
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBifunctional PdMoPt trimetallene boosts alcohol–water electrolysis†
Junfeng
Liu
 *a,
Tong
Li
a,
Qiuxia
Wang
a,
Haiting
Liu
a,
Jingjing
Wu
bc,
Yanping
Sui
b,
Huaming
Li
a,
Pengyi
Tang
*bcd and
Yong
Wang
*a
*a,
Tong
Li
a,
Qiuxia
Wang
a,
Haiting
Liu
a,
Jingjing
Wu
bc,
Yanping
Sui
b,
Huaming
Li
a,
Pengyi
Tang
*bcd and
Yong
Wang
*a
aInstitute for Energy Research, School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: jliu@ujs.edu.cn; wangyong@ujs.edu.cn
bNational Key Laboratory of Materials for Integrated Circuits, Shanghai Institute of Microsystem and Information Technology (SIMIT), Chinese Academy of Sciences (CAS), Shanghai, 200050, China. E-mail: py.tang@mail.sim.ac.cn
c2020 X-Lab, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China
dSchool of Graduate Study, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 16th September 2024
Abstract
Substituting oxygen evolution with alcohol oxidation is crucial for enhancing the cathodic hydrogen evolution reaction (HER) at low voltages. However, the development of high-performance bifunctional catalysts remains a challenge. In this study, an ultrathin and porous PdMoPt trimetallene is developed using a wet-chemical strategy. The synergetic effect between alloying metals regulates the adsorption energy of reaction intermediates, resulting in exceptional activity and stability for the electrooxidation of various alcohols. Specifically, the mass activity of PdMoPt trimetallene toward the electrooxidation of methanol, ethylene glycol, and glycerol reaches 6.13, 5.5, and 4.37 A mgPd+Pt−1, respectively. Moreover, the catalyst demonstrates outstanding HER activity, requiring only a 39 mV overpotential to achieve 10 mA cm−2. By employing PdMoPt trimetallene as both the anode and cathode catalyst, we established an alcohol–water hybrid electrolysis system, significantly reducing the voltage requirements for hydrogen production. This work presents a promising avenue for the development of bifunctional catalysts for energy-efficient hydrogen production.
Introduction
Hydrogen, with its high energy density and zero carbon emissions, stands out as a promising alternative to fossil fuels.1–5 Among various hydrogen production techniques, electrochemical water splitting emerges as particularly suitable and effective. However, the practical application of electrochemical water splitting encounters challenges attributed to the sluggish kinetics and high overpotential associated with the anodic oxygen evolution reaction (OER), resulting in substantial electricity consumption during hydrogen production.6–8 To address this challenge, a promising strategy involves replacing the OER with the more thermodynamically favorable electrooxidation of small molecule substrates.9–12 Alcohols such as methanol, ethylene glycol, and glycerol, which readily dissolve in aqueous media and exhibit favorable oxidation thermodynamics, hold significant potential as alternative substrates.13–15 Therefore, the integration of the anodic alcohol oxidation reaction (AOR) with the cathodic hydrogen evolution reaction (HER) presents an attractive approach for achieving energy-efficient hydrogen production.The rational design of efficient electrocatalysts for both the anode and cathode is crucial for implementing this strategy. Employing a bifunctional catalyst can simplify the design, operation, and recycling of coupled AOR-HER systems, leading to cost reductions, easier maintenance, and improved system stability. Therefore, developing catalysts with both AOR and HER functionalities is highly advantageous. While Pd- and Pt-based materials exhibit high activity in both the AOR and HER, persistent limitations such as high cost and susceptibility to poisoning remain.16–20 Structural engineering, involving the regulation of morphology, dimensionality, and size, has proven to be an effective approach.21–23 In particular, ultrathin two-dimensional (2D) structures offer exceptional atomic utilization, abundant active sites, and high-density unsaturated atoms, making them ideal for surface-related electrocatalytic applications.24,25 In this context, metallenes, an emerging class of 2D materials composed of one or a few atomic layers, have garnered considerable attention for enhancing catalytic performance.26–29 Beyond single-element metallenes, recent advances have focused on engineering bimetallenes, such as PdMo, PdCr, and PdIn, by alloying Pd with various metals.30–32 The unique combination of strain, quantum size, and alloying effects in bimetallenes influences the electronic structure of Pd sites, resulting in superior performance in the HER, the AOR, and various catalytic processes.33–37 Previous studies have also demonstrated that the introduction of a third metal into alloys can further enhance the activity and durability of catalysts by fine-tuning the electronic structure and harnessing synergistic effects among the alloying metals.38–41 However, synthesizing 2D metallenes with three metal elements poses a significant challenge due to the distinct nucleation and growth kinetics of different compositional metals.
Herein, we present a novel procedure for synthesizing a trimetallene catalyst, PdMoPt, utilizing PdMo bimetallene as a template. The inclusion of Pt in the trimetallene catalyst provides several advantages. First, Pt acts as an additional active site for both the AOR and HER. Additionally, the presence of Pt allows for precise modulation of the electronic structure of Pd, thereby enhancing catalytic performance. Furthermore, the introduction of Pt induces lattice expansion in the trimetallene catalyst, effectively eliminating reaction intermediates on the catalyst surface. As a result of these structural modifications, the PdMoPt trimetallene catalyst exhibits enhanced performance in the electrooxidation of various alcohols, including methanol, ethylene glycol, and glycerol. Moreover, the PdMoPt trimetallene catalyst demonstrates remarkable activity for the HER in alkaline media, surpassing the performance of its PdMo counterpart and even commercial Pt/C catalysts. Building upon these results, we establish and evaluate a novel AOR-HER hybrid electrolysis system for efficient hydrogen generation at low cell voltage.
Results and discussion
Fig. 1a illustrates the synthetic procedure employed for the production of PdMoPt trimetallene. Initially, PdMo bimetallene is synthesized using Pd(acac)2 and Mo(CO)6 as metal precursors, with ascorbic acid acting as the reducing agent (Fig. S1†).30 Subsequently, Pt(acac)2, serving as the Pt precursor, is introduced into the PdMo suspension and subjected to a reaction at a relatively high temperature. At this temperature, Pt seeds nucleate, undergo deposition, and diffuse into the PdMo lattice, resulting in the formation of the PdMoPt trimetallene. The decomposition of Mo(CO)6 releases carbon monoxide, which adsorbs onto the Pd (111) facet surface, facilitating lateral growth and guiding the formation of ultrathin 2D nanosheet structures. An adequate amount of Mo(CO)6 is crucial for the formation of the ultrathin 2D structure, as evidenced by the thicker morphology observed in PdMo bimetallene synthesized with insufficient Mo(CO)6 (Fig. S2†). Furthermore, compared to one-pot synthesis, this two-step procedure prevents the competitive reaction between Pt nuclei and CO on the Pd (111) surface, thereby ensuring the successful formation of ultrathin 2D PdMoPt trimetallene (Fig. S3†). | ||
| Fig. 1 Preparation and characterization of PdMoPt trimetallene. (a) Schematic illustration of the synthesis of PdMoPt trimetallene. (b) TEM micrograph of PdMoPt trimetallene and (c) magnified details of the yellow square area. (d) HAADF-STEM micrograph of PdMoPt trimetallene and (e) magnified details of the orange square area. (f) Representative HRTEM images of a PdMoPt trimetallene sheet, and (g) magnified view of its yellow square area and (h) the magnified view of the orange square area. (i) AC HAADF-STEM image of a representative PdMoPt trimetallene. (j) STEM image and corresponding elemental mappings of PdMoPt trimetallene. | ||
The PdMoPt trimetallene exhibits a 2D curved structure, with a thickness of 1.5 nm and a lateral dimension exceeding 100 nm, as determined from the transmission electron microscopy (TEM) images (Fig. 1b and c). High-angle annular dark-field scanning TEM (HAADF-STEM) images (Fig. 1d and e) reveal distinctive network pores on the nanosheets, likely induced by the rapid diffusion of Pt into the PdMo lattice at elevated temperatures. This porous morphology provides additional diffusion channels and active sites, potentially enhancing electrocatalytic performance. High-resolution TEM (HRTEM) images exhibit clear lattice fringes with interplanar spacings of 2.06 and 2.38 Å, corresponding to the (200) and (111) crystal planes of Pd/Pt, respectively (Fig. 1f–h). Significantly, these lattice spacings are larger than those observed in the PdMo bimetallene (Fig. S4†), indicating the integration of Pt atoms into the PdMo bimetallene and consequent expansion of the initial PdMo lattice. The X-ray diffraction (XRD) pattern of PdMoPt trimetallene shows a slightly negative shift and strong diffraction peaks in comparison to PdMo bimetallene, indicating lattice expansion and enhanced crystallinity caused by the incorporation of Pt atoms at high temperature (Fig. S5†). Aberration-corrected (AC) HAADF-STEM reveals well-dispersed Pt atoms on the surfaces, further confirming the formation of alloyed PdMoPt trimetallene (Fig. 1i). Energy-dispersive X-ray (EDX) analysis (Fig. S6†) and elemental mapping (Fig. 1j) demonstrate the homogeneous distribution of Pd, Mo, and Pt throughout the PdMoPt trimetallene, with a Pd/Mo/Pt atomic ratio of 72/4/24.
To explore the electronic structure of PdMoPt trimetallene, X-ray photoelectron spectroscopy (XPS) measurements were conducted. The high-resolution Pd 3d XPS spectra for both PdMoPt trimetallene and PdMo bimetallene are presented in Fig. S7.† Analysis of the Pd 3d spectra for both materials revealed dual doublets. In PdMoPt trimetallene, the primary Pd component appeared at a binding energy of 336.4 eV (Pd 3d5/2), denoting a Pd0 chemical environment.42 This component exhibited a slight blueshift relative to PdMo bimetallene (336.2 eV), indicating the regulated electronic structure of Pd by the introduction of Pt. A minor secondary doublet emerged at 337.8 eV (Pd 3d5/2), attributed to Pd2+ species resulting from slight surface oxidation upon exposure to air. Mo, as indicated by XPS analysis, displayed a single doublet at a binding energy of 227.6 eV, corresponding to the metallic Mo0 state in both PdMoPt trimetallene and PdMo bimetallene. Additionally, XPS analysis of PdMoPt trimetallene revealed two distinct Pt chemical states. The minor component at a higher binding energy (73.5 eV) suggested an oxidized Pt environment, while the predominant component at a lower binding energy (72.0 eV) was ascribed to the Pt0 state, confirming the incorporation of Pt into the PdMo lattice. The atomic ratio of Pd, Mo, and Pt determined by XPS was Pd/Mo/Pt = 70/4/26, showing a slightly higher amount of Pt compared to the EDX measurement, which suggests a Pt-rich surface for the PdMoPt trimetallene. This minor discrepancy is due to the synthesis process, where Pt deposition on the PdMo surface is followed by diffusion into the lattice, leading to the surface enrichment of Pt.
The electrocatalytic performance of PdMoPt trimetallene was investigated following the deposition of PdMoPt onto a carbon support and the subsequent coating of the catalyst ink onto a glassy carbon electrode. To provide a comparative assessment, PdMo bimetallene, along with commercial Pd/C and Pt/C catalysts, was evaluated under identical conditions. The cyclic voltammogram (CV) of these catalysts, recorded in the N2-saturated 1 M KOH solution, is shown in Fig. 2a. In contrast to the PdMo catalyst, the PdMoPt catalyst exhibits clear hydrogen adsorption/desorption peaks within the 0.05 to 0.5 V potential range. This difference is attributed to the presence of highly exposed Pt species on the PdMoPt surface, potentially serving as more reactive sites during the HER. The apparent cathodic peaks in the backward scans at approximately 0.7 V are associated with the reduction of the surface Pd/Pt oxide layer. Notably, this reduction peak for the PdMoPt catalyst exhibits a positive shift of 11, 13 and 34 mV in comparison to PdMo, Pt/C, and Pd/C catalysts, respectively. This shift indicates a diminished Pd–O binding affinity on the PdMoPt surface, facilitating the more efficient removal of reaction intermediates during the AOR process.
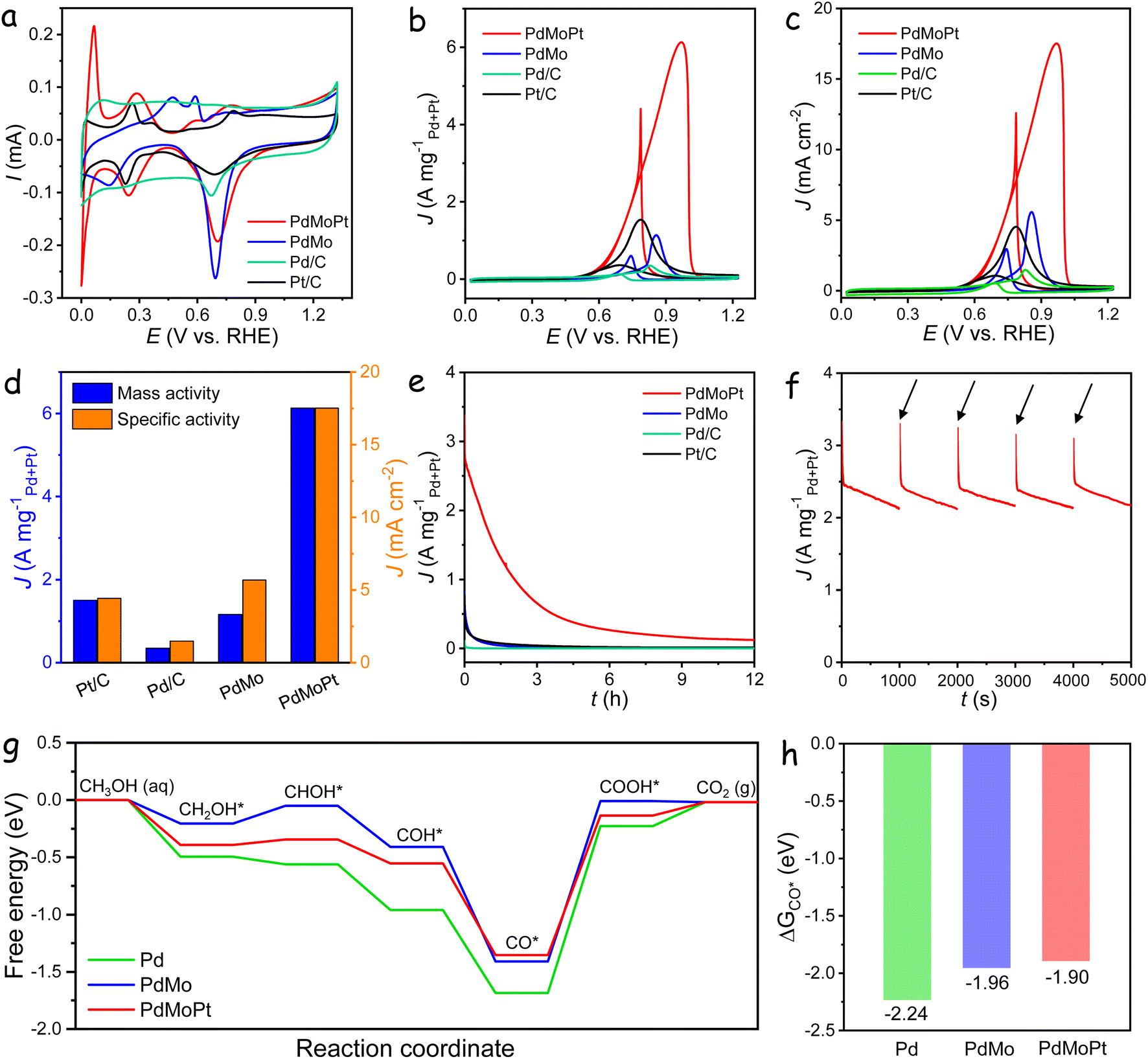 | ||
| Fig. 2 MOR performance. (a) CV curves of the catalysts in a 1 M KOH solution. (b) CV curves of the catalysts normalized to the loading amounts of Pd and Pt in a 1 M KOH with 1 M methanol solution. (c) CV curves of the catalysts normalized to ECSA in a 1 M KOH with 1 M methanol solution. (d) Comparison of specific and mass activities of the catalysts. (e) CA curves of the catalysts in a 1 M KOH with 1 M methanol solution. (f) CA curves of the catalysts with CV reactivation every 1000 s. (g) The calculated MOR free-energy profiles. (h) ΔGCO* on the surfaces of Pd, PdMo, and PdMoPt. | ||
The electrochemically active surface area (ECSA) normalized by the Pd + Pt content of the catalysts was calculated from the CO stripping curves using the integrated charge of the CO adsorption peak. As shown in Fig. S8,† the PdMoPt catalyst exhibited high ECSA values of 35 m2 gPd+Pt−1, surpassing those of PdMo (20 m2 gPd−1), Pd/C (24 m2 gPd−1) and Pt/C (33 m2 gPt−1) catalysts. The enhanced ECSA in PdMoPt trimetallene can be attributed to its ultrathin and porous 2D structure, along with the incorporation of Pt atoms, both of which increase accessibility to Pd/Pt active sites. Moreover, CO stripping experiments revealed a negative potential shift of approximately 30 mV for PdMo and further shift of 110 mV for PdMoPt, compared to the Pd/C catalyst. This shift indicates facile CO desorption and increased CO-poisoning resistance on PdMoPt trimetallene due to the combined effects of Mo and Pt on surface Pd atoms.
The electrocatalytic performance toward the methanol oxidation reaction (MOR) was investigated in a solution containing 1 M KOH with 1 M methanol at a sweep rate of 50 mV s−1. As shown in Fig. 2b, distinct oxidation peaks were identified in both the anodic and cathodic scans for all catalysts. The forward scan exhibited a peak attributed to methanol oxidation, while the backward scan peak corresponded to the removal of carbonaceous species generated during the forward scan. During the forward scan, the PdMoPt catalyst demonstrated a lower onset potential for methanol oxidation in comparison to other catalysts, indicating a reduced activation energy and heightened activity towards the MOR (Fig. S9†). The mass activities of the catalysts were normalized against the loading amount of Pd and Pt. Notably, PdMoPt exhibited a (Pd + Pt)-mass-normalized peak current density of 6.13 A mgPd+Pt−1, which is 5.3, 4.0 and 18 times higher than those of PdMo (1.16 A mgPd−1), Pt/C (1.50 A mgPt−1) and Pd/C (0.35 A mgPd−1), respectively. To evaluate intrinsic catalytic activities, specific activities were calculated by normalizing currents to the corresponding ECSA (Fig. 2c). As shown in Fig. 2d, the calculated specific activity for PdMoPt was 17.52 mA cm−2, higher than those of PdMo (5.68 mA cm−2), Pt/C (4.43 mA cm−2) and Pd/C (1.45 mA cm−2). To the best of our knowledge, these values exceed those of most reported state-of-the-art electrocatalysts (Table S1†).
The MOR activity of catalysts was further assessed using the turnover frequency values calculated based on the catalyst mass (TOFmass) and surface active sites (TOFsurface). As shown in Fig. S10,† PdMoPt achieves a TOFmass of 0.27 s−1 and a TOFsurface of 2.76 s−1 at 0.7 V, the highest among the tested catalysts, indicating that the introduction of Mo and Pt enhances the intrinsic activity of surface Pd sites. Additionally, PdMoPt exhibited the smallest Tafel slope of 135 mV dec−1 among the measured catalysts, indicating accelerated charge-transfer kinetics for the MOR in an alkaline medium (Fig. S11†). Electrochemical impedance spectroscopy (EIS) was conducted to assess electron transfer rates during the MOR (Fig. S12†). The Nyquist plots, simulated based on an equivalent circuit, highlighted the lowest charge-transfer resistance (Rct) of the PdMoPt catalyst, confirming its superior charge-transfer capacity during the MOR.
Catalyst stability was assessed through chronoamperometry (CA) tests at a constant potential of −0.1 V versus Hg/HgO (Fig. 2e). Despite significant activity decay for all catalysts, the PdMoPt catalyst exhibited remarkable durability, maintaining a high mass activity of 121 mA mgPd+Pt−1 even after 12 h of operation. In contrast, PdMo, commercial Pt/C, and Pd/C catalysts displayed negligible activities, underscoring the superior durability of the PdMoPt catalyst. Fig. 2f shows CA curves of the PdMoPt catalyst over five consecutive 1000 s periods, with five CV cycles conducted to reactivate the catalysts between each period, showing a substantial recovery of activity after each reactivation. This suggests that the observed stability decline during long-term measurements is primarily due to the temporary blockage of active sites by intermediates rather than structural deterioration, highlighting its superior durability.
To gain a comprehensive understanding of the influence of Mo and Pt on the MOR activity of PdMoPt trimetallene, models of Pd, PdMo, and PdMoPt were constructed by incorporating Mo and Pt into Pd (111). The optimized structures are shown in Fig. S13.† Density Functional Theory (DFT) calculations were conducted to determine the d-band center of Pd, PdMo, and PdMoPt. As presented in Fig. S14,† the results revealed a negative shift in the d-band center of Pd upon the integration of Mo and Pt atoms, transitioning from −1.59 eV for Pd to −1.87 eV for PdMo and further decreasing to −2.28 eV for PdMoPt. The reduced d-band center effectively mitigates the overbinding of reaction intermediates on Pd active sites, facilitating their desorption and thereby enhancing the AOR performance.
Fig. 2g displays the calculated free energy diagrams for the MOR on Pd, PdMo, and PdMoPt surfaces, with the corresponding geometric structures of key intermediates shown in Fig. S15–S17.† The methanol oxidation proceeds through a stepwise dehydrogenation process, resulting in the formation of a CO* intermediate, which is subsequently oxidized to CO2. These free energy diagrams emphasize this sequential dehydrogenation and oxidation sequence. The calculations reveal that the potential-determining step (PDS) for the MOR is the conversion of CO* to COOH* across all catalyst models. The PDS on PdMoPt requires an energy of 1.22 eV, which is lower than that on Pd (1.46 eV) and PdMo (1.40 eV). This reduced energy barrier for PdMoPt correlates with its superior MOR activity, as observed in electrocatalytic measurements. Furthermore, DFT calculations demonstrate a decrease in CO binding strength with the inclusion of Mo and Pt (Fig. 2h). The adsorption energies shift from −2.24 eV for Pd to −1.96 eV for PdMo and to −1.90 eV for PdMoPt. This reduced CO binding suggests improved resistance to CO poisoning due to the synergistic effects of Mo and Pt on Pd active sites, aligning with the results from CO stripping experiments.
In addition to the MOR, the electrocatalytic performance of the catalysts toward the ethylene glycol oxidation reaction (EGOR) and glycerol oxidation reaction (GOR) was also evaluated in the presence of 1 M KOH with 1 M ethylene glycol or glycerol. Remarkably, the PdMoPt catalyst consistently outperformed the other catalysts, exhibiting the highest mass activity and specific activity for both the EGOR and GOR among all tested catalysts. Specifically, PdMoPt exhibited a mass activity of 5.50 A mgPd+Pt−1 and a specific activity of 15.71 mA cm−2 for the EGOR (Fig. 3a and S18†), along with a mass activity of 4.37 A mgPd+Pt−1 and a specific activity of 12.49 mA cm−2 for the GOR (Fig. 3c and S19†). These values are significantly higher than those of PdMo, commercial Pt/C and Pd/C, and the majority of previously reported Pd- and Pt-based electrocatalysts (Tables S2 and S3†). Furthermore, PdMoPt displayed lower onset potentials, faster reaction kinetics, and better stability than other catalysts during both the EGOR and GOR (Fig. 3b, d and S20–S23†). It is important to note that these performances were recorded without iR compensation. When accounting for solution resistance in the electrolyte (Table S4†), the polarization curves corrected with 85% iR compensation reveal even better performance, maintaining the activity trend of PdMoPt > PdMo > Pt/C > Pd/C (Fig. S24†). When evaluating the performance of the PdMoPt catalyst with different alcohols, the highest activity was observed with the MOR, whereas the GOR exhibited relatively low activity and stability. The stability of the GOR, in particular, deteriorated rapidly during measurements, likely due to the larger molecular structure of glycerol. The oxidation of glycerol generates complex intermediates that strongly adsorb onto the catalyst surface, significantly reducing the number of active catalytic sites.
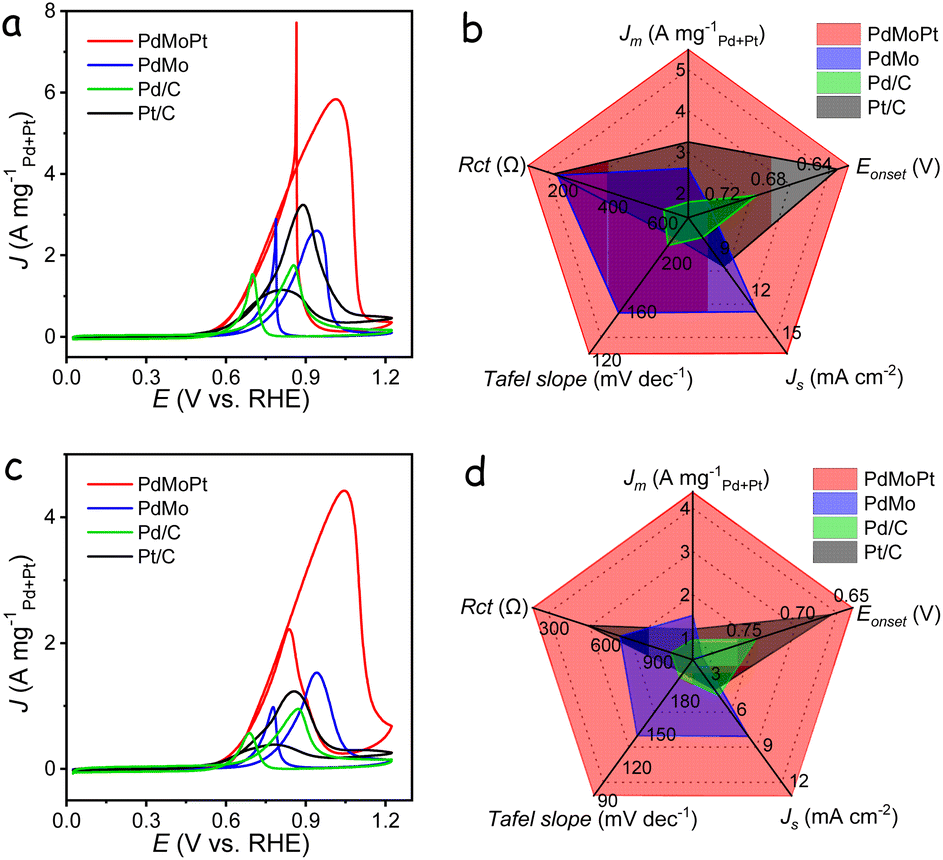 | ||
| Fig. 3 EGOR and GOR performance. (a) CV curves of the catalysts in a 1 M KOH with 1 M ethylene glycol solution. (b) Comprehensive comparison of the catalysts for EGOR measurements. (c) CV curves of the catalysts in a 1 M KOH with 1 M glycerol solution. (d) Comprehensive comparison of the catalysts for GOR measurements. | ||
To further investigate the role of Mo and Pt in the AOR, PdPt bimetallene and PdMoPt trimetallene with reduced Pt content were also produced (Fig. S25†), and their performance is shown in Fig. S26.† In the absence of either Mo or Pt, both PdPt and PdMo exhibited lower activity compared to PdMoPt across all AOR, indicating a synergistic interaction between Pd, Mo, and Pt. Furthermore, PdMoPt demonstrated enhanced performance with increasing amounts of Pt in the trimetallene, highlighting the crucial role of Pt in reducing the energy barrier for the oxidation of alcohols on the PdMoPt surface. Overall, these results underscore the significantly enhanced activity and durability of PdMoPt trimetallene in the electrooxidation of various alcohols. This superior performance can be attributed to the ultrathin and porous morphology of PdMoPt, combined with the synergistic effects resulting from the incorporation of Pt.
The electrocatalytic performance of the catalysts in the HER was evaluated using a three-electrode system in a 1 M KOH electrolyte. Linear sweep voltammetry (LSV) curves, normalized to the geometric area of the glassy carbon electrode, are presented in Fig. 4a. The PdMoPt catalyst demonstrated an exceptionally low overpotential of 39 mV to achieve a current density of 10 mA cm−2, outperforming commercial Pt/C (60 mV) by 21 mV and significantly surpassing PdMo (390 mV) and Pd/C (411 mV). Although lower overpotentials can be achieved with iR compensation (Fig. S27†), the uncorrected data are more relevant to practical applications and the potential for bubble interference during HER measurements.43 Assuming that all metal species are catalytically active, the TOFmass value of PdMoPt is 1.29 s−1 at −0.2 V, notably higher than that of PdMo and Pd/C, yet lower than that of the Pt/C catalyst. However, when accounting for only the surface active sites, the TOFsurface value of PdMoPt is substantially higher, reaching 13.25 s−1 at −0.2 V, which is comparable to that of the Pt/C catalyst (Fig. S28†). The corresponding Tafel slope for PdMoPt was measured to be 67 mV dec−1, which is notably smaller than those of PdMo (192 mV dec−1), commercial Pd/C (424 mV dec−1), and Pt/C (70 mV dec−1), confirming its rapid kinetic performance in the HER (Fig. 4b). Stability assessments through chronopotentiometry measurements revealed a minimal potential decrease over a 12 h test period at both 10 and 100 mA cm−2 (Fig. 4c). Additionally, accelerated durability tests showed negligible changes in overpotential on the LSV curves before and after 1000 and 3000 cycles (Fig. S29†). The exceptional activity and stability of PdMoPt are attributed to its ultrathin structure, along with the strain and synergistic effects between Pd and Pt atoms.
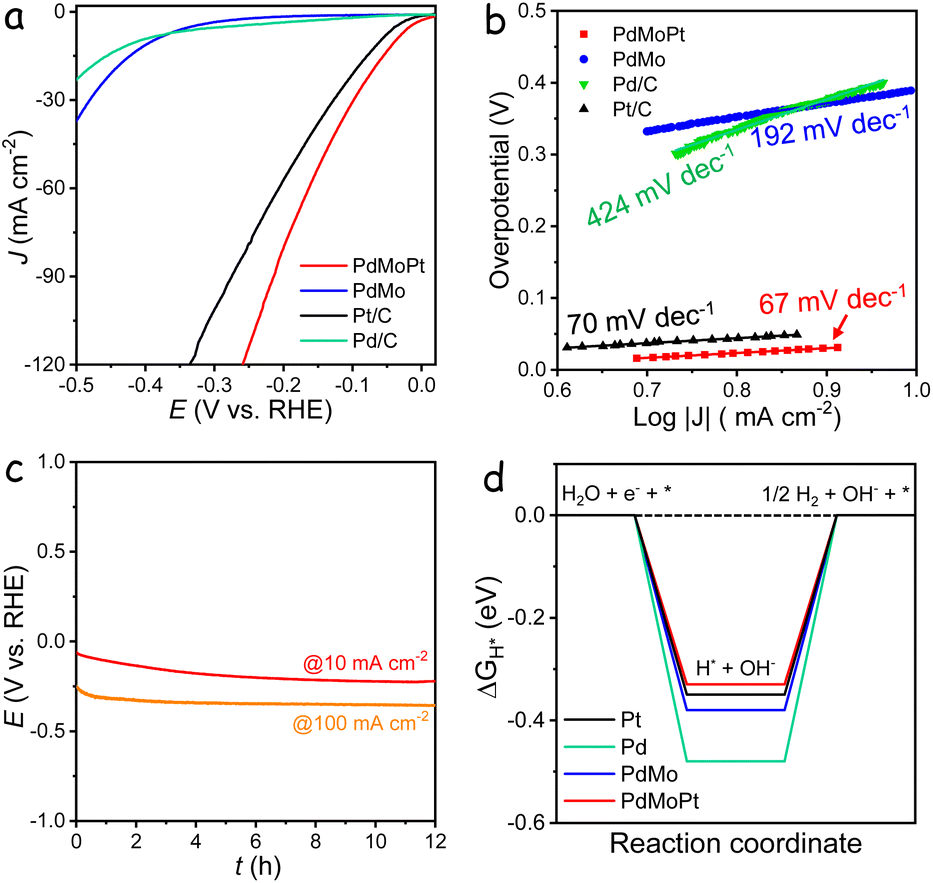 | ||
| Fig. 4 HER performance. (a) Polarization curves of the catalysts in a 1 M KOH solution. (b) Tafel plots of the catalysts for the HER. (c) Chronopotentiometric curves of the PdMoPt catalyst carried out in 1 M KOH with a constant current density of 10 and 100 mA cm−2. (d) Calculated free energy diagram for hydrogen adsorption on the Pd, Pt, PdMo, and PdMoPt surfaces. | ||
To gain deeper insights into the enhanced HER activity of PdMoPt, DFT calculations were conducted to determine the hydrogen adsorption energy (ΔGH*) on the surface of various catalysts (Fig. S30†). The incorporation of Mo and Pt into Pd resulted in smaller |ΔGH*| values, approaching zero, indicating a faster release of hydrogen and consequently an improved HER process (Fig. 4d). These results align with the experimentally measured HER performance of the catalysts, offering additional validation of the synergistic effect among the ternary metals on the energetic kinetics of the HER.
As corroborated by the aforementioned measurements, the PdMoPt trimetallene catalyst proves as a highly efficient bifunctional electrocatalyst for both the AOR and the HER in alkaline solution. Encouraged by these promising results, an alcohol–water hybrid electrolyzer was established utilizing PdMoPt as both the anodic and cathodic catalyst, as shown in Fig. 5a. To achieve a current density of 10 mA cm−2, the electrolyzer required remarkably low cell voltages of 0.67, 0.68, and 0.73 V when utilizing methanol, ethylene glycol, and glycerol as anodic substrates, respectively. These activity trends align with those observed in three-electrode AOR measurements, indicating the significant impact of anodic alcohols on the performance of the AOR-HER electrolyzer. These cell voltages of the hybrid electrolyzer are notably lower than the 1.67 V required to attain the same current density during water electrolysis in 1 M KOH, using commercial Pt/C as the cathode and commercial IrO2 as the anode (Fig. 5b). The reduction in cell voltages highlights the intrinsic energy efficiency of the hybrid electrocatalytic hydrogen production strategy.
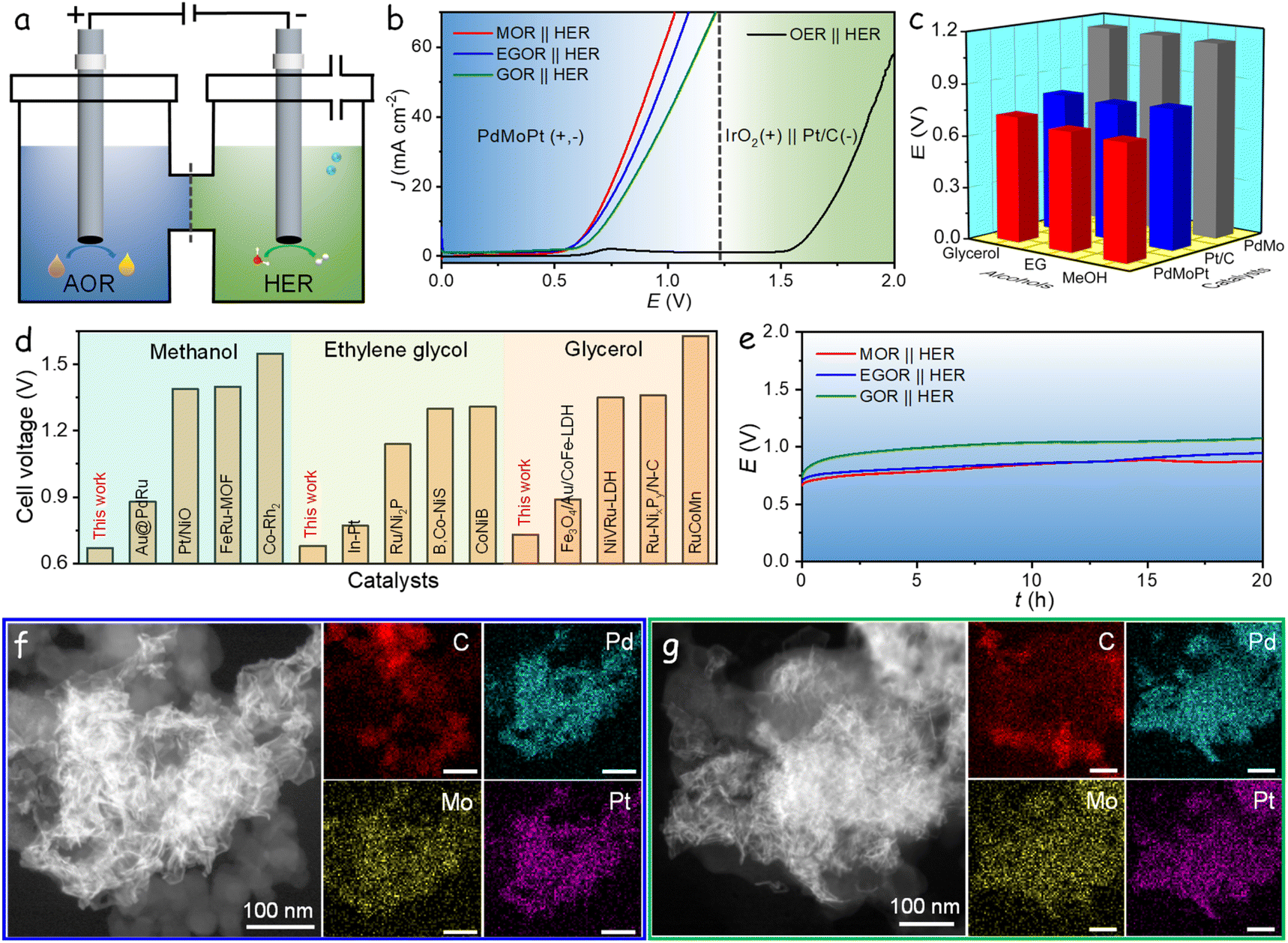 | ||
| Fig. 5 Alcohol–water electrolysis performance. (a) Schematic diagram of an alcohol–water hybrid electrolyzer. (b) LSV curves of the cells equipped with PdMoPt as both anode and cathode catalysts in the AOR-HER based electrolyzer and equipped with IrO2 as the anode and Pt/C as the cathode in alkaline solution. (c) Comparison of the cell voltage for Pt/C, PdMo and PdMoPt in the AOR-HER hybrid electrolyzers. (d) Comparison of recent electrolysis performances of catalysts for alcohol–water hybrid electrolyzers.44–55 (e) Chronopotentiometric curves of PdMoPt as both anode and cathode catalysts with a constant current density of 10 mA cm−2 in the AOR-HER based electrolyzers. (f) HAADF-STEM and elemental mappings of the PdMoPt catalyst at the anode after the stability measurement. (g) HAADF-STEM and elemental mappings of the PdMoPt catalyst at the cathode after the stability measurement. | ||
Furthermore, hybrid electrolyzers using PdMo, Pd/C, or Pt/C as catalysts in both electrodes were also evaluated (Fig. S31†). Due to the limited activity of Pd/C for both the AOR and HER, only minimal current density was obtained even at high cell voltages. Fig. 5c presents a comparative analysis of the cell voltages required to achieve 10 mA cm−2 for PdMoPt, PdMo, and Pt/C catalysts. Importantly, all these catalysts demonstrated lower cell voltages in the hybrid system compared to the conventional water-splitting system for hydrogen production, with PdMoPt exhibiting the highest activity. Compared to other catalysts reported in the literature for alcohol-assisted water electrolysis, the PdMoPt-based electrolyzer ranks among the top performances (Fig. 5d). Additionally, PdMoPt exhibits exceptional stability, maintaining an almost consistent potential throughout a continuous 20 h operation at 10 mA cm−2 in both the MOR-HER and EGOR-HER hybrid electrolysis cells. In contrast, a slight increase in potential observed during the initial stage of the GOR-HER system suggests a decline in activity, which we attribute to the low stability of the anodic GOR (Fig. 5e).
Subsequent to the stability assessment, PdMoPt was retrieved from both the anode and cathode and subjected to structural characterization through HAADF-STEM and EDX elemental maps, as shown in Fig. 5f, g, S32 and S33.† The analyses revealed a consistently curved and ultrathin structure, uniformly dispersed on the carbon support with no observable aggregation. The EDX elemental maps confirmed the homogeneous distribution of Pd, Mo, and Pt elements throughout the catalysts. Inductively coupled plasma analyses confirmed its stability, showing less than a 1% difference in element content before and after the reaction, indicating minimal dissolution of elements in the electrolyte during testing. The exceptional structural stability significantly contributes to the outstanding catalytic activity and durability exhibited by the PdMoPt catalyst. These results highlight PdMoPt trimetallene as a highly efficient and stable bifunctional catalyst for energy-efficient hydrogen production coupled with anodic alcohol oxidation.
Conclusions
In summary, we have successfully engineered a novel 2D PdMoPt trimetallene with a thickness of 1.5 nm by alloying Pt with a PdMo bimetallene. This unique structure provides highly exposed catalytically active sites and enhanced resistance to CO-poisoning during electrocatalysis. Consequently, the PdMoPt catalyst demonstrates superior activity and stability in the electrooxidation of various alcohols, including methanol, ethylene glycol, and glycerol, along with the HER in alkaline media. These performances surpass those of its PdMo counterpart, as well as commercial Pd/C and Pt/C catalysts, positioning PdMoPt among the best-reported catalysts. DFT calculations revealed a pronounced downward shift in the d-band center of Pd, promoting the release of adsorbed CO and H upon the introduction of Mo and Pt. This synergistic effect significantly contributes to the superior performance in both the AOR and the HER. By utilizing PdMoPt as both anodic and cathodic catalysts, a novel AOR-HER hybrid electrolysis system has been established for energy-efficient hydrogen production. Particularly notable is the remarkably low cell voltage of 0.67 V required to achieve a current density of 10 mA cm−2 in the MOR-HER system. Furthermore, PdMoPt demonstrates exceptional electrocatalytic and structural stability in both the anode and the cathode. This study introduces a promising strategy for designing trimetallenes with tailored electronic properties for diverse electrocatalytic applications.Data availability
The data supporting this study are available within the ESI.†Author contributions
J. L. conceived the project and guided and supervised the work. T. L. produced the materials, performed the electrochemical measurements, and wrote a first draft of the manuscript. Q. W., H. L., J. W., Y. S., H. L., P. T. and Y. W. joined the characterization data analysis and discussion. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22008091), the funding for scientific research startup of Jiangsu University (No. 19JDG044 and 4111510015), the Jiangsu Provincial Program for High-Level Innovative and Entrepreneurial Talents Introduction, the Jiangsu Distinguished Professors project (No. 1711510024), Chinese Academy of Sciences Pioneer Hundred Talents Program B (E2XBRD1001), and the Young Talents Cultivation Program from the Shanghai Branch of the Chinese Academy of Sciences (E2XYRD1).Notes and references
- M. Luo, J. Yang, X. Li, M. Eguchi, Y. Yamauchi and Z. Wang, Chem. Sci., 2023, 14, 3400–3414 RSC
.
- J. Li, L. Li, J. Wang, A. Cabot and Y. Zhu, ACS Energy Lett., 2024, 9, 853–879 CrossRef
- D. Chen, H. Bai, J. Zhu, C. Wu, H. Zhao, D. Wu, J. Jiao, P. Ji and S. Mu, Adv. Energy Mater., 2023, 13, 2300499 CrossRef CAS
- Z. Chen, J. Dong, J. Wu, Q. Shao, N. Luo, M. Xu, Y. Sun, Y. Tang, J. Peng and H. Cheng, Nat. Commun., 2023, 14, 4210 CrossRef CAS
- M. Gao, J. Fan, X. Li, Q. Wang, D. Li, J. Feng and X. Duan, Angew. Chem., Int. Ed., 2023, 62, e202216527 CrossRef CAS PubMed
- E. Romeo, F. Illas and F. Calle-Vallejo, Chem. Sci., 2023, 14, 3622–3629 RSC
- E. Gioria, S. Li, A. Mazheika, R. Naumann d'Alnoncourt, A. Thomas and F. Rosowski, Angew. Chem., Int. Ed., 2023, 62, e202217888 CrossRef CAS
- B. Deng, G. Yu, W. Zhao, Y. Long, C. Yang, P. Du, X. He, Z. Zhang, K. Huang, X. Li and H. Wu, Energy Environ. Sci., 2023, 16, 5210–5219 RSC
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS
- Z. Ge, Y. Ding, T. J. Wang, F. Shi, P. Jin, P. Chen, B. He, S. Yin and Y. Chen, J. Energy Chem., 2023, 77, 209–216 CrossRef
- D. Du, Q. Geng, L. Ma, S. Ren, J. X. Li, W. Dong, Q. Hua, L. Fan, R. Shao, X. Wang, C. Li and Y. Yamauchi, Chem. Sci., 2022, 13, 3819–3825 RSC
- Y. Liu, E. Zhu, J. Huang, A. Zhang, A. H. Shah, Q. Jia, M. Xu, E. Liu, Q. Sun, X. Duan and Y. Huang, Nano Lett., 2023, 23, 2758–2763 CrossRef
- X. Zhang, L. Hui, D. Yan, J. Li, X. Chen, H. Wu and Y. Li, Angew. Chem., Int. Ed., 2023, 62, e202308968 CrossRef PubMed
- Z. Lin, Z. Wang, J. Gong, T. Jin, S. Shen, Q. Zhang, J. Wang and W. Zhong, Adv. Funct. Mater., 2023, 33, 2307510 CrossRef
- Q. Xue, Z. Wang, Y. Ding, F. Li and Y. Chen, Chin. J. Catal., 2023, 45, 6–16 CrossRef CAS
- Q. Wang, J. Liu, T. Li, T. Zhang, J. Arbiol, S. Yan, Y. Wang, H. Li and A. Cabot, Chem. Eng. J., 2022, 446, 136878 CrossRef CAS
- T. Li, Q. Wang, W. Zhang, H. Li, Y. Wang and J. Liu, Chem. Sci., 2023, 14, 9488–9495 RSC
- K. Wei, H. Lin, X. Zhao, Z. Zhao, N. Marinkovic, M. Morales, Z. Huang, L. Perlmutter, H. Guan, C. Harris, M. Chi, G. Lu, K. Sasaki and S. Sun, J. Am. Chem. Soc., 2023, 145, 19076–19085 CrossRef CAS PubMed
- T. Li, Q. Wang, J. Wu, Y. Sui, P. Tang, H. Liu, W. Zhang, H. Li, Y. Wang, A. Cabot and J. Liu, Small, 2023, 20, 2306178 CrossRef
- S. Yang, Z. Si, G. Li, P. Zhan, C. Liu, L. Lu, B. Han, H. Xie and P. Qin, Small, 2023, 19, 2207651 CrossRef CAS PubMed
- C. Wang, W. Gao, X. Wan, B. Yao, W. Mu, J. Gao, Q. Fu and D. Wen, Chem. Sci., 2022, 13, 13956–13965 RSC
- H. Wang, J. Chen, Y. Lin, X. Wang, J. Li, Y. Li, L. Gao, L. Zhang, D. Chao, X. Xiao and J. M. Lee, Adv. Mater., 2021, 33, 2008422 CrossRef CAS
- L. Ji, H. Che, N. Qian, J. Li, S. Luo, X. Li, X. Wu, Q. Xu, X. Gong, X. Cui, H. Zhang and D. Yang, Appl. Catal., B, 2023, 328, 122521 CrossRef CAS
- H. Kandambige, J. Bai, J. Mei, D. Qi, T. Liao and Z. Sun, ACS Mater. Lett., 2023, 5, 2216–2225 CrossRef CAS
- F. Lin, F. Lv, Q. Zhang, H. Luo, K. Wang, J. Zhou, W. Zhang, W. Zhang, D. Wang, L. Gu and S. Guo, Adv. Mater., 2022, 34, 2202084 CrossRef CAS PubMed
- J. Liu, H. Liu, Q. Wang, T. Li, T. Yang, W. Zhang, H. Xu, H. Li, X. Qi, Y. Wang and A. Cabot, Chem. Eng. J., 2024, 486, 150258 CrossRef CAS
- M. Xie, B. Zhang, Z. Jin, P. Li and G. Yu, ACS Nano, 2022, 16, 13715–13727 Search PubMed
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 Search PubMed
- J. Zhang, F. Lv, Z. Li, G. Jiang, M. Tan, M. Yuan, Q. Zhang, Y. Cao, H. Zheng, L. Zhang, C. Tang, W. Fu, C. Liu, K. Liu, L. Gu, J. Jiang, G. Zhang and S. Guo, Adv. Mater., 2022, 34, 2105276 CrossRef CAS
- S. Yin, S. Liu, Z. Wang, Y. Xu, X. Li, H. Wang and L. Wang, Chem. Eng. J., 2022, 435, 134711 CrossRef CAS
- H. Wang, W. Wang, Q. Mao, H. Yu, K. Deng, Y. Xu, X. Li, Z. Wang and L. Wang, Chem. Eng. J., 2022, 450, 137995 CrossRef CAS
- S. Huang, S. Lu, S. Gong, Q. Zhang, F. Duan, H. Zhu, H. Gu, W. Dong and M. Du, ACS Nano, 2022, 16, 522–532 CrossRef CAS
- K. Zhang, Y. He, R. Guo, W. Wang, Q. Zhan, R. Li, T. He, C. Wu and M. Jin, ACS Energy Lett., 2022, 7, 3329–3336 CrossRef CAS
- X. Liu, Y. Jiang, J. Huang, W. Zhong, B. He, P. Jin and Y. Chen, Carbon Energy, 2023, 5, e367 CrossRef CAS
- F. Lv, B. Huang, J. Feng, W. Zhang, K. Wang, N. Li, J. Zhou, P. Zhou, W. Yang, Y. Du, D. Su and S. Guo, Natl. Sci. Rev., 2021, 8, nwab019 CrossRef CAS PubMed
- L. Chen, Z. Song, S. Zhang, C. K. Chang, Y. C. Chuang, X. Peng, C. Dun, J. J. Urban, J. Guo, J. L. Chen, D. Prendergast, M. Salmeron, G. A. Somorjai and J. Su, Science, 2023, 381, 857–861 CrossRef CAS
- J. Liu, Q. Wang, T. Li, Y. Wang, H. Li and A. Cabot, Nano Res., 2023, 16, 2041–2048 CrossRef
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef
- Q. Wang, T. Li, S. Yan, W. Zhang, G. Lv, H. Xu, H. Li, Y. Wang and J. Liu, Inorg. Chem., 2022, 61, 16211–16219 CrossRef PubMed
-
J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, 1992 Search PubMed
- M. Bae, Y. Kang, D. W. Lee, D. Jeon and J. Ryu, Adv. Energy Mater., 2022, 12, 2201452 CrossRef
- H. Wang, L. Cui, S. Yin, H. Yu, K. Deng, Y. Xu, X. Wang, Z. Wang and L. Wang, J. Mater. Chem. A, 2022, 10, 18889–18894 RSC
- G. Ma, X. Zhang, G. Zhou and X. Wang, Chem. Eng. J., 2021, 411, 128292 CrossRef CAS
- Q. Ling, Z. Zhao, Z. Li, K. Yan, C. Ding, P. Chen, Z. Sun, G. He, J. Lv and M. Zhang, J. Mater. Chem. A, 2023, 11, 2876–2888 RSC
- Y. Guo, X. Yang, X. Liu, X. Tong and N. Yang, Adv. Funct. Mater., 2023, 33, 2209134 CrossRef CAS
- Y. Zhu, X. Zhu, L. Bu, Q. Shao, Y. Li, Z. Hu, C.-T. Chen, C.-W. Pao, S. Yang and X. Huang, Adv. Funct. Mater., 2020, 30, 2004310 Search PubMed
- G. Ma, N. Yang, Y. Xue, G. Zhou and X. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 42763–42772 Search PubMed
- Z. Chen, W. Wei, Y. Shen and B. J. Ni, Green Chem., 2023, 25, 5979–5988 RSC
- M. Braun, M. Chatwani, P. Kumar, Y. Hao, I. Sanjuán, A. A. Apostoleri, A. C. Brix, D. M. Morales, U. Hagemann, M. Heidelmann, J. Masa, W. Schuhmann and C. Andronescu, JPhys Energy, 2023, 5, 024005 Search PubMed
- Y. Xu, T. Liu, K. Shi, H. Yu, K. Deng, X. Wang, Z. Wang, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 20365–20374 Search PubMed
- S. Liu, E. Zhang, X. Wan, R. Pan, Y. Li, X. Zhang, M. Su, J. Liu and J. Zhang, Sci. China Mater., 2022, 65, 131–138 Search PubMed
- Q. Qian, X. He, Z. Li, Y. Chen, Y. Feng, M. Cheng, H. Zhang, W. Wang, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, 35, 2300935 Search PubMed
- F. Sun, Y. Zhou, Z. You, H. Xia, Y. Tuo, S. Wang, C. Jia and J. Zhang, Small, 2021, 17, 2103307 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04764h |
| This journal is © The Royal Society of Chemistry 2024 |