, e202115854 CrossRef CAS PubMed.
- (a) C. Röger, Y. Miloslavina, D. Brunner, A. R. Holzwarth and F. Würthner, J. Am. Chem. Soc., 2008, 130, 5929 CrossRef PubMed; (b) S. Ganapathy, S. Sengupta, P. K. Wawrzyniak, V. Huber, F. Buda, U. Baumeister, F. Würthner and H. J. M. de Groot, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11472 CrossRef CAS PubMed.
- X. Zhang, M. C. Wasson, M. Shayan, E. K. Berdichevsky, J. Ricardo-Noordberg, Z. Singh, E. K. Papazyan, A. J. Castro, P. Marino, Z. Ajoyan, Z. Chen, T. Islamoglu, A. J. Howarth, Y. Liu, M. B. Majewski, M. J. Katz, J. E. Mondloch and O. K. Farha, Coord. Chem. Rev., 2021, 429, 213615 CrossRef CAS PubMed.
- L. Yang, P. Cai, L. Zhang, X. Xu, A. A. Yakovenko, Q. Wang, J. Pang, S. Yuan, X. Zou, N. Huang, Z. Huang and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 12129 CrossRef CAS PubMed.
- (a) Z. Wang, Z. Zeng, H. Wang, G. Zeng, P. Xu, R. Xiao, D. Huang, S. Chen, Y. He, C. Zhou, M. Cheng and H. Qin, Coord. Chem. Rev., 2021, 439, 213902 CrossRef CAS; (b) E. P. Gómez-Oliveira, D. Reinares-Fisac, L. M. Aguirre-Díaz, F. Esteban-Betegón, M. Pintado-Sierra, E. Gutiérrez-Puebla, M. Iglesias, M. Ángeles Monge and F. Gándara, Angew. Chem., Int. Ed., 2022, 61, e202209335 CrossRef PubMed.
- K. Ni, T. Luo, G. Lan, A. Culbert, Y. Song, T. Wu, X. Jiang and W. Lin, Angew. Chem., Int. Ed., 2020, 59, 1108 CrossRef CAS PubMed.
- M. M. Olmstead, D. A. Costa, K. Maitra, B. C. Noll, S. L. Phillips, P. M. Van Calcar and A. L. Balch, J. Am. Chem. Soc., 1999, 121, 7090 CrossRef CAS.
- M. Rossi, J. P. Glusker, L. Randaccio, M. F. Summers, P. J. Toscano and L. G. Marzilli, J. Am. Chem. Soc., 1985, 107, 1729 CrossRef CAS.
- (a) D. V. Konarev, S. S. Khasanov, A. Otsuka, Y. Yoshida and G. Saito, J. Am. Chem. Soc., 2002, 124, 7648 CrossRef CAS PubMed; (b) P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487 CrossRef CAS.
- D. Xie, S. Wang, S. Li, W. Yang and Y.-S. Feng, Catal. Sci. Technol., 2022, 12, 3254 RSC.
- I. Strauss, A. Mundstock, D. Hinrichs, R. Himstedt, A. Knebel, C. Reinhardt, D. Dorfs and J. Caro, Angew. Chem., Int. Ed., 2018, 57, 7434 CrossRef CAS PubMed.
- (a) S. Van Doorslaer and A. Schweiger, Phys. Chem. Chem. Phys., 2001, 3, 159 RSC; (b) T. Yamabayashi, M. Atzori, L. Tesi, G. Cosquer, F. Santanni, M.-E. Boulon, E. Morra, S. Benci, R. Torre, M. Chiesa, L. Sorace, R. Sessoli and M. Yamashita, J. Am. Chem. Soc., 2018, 140, 12090 CrossRef CAS PubMed.
- A. T. Gallagher, C. D. Malliakas and T. D. Harris, Inorg. Chem., 2017, 56, 4654 CrossRef PubMed.
- F. Ann Walker, J. Magn. Reson., 1974, 15, 201 Search PubMed.
- (a) J. A. de Bolfo, T. D. Smith, J. F. Boas and J. R. Pilbrow, J. Chem. Soc., Dalton Trans., 1976, 1495 RSC; (b) Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723 CrossRef CAS PubMed.
- (a) X. Ye, L.-H. Chung, K. Li, S. Zheng, Y.-L. Wong, Z. Feng, Y. He, D. Chu, Z. Xu, L. Yu and J. He, Nat. Commun., 2022, 13, 6116 CrossRef CAS PubMed; (b) J. Su, P. Cai, T. Yan, Z.-M. Yang, S. Yuan, J.-L. Zuo and H.-C. Zhou, Chem. Sci., 2022, 13, 1657 RSC; (c) J. Su, N. Xu, R. Murase, Z.-M. Yang, D. M. D'Alessandro, J.-L. Zuo and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 4789 CrossRef CAS PubMed; (d) B. Lü, Y. Chen, P. Li, B. Wang, K. Müllen and M. Yin, Nat. Commun., 2019, 10, 767 CrossRef PubMed; (e) Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963 CrossRef CAS PubMed; (f) S. Zhou, X. Kong, M. Strømme and C. Xu, ACS Mater. Lett., 2022, 4, 1058 CrossRef CAS.
- X. Q. Li, J. L. Li, J. Y. Lu, N. Xu, C. L. Chen, X. Z. Min, B. Zhu, H. X. Li, L. Zhou, S. N. Zhu, T. J. Zhang and J. Zhu, Joule, 2018, 2, 1331 CrossRef CAS.
- (a) X. Yan, S. Lyu, X.-Q. Xu, W. Chen, P. Shang, Z. Yang, G. Zhang, W. Chen, Y. Wang and L. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201900 CrossRef CAS PubMed; (b) J. Liu, Y. Cui, Y. Pan, Z. Chen, T. Jia, C. Li and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117087 CrossRef CAS PubMed; (c) X. Tang, Z. Chen, Q. Xu, Y. Su, H. Xu, S. Horike, H. Zhang, Y. Li and C. Gu, CCS Chem., 2021, 4, 2842 CrossRef; (d) Q. Ma, P. Yin, M. Zhao, Z. Luo, Y. Huang, Q. He, Y. Yu, Z. Liu, Z. Hu, B. Chen and H. Zhang, Adv. Mater., 2019, 31, 1808249 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, X-ray crystallographic data, and additional structural and characterizing figures (PXRD, TGA, FT-IR, SEM, EPR, stability analysis, adsorption–desorption isotherms, UV-Vis DRS, photoluminescence, photothermal conversion experiments, etc.). CCDC 2323214 and 2356520. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04063e |
| This journal is © The Royal Society of Chemistry 2024 |