
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly enantioselective synthesis of both enantiomers of tetrahydroquinoxaline derivatives via Ir-catalyzed asymmetric hydrogenation†
Ana
Xu
a,
Lanxing
Ren
ae,
Junrong
Huang
 a,
Yuxiang
Zhu
d,
Gang
Wang
a,
Chaoyi
Li
a,
Yongqiang
Sun
a,
Lijuan
Song
*a,
Hengzhi
You
*ab and
Fen-Er
Chen
*abc
a,
Yuxiang
Zhu
d,
Gang
Wang
a,
Chaoyi
Li
a,
Yongqiang
Sun
a,
Lijuan
Song
*a,
Hengzhi
You
*ab and
Fen-Er
Chen
*abc
aSchool of Science, Harbin Institute of Technology (Shenzhen), Taoyuan Street, Nanshan District, Shenzhen, 518055, China. E-mail: songlijuan@hit.edu.cn; youhengzhi@hit.edu.cn; rfchen@fudan.edu.cn
bGreen Pharmaceutical Engineering Research Center, Harbin Institute of Technology (Shenzhen), Taoyuan Street, Nanshan District, Shenzhen, 518055, China
cDepartment of Chemistry, Engineering Center of Catalysis and Synthesis for Chiral Molecules, Fudan University, Shanghai, 200433, China
dSchool of Pharmaceutical Sciences (Shenzhen), Shenzhen Campus of Sun Yat-sen University, Shenzhen, 518107, China
eSchool of Chemistry and Chemical Engineering, Hunan Province Key Laboratory for the Design and Application of Actinide Complexes, University of South China, Hengyang City, Hunan Province 421001, P.R. China
First published on 23rd August 2024
Abstract
A novel Ir-catalyzed asymmetric hydrogenation protocol for the synthesis of chiral tetrahydroquinoxaline (THQ) derivatives has been developed. By simply adjusting the reaction solvent, both enantiomers of mono-substituted chiral THQs could be selectively obtained in high yields with excellent enantioselectivities (toluene/dioxane: up to 93% yield and 98% ee (R); EtOH: up to 83% yield and 93% ee (S)). For 2,3-disubstituted chiral THQs, the cis-hydrogenation products were obtained with up to 95% yield, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, and 94% ee. Remarkably, this methodology was also applicable under continuous flow conditions, yielding gram-scale products with comparable yields and enantioselectivities (dioxane: 91% yield and 93% ee (R); EtOH: 90% yield and 87% ee (S)). Unlike previously reported Ir-catalyzed asymmetric hydrogenation protocols, this system exhibited a significant improvement as it required no additional additives. Furthermore, comprehensive mechanistic studies including deuterium-labeling experiments, control experiments, kinetic studies, and density functional theory (DFT) calculations were conducted to reveal the underlying mechanism of enantioselectivities for both enantiomers.
:1 dr, and 94% ee. Remarkably, this methodology was also applicable under continuous flow conditions, yielding gram-scale products with comparable yields and enantioselectivities (dioxane: 91% yield and 93% ee (R); EtOH: 90% yield and 87% ee (S)). Unlike previously reported Ir-catalyzed asymmetric hydrogenation protocols, this system exhibited a significant improvement as it required no additional additives. Furthermore, comprehensive mechanistic studies including deuterium-labeling experiments, control experiments, kinetic studies, and density functional theory (DFT) calculations were conducted to reveal the underlying mechanism of enantioselectivities for both enantiomers.
Introduction
Chiral N-heteroaromatic compounds have received increasing attention in recent decades, mainly because chiral products often form the core structures of numerous bioactive molecules.1–4 Typically, a chiral THQ fragment is the key structural unit of cholesterol ester transfer protein (CETP) inhibitors and BD-2 selective BET inhibitors, which are pivotal in treating atherosclerosis and obesity5 or exhibiting anti-inflammatory activity in vivo.6,7 Asymmetric catalysis, primarily employing chiral transition-metal catalysts, enzymatic catalysts and organocatalysts, is an efficient and powerful strategy to obtain enantiopure molecules.8–12 Among them, asymmetric hydrogenation of quinoxalines represents one of the most straightforward and efficient approaches for the synthesis of chiral THQs, and offers excellent atom economy, with almost no waste or byproducts, and thus is a highly sustainable and green strategy.13–15 In 2009, both Chan and Vries reported a highly enantioselective iridium-catalyzed asymmetric hydrogenation of alkyl-substituted quinoxalines, employing the diphosphinite ligand (R)-H8-binapo and the monodentate phosphoramidite ligand (S)-PipPhos, respectively (Scheme 1A).16,17 Recently, Chen reported an Rh-catalyzed asymmetric hydrogenation of alkyl-substituted quinoxalines with a chiral ferrocene-based bisphosphine–thiourea ligand, leading to the production of the desired products with high enantioselectivities (Scheme 1A).18 Additionally, Fan and Rueping developed a relay catalyzed asymmetric hydrogenation and a transfer hydrogenation approach, respectively, to achieve the reduction of aryl-substituted quinoxalines with good enantioselectivity (Scheme 1B).19,20 Furthermore, the more challenging asymmetric hydrogenation system of 2,3-disubstituted quinoxalines has been successfully developed using a chiral cationic ruthenium diamine catalyst or frustrated Lewis pair catalyst to afford the desired products with moderate diastereoselectivities and high enantioselectivities (Scheme 1C).21,22 Liu disclosed a novel Mn-catalyzed stereodivergent asymmetric hydrogenation of di-substituted quinoxalines, providing cis- or trans-chiral THQs with high yields and good stereoselectivities (Scheme 1C).23 Despite these advancements, the synthesis of both enantiomers of chiral THQ with the same catalyst has remained challenging until now. Herein, we report the first general and efficient Ir-catalyzed protocol for the synthesis of chiral aryl-substituted THQs. By simply adjusting the reaction solvent, both enantiomers of chiral THQ derivatives could be selectively obtained in high yields with excellent enantioselectivities (toluene/dioxane: up to 93% yield and 98% ee (R); EtOH: up to 83% yield and 93% ee (S)). Furthermore, the more challenging 2,3-disubstituted quinoxalines were smoothly hydrogenated with up to 95% yield, 20:1 dr, and 94% ee. In addition, both enantiomers of chiral THQ could be obtained under continuous flow conditions, yielding gram-scale products without a noticeable decrease in yields and enantioselectivities (dioxane: 91% yield and 93% ee (R); EtOH: 90% yield and 87% ee (S)) (Scheme 1D).
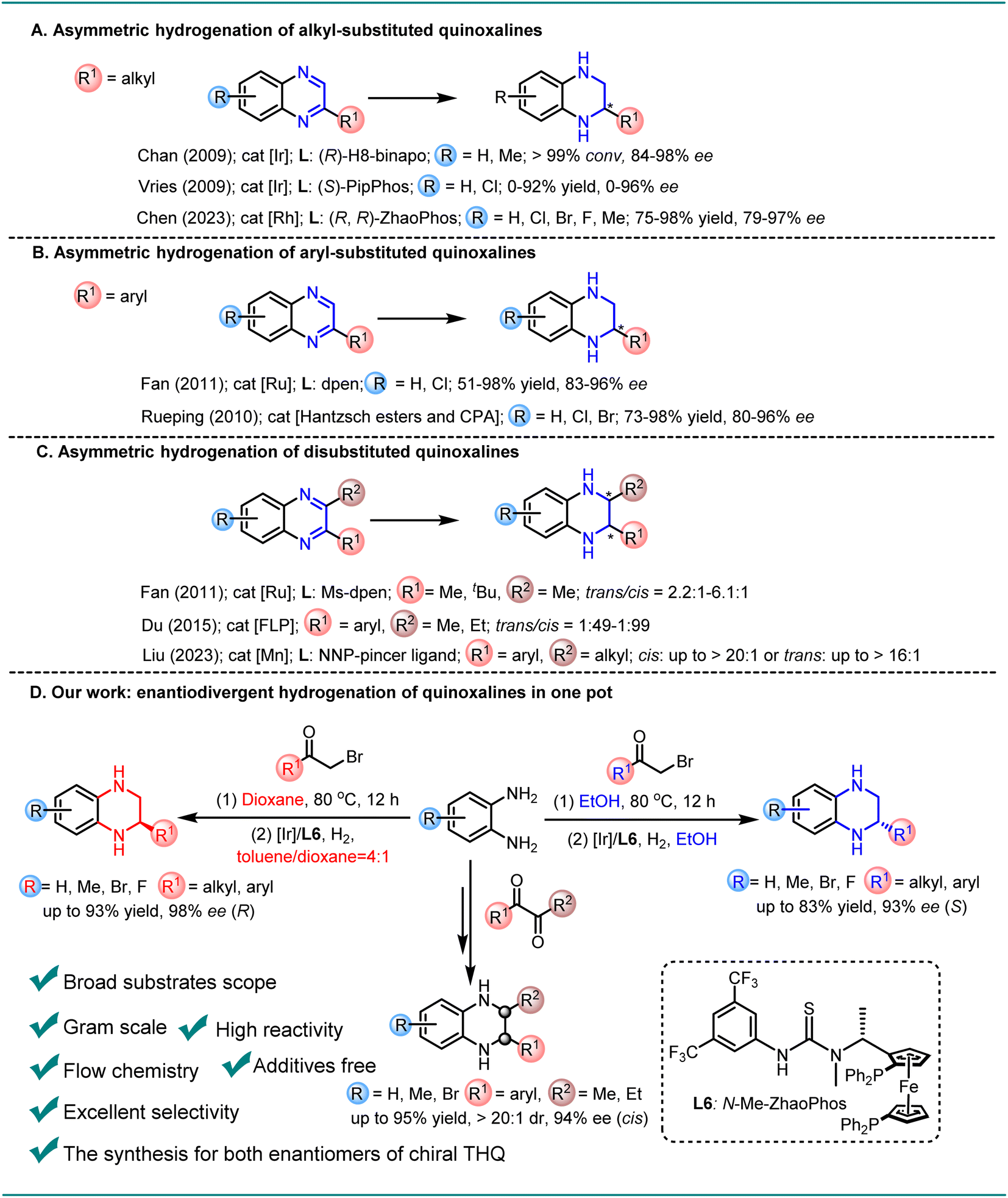 | ||
| Scheme 1 Asymmetric synthesis of chiral THQs. | ||
Results and discussion
Based on our earlier work on Rh-catalyzed asymmetric hydrogenation of alkyl-substituted quinoxalines and quinoxalinones,18 2-phenylquinoxaline 1aa′ was chosen as the model substrate for further optimization with an Ir-catalyzed asymmetric hydrogenation reaction (Scheme 2). To our delight, the desired product 2a was obtained with a full conversion and moderate enantioselectivity using ligand L1 (99% conversion and 65% ee), while only 19% ee was obtained in our earlier work. Encouraged by this promising outcome, we further explored a series of ferrocene-based chiral phosphine ligands. It was observed that ligands L2–L5, each containing a thiourea motif, consistently yielded the desired product with high conversions but moderate enantioselectivities (91–99% conversion and 43–65% ee). Pleasingly, ligand L6, featuring an N-methyl substitute, delivered the hydrogenation product 2a with 99% conversion and an improved ee value of 85%. This observed enhancement could be attributed to the favorable interaction between the catalyst and substrates facilitated by the N-methyl substitute. In addition, several other ferrocene-based chiral phosphine ligands (L7–L9), lacking either the thiourea or the aromatic unit, failed to provide satisfactory enantioselectivity (0–20% ee). This suggests that the presence of both thiourea and aromatic units is indispensable for high conversion and excellent enantioselectivity. Consequently, ligand L6 emerged as a promising ligand for the asymmetric hydrogenation of quinoxaline derivatives.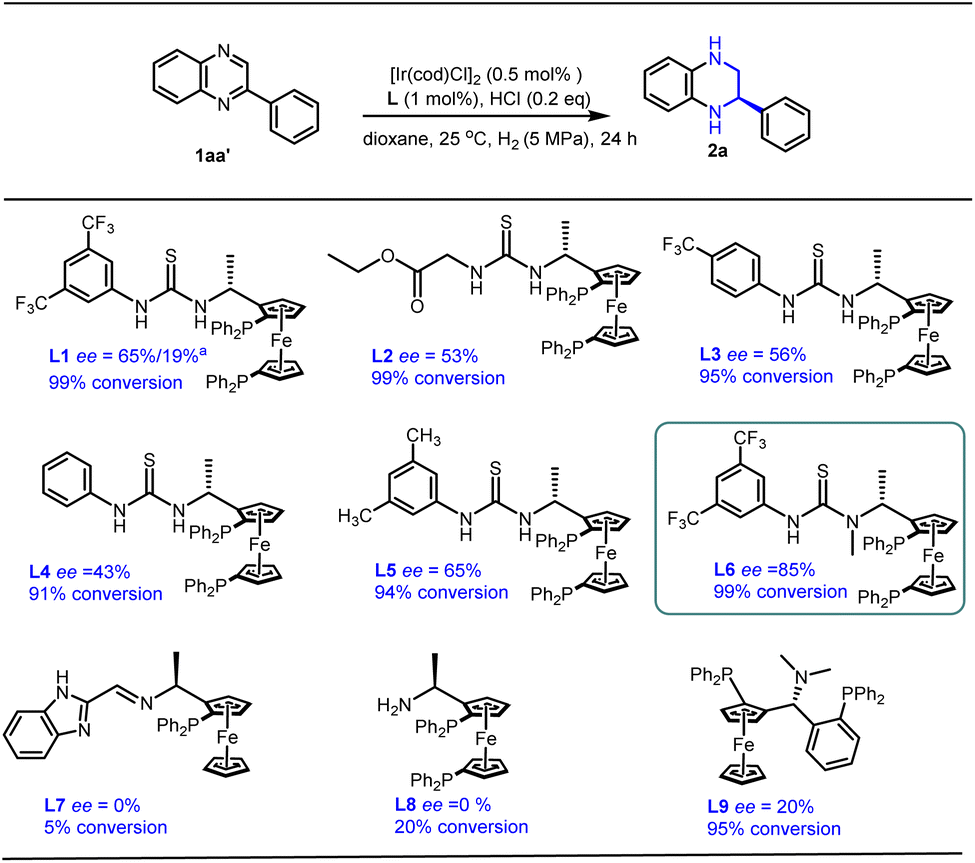 | ||
| Scheme 2 Ligand screening for the asymmetric hydrogenation of 2-phenylquinoxaline. Reaction conditions: 1aa′ (0.25 mmol) in 2 mL dioxane and 1aa′/[Ir(cod)Cl]2/ligand ratio = 100/0.5/1; a1aa′ hydrochloride/[Rh(cod)Cl]2/ligand ratio = 100/0.5/1 and DCM solvent. Conversions were determined by 1H NMR analysis. The ee values were determined by HPLC. | ||
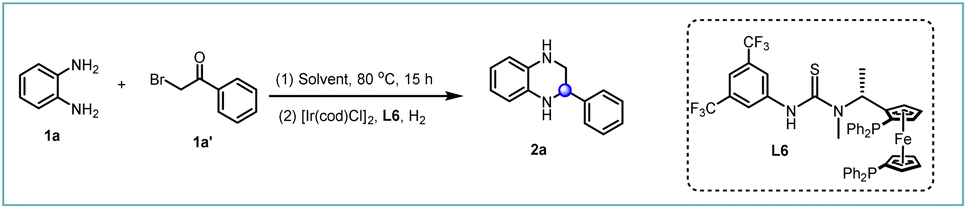
In an effort to enhance step economy, we aimed to synthesize chiral THQs from simple starting materials.24 Commercially available o-phenylene diamine 1a and 2-bromoacetophenones 1a′ were chosen as the model substrates.25 These compounds successfully underwent cyclization to generate 2-phenylquinoxaline without the use of additives,26–28 which avoids the risk of catalyst poisoning in the subsequent hydrogenation step. Encouragingly, the cyclization reaction proceeded smoothly and, following a simple post-processing step, the hydrogenation reaction was carried out to yield the desired chiral product 2a with high conversion and good enantioselectivity (Table 1, entry 1). Generally, the addition of hydrochloric acid (HCl) accelerates the reaction rate and controls the enantioselectivity in the asymmetric hydrogenation of imines by forming a salt bridge between the catalyst and the substrates.29–31 However, as the amount of HCl was reduced, an increase in ee values was observed in this system (Table 1, entries 1–3), with the best result obtained in the absence of HCl (99% conversion and 97% ee). Further investigation with other acids and KCl showed that the decreased enantioselectivity was attributed to the acidity rather than the chloride counterion (Table 1, entries 4–6). After screening various solvents, no further improvement was observed (Table 1, entries 7–13). However, comparable yields and enantioselectivities were achieved in single dioxane and toluene solvents (Table 1, entries 7 and 8). Surprisingly, the reversed configuration hydrogenation product (S)-2a was observed in MeOH with 99% conversion and 85% ee (Table 1, entries 14). This might be attributed to the participation of alcoholic solvent molecules in the enantiocontrol step. Coincidentally, Zhang's group also observed this interesting phenomenon in asymmetric hydrogenation of quinolines using the strong Brønsted acid HCl or CH3CO2H as the additive.32 Further screening of other alcoholic solvents and additives, including trifluoroethanol, was performed (Table 1, entries 15–19), resulting in a satisfactory conversion and enantioselectivity in ethanol solvent (99% conversion and 89% ee). In addition, this reversed product can also be observed in toluene systems by adding an appropriate amount of ethanol solvent (Table S4†). The transfer hydrogenation pathway was excluded in the EtOH system because the reaction did not occur in the absence of H2 (Table 1, entry 20). Notably, high reactivity and good enantioselectivity were maintained even at a molar substrate-to-catalyst (S/C) ratio of up to 1500 (toluene/dioxane) and 1000 (ethanol) (Table 1, entries 21–22). Consequently, toluene/dioxane and ethanol were selected as the optimal solvents for the synthesis of (R)- and (S)-THQs, respectively.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Additive | S/C | Conv. (%) | ee (%) |
| a Reaction conditions: 1a and 1a′ (0.25 mmol) in 1 mL solvent, 1a/[Ir(cod)Cl]2/ligand ratio = 100/0.5/1, room temperature, 18 h and H2 (2 MPa), and Tol/dio = toluene/dioxane. b The reaction temperature was 45 °C. c The reaction temperature was 60 °C. d Additive 0.2 eq. (substrate at 1 equivalent). e Without the use of H2. Conversions were determined by 1H NMR analysis. The ee values were determined by HPLC. | |||||
| 1 | Tol/dio = 4:1 |
0.2 eq. HCl | 100 | 99 | 85 (R) |
| 2 | Tol/dio = 4:1 |
0.1 eq. HCl | 100 | 99 | 94 (R) |
| 3 | Tol/dio = 4:1 |
— | 100 | 99 | 97 (R) |
| 4d | Tol/dio = 4:1 |
KCl | 100 | 99 | 97 (R) |
| 5d | Tol/dio = 4:1 |
CF3CO2H | 100 | 99 | 82 (R) |
| 6d | Tol/dio = 4:1 |
CF3SO3H | 100 | 99 | 61 (R) |
| 7 | Dioxane | — | 100 | 99 | 93 (R) |
| 8 | Toluene | — | 100 | 99 | 96 (R) |
| 9 | CH3CN | — | 100 | 73 | 67 (R) |
| 10 | THF | — | 100 | 88 | 59 (R) |
| 11 | Et2O | — | 100 | 34 | 61 (R) |
| 12 | CHCl3 | — | 100 | 27 | 63 (R) |
| 13 | DMSO | — | 100 | Trace | — |
| 14 | MeOH | — | 100 | 99 | 85 (S) |
| 15 | (CH3)2CHCH2OH | — | 100 | 99 | 79 (S) |
| 16 | iPrOH | — | 100 | 67 | 50 (S) |
| 17 | EtOH | — | 100 | 99 | 89 (S) |
| 18 | CF3CO2H | — | 100 | 86 | 55 (S) |
| 19d | EtOH | CH3CO2H | 100 | 96 | 85 (S) |
| 20e | EtOH | — | 100 | — | — |
| 21 | Tol/dio = 4:1 |
— | 1500 | 15b/97c | 90b/90c (R) |
| 22 | EtOH | — | 1000 | 99 | 81 (S) |
Having established the two optimized reaction conditions, we proceeded to evaluate the scope of the hydrogenation reaction. As shown in Table 2, a broad range of both enantiomers of chiral THQs were synthesized with high yields and good to excellent enantioselectivities in both solvent systems. The electron-donating groups introduced onto the C2-phenyl ring were well tolerated in toluene/dioxane and ethanol systems, giving the products 2a–2f with high yields and excellent enantioselectivities. Interestingly, there was a slight decrease in ee values in the EtOH system when a meta- or ortho-methyl substituent was introduced onto the phenyl ring (EtOH system: 2c, 79% and 81% ee; 2d, 75% and 87% ee). This might indicate that the methyl substitution possibly affects the transition state of the enantio-determining step, leading to a decrease in ee. To our delight, challenging substituents, including –N(Me)2 and –OH, could be well tolerated, despite the fact that heteroatoms could easily interrupt the coordination between the substrate and the catalyst (toluene/dioxane system: 2g, 85% yield and 94% ee, and 2h, 79% yield and 91% ee; EtOH system: 2g, 72% yield and 92% ee, and 2h, 76% yield and 91% ee). Subsequently, the effect of electron-withdrawing groups was investigated, and comparable results were obtained (2i–2o). Pleasingly, the enantioselectivity was only affected by the strong electron-withdrawing groups attached, resulting in a slight decrease in ee (2o). It is noteworthy that the uncommon para-ester substituent and pharmaceutically useful CF3 substituent33 were compatible with both solvent systems, affording products 2m and 2n with good yield and enantioselectivities. The screening of other aromatic substituents at the C2-position, including biphenyl, naphthyl and thiophene groups (2p–2r), led to the desired products in good yields with good to excellent enantioselectivities (toluene/dioxane system: 80–93% yield and 88–97% ee and EtOH system: 76–82% yield and 77–92% ee). Interestingly, the use of secondary alkyl substituents gave the products with comparable results in the toluene/dioxane system (2s, 88% yield and 94% ee; 2t, 86% yield and 97% ee), while the enantioselectivities in the EtOH system dramatically decreased (EtOH system: 2s, 82% yield and 24% ee; 2t, 75% yield and 30% ee). These results indicate that the aromaticity at the C2-position can potentially play an important role in the enantioselectivity in the EtOH system through π–π interactions between the quinoxaline and the catalyst/ligand. Using more sterically hindered tert-butyl substituents generated the products 2u in good yield, but with a much lower ee in both solvent systems. Finally, the effects of substituents at the C6- and C7-positions of the quinoxaline were also evaluated (2v–2y), leading to the synthesis of products with good to excellent yields and enantioselectivities, with the exception of (S)-2x in the EtOH system (EtOH system: 78% yield and 56% ee).
| a Reaction conditions: 1a and 1a′ (0.25 mmol) in 1 mL solvent, 80 °C, and 15 h. 1a/[Ir(cod)Cl]2/ligand ratio = 100/0.5/1, room temperature, 18 h and 2 MPa H2. b The 2-phenylquinoxaline intermediate was prepared through a separate procedure. Yields were all isolated yields. The ee values were determined by HPLC. |
|---|
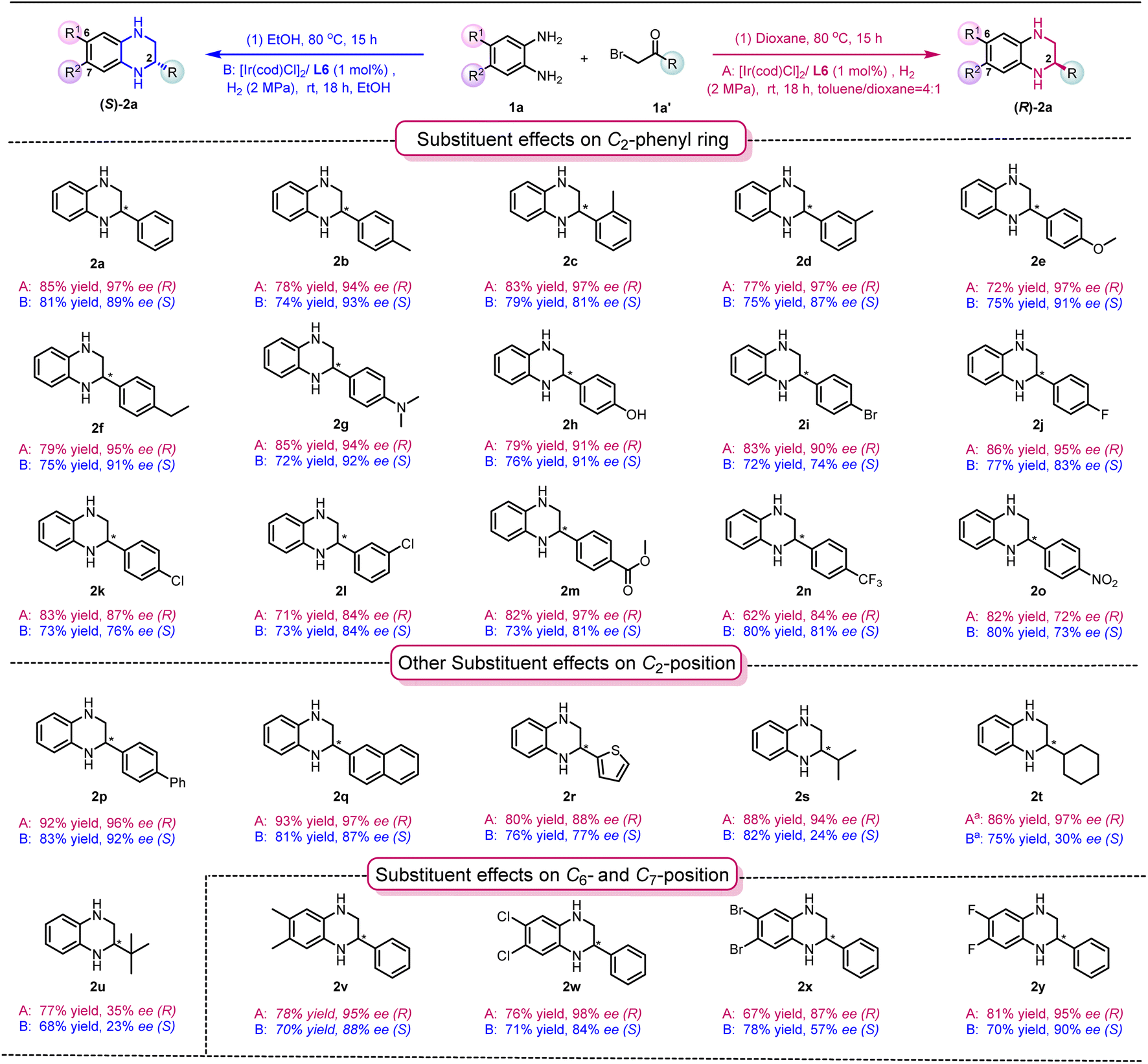
|
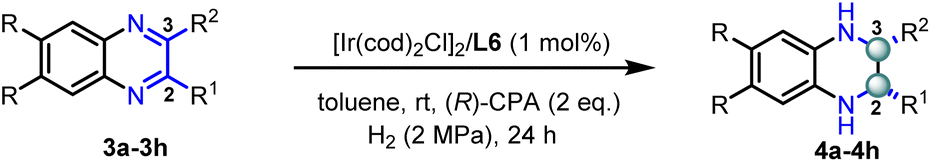
Encouraged by the excellent results obtained for the mono-substituted quinoxaline derivatives, we further expanded this methodology to the more challenging 2-aryl-3-methyl quinoxalines (see Table S3† for optimization details). As shown in Table 3, a series of 2,3-disubstituted quinoxalines were smoothly hydrogenated to generate enantiopure cis-THQs with 77–94% yields, >20:1 dr, and 80–94% ee. Notably, functional groups such as halides (3b and 3c) and methyl (3d) on the quinoxaline aromatic ring were well-tolerated, affording the corresponding hydrogenated products with excellent enantioselectivities (90–94% ee). The substrates bearing an electron-withdrawing group (3e) or a strong electron-donating group (3f) on the 2-phenyl group proceeded efficiently to produce the desired products with good yields and excellent enantioselectivities. In addition, the substrate with a strong electron-withdrawing group (3g) was also tested and the corresponding chiral product was obtained with good yield and moderate enantioselectivity. Finally, replacing methyl with an ethyl substituent at the C3-position (3h) led to a slight decrease in reactivity and enantioselectivity, resulting in the product in 77% yield and 80% ee.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | R1 | R2 | 4 | Yield (%) | cis/trans | ee (%) |
| a Reaction conditions: 0.25 mmol substrate 3, 1 mol% [Ir (COD)Cl]2/L6, and (R)-4-hydroxydinaphtho[2,1-d:1′,2′-f][1,3,2]dioxaphosphepine 4-oxide ((R)-CPA) (2 eq.). Yields were all isolated yields. The ee values were determined by HPLC. | |||||||
| 1 | H | Ph | Me | 4a | 94 | >20:1 |
91 |
| 2 | Br | Ph | Me | 4b | 90 | >20:1 |
94 |
| 3 | F | Ph | Me | 4c | 86 | >20:1 |
90 |
| 4 | Me | Ph | Me | 4d | 82 | >20:1 |
92 |
| 5 | H | 4-Cl-Ph | Me | 4e | 90 | >20:1 |
90 |
| 6 | H | 4-OMe-Ph | Me | 4f | 87 | >20:1 |
92 |
| 7 | H | 4-CF3-Ph | Me | 4g | 91 | >20:1 |
70 |
| 8 | H | Ph | Et | 4h | 77 | >20:1 |
80 |
Flow chemistry, characterized by a short residence time, high surface area-to-volume ratio, excellent reproducibility, and ease of scale-up, has emerged as an advantageous approach in organic synthesis.34–39 Asymmetric hydrogenation is notably well-suited for flow conditions owing to the ease of pressurization and enhanced safety.40,41 In line with our interest in flow chemistry,42–44 we successfully applied this methodology under continuous flow conditions. As shown in Scheme 3, we first prepared a solution of substrate 1aa′ and the Ir catalyst. This solution was then pumped into a mixer. Concurrently, hydrogen gas was released through a mass flow controller in another stream, converging with the reaction solution in the mixer. Subsequently, the relative parameters were optimized, including the concentration of substrate, gas velocity, liquid velocity, the pressure of H2, and the residence time (see Table S2† for optimization details). Under the optimized reaction conditions, the desired products (R)-2a and (S)-2a were obtained with high yields and excellent enantioselectivities (dioxane system: 91% yield and 93% ee (R); EtOH system: 90% yield and 87% ee (S)). Surprisingly, we found that the addition of HCl is essential for this transformation in dioxane, which could be due to insufficient mixing of the gas–liquid phase under continuous flow conditions. The use of acid enables the breaking of the aromaticity, thereby facilitating the hydrogenation process. Finally, a gram-scale reaction was conducted in both solvent systems to generate both enantiomers of chiral 2a with comparable yields and enantioselectivities ((S)-2a, 2.75 g, 90% yield, and 87% ee, over 27 h in the EtOH system; (R)-2a, 2.78 g, 91% yield, and 93% ee, over 60 h in the dioxane system).
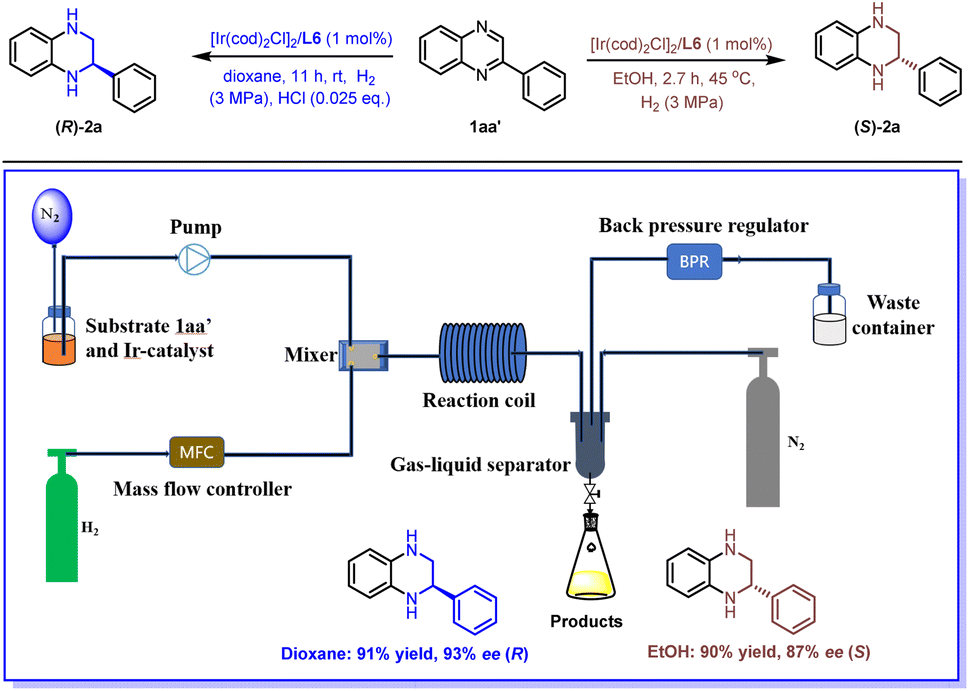 | ||
| Scheme 3 Ir-catalyzed asymmetric hydrogenation of 2-phenylquinoxaline (1aa′) under continuous flow conditions. Reaction conditions: (R)-2a: 2-phenylquinoxaline (1aa′, 0.05 M) in dioxane, 1.0 mol% [Ir (COD)Cl]2/L6, H2 (3 MPa), rt, HCl (0.025 eq.), and residence time 11 h. (S)-2a: 2-phenylquinoxaline (1aa′, 0.05 M) in EtOH, 1.0 mol% [Ir (COD)Cl]2/L6, H2 (3 MPa), 45 °C, and residence time 2.7 h. Yields were all isolated yields. The ee values were determined by HPLC. | ||
To further explore the mechanism of this transformation, a series of isotope-labeling experiments of 2-phenylquinoxaline (1aa′) were performed, as shown in Scheme 4a. First, the Ir-catalyzed asymmetric hydrogenation of 2-phenylquinoxaline (1aa′) was carried out under 1 MPa D2 in the toluene system. It was observed that the hydrogenation product was obtained with 98% and 90% deuterium atoms in the C2- and C3-positions (Scheme 4a (1)). Next, we repeated this reaction under 1 MPa D2 in ethanol-d1 solvent and the result showed that the deuterium atoms were added in the C2- and C3-positions (Scheme 4a (2)). Furthermore, this hydrogenation reaction was performed at 1 MPa D2 in ethanol solvent and found that only a small number of deuterium atoms were detected in the corresponding product (Scheme 4a (3)), which means that the hydrogen atoms were almost all from ethanol in the EtOH reaction system. In contrast, we continued this hydrogenation reaction under 1 MPa H2 in ethanol-d1 solvent (Scheme 4a (4)). The desired product was isolated with 52% deuterium atoms at the C2-position and some of the deuterium atoms were present in the C3-position (20% and 32%, respectively). These results showed that the hydrogen source in this transformation was derived from hydrogen gas in toluene and dioxane systems, whereas it was predominantly derived from ethanol in the EtOH system. This suggested that the Ir-catalyzed asymmetric hydrogenation of quinoxalines may follow distinct reaction pathways.
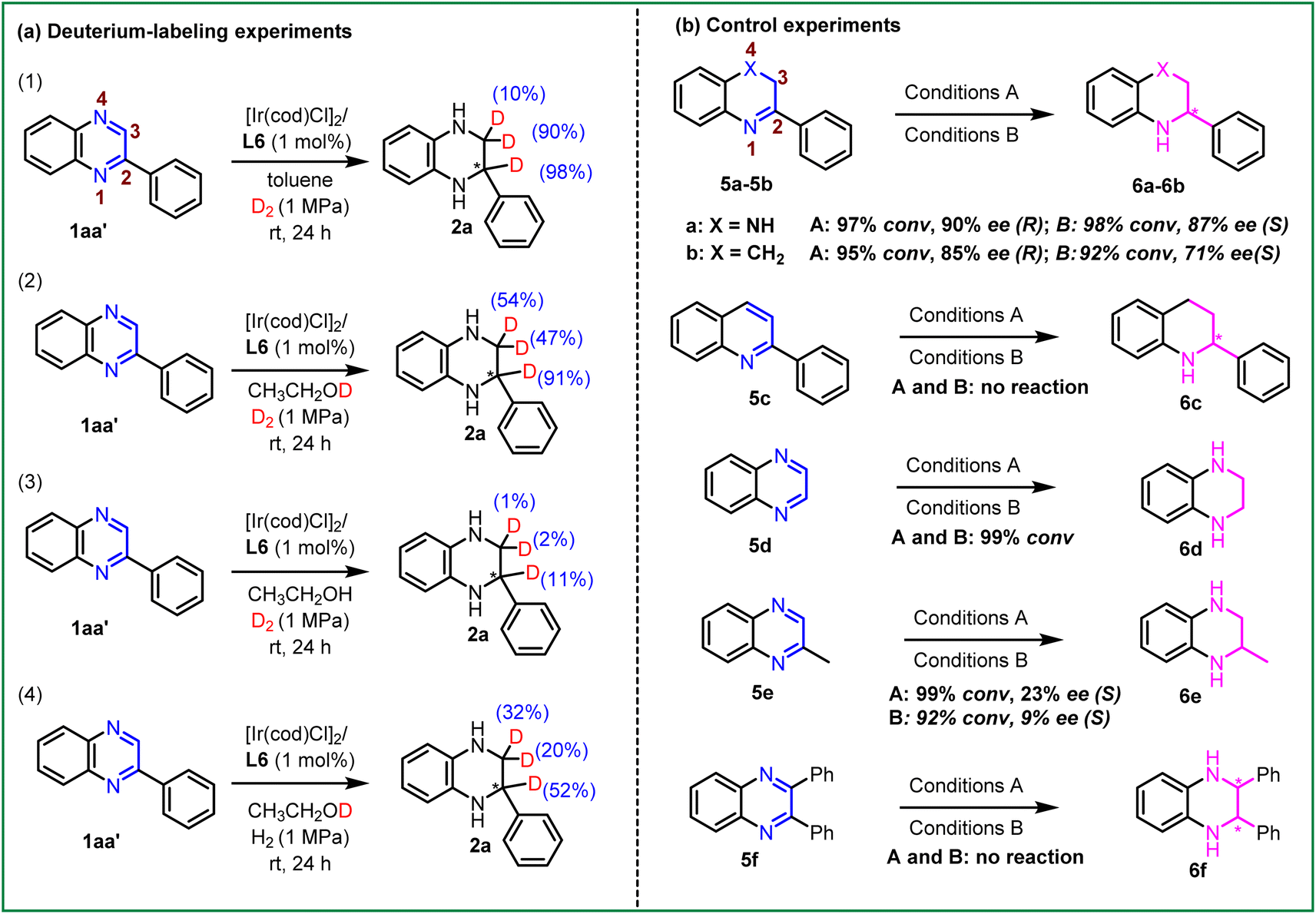 | ||
| Scheme 4 (a) Deuterium-labeling experiments of 1aa′; (b) control experiments of various N-heteroaromatic compounds. Conditions A; substrate/[Ir(cod)Cl]2/L6 = 100/0.5/1 in 2 mL toluene/dioxane = 4:1, room temperature, 18 h and 2 MPa H2. Conditions B; substrate/[Ir(cod)Cl]2/L6 = 100/0.5/1 in 2 mL EtOH, room temperature, 18 h and 2 MPa H2. Yields were all isolated yields. The ee values were determined by HPLC. | ||
Subsequently, control experiments with various N-heteroaromatic compounds were conducted. As shown in Scheme 4b, re-subjection of the intermediate 5a under the standard reaction conditions in both solvent systems led to the desired product 6a with 90% ee and 87% ee, respectively. This suggested that the hydrogenation could be a stepwise process, involving two sequential 3,4- and 1,2-hydrogenation processes. The substrate 5b, without N4-heteroatoms, also displayed high reactivities and good enantioselectivities in both solvent systems. This result indicated that the Ir–H complexes could directly recognize the unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the substrate without the aid of the N4-heteroatoms in the second hydrogenation step. Moreover, it was observed that 5c failed to undergo the anticipated hydrogenation. This lack of reactivity could be attributed to its stronger aromaticity, highlighting the significance of N4-heteroatoms during the first hydrogenation step. As we expected, substrates 5d and 5e afforded the products in excellent conversion, but the enantioselectivity of 6e was not ideal. This observation suggests that the benzene substituent or the appropriate alkyl substituents may be essential in the enantio-determining step. The challenging disubstituent-substrate 2,3-diphenylquinoxaline (5f) could not be hydrogenated in this system, which may be attributed to its stronger steric hindrance or aromaticity.
N bond in the substrate without the aid of the N4-heteroatoms in the second hydrogenation step. Moreover, it was observed that 5c failed to undergo the anticipated hydrogenation. This lack of reactivity could be attributed to its stronger aromaticity, highlighting the significance of N4-heteroatoms during the first hydrogenation step. As we expected, substrates 5d and 5e afforded the products in excellent conversion, but the enantioselectivity of 6e was not ideal. This observation suggests that the benzene substituent or the appropriate alkyl substituents may be essential in the enantio-determining step. The challenging disubstituent-substrate 2,3-diphenylquinoxaline (5f) could not be hydrogenated in this system, which may be attributed to its stronger steric hindrance or aromaticity.
To gather further evidence for identifying the key intermediates in the asymmetric hydrogenation process, we compared the reactivities of CN bonds in different quinoxalines (Scheme 5A). An equimolar solution of 5d and 1aa′ was treated under the standard hydrogenation conditions in toluene solvent. The result suggested that 5d rapidly converted to 6d with 76% conversion in 2 h, while only 45% conversion was observed for 1aa′. These findings further support the hypothesis that the non-substituted CN bond is more readily reduced, suggesting that 5a can be the key intermediate in the asymmetric hydrogenation of 2-phenylquinoxaline. To investigate the kinetic profile of both enantiomers, the asymmetric hydrogenation of 1aa′ was monitored in both toluene/dioxane and EtOH solvent systems (Scheme 5B). In the toluene/dioxane system, a gradual increase of the conversion rate was observed, along with the slow formation of product (R)-2a in 10 h. The intermediate 5a reached a maximum yield of 18% after 8 h, after which a rapid second hydrogenation process was observed. These results supported a stepwise hydrogenation process, with selective hydrogenation of the non-substituted CN bond occurring first. In contrast, the conversion rate of 1aa′ in the EtOH system was faster compared to that in the toluene/dioxane system, achieving full conversion within 11 h. Surprisingly, the formation of 5a was scarcely observed during the reaction, likely due to its high reactivity towards hydrogenation in the EtOH system. Consequently, these results indicated that the ethanol solvent was able to activate the substrate, thereby accelerating the hydrogenation process.
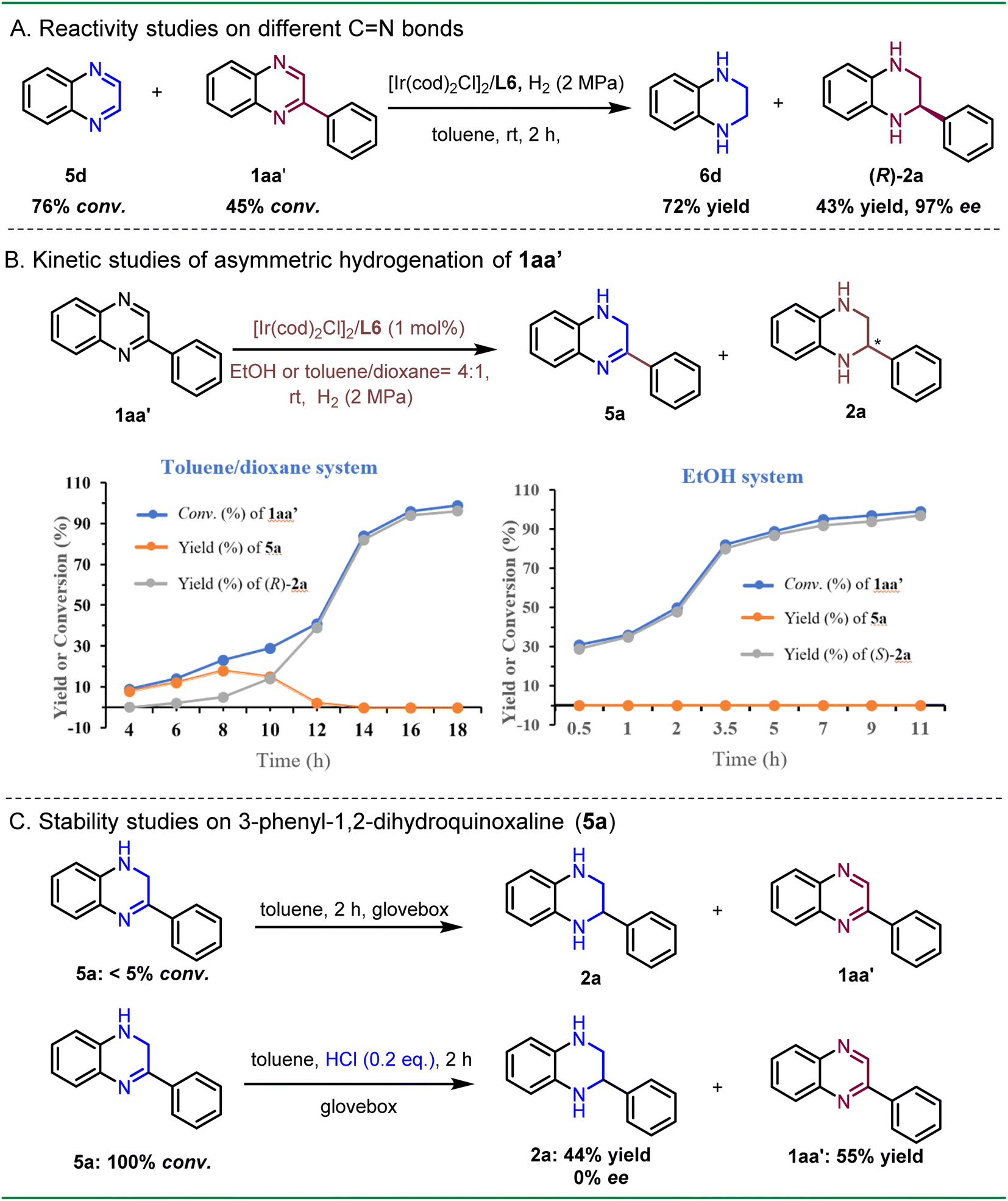 | ||
| Scheme 5 Mechanistic studies of the asymmetric hydrogenation reaction process. | ||
To explore whether the intermediate 5a is prone to disproportionation,20 we dissolved the intermediate 5a in toluene and placed it in a glove box for 2 h. The same procedure was applied to another sample, but with the addition of 0.2 equivalents of HCl (Scheme 5C). The above control experiments revealed that intermediate 5a remained almost unreacted in the absence of HCl. However, it underwent a full conversion with the addition of a catalytic amount of HCl, generating racemates 2a and 1aa′. This was consistent with the findings during optimization that the presence of HCl led to a negative effect on the enantioselectivity (see Table 1, entries 1–3).
Density functional theory (DFT) calculations were conducted to explore the mechanism of this process. From literature reports on transition-metal-catalyzed asymmetric hydrogenation, a similar reaction pathway in toluene/dioxane has been calculated (Scheme 6).45,46 Initially, oxidative addition of H2 to the Ir-complex A-1via a barrierless transition state A-TS1 generates a stable Ir(III) intermediate A-3 (−5.7 kcal mol−1). Based on the finding that the N4 heteroatom is critical in the first hydrogenation step (Scheme 4b) and combining the different reactivities of the two CN bonds, four potential sites for hydrogen transfer—N1, C2, C3, and N4—are identified, where the substrate coordinates with iridium via the N4–Ir bond. Computational results suggest that the hydrogen transferred to the C3-position viaA-TS2-C3 is the most favorable, with the lowest energy barrier of 11.0 kcal mol−1. It is much lower than that of other competing transition states (A-TS2-N4, A-TS2-C2, and A-TS2-N1). The generated intermediate A-4 then undergoes a second hydrogen transfer to the N4-position through transition state A-TS3, with an activation free energy of 16.1 kcal mol−1, which results in the formation of intermediate A-5. Next, the semireduction intermediate 5a can be obtained through rapid dissociation from complex A-5. This is in good agreement with the control experiments and kinetic studies shown in Schemes 4b and 5B. Meanwhile, another H2 molecule could coordinate with complex A-6 to initiate the second hydrogenation. The hydrogen transfer from A-7 involves three competing pathways. The most favored pathway occurs through transition state A-TS4, with hydrogen transferring to the N4-position. It has an activation free energy of 15.3 kcal mol−1, significantly lower than that of transition states A-TS4-1 and A-TS4-2 (−26.6 kcal mol−1 and 67.9 kcal mol−1, respectively). The intermediate A-9 is obtained through a proton transfer between N1 and N4 positions. According to the result of the control experiment, the Ir–H complexes could directly recognize the unsaturated CN bond without the aid of the N4-heteroatoms in the second hydrogenation step, and the transition state A-TS5OR or A-TS5OS was proposed. The second hydrogen transfer to the C2-position from the Re or Si face via the transition state A-TS5OR or A-TS5OS is the enantio-determining step. The favored transition state A-TS5OR yields the final R-products with an energy barrier of 18.8 kcal mol−1, which is significantly lower than that of the S- transition state (22.0 kcal mol−1). Furthermore, the inner-sphere pathway with the substrate coordination to iridium is calculated (see Scheme S2† for more details), indicating that the energy barrier of the rate-determining step is 30.2 kcal mol−1, which is much higher than that of this pathway (18.8 kcal mol−1).
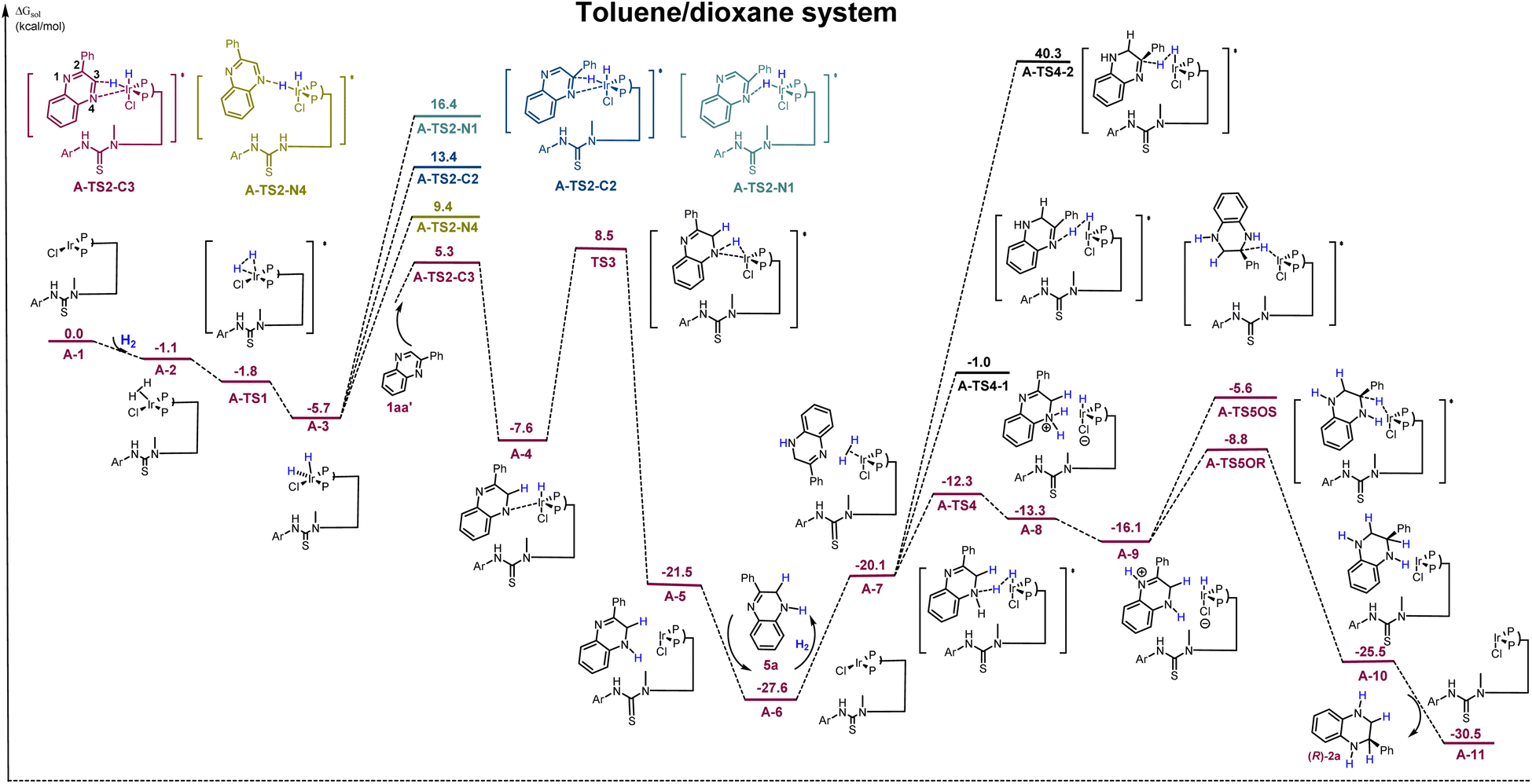 | ||
| Scheme 6 DFT calculated energy potential surface for the proposed pathway in toluene/dioxane systems. (Ar = 3,5-di-CF3Ph; the phenyl groups on the phosphorus atoms are omitted for clarity; the relative Gibbs energies are labelled in kcal mol−1). | ||
It is widely acknowledged that solvent can affect the rate, mechanism, and stereochemistry of the reactions.47 However, comprehensively illustrating the role of solvents remains challenging.48,49 Although Zhang and colleagues have reported the enantiodivergent synthesis of chiral tetrahydroquinoline derivatives using a solvent-dependent enantioselective system, the specific role of the solvent was not thoroughly elucidated.32 The results of optimization of reaction conditions and kinetic studies indicated that ethanol molecules could participate in the enantiocontrol step to realize reversed enantioselectivity, while activating the substrate in our catalytic system. These suggested that the ethanol molecule functions as a hydrogen-bond donor, which can activate the quinoxaline ring by the formation of a hydrogen bond. Hence, in order to gain insight into this interesting reversed configuration phenomenon, the transition states TS5OR-ET and TS5OS-ET were proposed to be the enantio-determining step in the EtOH system (Fig. 1). To our delight, the calculation suggests that the energy barrier for the formation of the final S-products viaTS5OS-ET is 3.6 kcal mol−1 lower than that for the R-product, which is consistent with our experiment. Furthermore, distortion/interaction analysis was conducted for the transition states A-TS5OR, A-TS5OS, TS5OR-ET and TS5OS-ET (Fig. 1).50–52 The complex is divided into 2-phenylquinoxaline 1aa′ and catalyst fragments. The analysis suggests that the interaction energy between the catalyst and substrate controls the enantio-selectivity. The interaction energy of A-TS5OR is 9.5 kcal mol−1 higher than that of A-TS5OS, which can be attributed to the favorable π–π interaction between the substrate and ligand (the distance between two benzene rings in A-TS5OR and A-TS5OS is 3.75 Å and 4.66 Å, respectively). This is the origin of the enantioselectivity in the toluene/dioxane system. In contrast, the interaction energy of TS5OS-ET is 6.6 kcal mol−1 higher than that of TS5OR-ET in the EtOH system, owing to the more favorable face-to-face interactions. Stronger interactions between the benzene ring on the substrate and the trifluoromethyl-substituted benzene ring on the ligand (distance of 3.48 Å), as well as between the benzene ring on the substrate and benzene ring on the ligand (distance of 4.72 Å), are found in TS5OS-ET. In addition, hydrogen bonding interactions between the substrate and ethanol in TS5OS-ET contribute to the lower energy barrier, as indicated by the O⋯H distance of 1.79 Å and Cl⋯H distance of 2.20 Å. Taken together, π–π interactions and hydrogen bonding interactions make TS5OS-ET significantly more stable than TS5OR-ET, which accounts for the origin of the enantioselectivity in the EtOH system. Especially, the 2-phenyl substituent on the substrate is essential for the high enantioselectivity in the EtOH system. The absence of the 2-phenyl substituent afforded the products with poor ee values (Table 2, 2s and 2t).
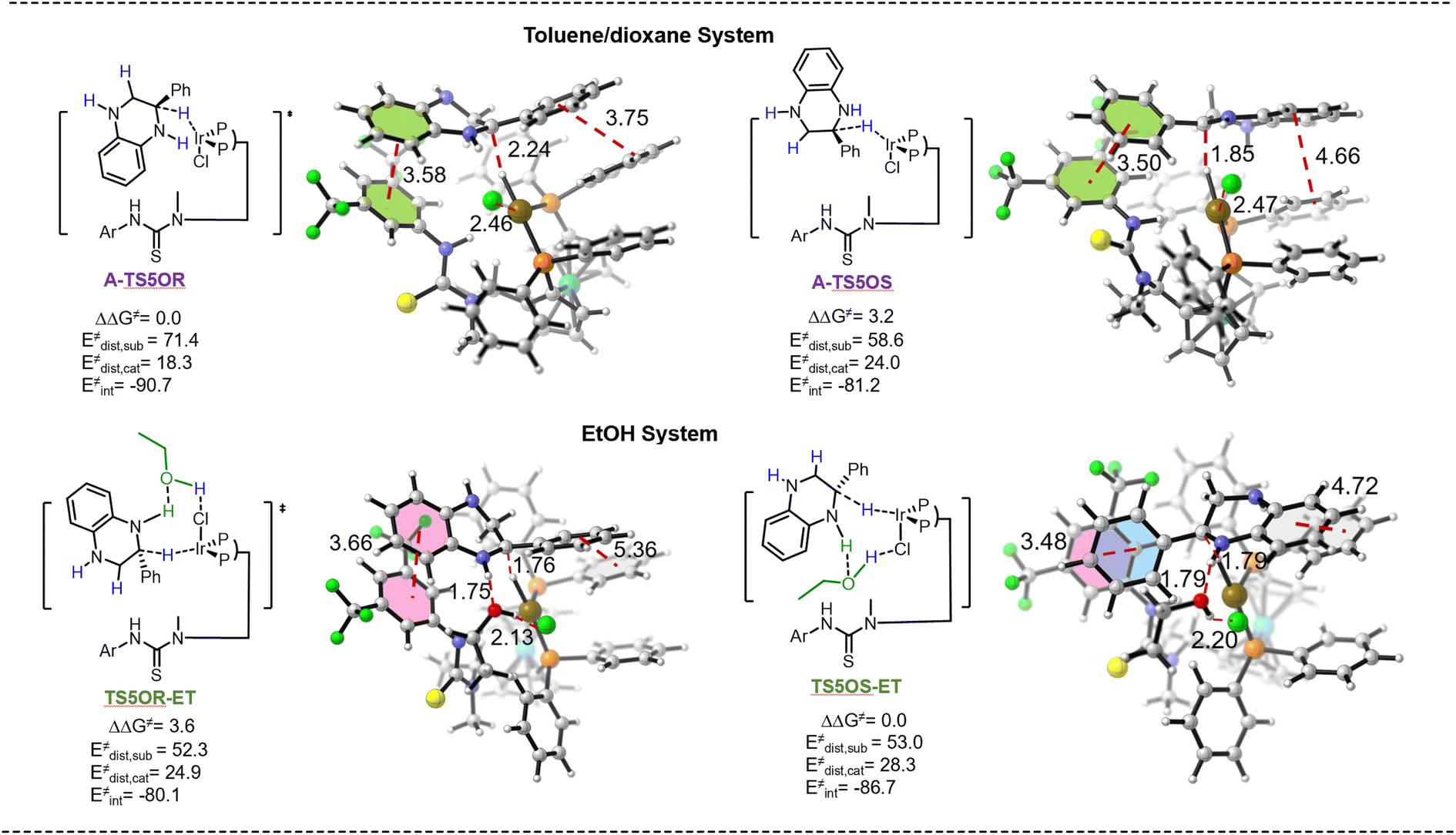 | ||
| Fig. 1 Distortion–interaction analysis method in the enantio-determining step for the single-point energy calculations; the relative Gibbs energies are labeled in kcal mol−1. | ||
Conclusion
In conclusion, we have developed a novel Ir-catalyzed asymmetric hydrogenation method for the synthesis of chiral tetrahydroquinoxaline under mild and green conditions. Both enantiomers of chiral THQs could be efficiently obtained by adjusting the reaction solvent with excellent yields and enantioselectivities. Pleasingly, this methodology shows great tolerance to a board range of substrates, including 2-substituted and 2,3-disubstituted quinoxalines. Notably, this methodology has been successfully applied under continuous flow conditions, enabling the gram-scale synthesis of enantiopure THQs in both solvent systems. Mechanistic studies, including deuterium labeling experiments, control experiments, kinetic experiments and DFT calculations, were conducted. These studies revealed that the non-covalent π–π and O–H interactions between the chiral catalyst, solvent and substrates are essential for controlling the enantioselectivity. It represents a valuable advance in solvent-controlled enantioselectivity synthesis for transition-metal-catalyzed asymmetric hydrogenation reactions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Shenzhen Science and Technology Research Fund No. JSGG20201103153807021; GXWD20220811173736002; KCXFZ20230731094904009. We are also grateful to the Natural Science Foundation of Guangdong Province (No. 2021A1515110366; No. 2022A1515011859), National Natural Science Foundation of China (No. 22302048; No. 82204231; No. 22203023), Shenzhen Key Laboratory of Advanced Functional Carbon Materials Research and Comprehensive Application, and Shenzhen Bay Laboratory Supercomputing Center. We appreciate the guidance and insights provided by Professor Xu-Mu Zhang.Notes and references
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, Rings in Drugs, J. Med. Chem., 2014, 57(14), 5845–5859 CrossRef CAS PubMed.
- J. Wen, X. Fan, R. Tan, H.-C. Chien, Q. Zhou, L. W. Chung and X. Zhang, Brønsted-Acid-Promoted Rh-Catalyzed Asymmetric Hydrogenation of N-Unprotected Indoles: A Cocatalysis of Transition Metal and Anion Binding,, Org. Lett., 2018, 20(8), 2143–2147 CrossRef CAS PubMed.
- A. N. Kim and B. M. Stoltz, Recent Advances in Homogeneous Catalysts for the Asymmetric Hydrogenation of Heteroarenes, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed.
- S. Wang, C. Xie, Y. Zhu, G. Zi, Z. Zhang and G. Hou, Enantioselective Synthesis of Chiral Cyclic Hydrazines by Ni-Catalyzed Asymmetric Hydrogenation,, Org. Lett., 2023, 25(20), 3644–3648 CrossRef CAS PubMed.
- G. Chang; M. T. Didiuk; J. I. Finneman; R. S. Garigipati; R. M. Kelley, D. A. Perry,; R. B. Ruggeri; B. M. Bechle, Preparation of 1,2,4-Substituted 1,2,3,4-Tetrahydro-and 1,2-Dihydro-Quinoline and 1,2,3,4-Tetrahydro-Quinoxaline Derivatives as Cetp Inhibitors for the Treatment of Atherosclerosis and Obesity, WIPO Pat., WO2004085401A1, 2004.
- R. P. Law, S. J. Atkinson, P. Bamborough, C.-w. Chung, E. H. Demont, L. J. Gordon, M. Lindon, R. K. Prinjha, A. J. B. Watson and D. J. Hirst, Discovery of Tetrahydroquinoxalines as Bromodomain and Extra-Terminal Domain (Bet) Inhibitors with Selectivity for the Second Bromodomain, J. Med. Chem., 2018, 61(10), 4317–4334 CrossRef CAS PubMed.
- A. Ansari; S. J. Jeffries; P. Shah; C. Zhang, Methods and Compounds for the Treatment of Genetic Disease, WIPO Pat., WO2021158707A1, 2021.
- F. H. Zhang, F. J. Zhang, L. Li Mao, J. H. Xie and Q. L. Zhou, Enantioselective Hydrogenation of Dialkyl Ketones, Nat. Catal., 2020, 3, 621–627 CrossRef CAS.
- R. A. A. Abdine, G. Hedouin, F. Colobert and J. Wencel-Delord, Metal-Catalyzed Asymmetric Hydrogenation of CN Bonds, ACS Catal., 2020, 11, 215–247 CrossRef.
- Y. H. Hu, Z. F. Zhang, Y. G. Liu and W. B. Zhang, Cobalt-Catalyzed Chemo- and Enantioselective Hydrogenation of Conjugated Enynes, Angew. Chem., Int. Ed., 2021, 60, 16989–16993 CrossRef CAS PubMed.
- R. Gunasekar, R. L. Goodyear, P. I. Silvestri and L. J. Xiao, Recent Developments in Enantio- and Diastereoselective Hydrogenation of N -Heteroaromatic Compounds, Org. Biomol. Chem., 2022, 20, 1794–1827 RSC.
- G. Liu, L. Zheng, K. Tian, H. Wang, L. Wa Chung, X. Zhang and X.-Q. Dong, Ir-Catalyzed Asymmetric Hydrogenation of Unprotected Indoles: Scope Investigations and Mechanistic Studies, CCS Chem., 2023, 5(6), 1398–1410 CrossRef CAS.
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Asymmetric Hydrogenation of Heteroarenes and Arenes, Chem. Rev., 2011, 112(4), 2557–2590 CrossRef PubMed.
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Selective Arene Hydrogenation for Direct Access to Saturated Carbo- and Heterocycles, Angew. Chem., Int. Ed., 2019, 58(31), 10460–10476 CrossRef CAS PubMed.
- Z. Yang, Y. Chen, L. Wan, Y. Li, D. Chen, J. Tao, P. Tang and F.-E. Chen, Iridium-Catalyzed Asymmetric, Complete Hydrogenation of Pyrimidinium Salts under Batch and Flow, Green Chem., 2024, 26(1), 317–322 RSC.
- N. Mršić, T. Jerphagnon, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Asymmetric Hydrogenation of Quinoxalines Catalyzed by Iridium/Pipphos, Adv. Synth. Catal., 2009, 351(16), 2549–2552 CrossRef.
- W. Tang, L. Xu, Q. H. Fan, J. Wang, B. Fan, Z. Zhou, K. h. Lam and A. S. C. Chan, Asymmetric Hydrogenation of Quinoxalines with Diphosphinite Ligands: A Practical Synthesis of Enantioenriched, Substituted Tetrahydroquinoxalines, Angew. Chem., Int. Ed., 2009, 48(48), 9135–9138 CrossRef CAS.
- A. Xu, C. Li, J. Huang, H. Pang, C. Zhao, L. Song, H. You, X. Zhang and F.-E. Chen, Highly Enantioselective Synthesis of Both Tetrahydroquinoxalines and Dihydroquinoxalinones Via Rh–Thiourea Catalyzed Asymmetric Hydrogenation, Chem. Sci., 2023, 14(34), 9024–9032 RSC.
- M. Rueping, F. Tato and F. R. Schoepke, The First General, Efficient and Highly Enantioselective Reduction of Quinoxalines and Quinoxalinones, Chem.–Eur. J., 2010, 16(9), 2688–2691 CrossRef CAS PubMed.
- Q.-A. Chen, D.-S. Wang, Y.-G. Zhou, Y. Duan, H.-J. Fan, Y. Yang and Z. Zhang, Convergent Asymmetric Disproportionation Reactions: Metal/Brønsted Acid Relay Catalysis for Enantioselective Reduction of Quinoxalines, J. Am. Chem. Soc., 2011, 133(16), 6126–6129 CrossRef CAS PubMed.
- J. Qin, F. Chen, Z. Ding, Y. M. He, L. Xu and Q. H. Fan, Asymmetric Hydrogenation of 2- and 2,3-Substituted Quinoxalines with Chiral Cationic Ruthenium Diamine Catalysts,, Org. Lett., 2011, 13, 6568–6571 CrossRef CAS PubMed.
- Z. Zhang and H. Du, A Highly Cis-Selective and Enantioselective Metal-Free Hydrogenation of 2,3-Disubstituted Quinoxalines, Angew. Chem., Int. Ed., 2014, 127(2), 633–636 CrossRef.
- C. Liu, X. Liu and Q. Liu, Stereodivergent Asymmetric Hydrogenation of Quinoxalines, Chem, 2023, 9(9), 2585–2600 CAS.
- S. Fleischer, S. Zhou, S. Werkmeister, K. Junge and M. Beller, Cooperative Iron–Brønsted Acid Catalysis: Enantioselective Hydrogenation of Quinoxalines and 2 H-1,4-Benzoxazines, Chem.–Eur. J., 2013, 19(16), 4997–5003 CrossRef CAS PubMed.
- Z. Luo, Z. Li, H. Zhao, J. Yang, L. Xu, M. Lei, Q. Fan and P. J. Walsh, Borane-Catalyzed Tandem Cyclization/Hydrosilylation Towards Enantio- and Diastereoselective Construction of Trans-2,3 Disubstituted-1,2,3,4-Tetrahydroquinoxalines, Angew. Chem., Int. Ed., 2023, 62(32), e202305449 CrossRef CAS.
- S. Alavi, M. H. Mosslemin, R. Mohebat and A. R. Massah, Green Synthesis of Novel Quinoxaline Sulfonamides with Antibacterial Activity, Res. Chem. Intermed., 2017, 43(8), 4549–4559 CrossRef CAS.
- M. Di Filippo and M. Baumann, Continuous Flow Synthesis of Quinolines Via a Scalable Tandem Photoisomerization-Cyclization Process, Eur. J. Org Chem., 2020, 2020(39), 6199–6211 CrossRef CAS.
- B. Borah and L. R. Chowhan, Recent Advances in the Transition-Metal-Free Synthesis of Quinoxalines, RSC Adv., 2021, 11(59), 37325–37353 RSC.
- Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Rhodium-Catalyzed Asymmetric Hydrogenation of Unprotected Nh Imines Assisted by a Thiourea, Angew. Chem., Int. Ed., 2014, 53(32), 8467–8470 CrossRef CAS PubMed.
- J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Strong Brønsted Acid Promoted Asymmetric Hydrogenation of Isoquinolines and Quinolines Catalyzed by a Rh–Thiourea Chiral Phosphine Complex Via Anion Binding, Chem. Sci., 2016, 7(5), 3047–3051 RSC.
- Z. Han, G. Liu, R. Wang, X.-Q. Dong and X. Zhang, Highly Efficient Ir-Catalyzed Asymmetric Hydrogenation of Benzoxazinones and Derivatives with a Brønsted Acid Cocatalyst, Chem. Sci., 2019, 10(15), 4328–4333 RSC.
- Z. Y. Han, G. Liu, X. L. Yang, X. Q. Dong and X. M. Zhang, Enantiodivergent Synthesis of Chiral Tetrahydroquinoline Derivatives Via Ir-Catalyzed Asymmetric Hydrogenation: Solvent-Dependent Enantioselective Control and Mechanistic Investigations, ACS Catal., 2021, 11, 7281–7291 CrossRef CAS.
- M.-W. Chen, Z. Deng, Q. Yang, J. Huang and Y. Peng, Enantioselective Synthesis of Trifluoromethylated Dihydroquinoxalinones Via Palladium-Catalyzed Hydrogenation, Org. Chem. Front., 2019, 6(6), 746–750 RSC.
- P. T. Baraldi and V. Hessel, Micro Reactor and Flow Chemistry for Industrial Applications in Drug Discovery and Development,, Green Process. Synth., 2012, 1, 149–167 CAS.
- P. D. Morse, R. L. Beingessner and T. F. Jamison, Enhanced Reaction Efficiency in Continuous Flow, Isr. J. Chem., 2016, 57(3–4), 218–227 Search PubMed.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, A Perspective on Continuous Flow Chemistry in the Pharmaceutical Industry, Org. Process Res. Dev., 2020, 24(10), 1802–1813 CrossRef CAS.
- M. Guidi, P. H. Seeberger and K. Gilmore, How to Approach Flow Chemistry, Chem. Soc. Rev., 2020, 49(24), 8910–8932 RSC.
- J. Liao, X. Jia, F. Wu, J. Huang, G. Shen, H. You and F.-E. Chen, Rapid Mild Macrocyclization of Depsipeptides under Continuous Flow: Total Syntheses of Five Cyclodepsipeptides, Org. Chem. Front., 2022, 9(23), 6640–6645 RSC.
- J. Li, X. Song, Y. Wang, J. Huang, H. You and F.-E. Chen, Copper-Catalyzed Asymmetric Allylic Alkylation of Racemic Inert Cyclic Allylic Ethers under Batch and Flow Conditions, Chem. Sci., 2023, 14(16), 4351–4356 RSC.
- F. Guan, A. J. Blacker, B. Hall, N. Kapur, J. Wen and X. Zhang, High-Pressure Asymmetric Hydrogenation in a Customized Flow Reactor and Its Application in Multi-Step Flow Synthesis of Chiral Drugs, J. Flow Chem., 2021, 11(4), 763–772 CrossRef CAS.
- L. Xu, G. Wang, N. Rong, Y. Gu, L. A. Hu, H. You and Q. Yin, Enantioselective and Step-Economic Synthesis of the Chiral Amine Fragment in the Tyrosine Kinase Inhibitor Repotrectinib by Direct Asymmetric Reductive Amination under Batch and Flow, Org. Process Res. Dev., 2023, 1539–1545 Search PubMed.
- J. Li, X. Song, F. Wu, H. You and F. E. Chen, Copper-Catalyzed Asymmetric Allylic Alkylation of Racemic Cyclic Allyl Bromides with Organolithium Compounds, Eur. J. Org Chem., 2022, 2022(34), e202200860 CrossRef CAS.
- D. Zhang, F. Wu, Z. Wan, Y. Wang, X. He, B. Guo, H. You and F.-E. Chen, A Palladium Polyaniline Complex: A Simple and Efficient Catalyst for Batch and Flow Suzuki–Miyaura Cross-Couplings, Chem. Commun., 2022, 58(77), 10845–10848 RSC.
- H. Pang, J. Huang, J. Wang, G. Wang, A. Xu, L. Luo, Q. Yuan, H. You and F.-E. Chen, Practical Synthesis of Tetrahydrofolate by Highly Efficient Catalytic Hydrogenation in Continuous Flow, J. Flow Chem., 2024, 14, 427–435 CrossRef CAS.
- T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Transition-Metal-Catalyzed Asymmetric Hydrogenation and Transfer Hydrogenation: Sustainable Chemistry to Access Bioactive Molecules, Chem. Rec., 2016, 16(6), 2754–2771 CrossRef PubMed.
- F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56(34), 10070–10086 CrossRef CAS.
- P. J. Dyson and P. G. Jessop, Solvent Effects in Catalysis: Rational Improvements of Catalysts Via Manipulation of Solvent Interactions, Catal. Sci. Technol., 2016, 6(10), 3302–3316 RSC.
- T. Ishii, R. Watanabe, T. Moriya, H. Ohmiya, S. Mori and M. Sawamura, Cooperative Catalysis of Metal and O–H⋯O/Sp3–C–H⋯O Two-Point Hydrogen Bonds in Alcoholic Solvents: Cu-Catalyzed Enantioselective Direct Alkynylation of Aldehydes with Terminal Alkynes, Chem.–Eur. J., 2013, 19(40), 13547–13553 CrossRef CAS PubMed.
- S. Sakai, A. Fujioka, K. Imai, K. Uchiyama, Y. Shimizu, K. Higashida and M. Sawamura, Silver-Catalyzed Asymmetric Aldol Reaction of Isocyanoacetic Acid Derivatives Enabled by Cooperative Participation of Classical and Nonclassical Hydrogen Bonds, Adv. Synth. Catal., 2022, 364(14), 2333–2339 CrossRef CAS.
- W. Y. Tong, X. Su, P. Sun, S. Xu, S. Qu and X. Wang, Understanding the Reaction Mechanism of Ni-Catalyzed Regio- and Enantioselective Hydroalkylation of Enamines: Chemoselectivity of (Bi-Oxazoline)Nih, J. Org. Chem., 2023, 88(21), 15404–15413 CrossRef CAS PubMed.
- S. Lan, H. Huang, W. Liu, C. Xu, X. Lei, W. Dong, J. Liu, S. Yang, A. E. Cotman, Q. Zhang and X. Fang, Asymmetric Transfer Hydrogenation of Cyclobutenediones, J. Am. Chem. Soc., 2024, 146(7), 4942–4957 CrossRef CAS PubMed.
- G. Liu, X. Yang, P. Gu, M. Wang, X. Zhang and X.-Q. Dong, Challenging Task of Ni-Catalyzed Highly Regio-/Enantioselective Semihydrogenation of Racemic Tetrasubstituted Allenes Via a Kinetic Resolution Process, J. Am. Chem. Soc., 2024, 146(11), 7419–7430 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04222k |
| This journal is © The Royal Society of Chemistry 2024 |