
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous modulation of cathode/anode and electrolyte interfaces via a nitrile additive for high-energy-density lithium-metal batteries†
Ziye
Wang
a,
Yingshuai
Wang
 a,
Yuhang
Xin
a,
Qingbo
Zhou
a,
Xiangyu
Ding
a,
Lei
Liu
a,
Tinglu
Song
a,
Feng
Wu
a,
Zhongbao
Wei
*b and
Hongcai
Gao
*acd
a,
Yuhang
Xin
a,
Qingbo
Zhou
a,
Xiangyu
Ding
a,
Lei
Liu
a,
Tinglu
Song
a,
Feng
Wu
a,
Zhongbao
Wei
*b and
Hongcai
Gao
*acd
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, P.R. China. E-mail: gaohc@bit.edu.cn
bSchool of Mechanical Engineering, Beijing Institute of Technology, Beijing 100081, P.R. China
cYangtze Delta Region Academy of Beijing Institute of Technology, Jiaxing, 314019, P.R. China
dBeijing Institute of Technology Chongqing Innovation Center, Chongqing 401120, P.R. China
First published on 13th September 2024
Abstract
Nickel-rich layered oxides have great potential for commercial development applications, so it is critical to address their stability over long cycles. Ensuring long-term cycle stability relies heavily on the stability of the interface between the electrode and electrolyte in Li|LiNi0.8Co0.1Mn0.1O2 (NCM811) batteries. In this work, a denser, more stable and thinner nickel-rich cathode/electrolyte interface was constructed by electrolyte engineering with succinonitrile (SN) as an additive. The increase of organic compound content in the formed Ni-rich cathode/electrolyte interface can fully release the stress and strain generated during repetitive charge–discharge processes, and significantly reduce the irreversible phase transition during the nickel-rich cathode charge–discharge processes. Additionally, this interface impedes the breakdown of electrolytes and the dissolution of transition metals. Furthermore, the addition of SN additives also forms a more stable lithium metal anode/electrolyte interface. Notably, batteries containing SN additives (0.5, 1.0 and 1.5 wt%) show excellent electrochemical performance compared to base electrolytes. Particularly, the improvement is most significant with an SN addition of 1.0 wt%. After 250 cycles at 1C rate, the capacity retention rate of the battery improved by 32.8%. Thus, this work provides a new perspective for simultaneously constructing a stable interface of nickel-rich cathode and lithium metal anode with a high energy density in lithium metal batteries.
1 Introduction
In recent years, alongside the rapid growth of portable electronic devices and electric vehicles (EVs), there has been a significant increase in the demand for rechargeable batteries with high energy density and excellent safety performance.1–5 Lithium metal batteries are expected to be widely used in various fields because of their high theoretical specific capacity and incredibly low redox potential.6,7 Among them, the layered nickel-rich cathode material (LiCoxMnyNi1−x−yO2, where x + y < 0.2) is the preferred material for the next generation of lithium metal batteries with high energy density due to the high energy storage capacity.8,9 LiNi0.8Co0.1Mn0.1O2 (NCM811) has excellent energy density and is a promising cathode material.10,11 However, NCM811 suffers from inherent drawbacks, such as poor cycling stability12 and unstable surface chemistry.13 Among them, with the increase of discharge times, the oxygen released from the material will react with the electrolyte, damaging the electrolyte structure and causing safety hazards.14 In addition, lithium metal anodes also undergo large changes in volume and shape during repeated plating and stripping, and a stable lithium metal anode/electrolyte interface layer cannot be formed.6 Additionally, traditional electrolytes exhibit poor cycling stability,15 leading to the inability to form a stable electrode/electrolyte passivation layer, thus limiting the further development of LMBs.16 Various methods have been applied to improve battery capacity retention,17–20 with the addition of additives to the electrolyte being a crucial improvement approach.21 This method can alter the structure of the electrode/electrolyte interface layer to form a more stable interface, while reducing the continuous decomposition of the electrolyte and further damage to the electrode materials.22To date, a variety of additives have been used in lithium metal electrolytes to stabilize the electrode/electrolyte interface.23 Among these additives, fluorine-containing ones are particularly noted for their role in improving the electrode/electrolyte interface.24,25 Fluoroethylene carbonate (FEC) is acknowledged as a film forming additive with excellent properties and is also a well-established fluorine-containing additive.26–29 FEC could improve the low-temperature cycling stability of mesocarbon microbead (MCMB) anodes to some extent, reduce the formation of LiF in the solid electrolyte interphase (SEI) layer, and accelerate the transmission performance of Li+.30 A series of fully fluorinated alkyl-substituted ethylene carbonate esters were synthesized, and a small amount of 4-(perfluorooctyl)-1,3-dioxazolam-2-one (PFO-EC) could decompose to form a dual passivation layer, acting as an effective additive to maintain the cycle stability of lithium-ion batteries.31 However, additives containing fluorine may react with water to generate hazardous gases, and there is significant potential for improvement in their environmental performance.32
Phosphorus-containing additives are also familiar electrolyte additives, promoting the formation of the battery cathode/electrolyte interface (CEI) film, and widely applied as flame retardants.33–35 Three phosphorus molecules, trifluoro propyl phosphate (5F-TPrP), hexafluoro-isopropyl phosphate (HFip) and tri(hexafluoro isopropyl) phosphite (THFPP), can be used as high-pressure flame retardant electrolyte additives.36 Diethyl (2-(trimethylsilyl) ethyl phosphonate) (DETSP) was investigated as a multi-functional additive, which not only improves the flame retardancy of the electrolyte, but also enhances the cycling stability of NCM811.13 Additionally, tri(trimethylsilyl) phosphite (TMSP),37 phosphor nitrile compounds,38 and triethyl phosphate (TEP)39 have all been found to enhance the cycle stability of the cells. However, excessive addition of phosphorus-containing additives will cause the decrease of electrochemical performance.40
Nitrogen additives are also widely used in electrolytes for lithium batteries.41 Among them, utilizing N,N-4,4-diphenylmethane-bis-maleimide (BMI) can increase the electrochemical characteristics of batteries under high voltage.42 Tris(trimethylsilyl) isothiocyanate (TMSNCS) is an electrolyte additive based on amino alkyl silane that effectively passivates active substances and forms a stable interface layer at the electrode/electrolyte interface.43 Introducing acetonitrile into the electrolyte can remarkably raise the cycling capability of batteries and modify the electrode/electrolyte interface due to the robust interaction between the nitrile functional group (–CN) and the transition metal ions within the positive electrode material.44 Among them, 1,4-diaminobenzene (DCB) (acetonitrile terephthalate) is applied to lithium metal batteries, which can optimize the Li+ coordination solvation structure in the electrolyte.45 1,3,6-Hexanotricarbonitrile (HTCN) plays an important role in changing the composition, thickness and formation mechanism of the CEI layer, thereby improving cycle stability.46 The application of isobutyronitrile (IBN) and trimethylacetonitrile (TMAN) to the electrolyte composition has significantly improved the cycle stability of the battery.47 Nitriles are an important class of nitrogen-containing additives.48 Therefore, exploring superior nitrile-based electrolyte additives to enhance the electrochemical performance of lithium metal batteries is crucial.
In this work, we attempted to introduce succinonitrile (SN) as an effective and structurally simple multifunctional additive. The additive can participate in the construction of the CEI layer, forming a more uniform, thinner and stronger CEI layer, and can improve the transmission rate of Li+, while effectively inhibiting the dissolution loss of the nickel-rich cathode during the cycle. Furthermore, compared with the batteries without SN additives, the Li|NMC811 batteries with added SN exhibited superior cycle stability, with the best performance observed when 1.0 wt% SN is added. This is attributed to the formation of a more stable electrode/electrolyte interface layer on both the anode and cathode surfaces simultaneously.
2 Results and discussion
Firstly, the cycling performance of Li|NCM811 batteries containing different contents of SN additives was investigated to determine the function of SN additives within the system and to find a more suitable addition ratio. In Fig. 1(a), the current density of the first three laps was 0.1C, 0.2C, 0.5C (1C = 180 mA h g−1) under the electrochemical window of 3.0–4.3 V and 25 °C, and the current density was 1C from the fourth lap. After 250 cycles, the discharge capacity of the cells containing SN with 1.0 wt% (abbreviated as 1.0 wt%-SN) remained at 115.3 mA h g−1, and the discharge capacity of the cells containing SN with 0.5 wt% (abbreviated as 0.5 wt%-SN) and 1.5 wt% (abbreviated as 1.5 wt%-SN) was lower than that of 1.0 wt%-SN, but was much higher than that of the blank electrolyte cell (abbreviated as LE). This experimental result obviously demonstrates that the addition of SN in the electrolyte makes the cycling performance of the nickel-rich cathode significantly improved. According to the comparison results in Fig. 1(b), this study has the most obvious improvement effect on the capacity retention rate of the battery, with the increase amount exceeding 30%. Therefore, it is evident that this work provides an idea for finding ways to improve the capacity retention rate.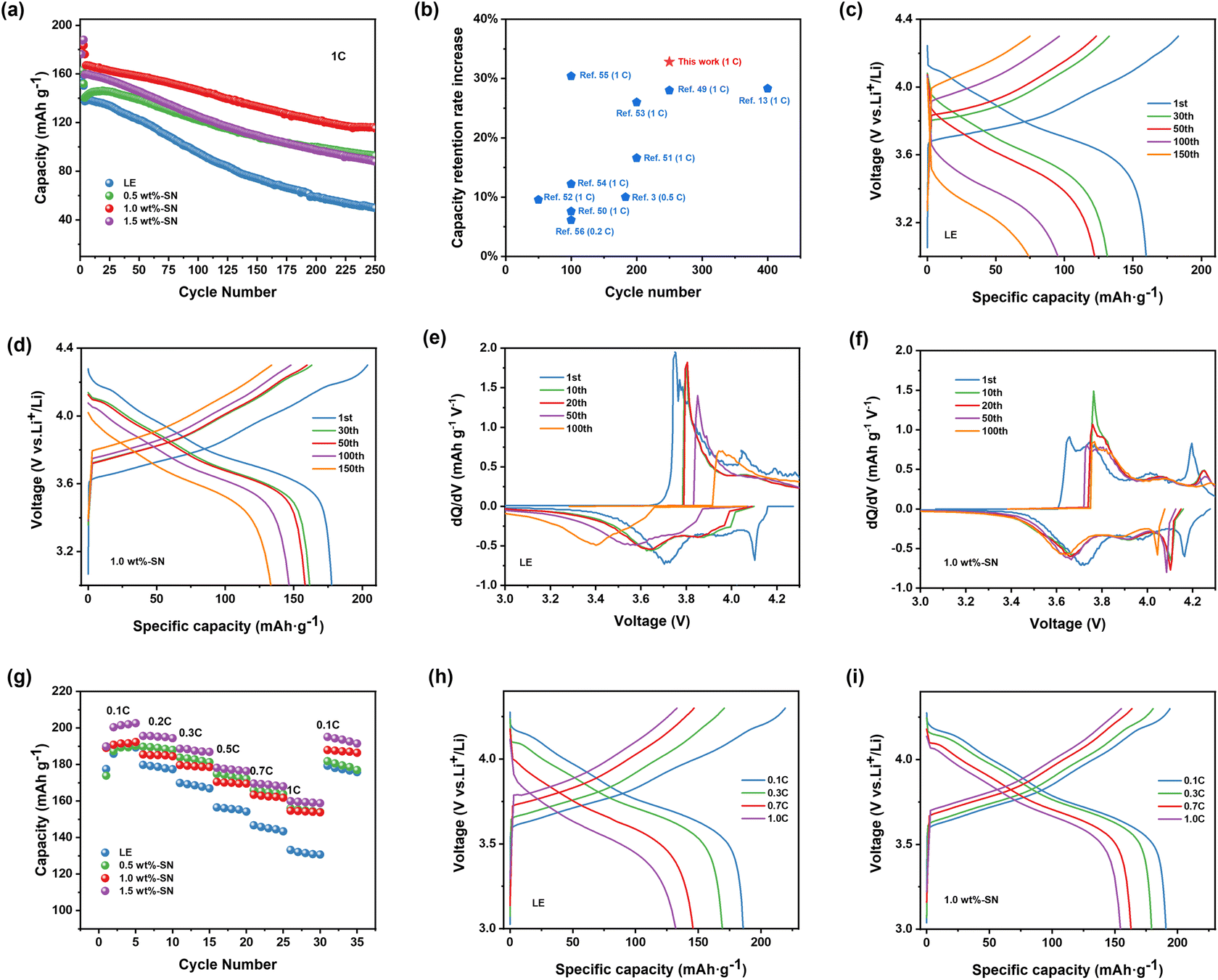 | ||
| Fig. 1 (a) Cycling performance of Li|NCM811 cells at 1C after three formation cycles at 0.1C, 0.2C, 0.5C in the voltage range of 3.0–4.3 V with different amounts of the SN additive at 25 °C. (b) Comparison of the electrochemical performance for Li|NCM cells in this work with previously reported reports.3,13,49–56 Voltage profiles of selected cycles for cells (c) without SN (LE) and (d) with 1.0 wt% SN (1.0 wt%-SN). (dQ/dV) curves of selected cycles for cells (e) without SN and (f) with 1.0 wt% SN. (g) Rate performance of Li|NCM811 cells in LE and 1.0 wt%-SN. (h and i) Charge–discharge profiles of Li|NCM811 cells with (h) LE and (i) 1.0 wt%-SN. | ||
Among them, the concentration of electrolyte additives is a significant element affecting the electrochemical capability of the cell. When the concentration is too low or too high, the effect may not be ideal.49 According to the above results, when the addition amount of SN was 1.0 wt%, the electrochemical performance was the best. The discharge capacity of the blank electrolyte cell was the lowest, only 49.8 mA h g−1. It can be speculated that the CEI film formed by LE was not stable enough to protect the nickel-rich cathode from the erosion of the electrolyte.49 After the nickel-rich cathode was eroded, its structure would deteriorate rapidly.
In this discussion, our main focus is on enhancing the Li|NCM811 cell performance using 1.0 wt%-SN compared with LE.
By analyzing the voltage curves of different cycle data in Fig. 1(c) and (d), it can be observed that the charge–discharge curve of the cell with the blank electrolyte changed sharply from the 30th cycle. However, the charge–discharge curve of the cell containing 1.0 wt%-SN did not change sharply, and the curve shape was basically the same. At the same time, compared with the voltage curves in Fig. S1(a) and (b),† the charge–discharge curve of the cell containing 1.0 wt%-SN was the most stable. In fact, the cell without SN lost a significant number of active sites. Therefore, in the presence of SN, the loss of active sites was significantly inhibited, which was conducive to improving the cycle performance of the Li|NCM811 cell. Fig. 1(e) and (f) show the dQ/dV curves of the cell after a specific cycle, which can reflect the changes of phase change reactions (peaks) occurring in NCM811 during the charge–discharge process.50 Through comparison, it was found that in the blank electrolyte cell, with increasing number of cycles, the oxidation–reduction strength of the second hexagonal phase (H2) → the third hexagonal phase (H3) rapidly decreases, leading to increasingly intense polarization and a sharp reduction in reaction area, indicating irreversible structural degradation during the cycling process. In contrast, with the increase of the number of cycle loops, the voltage and reaction area of the 1.0 wt%-SN cell changed much less dramatically than those of the blank electrolyte cell. The dQ/dV curves in Fig. S2(a) and (b)† also show that the cell containing the SN additive can effectively inhibit electrode polarization and maintain the activity of active sites, which plays a significant role in maintaining the stability of the NCM811 structure.
Fig. 1(g)–(i) depict the cycling rate performance of Li|NCM811 batteries at current densities ranging from 0.1C to 1C. As the current density increased, the discharge specific capacity of the blank electrolyte cell decreased rapidly. However, the addition of SN significantly reduced this decrease in discharge specific capacity. Compared with the cell without SN, incorporating 1.0 wt%-SN results in a significant increase in its 1C/0.1C capacity from 69.0% to 80.0%, indicating that SN additives can improve the rate performance of Li|NCM811 batteries by participating in CEI film construction and maintaining excellent electrochemical performance while also improving Li+ transfer rates.
The wettability of the electrolyte on the electrode and separator surfaces reflects its diffusion ability on these substrates, which is crucial for achieving excellent electrochemical performance.51,57 The contact angle is a technical means used to characterize substrate wettability. Fig. 2(a) shows that LE electrolyte and 1.0 wt%-SN had good wettability to cathodes but differ greatly in their wetting abilities towards separators. Specifically, compared with LE electrolyte (45.03°), the contact angle of separator-wetting for 1.0 wt%-SN (33.53°) indicated better permeability. Insufficient or uneven wetting by electrolytes can lead to local polarization within batteries, exacerbating decomposition reactions within them as well as causing localized cathode reactions that result in unstable CEI layers.51,58 Therefore, improving wetting properties is critical for enhancing electrochemical performance; fortunately, the results suggest that adding 1.0 wt%-SN considerably improves overall wetting properties.
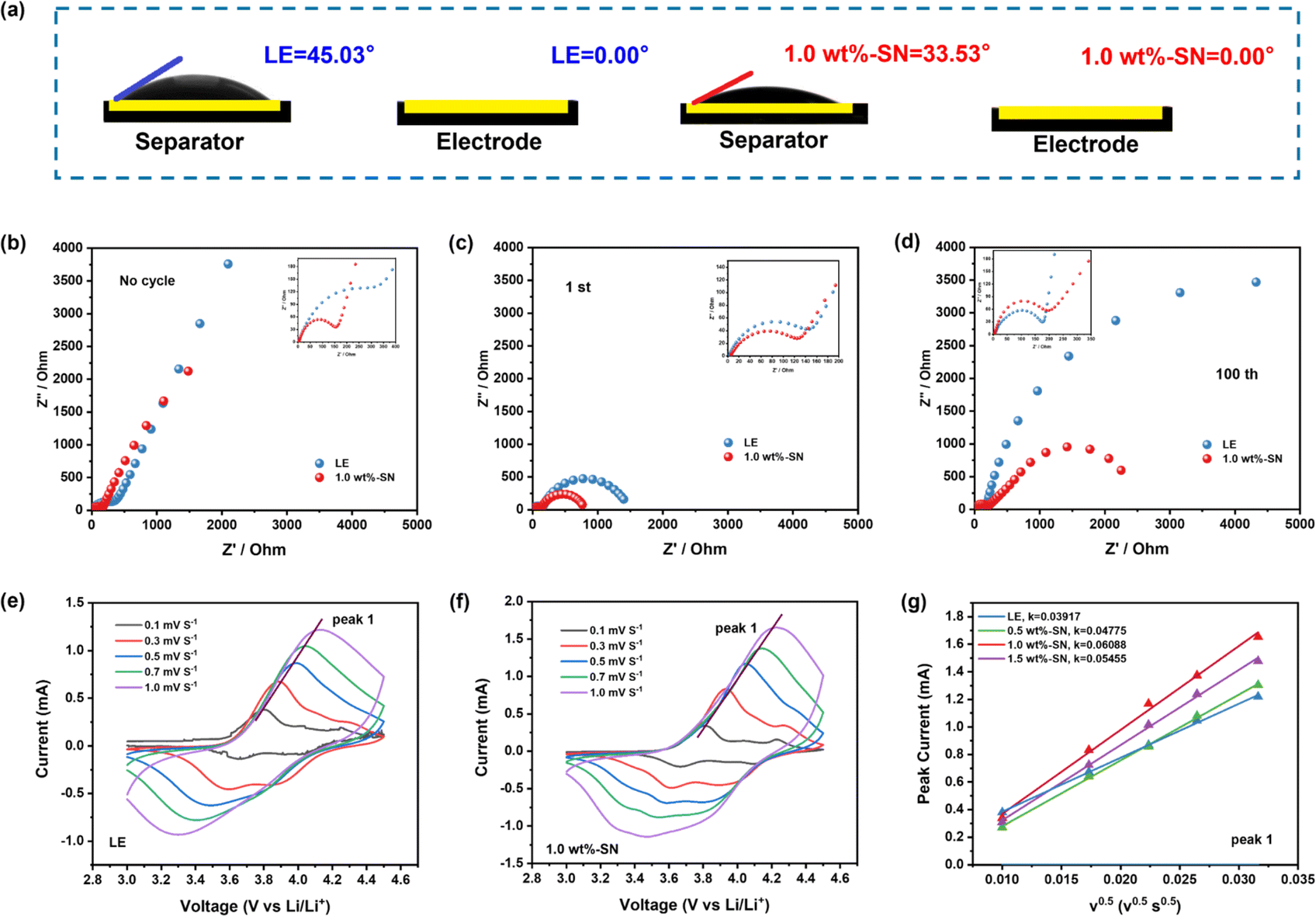 | ||
| Fig. 2 (a) Contact angle of the electrolyte on the separator and electrode. Nyquist plots of the cycled cathodes (b) without cycles, at (c) 1st and (d) 500th cycles. CV curves recorded at various scan rates (0.1–1.0 mV s−1) within a potential window of 3.0–4.5 V for (e) LE and (f) 1.0 wt%-SN. (g) Maximum peak currents for the electrolyte without SN and with SN. | ||
In addition, electrochemical impedance spectroscopy (EIS) of LE and 1.0 wt%-SN was conducted to investigate their CEI membrane formation behavior. The equivalent circuits and fitting results of the electrochemical impedance spectroscopy system are shown in Fig. S3.† Before cycling (Fig. 2(b)), the impedance of LE (357 Ω) was significantly higher compared to that of 1.0 wt%-SN (155.3 Ω). After the initial cycle (Fig. 2(c)), the surface film resistance (Rf) of 1.0 wt%-SN was smaller than that of LE, and the electrode/electrolyte interface charge transfer (Rct) was significantly lower compared to LE. This trend becomes more pronounced with the increase of the cycle numbers. It is worth noting that Rf represents the impedance of the CEI film deposited between the electrolyte and the cathode interface, while Rct represents the impedance of charge transfer between the electrode and electrolyte, and a smaller Rct represents the superior stability of the CEI layer during the cycle.52,59 After 100 cycles (Fig. 2(d)), Rf of 1.0 wt%-SN is slightly smaller than that of LE, and its Rct is significantly smaller than that of LE, which means that the rate of Li+ transport through the CEI layer in 1.0 wt%-SN is faster, and the electrochemical reaction kinetics is accelerated.60 Furthermore, the Li+ diffusion coefficients of electrodes under LE and 1.0 wt%-SN were studied using cyclic voltammetry (CV) curves in the range of 3.0–4.5 V vs. Li/Li+ potential window and 0.1–1.0 mV s−1 scanning rate. No additional oxidation peak appeared in electrolytes with the SN additive (Fig. 2(e) and (f) and S4(a) and (b)†). At the same time, as the scanning rate increased, the peak current gradually increased, and the oxidation/reduction peak also moved towards the higher/lower potential.61Fig. 2(g) was plotted as the square root of the maximum current and the associated scan rate. A comparison reveals that the addition of SN additives resulted in decreased potential shift and enhanced current magnitude. Among them, 1.0 wt%-SN showed the most significant reduction in potential shift and increase in current. These results indicate that the addition of the SN additive can enhance the transmission capacity of Li+, with the 1.0 wt%-SN exhibiting the strongest Li+ transmission capacity. These conclusions prove that the addition of the SN additive can indeed improve the electrochemical kinetics of the CEI layer.
Inspired by the molecular orbital theory, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels and intermolecular binding energy of all electrolyte molecules were calculated using the quantization software Gaussian. The generalized function and basis set for DFT calculation was chosen as B3lyp/6-311 + g**, and Gd3bj dispersion correction was added.62 As shown in Fig. 3(a), the LUMO and HOMO of the additive dominate the deposition behavior on the Li side. For LOMO, EC, DEC and DMC were −0.585 eV, −0.364 eV and −0.227 eV, respectively, and the LOMO of additive SN (−1.06 eV) was smaller than that of each electrolyte component. Therefore, the SN additive has strong electron acquisition ability and is preferentially decomposed before the solvent to form SEI rich in organic nitride, which means that the additive can reduce the consumption of carbonate solvent and significantly improve the stability of the lithium anode.63 For HOMO, additives can also decompose and participate in the construction of the CEI layer, showing excellent performance in the battery cycle process.
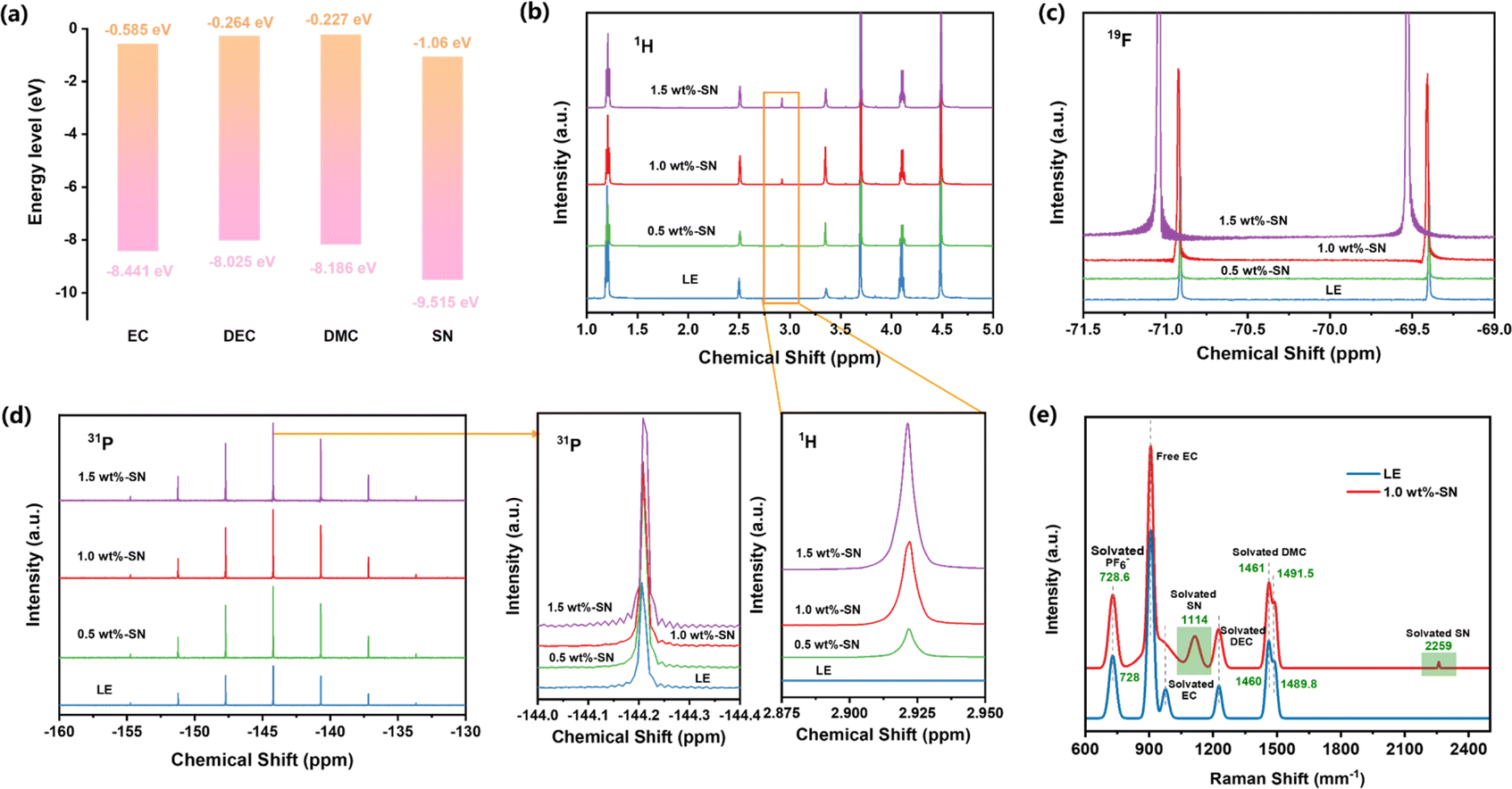 | ||
| Fig. 3 (a) The values of LUMO–HOMO of EC, DEC, DMC and SN molecules. (b) 1H, (c) 19F and (d) 31P solution NMR spectra were collected from electrolytes without SN (LE) and with SN. (e) Raman spectra of the LE and 1.0 wt%-SN electrolytes. | ||
In order to investigate the effect of the SN additive on electrolyte solvation, various electrolytes were analysed by nuclear magnetic resonance (NMR). Firstly, the electrolyte was analysed using the 1H NMR spectrum. As can be seen from Fig. 3(b), the spectrograms of the electrolyte with SN as the additive and the blank electrolyte are basically the same. However, the spectrogram with SN added has an extra peak at about 2.92 ppm, which does not exist in the blank electrolyte. With increasing intensity of the peak due to SN addition, it can be seen that the appearance of this peak is due to the formation of a new solvent pair. It is worth noting that LiPF6 is widely used in the electrolyte of lithium batteries.64,65 In order to investigate the effect of SN on the coupling of Li+ and PF6− ions as well as electrolyte transmission characteristics, 19F and 31P nuclear magnetic resonance (NMR) detections were performed on the blank electrolyte and electrolytes with added SN. Initially, it can be observed that the 31P NMR spectrum (Fig. 3(d)) is composed of a septet, with adjacent peak pairs isolated by the P–F coupling constant (in this case ≈710 Hz); meanwhile, the 19F NMR spectrum (Fig. 3(c)) displays a doublet, indicating the resonance of PF6− ions.66 Further detailed analysis reveals that in the 31P NMR spectrum, each electrolyte exhibits a septet, indicating that the addition of SN does not disrupt the solvent pair structure. However, due to the internal position of P within ion clusters, the addition of a small amount of SN does not cause a significant shift in the peaks in the 31P NMR spectrum. Nevertheless, electrolytes containing SN additives exhibit more negative chemical shifts in the 19F NMR spectrum compared to blank electrolytes. Additionally, with increasing SN content, the chemical shift moves in a more negative direction. These negative chemical shifts indicate an increased shielding effect of the nucleus in the existence of SN, suggesting a weakened coupling between Li+ and PF6− ions, thereby increasing the independence of each ion and aiding in the enhancement of Li+ transport capability.67,68
Furthermore, Raman spectroscopy (Fig. 3(e)) was used to detect LE and 1.0 wt%-SN. At 2259 cm−1, the stretching vibration of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond shows a distinct Raman peak, proving the solvation of SN. At the same time, C–C stretching vibration appeared at 1114 cm−1, which further proved the solvation of SN.44 Both LE and 1.0 wt%-SN electrolytes showed C–O stretching vibration peaks and C–O–C bending vibration peaks of solvated DMC. In contrast, the two peaks produced by solvated DMC (1461, 1491.5 cm−1) in 1.0 wt%-SN were redshifted compared with LE (1460, 1489.8 cm−1). The results indicated that the solvation of DMC was weakened after SN was added. The P–F stretching vibration peak of PF6− appears near 728 cm−1. Compared with LE (728 cm−1), the peak position of 1.0 wt%-SN (728.6 cm−1) showed an obvious redshift. The occurrence of redshift indicates that the interaction between Li+ and PF6− is weakened, which is more conducive to the free movement of Li+.45 This is consistent with the results of NMR spectra analysis. Moreover, the conductivity measurement results at 25 °C also prove that the conductivity of 1.0 wt%-SN (9.59 ms cm−1) is significantly higher than that of LE (9.56 ms cm−1), which also confirms that the movement rate of Li+ is accelerated after the addition of SN, which is also consistent with the above analysis results.46 Not only that, at 25 °C, the analysis of the viscosity of the two electrolytes also revealed that the viscosity of 1.0 wt%-SN (1.92 cP) was significantly lower than that of LE (3.04 cP) after adding SN. The decrease in viscosity reduces the impedance of the electrolyte, which is consistent with the impedance comparison detected in Fig. 2(b)–(d).62,69 At the same time, the viscosity reduction is also conducive to the movement of Li+. These characteristics all confirm that the addition of SN can accelerate the transmission rate of Li+.
N triple bond shows a distinct Raman peak, proving the solvation of SN. At the same time, C–C stretching vibration appeared at 1114 cm−1, which further proved the solvation of SN.44 Both LE and 1.0 wt%-SN electrolytes showed C–O stretching vibration peaks and C–O–C bending vibration peaks of solvated DMC. In contrast, the two peaks produced by solvated DMC (1461, 1491.5 cm−1) in 1.0 wt%-SN were redshifted compared with LE (1460, 1489.8 cm−1). The results indicated that the solvation of DMC was weakened after SN was added. The P–F stretching vibration peak of PF6− appears near 728 cm−1. Compared with LE (728 cm−1), the peak position of 1.0 wt%-SN (728.6 cm−1) showed an obvious redshift. The occurrence of redshift indicates that the interaction between Li+ and PF6− is weakened, which is more conducive to the free movement of Li+.45 This is consistent with the results of NMR spectra analysis. Moreover, the conductivity measurement results at 25 °C also prove that the conductivity of 1.0 wt%-SN (9.59 ms cm−1) is significantly higher than that of LE (9.56 ms cm−1), which also confirms that the movement rate of Li+ is accelerated after the addition of SN, which is also consistent with the above analysis results.46 Not only that, at 25 °C, the analysis of the viscosity of the two electrolytes also revealed that the viscosity of 1.0 wt%-SN (1.92 cP) was significantly lower than that of LE (3.04 cP) after adding SN. The decrease in viscosity reduces the impedance of the electrolyte, which is consistent with the impedance comparison detected in Fig. 2(b)–(d).62,69 At the same time, the viscosity reduction is also conducive to the movement of Li+. These characteristics all confirm that the addition of SN can accelerate the transmission rate of Li+.
In order to further clarify the effect of SN addition on the Li|NCM811 cell, the NCM811 cathodes were analyzed by scanning electron microscopy (SEM) after 100 cycles. The surface of the original electrode was relatively smooth (Fig. S5†). After 100 cycles, it was obvious that the surface topography of the nickel-rich cathode material without the SN additive had greatly changed, with obvious dissolution, obvious deposition on the surface, and large and deep cracks in the particles (Fig. 4(a)). The energy spectrum (EDS) analysis showed that the surface had a high content of P and F elements, while P and F elements only existed in the LiPF6 salt of the electrolyte, so it was speculated that the decomposition of the blank electrolyte after cycling was serious, and the CEI layer formed was thick. In contrast, after the same number of cycles, the surface topography of the NCM811 electrode in the 1.0 wt%-SN was still very similar to that of the original electrode (Fig. 4(b)), with a smooth surface and no obvious deposition, and no microcracks were found. At the same time, the energy dispersive spectroscopy (EDS) analysis revealed that compared with the LE electrolyte, the surface P and F element contents in the 1.0 wt%-SN after circulation were significantly reduced, indicating that after the circulation of 1.0 wt%-SN, the electrolyte decomposition was reduced and the CEI layer formed was thinner. This will be conducive to improving the transmission rate of Li+ through the CEI film.70 In order to further explore the thickness of the CEI layer, the battery after 20 cycles at 1C was disassembled and the cross-section of NCM811 was analyzed by transmission electron microscopy (TEM). The image shows the difference in CEI thickness. Through comparison, it can be clearly seen that the CEI layer thickness of LE (Fig. 4(b)) is significantly greater than 1.0 wt%-SN (Fig. 4(d)). Moreover, the thickness of the CEI layer of LE varies greatly, while the thickness of the CEI layer of 1.0 wt%-SN varies little. This indicates that the CEI layer formed by 1.0 wt%-SN on the cathode side of NCM811 is uniform and thin. This is consistent with the analysis of SEM test results. These results show that in the blank electrolyte and the electrolyte with the SN additive, the electrolyte decomposes on the cathode surface during the cycle, but it decomposes more violently in the LE and deposits more on the surface.
 | ||
| Fig. 4 SEM and EDS images of NCM811 cathodes after 100 cycles and TEM images of NCM811 cathodes after 20 cycles in (a, c) LE and (b, d) 1.0 wt%-SN. Lithium metal anode and diaphragm pictures and SEM images of lithium metal anodes after 20 cycles in (e, f, g) LE and (h, i, j) 1.0 wt%-SN. | ||
Not only the nickel-rich cathode was observed, but also the lithium anode side was tested by SEM. After cycling at 1C for 20 cycles, the morphologies of the lithium metal anode and diaphragm were observed (Fig. 4(e)–(i)). After cycling in LE, the surface of the lithium anode and diaphragm has obvious black material deposition (Fig. 4(e) and (f)), while the surface of the lithium metal anode and diaphragm circulating in 1.0 wt%-SN (Fig. 4(h) and (i)) remains clean and maintains a good initial morphology. The results showed that the active substances in SN dissolved and reached the lithium metal side through the diaphragm, and the loss of active substances led to the attenuation of LE capacity (Fig. 1(a)). Through SEM detection, there were still obvious lithium dendrites and cracks in the lithium metal anode after cycling in LE (Fig. 4(g)). However, the surface of the lithium metal negative electrode remains smooth and clean after cycling in 1.0 wt%-SN (Fig. 4(j)), and the appearance of the uncycled lithium sheet is little different (Fig. S5(b)†). This shows that on the lithium metal side, the addition of SN can effectively maintain the morphology of lithium metal, maintain the stability of the active substance, and thus maintain the stability of the electrochemical cycle.70
At the same time, in order to explore the reasons for the performance improvement after the addition of SN, the nickel-rich cathode after 15 cycles was subjected to XRD test. The XRD results (Fig. S6†) showed that both the LE and 1.0 wt%-SN can maintain a good layered structure after cycling, and no other impurity peaks appeared. In the cycling process of the NCM811 material, the (003) crystal plane will have a small angle shift with the insertion and precipitation of Li+.71 By comparing the NCM811 material after cycling in the two electrolytes and the NCM811 material without cycling, and correcting with the Al peak as the benchmark, the results indicate that after cycling in the LE, the rightward shift of the (003) crystal plane of NCM811 was more pronounced compared to the shift observed after cycling in the 1.0 wt%-SN. This shows that the structure of NCM811 can be better preserved after cycling in the 1.0 wt%-SN. It prevents the collapse of its structure, which is conducive to improving the electrochemical cycling stability performance.72
To investigate the evolution of the CEI layer in the presence of the SN additive during the electrochemical cycle, the nickel-rich cathode and lithium side of the LE and SN cells after 100 cycles were respectively analyzed by high-resolution XPS. For the nickel-rich cathode side, it can be seen from the C 1s spectra (Fig. 5(a)) that abundant carbon-containing substances are detected on the NCM811 electrode surface of both electrolyte batteries after cycling, regardless of SN addition. Among them, C–F, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, C–H and C–C carbon containing substances primarily originate from acetylene black, PVDF binder and the decomposition of carbonate electrolytes.73
O, C–O, C–H and C–C carbon containing substances primarily originate from acetylene black, PVDF binder and the decomposition of carbonate electrolytes.73
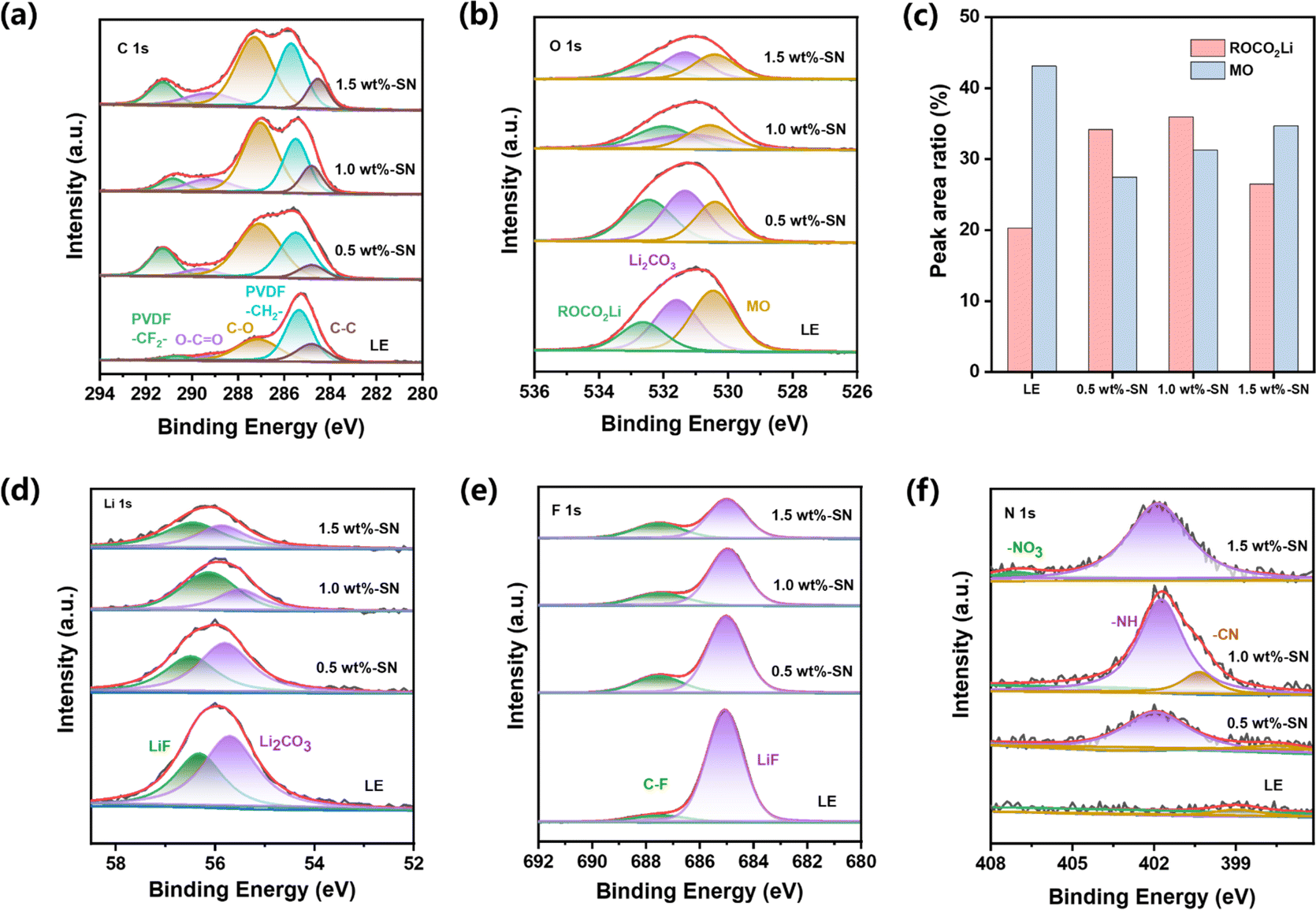 | ||
| Fig. 5 XPS spectra of C 1s (a), O 1s (b), Li 1s (d), F 1s (e), N 1s (f) and the relative contents of different constituents in O 1s (c) of NCM811 cathodes from the cells cycled for 100 cycles without SN (LE) and with SN. | ||
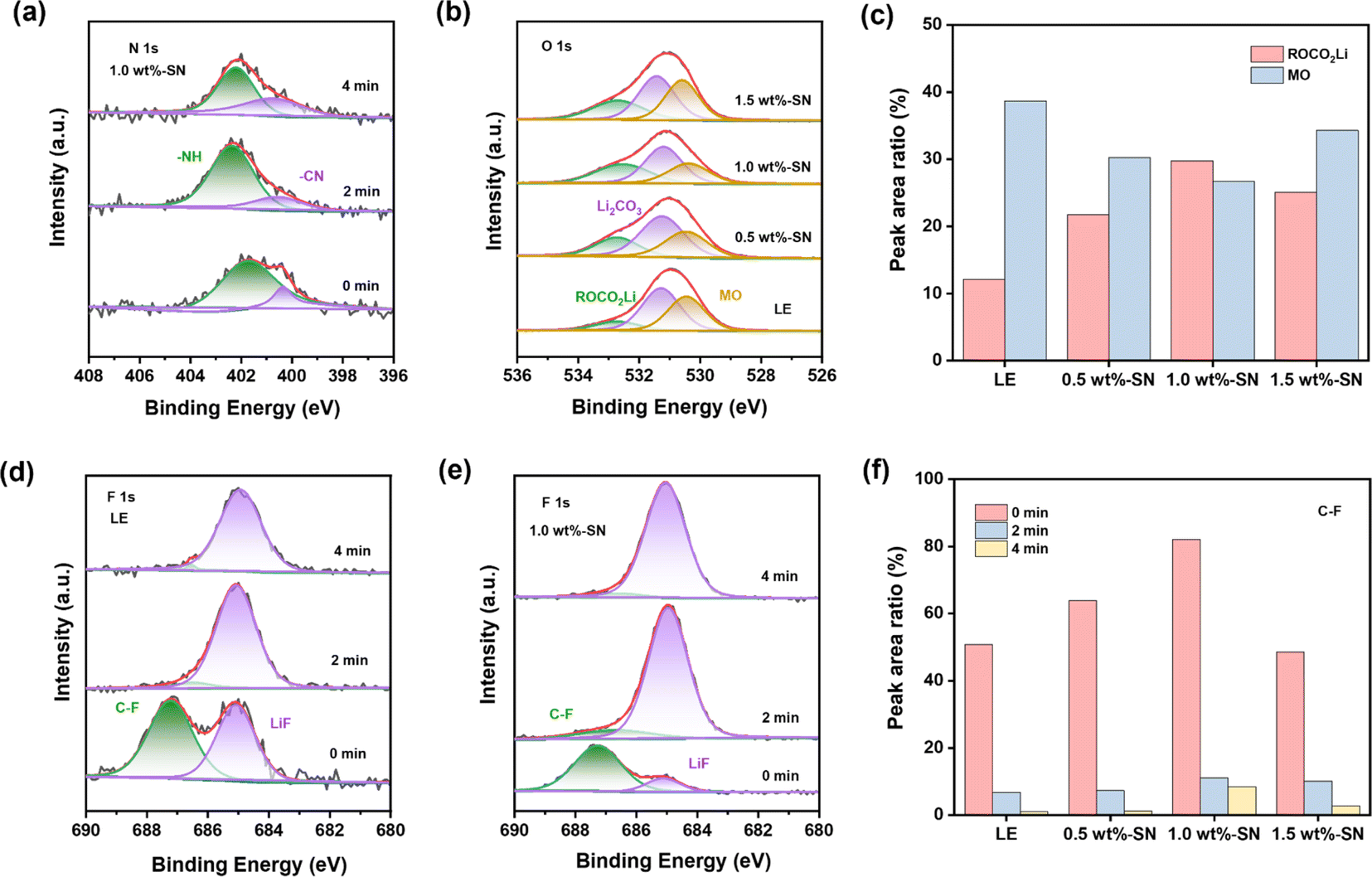
Compared with LE, higher C–C content on the nickel-rich cathode side after cycles in 1.0 wt%-SN and 1.5 wt%-SN was revealed, where the CEI layer was more stable. In the O 1s spectrum (Fig. 5(b)), there were two peaks near 532.4 eV and 531.3 eV, due to the formation of ROCO2Li and Li2CO3 with the increase of cycling. At the same time, an obvious peak was also discovered near 530.4 eV, related to the precipitation of metal oxides in the active material. In contrast (Fig. 5(c)), the content of ROCO2Li was distinctly enhanced in cells with the SN additive. It was not difficult to find that in the electrode/electrolyte interface layer generated by NCM811, the content of organic oxygen was significantly increased in the presence of the SN additive, pretty beneficial to the cycling stability of the interface layer. When the addition of SN was 1.0 wt%, the organic oxygen content was the highest, showing that the CEI layer is the most stable in 1.0 wt%-SN. In addition, the peak strength of metal oxide with SN is significantly weaker than that of LE after cycling, demonstrating that the TM's loss on the cathode surface is greatly reduced after SN is added.74
The Li 1s spectrum (Fig. 5(d)) showed that compared with LE the content of Li2CO3 was reduced deeply in the cells with LE, meanwhile, the decrease of 1.0 wt%-SN was the most obvious. The phenomenon about the change of the Li2CO3 content can also be observed in the O 1s spectrum (Fig. 5(b)). The introduction of SN effectively reduced the decomposition of carbonate electrolyte. In the F 1s spectrum (Fig. 5(e)), the content of LiF in the cells with SN was obviously lower than that of LE, showing that the addition of SN effectively reduced the decomposition of electrolyte salts and was conducive to the stability of the electrolyte. At the same time, it was not difficult to observe that the content of organic fluorine in the electrolyte added with SN was significantly increased, enhancing the stability of the interface film.
By analysing the N 1s spectrum (Fig. 5(f)), nitrogen-containing substances can be detected on the NCM811 side interface after adding the SN additive. With the increase of SN addition, the detection amount of –NH also increased gradually. For other nitrogen-containing substances, only 1.0 wt%-SN showed obvious CN nitrogen-containing organic matter. It showed that SN can be effectively decomposed only in 1.0 wt%-SN and participate in the stability of the stable interface layer. In addition, the existence of –NO3 was also detected in 1.5 wt%-SN, which indicated that SN in the interface was completely decomposed and organic nitride could not be formed to support the stability of the interface layer. On the whole, SN can participate in the construction of the interface layer, and the decomposition of SN replaces the decomposition of some electrolytes, which reduces the loss of electrolytes and is beneficial to the long-term cycle stability of electrolytes. However, excessive addition of SN will lead to excessive decomposition on the nickel-rich cathode side, which cannot support the construction of a more stable CEI layer.
After 100 cycles, the high-resolution XPS analysis of LE and batteries containing SN was carried out on the anode side of the lithium metal. As for the C 1s spectrum (Fig. S7†), similar to the C 1s spectrum on the nickel-rich cathode side (Fig. 5(a)), on the surface of the lithium metal anode after cycling, both LE and the batteries containing SN showed rich carbonaceous substances. However, compared with the NCM811 side, the C–C content on the anode side of lithium metal was obviously increased, indicating that the electrode/electrolyte interface film formed on the anode side of lithium metal was more stable than the nickel-rich cathode.50 In addition, for the N 1s spectrum (Fig. 6(a)), nitrogen-containing substances can also be observed on the lithium metal anode.
 | ||
| Fig. 6 XPS spectra of N 1s (a), O 1s (b), F 1s (d, e) and the relative content of different constituents in O 1s (c) and F 1s (f) of lithium metal anodes from the cells cycled for 100 cycles without SN (LE) and with SN. | ||
For the surface of the lithium metal anode/electrolyte transition layer, the presence of CN-containing organonitride was not detected in both 0.5 wt%-SN and 1.5 wt%-SN, but only on the surface of the 1.0 wt%-SN interface layer. At the same time, with increasing detection depth, organic nitrides were never detected in 0.5 wt%-SN, but –NO3 was detected (Fig. S8(a)†). It can be deduced that insufficient SN addition may lead to its excessive decomposition on the lithium anode side with the increase of the number of cycles, and it cannot provide sufficient organic nitrides. For 1.5 wt%-SN (Fig. S8(b)†), the presence of organic nitrides was detected under deeper detection conditions, but not at the surface of the transition layer. Therefore, the surface SEI layer is more likely to be destroyed than the 1.0 wt%-SN layer. In contrast, with the increase of detection depth of 1.0 wt%-SN, the content of organic nitride increased rather than decreasing, which fully supported the stability of the SEI film. In the O 1s spectrum (Fig. 6(b)), the results detected were highly consistent with those detected at the nickel-rich cathode (Fig. 5(b)). On the lithium anode side, due to the generation of ROCO2Li and Li2CO3 during the cycle, two peaks appeared near 532.7 eV and 531.4 eV. Peaks associated with metal oxidation bonds in the active material were also detected near 530.5 eV. By comparison (Fig. 6(c)), on the lithium anode side, the content of ROCO2Li is higher and the content of metal oxide (mainly lithium oxide) is lower in the battery containing the SN additive. When the SN addition amount is 1.0 wt%, the effect is most significant. These results indicate that the addition of SN also contributes significantly to the lithium anode side, stabilizing the SEI layer while reducing the erosion of the lithium anode itself during the cycle.
For the F 1s spectrum (Fig. 6(d)–(f) and S9†), it was evident that with the increase of sputtering depth, the C–F organic fluorine content containing the SN additive is markedly higher compared to the LE, and the 1.0 wt%-SN had the highest content at each depth, further indicating that the SEI layer formed by the 1.0 wt%-SN on the lithium anode side is more stable. Overall, the addition of SN in the nickel-rich cathode and lithium anode is conducive to the formation of a stable electrode/electrolyte interface layer, and the SN decomposition in the electrode/electrolyte interface layer also reduces the loss of the electrolyte and the erosion of the electrode.
To further analyze the distribution and strength of the CEI film formed on the surface of the NCM811 nickel-rich cathode during charge–discharge processes, the Li|NCM811 cells after 100 cycles at 1C rate were dismantled and measured using the Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) technique. The relevant depth profile, 3D reconstructed images, and surface distribution maps are shown in Fig. 7(a)–(e), respectively.75–77
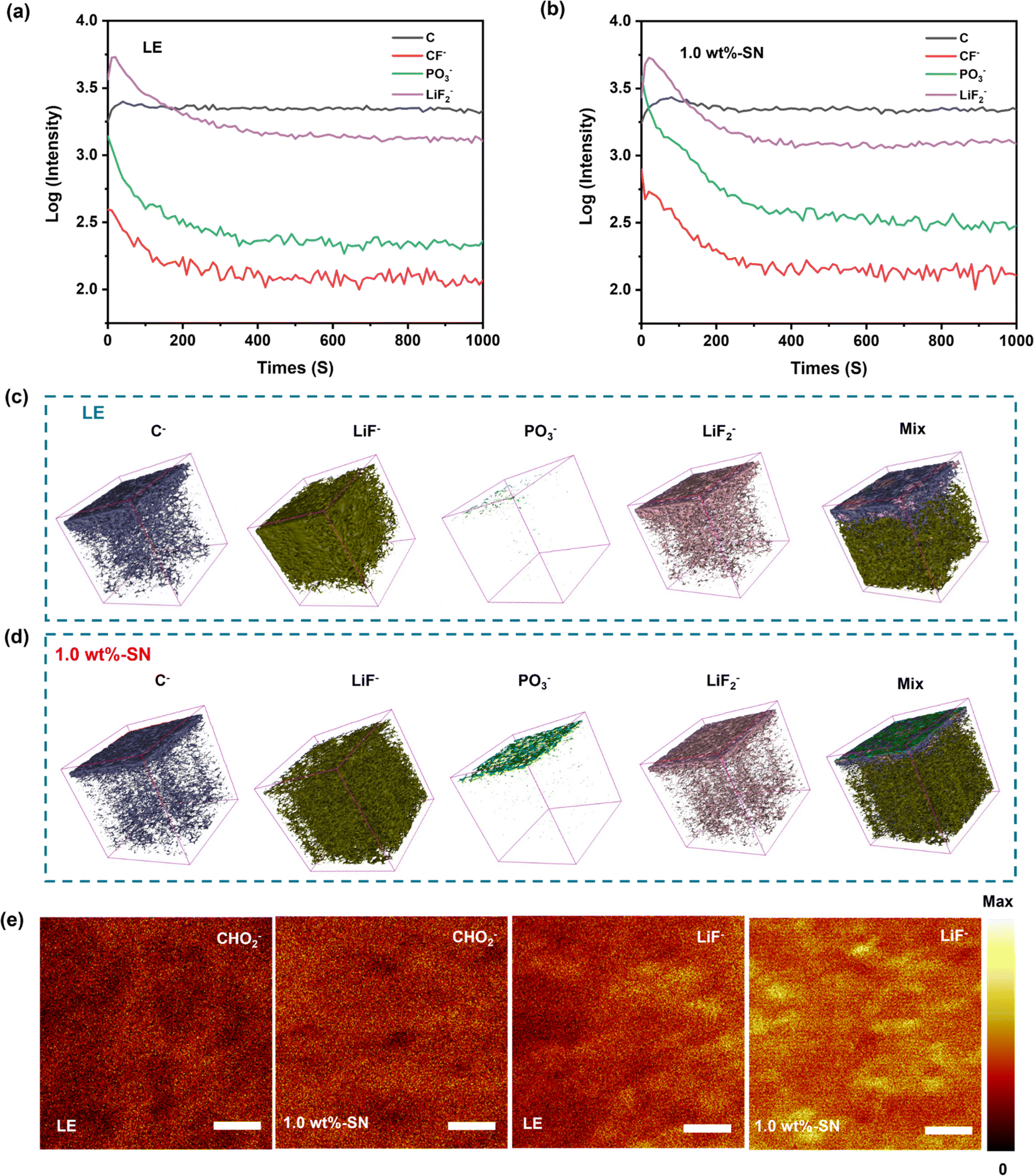 | ||
| Fig. 7 TOF-SIMS depth profiles of partial species (a, b), corresponding 3D-reconstructed images (c, d) and secondary ion images under negative (−) ion mode surface profile (the length of the scale bar is 10 μm) (e) for the cycled NMC811 electrodes with or without the SN additive after long-term cycling, respectively. | ||
In the TOF-SIMS spectra, LiF− and LiF2− are attributed to the LiF species, possibly originating from the decomposition of LiPF6 salt. CHO2− and C− are likely generated from the decomposition of ester-based electrolytes. From the depth profiles (Fig. 7(a) and (b) and S10†), three-dimensional reconstruction images (Fig. 7(c) and (d)), and surface distribution maps (Fig. 7(e)), it can be observed that the CEI layers formed by the blank electrolyte and SN additive-containing electrolyte exhibit a multi-layered structure, with interpenetration of organic and inorganic species.78
The profiles (Fig. 7(a) and (b) and S10†) showed that the contents of C− and CF− on the surface of LE were obviously less than 1.0 wt%-SN, but they were basically the same with the increase of depth. The surface analysis diagram also showed that the distribution of CHO2− on the surface of LE was obviously less than 1.0 wt%-SN. However, in the three-dimensional reconstructed image, the C− depth of LE was obviously higher. In combination, in the CEI layer formed by LE, the surface distribution of carbonaceous organic matter and organic fluoride was loose and the longitudinal distribution was deep, indicating that the formed interface layer was fluffy and thick. On the other hand, the carbonaceous organic matter in the interface layer formed by 1.0 wt%-SN was more concentrated in the surface layer, dense and thin. These phenomena are consistent with the XPS results of the nickel-rich cathode side. In addition, from the figure, it was found that in 1.0 wt%-SN the density of PO3− distributed on the surface and the content at all depths was higher than that of LE. This indicates that in LE, the fluoride generated by the decomposition of lithium salt tends to be dispersed in the whole electrolyte, meanwhile, in 1.0 wt%-SN, the fluorine-containing compounds preferentially form a strong protective film on most of the surface of the electrode, thereby inhibiting the continued decomposition of the electrolyte and protecting the transition metal from dissolution. This is conducive to enhancing the structural stability and cycle stability of the material.79
From the analysis of three-dimensional reconstructed images of different components, it was clearly found that the CEI film formed in LE was thicker and more porous, while a thinner and denser film was formed in 1.0 wt%-SN. The fluorine-containing compounds were evenly distributed on the top layer in 1.0 wt%-SN. The compact CEI film significantly maintains the structural integrity of the nickel-rich cathode and mitigates further electrolyte decomposition.80,81 In addition, the thin and dense nickel-rich cathode/electrolyte interface layer is stable enough to buffer and release the stress and strain generated during repeated charging and discharging, preventing the destruction of the membrane structure and achieving electrochemical cycle stability.78
Based on all the above characterization and testing, we summarize the possible mechanism of the SN additive, which is applicable to both CEI layer and SEI layer (Fig. 8). First of all, the addition of SN is conducive to improving the migration ability of Li+, which is helpful to the improvement of electrochemical stability. The construction of the electrode/electrolyte interface layer includes the participation of the SN additive, and SN plays a good role in the construction of the transition layer of the cathode and anode at the same time. With the participation of SN, the CF content of the formed electrode/electrolyte interface layer increases, and the addition of SN replaces the decomposition of part of the electrolyte to form nitrogen-containing organic matter. The increase of organic fluoride/nitride significantly improves the structural stability of the CEI/SEI layer. At the same time, the interface layer formed by the blank electrolyte is thick and uneven, which leads to the inability to protect the electrode well, and a large amount of metal oxide MO will be precipitated with the increase of the cycle, and the cathode and anode continue to decompose, resulting in greatly reduced cycle stability. With the participation of SN, the interface layer formed is thin and dense, and the dense transition layer can prevent the further decomposition of the electrode, thus greatly improving the cycle stability.82,83
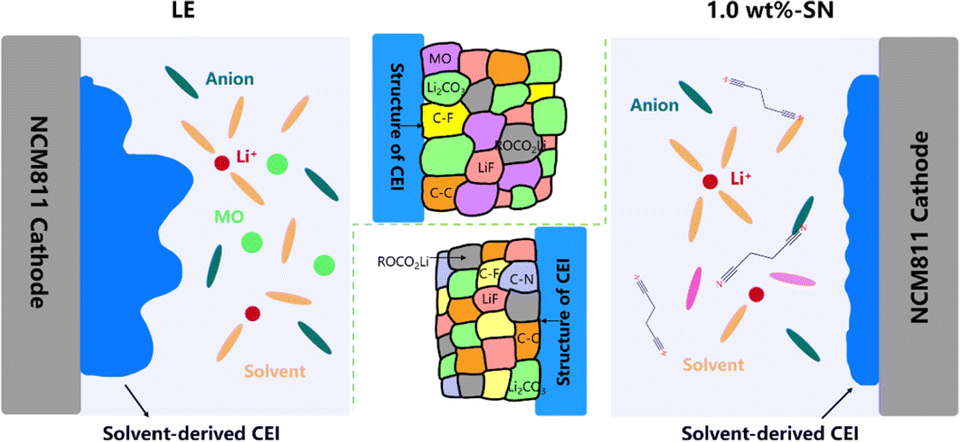 | ||
| Fig. 8 Schematic illustration of the NCM811 cathode CEI generation on LE and 1.0 wt%-SN electrolytes. | ||
3 Conclusion
In summary, by using electrolyte engineering, we have successfully explored and designed with SN as an additive ways to improve the cycle stability performance of Li|NCM811 batteries, and we investigated the contribution of SN to improving the electrochemical cycle performance, as well as the impact on CEI and SEI. SN can enhance the infiltration of electrolyte to the nickel-rich cathode, and at the same time facilitate easier detachment of Li+ ions, thereby enabling Li|NCM811 batteries with a higher rate capacity. A uniform, stable and thin CEI layer is formed on the cathode surface, which greatly reduces the irreversible phase transition of the nickel-rich cathode during the charge–discharge process, while inhibiting the decomposition of the electrolyte and the dissolution of the transition metal. In addition, a stable SEI layer is formed on the anode surface, effectively stabilizing the electrode/electrolyte interface. This work provides an efficient approach to improve the performance of the cathode/electrolyte interface and the anode/electrolyte interface using a single electrolyte additive.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ziye Wang: methodology, formal analysis, investigation, writing – original draft. Yingshuai Wang: data curation, formal analysis, writing – review & editing. Yuhang Xin: formal analysis, writing – review & editing. Qingbo Zhou: methodology, formal analysis, investigation. Xiangyu Ding: methodology, formal analysis. Lei Liu: methodology, formal analysis. Tinglu Song: formal analysis. Feng Wu: investigation. Zhongbao Wei: investigation. Hongcai Gao: conceptualization, methodology, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key R&D Program of China (2022YFB3803501), Natural Science Foundation of China (52272188 and U22A20227), Natural Science Foundation of Beijing (2232025), Natural Science Foundation of Chongqing (2022NSCQ-MSX2179), Department of Science and Technology of Henan Province (Z20221343029) and the Experimental Center of Advanced Materials in Beijing Institute of Technology.Notes and references
- C. Zhang, S. Chou, Z. Guo and S. Dou, Adv. Funct. Mater., 2024, 34, 2308001 CrossRef.
- Y. Zhu, M. Ge, F. Ma, Q. Wang, P. Huang and C. Lai, Adv. Funct. Mater., 2024, 34, 2301964 CrossRef.
- V. A. K. Adiraju, O. B. Chae, J. R. Robinson and B. L. Lucht, ACS Energy Lett., 2023, 8, 2440–2446 CrossRef.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X.-Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef PubMed.
- Y. Wang, R. Ou, J. Yang, Y. Xin, P. Singh, F. Wu, Y. Qian and H. Gao, J. Energy Chem., 2024, 95, 407–427 CrossRef.
- S. K ainat, J. Anwer, A. Hamid, N. Gull and S. M. Khan, Mater. Chem. Phys., 2024, 313, 128796 CrossRef CAS.
- Q. Li, G. Liu, H. Cheng, Q. Sun, J. Zhang and J. Ming, Chem.–Eur. J., 2021, 27, 15842–15865 CrossRef CAS PubMed.
- Y. Qiu, D. Lu, Y. Gai and Y. Cai, ACS Appl. Mater. Interfaces, 2022, 14, 11398–11407 CrossRef CAS.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- Y. Wang, Q. Wang, X. Ding, M. Wang, Y. Xin and H. Gao, Appl. Surf. Sci., 2022, 601, 154218 CrossRef.
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132–A141 CrossRef.
- N. Khosla, J. Narayan, R. Narayan, X.-G. Sun and M. P. Paranthaman, J. Electrochem. Soc., 2023, 170, 030520 CrossRef.
- M.-C. Liu, Q.-S. Liu, Y.-Z. Quan, J.-L. Yu, G. Wu, X.-L. Wang and Y.-Z. Wang, Chin. Chem. Lett., 2023, 109123 Search PubMed.
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef PubMed.
- H. Zhang and J. Zhang, eTransportation, 2021, 7, 100105 CrossRef.
- S. Kim, G. Park, S. J. Lee, S. Seo, K. Ryu, C. H. Kim and J. W. Choi, Adv. Mater., 2023, 35, 2206625 CrossRef CAS PubMed.
- L. He, J. Wu, D. Lau, C. Hall, Y. Jiang, B. Thapa, Y. Gao, T. Yeung, A. Wotton, A. Lennon, N. Sharma and R. J. Patterson, Mater. Lett., 2021, 304, 130659 CrossRef CAS.
- X. Chen, M. Shang and J. Niu, Nano Lett., 2020, 20, 2639–2646 CrossRef CAS PubMed.
- C. Sengottaiyan, K. Kubota, S. Kumakura, Y. TaeHyeon, T. Hosaka and S. Komaba, J. Electrochem. Soc., 2021, 168, 110505 CrossRef CAS.
- H. Lv, X. Huang and Y. Liu, Ionics, 2020, 26, 1749–1770 CrossRef CAS.
- S. Zhang, M. He, C.-C. Su and Z. Zhang, Curr. Opin. Chem. Eng., 2016, 13, 24–35 CrossRef.
- T. van Ree, Curr. Opin. Electrochem., 2020, 21, 22–30 CrossRef CAS.
- H. Zhao, X. Yu, J. Li, B. Li, H. Shao, L. Li and Y. Deng, J. Mater. Chem. A, 2019, 7, 8700–8722 RSC.
- E. Markevich, G. Salitra, F. Chesneau, M. Schmidt and D. Aurbach, ACS Energy Lett., 2017, 2, 1321–1326 CrossRef CAS.
- X. Zhang, X. Cheng, X. Chen, C. Yan and Q. Zhang, Adv. Funct. Mater., 2017, 27, 1605989 CrossRef.
- K. Yao, J. P. Zheng and R. Liang, J. Power Sources, 2018, 381, 164–170 CrossRef CAS.
- L. Ma, S. L. Glazier, R. Petibon, J. Xia, J. M. Peters, Q. Liu, J. Allen, R. N. C. Doig and J. R. Dahn, J. Electrochem. Soc., 2017, 164, A5008–A5018 CrossRef CAS.
- A. Bordes, K. Eom and T. F. Fuller, J. Power Sources, 2014, 257, 163–169 CrossRef CAS.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, J. Electrochem. Soc., 2015, 162, A1094–A1101 CrossRef CAS.
- L. Liao, P. Zuo, Y. Ma, Y. An, G. Yin and Y. Gao, Electrochim. Acta, 2012, 74, 260–266 CrossRef CAS.
- Y. Zhu, M. D. Casselman, Y. Li, A. Wei and D. P. Abraham, J. Power Sources, 2014, 246, 184–191 CrossRef CAS.
- Y.-M. Lee, K.-M. Nam, E.-H. Hwang, Y.-G. Kwon, D.-H. Kang, S.-S. Kim and S.-W. Song, J. Phys. Chem. C, 2014, 118, 10631–10639 CrossRef CAS.
- W. Wang, C. Liao, L. Liu, W. Cai, Y. Yuan, Y. Hou, W. Guo, X. Zhou, S. Qiu, L. Song, Y. Kan and Y. Hu, J. Power Sources, 2019, 420, 143–151 CrossRef CAS.
- Y. E. Hyung, D. R. Vissers and K. Amine, J. Power Sources, 2003, 119–121, 383–387 CrossRef CAS.
- N. Xu, J. Shi, G. Liu, X. Yang, J. Zheng, Z. Zhang and Y. Yang, J. Power Sources Adv., 2021, 7, 100043 CrossRef.
- N. von Aspern, S. Röser, B. Rezaei Rad, P. Murmann, B. Streipert, X. Mönnighoff, S. D. Tillmann, M. Shevchuk, O. Stubbmann-Kazakova, G.-V. Röschenthaler, S. Nowak, M. Winter and I. Cekic-Laskovic, J. Fluorine Chem., 2017, 198, 24–33 CrossRef.
- Y.-M. Song, J.-G. Han, S. Park, K. T. Lee and N.-S. Choi, J. Mater. Chem. A, 2014, 2, 9506–9513 RSC.
- Y. Ji, P. Zhang, M. Lin, W. Zhao, Z. Zhang, Y. Zhao and Y. Yang, J. Power Sources, 2017, 359, 391–399 CrossRef.
- W. Tu, P. Xia, X. Zheng, C. Ye, M. Xu and W. Li, J. Power Sources, 2017, 341, 348–356 CrossRef.
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- S. Kim, S. O. Park, M.-Y. Lee, J.-A. Lee, I. Kristanto, T. K. Lee, D. Hwang, J. Kim, T.-U. Wi, H.-W. Lee, S. K. Kwak and N.-S. Choi, Energy Storage Mater., 2022, 45, 1–13 CrossRef.
- J. Yang, P. Zhao, Y. Shang, L. Wang, X. He, M. Fang and J. Wang, Electrochim. Acta, 2014, 121, 264–269 CrossRef CAS.
- J.-G. Han, M.-Y. Jeong, K. Kim, C. Park, C. H. Sung, D. W. Bak, K. H. Kim, K.-M. Jeong and N.-S. Choi, J. Power Sources, 2020, 446, 227366 CrossRef CAS.
- Q. Liu, F. Wu, D. Mu and B. Wu, Chin. Phys. B, 2020, 29, 048202 CrossRef CAS.
- Y. Hu, L. Li, H. Tu, X. Yi, J. Wang, J. Xu, W. Gong, H. Lin, X. Wu and M. Liu, Adv. Funct. Mater., 2022, 32, 2203336 CrossRef CAS.
- Y. Chen, Y. Zhao, A. Wang, D. Zhang, B. Li, X. He, X. Fan and J. Liu, Energy Environ. Sci., 2024, 17, 6113–6126 RSC.
- W. Li, S. Han, C. Xiao, J. Yan, B. Wu, P. Wen, J. Lin, M. Chen and X. Lin, Angew. Chem., Int. Ed., 2024, e202410392 Search PubMed.
- Y.-S. Kim, H. Lee and H.-K. Song, ACS Appl. Mater. Interfaces, 2014, 6, 8913–8920 CrossRef CAS PubMed.
- H. Duncan, N. Salem and Y. Abu-Lebdeh, J. Electrochem. Soc., 2013, 160, A838 CrossRef CAS.
- R. Xue, N. Liu, L. Bao, L. Chen, Y. Su, Y. Lu, J. Dong, S. Chen and F. Wu, J. Energy Chem., 2020, 50, 378–386 CrossRef.
- Y. Zheng, N. Xu, S. Chen, Y. Liao, G. Zhong, Z. Zhang and Y. Yang, ACS Appl. Energy Mater., 2020, 3, 2837–2845 CrossRef CAS.
- Q.-S. Liu, Y.-Z. Quan, M.-C. Liu, G.-R. Zhu, X.-L. Wang, G. Wu and Y.-Z. Wang, J. Energy Chem., 2023, 83, 239–246 Search PubMed.
- S. She, Y. Zhou, Z. Hong, Y. Huang and Y. Wu, ACS Appl. Energy Mater., 2023, 6, 7289–7297 CrossRef CAS.
- K. Jung, S. H. Oh and T. Yim, J. Electrochem. Sci. Technol., 2020, 12, 67–73 CrossRef.
- L. Zhou, Y. Zheng, T. Qian, Y. Gan, Y. Wang, J. Zhou, F. Cai, X. Zhou, J. Liu and L. Li, Inorg. Chem., 2024, 63, 5727–5733 CrossRef CAS PubMed.
- Z. Wang, L. Jiang, C. Liang, C. Zhao, Z. Wei and Q. Wang, J. Electrochem. Soc., 2020, 167, 130517 CrossRef CAS.
- C.-G. Shi, C.-H. Shen, X.-X. Peng, C.-X. Luo, L.-F. Shen, W.-J. Sheng, J.-J. Fan, Q. Wang, S.-J. Zhang, B.-B. Xu, J.-J. Xian, Y.-M. Wei, L. Huang, J.-T. Li and S.-G. Sun, Nano Energy, 2019, 65, 104084 CrossRef CAS.
- C. S. Yoon, D.-W. Jun, S.-T. Myung and Y.-K. Sun, ACS Energy Lett., 2017, 2, 1150–1155 CrossRef CAS.
- J. Kim, J. Lee, H. Ma, H. Y. Jeong, H. Cha, H. Lee, Y. Yoo, M. Park and J. Cho, Adv. Mater., 2018, 30, 1704309 CrossRef PubMed.
- F. Wu, J. Tian, N. Liu, Y. Lu, Y. Su, J. Wang, R. Chen, X. Ma, L. Bao and S. Chen, Energy Storage Mater., 2017, 8, 134–140 CrossRef.
- Z. Liang, K. Yan, G. Zhou, A. Pei, J. Zhao, Y. Sun, J. Xie, Y. Li, F. Shi, Y. Liu, D. Lin, K. Liu, H. Wang, H. Wang, Y. Lu and Y. Cui, Sci. Adv., 2019, 5, eaau5655 CrossRef CAS.
- D. Wu, C. Zhu, H. Wang, J. Huang, G. Jiang, Y. Yang, G. Yang, D. Tang and J. Ma, Angew. Chem., Int. Ed., 2024, 63, e202315608 CrossRef.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef.
- J. Ming, M. Li, P. Kumar and L.-J. Li, ACS Nano, 2016, 10, 6037–6044 CrossRef.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef.
- M. McEldrew, Z. A. H. Goodwin, S. Bi, M. Z. Bazant and A. A. Kornyshev, J. Chem. Phys., 2020, 152, 234506 CrossRef.
- K. Huang, S. Bi, B. Kurt, C. Xu, L. Wu, Z. Li, G. Feng and X. Zhang, Angew. Chem., 2021, 133, 19381–19389 CrossRef.
- S. Li, W. Zhang, Q. Wu, L. Fan, X. Wang, X. Wang, Z. Shen, Y. He and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 14935–14941 CrossRef CAS.
- K.-S. Oh, J.-H. Kim, S.-H. Kim, D. Oh, S.-P. Han, K. Jung, Z. Wang, L. Shi, Y. Su, T. Yim, S. Yuan and S.-Y. Lee, Adv. Energy Mater., 2021, 11, 2101813 CrossRef CAS.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- G. Jiang, J. Liu, Z. Wang and J. Ma, Adv. Funct. Mater., 2023, 33, 2300629 CrossRef CAS.
- X. Kong, S. Peng, J. Li, Z. Chen, Z. Chen, J. Wang and J. Zhao, J. Power Sources, 2020, 448, 227445 Search PubMed.
- S. Tan, Z. Shadike, J. Li, X. Wang, Y. Yang, R. Lin, A. Cresce, J. Hu, A. Hunt, I. Waluyo, L. Ma, F. Monaco, P. Cloetens, J. Xiao, Y. Liu, X.-Q. Yang, K. Xu and E. Hu, Nat. Energy, 2022, 7, 484–494 CrossRef CAS.
- F. Wu, W. Li, L. Chen, Y. Su, L. Bao, W. Bao, Z. Yang, J. Wang, Y. Lu and S. Chen, Energy Storage Mater., 2020, 28, 383–392 CrossRef.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149–8159 Search PubMed.
- H. Sheng, X.-H. Meng, D.-D. Xiao, M. Fan, W.-P. Chen, J. Wan, J. Tang, Y.-G. Zou, F. Wang, R. Wen, J.-L. Shi and Y.-G. Guo, Adv. Mater., 2022, 34, 2108947 Search PubMed.
- M. Malaki, A. Pokle, S.-K. Otto, A. Henss, J. P. Beaupain, A. Beyer, J. Müller, B. Butz, K. Wätzig, M. Kusnezoff, J. Janek and K. Volz, ACS Appl. Energy Mater., 2022, 5, 4651–4663 CrossRef CAS.
- H. Xu, Z. Li, T. Liu, C. Han, C. Guo, H. Zhao, Q. Li, J. Lu, K. Amine and X. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202202894 CrossRef CAS.
- H. Yan, T. Wang, L. Liu, T. Song, C. Li, L. Sun, L. Wu, J.-C. Zheng and Y. Dai, J. Power Sources, 2023, 557, 232559 Search PubMed.
- W. Zhao, Y. Ji, Z. Zhang, M. Lin, Z. Wu, X. Zheng, Q. Li and Y. Yang, Curr. Opin. Electrochem., 2017, 6, 84–91 CrossRef CAS.
- Z. Chen, Y. Chao, W. Li, G. G. Wallace, T. Bussell, J. Ding and C. Wang, Adv. Sci., 2021, 8, 2003694 Search PubMed.
- C. Li, X. Zhang, Z. Yang, H. Lv, T. Song, S. Lu, Y. Zhang, T. Yang, F. Xu, F. Wu and D. Mu, J. Energy Chem., 2023, 87, 342–350 CrossRef CAS.
- D. Gao, J. B. Xu, M. Lin, Q. Xu, C. F. Ma and H. F. Xiang, RSC Adv., 2015, 5, 17566–17571 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04122d |
| This journal is © The Royal Society of Chemistry 2024 |