
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugated polymers of an oxa[5]helicene-derived polycyclic heteroaromatic: tailoring energy levels and compatibility for high-performance perovskite solar cells†
Yaohang
Cai‡
a,
Yuyan
Zhang‡
a,
Lingyi
Fang
a,
Yutong
Ren
a,
Jidong
Zhang
b,
Yi
Yuan
a,
Jing
Zhang
a and
Peng
Wang
 *a
*a
aState Key Laboratory of Silicon and Advanced Semiconductor Materials, Department of Chemistry, Zhejiang University, Hangzhou 310058, China. E-mail: pw2015@zju.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China
First published on 28th August 2024
Abstract
In the quest to enhance the efficiency and durability of n–i–p perovskite solar cells (PSCs), engineering hole-transporting conjugated polymers with well-matched energy levels, exceptional film-forming properties, rapid hole transport, and superior moduli is paramount. Here, we present a novel approach involving the customization of a conjugated polymer, designated as p-DTPF4-EBEH, comprising alternating units of an oxa[5]helicene-based polycyclic heteroaromatic (DTPF4) and 5,5′-(2,5-di(hexyloxy)-1,4-phenylene)bis(3,4-ethylenedioxythiophene) (EBEH), synthesized through palladium-catalyzed direct arylation. Relative to homopolymers p-DTPF4 and p-EBEH, p-DTPF4-EBEH demonstrates a proper HOMO energy level, hole density, and hole mobility, alongside superior film-forming capabilities. Remarkably, compared to the commonly used hole transport material spiro-OMeTAD, p-DTPF4-EBEH not only exhibits superior film-forming property and hole mobility but also offers increased modulus and improved waterproofing. Incorporating p-DTPF4-EBEH as the hole transport material in PSCs results in an average power conversion efficiency of 25.8%, surpassing the 24.3% achieved with spiro-OMeTAD. Importantly, devices utilizing p-DTPF4-EBEH demonstrate enhanced thermal storage stability at 85 °C, along with operational robustness.
1 Introduction
Solution-processed metal halide perovskite films present promising opportunities for electronic device applications.1 These polycrystalline films, boasting tunable bandgaps, high absorption coefficients, and prolonged lifetimes of photogenerated charge carriers, are favored as the light-absorbing layer in photodiodes.2,3 Presently, single-junction perovskite solar cells (PSCs) have achieved a certified power conversion efficiency (PCE) record of up to 26.1%.4 The efficiency and stability of PSCs hinge not only on the quality of the perovskite films but also on the excellence of the charge transport materials.5,6In n–i–p type PSCs, low-cost colloidal oxides such as titanium dioxide and tin dioxide are commonly employed in electron transport layers, while the molecular semiconductor spiro-OMeTAD is a prevalent choice as the hole transport material (refer to the chemical structure in Fig. 1A).2,3,7–9 Spiro-OMeTAD's relatively shallow highest occupied molecular orbital (HOMO) energy level enables facile air doping with the aid of imide salts, augmenting its conductivity.10 Nevertheless, composite films derived from spiro-OMeTAD are susceptible to significant morphological degradation under prolonged exposure to high temperatures, attributable to spiro-OMeTAD's low glass transition temperature (Tg), culminating in diminished PCE.11–13
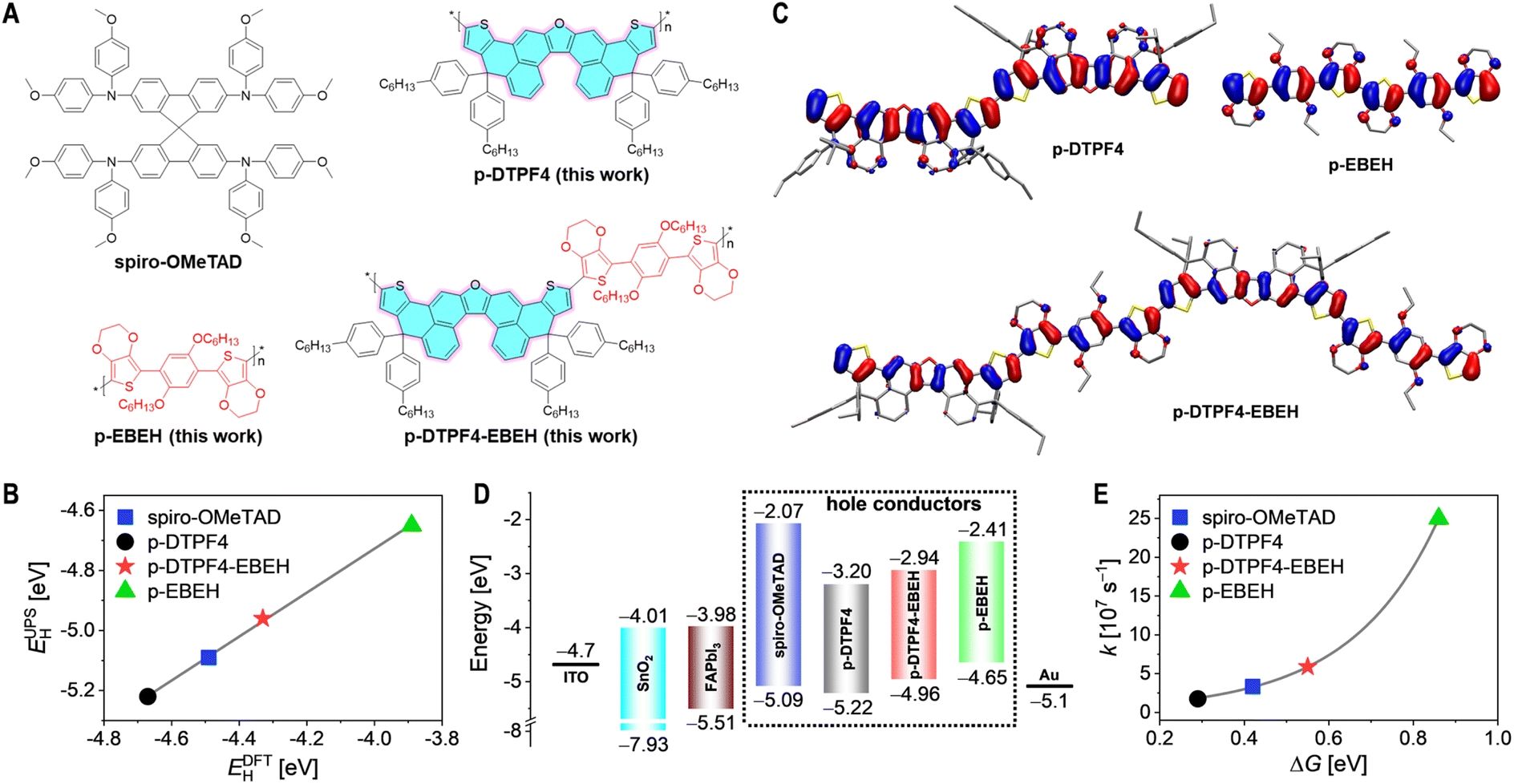 | ||
| Fig. 1 (A) Chemical structures of the conjugated polymers examined in this investigation alongside the standard molecular semiconductor employed in PSCs. (B) Correlation between the HOMO energy levels acquired through ultraviolet photoelectron spectroscopy measurements (EUPSH) and those computed via the density functional theory method (EDFTH). The solid gray line signifies the outcome of linear fitting. (C) Conformations and HOMO contour plots of the conjugated polymers computed employing periodic boundary conditions-density functional theory method. To enhance computational efficiency, the n-hexyl group was replaced by ethyl, with hydrogen atoms omitted for clarity. (D) Schematic energy diagram outlining components of PSCs, encompassing the electron transport layer (SnO2), light-absorbing layer (FAPbI3), hole conductors, and electrical contacts (ITO and Au). The HOMO (valence band) and LUMO (conduction band) energy levels are noted below and above the color bars, respectively. (E) Relationship between the rate constant (k) and the driving force (ΔG) pertinent to hole extraction. The discrepancy between the FAPbI3 valence band and the HOMO energy level of organic semiconductor is represented by ΔG. The solid gray line represents the fitting outcome based on Marcus electron transfer theory. | ||
It is paramount to underscore that in PSCs employing electron transport layers with relatively high conduction bands like titanium dioxide or tin dioxide, the selection of a hole transport layer with a sufficiently shallow HOMO energy level is imperative for efficient long-range charge separation. The ideal organic semiconductor for n–i–p type PSCs should exhibit rapid hole conduction, good film-forming ability, maintain intact morphology under prolonged heating, and effectively suppress species diffusivities. In contrast to molecular semiconductors, polymeric semiconductors feature elongated main chains and potential for interchain intertwining, advantageous for charge transport. Moreover, polymer films exhibit unique viscoelastic properties, affording enhanced resistance to stretch and impact. Although numerous semiconducting polymers have been scrutinized as hole transport materials in n–i–p type PSCs, high-performance candidates remain relatively scarce.14–26
Polycyclic heteroaromatics exhibit distinctive π-conjugated skeletons, showcasing remarkable performance in various optoelectronic devices.27,28 Particularly noteworthy are those with a reduced C/H ratio, pivotal for high-mobility organic field-effect transistors due to enhanced intermolecular charge transfer integrals and reduced reorganization energies.29–31 As early as 1881, Graebe et al. synthesized oxa[5]helicene by heating naphthol with lead oxide.32 Presently, oxa[5]helicene can be readily prepared from inexpensive 1,1′-binaphthol through a simple, quantitative dehydration reaction.33 In 2013, Nakahara et al. pioneered the application of oxa[5]helicene in organic single-crystal field-effect transistors, exhibiting hole mobilities of up to 1.0 cm2 V−1 s−1.34 In 2019, Xu et al. derivatized oxa[5]helicene with two electron-rich dimethoxydiphenylamines, yielding a molecular semiconductor for n–i–p type PSCs with a PCE of 21.0%.35 Subsequently, Wei et al. developed a molecular semiconductor based on pyrrole-bridged bis(oxa[5]helicene), suitable for fabricating 21.3%-efficiency PSCs with good thermal stability at 60 °C.36 Recently, He et al. synthesized a semiconducting polymer, p-O5H-E-POZ-E, comprising alternating oxa[5]helicene, 3,4-ethylenedioxythiophene, phenoxazine, and 3,4-ethylenedioxythiophene units, and employed it in PSCs, achieving an average PCE of 24.9% alongside excellent stability.37
In this investigation, we devised an oxa[5]helicene-embedded polycyclic heteroaromatic, 4,11-dihydrothieno[2′,3′:4,5]phenaleno[2,1-b]thieno[2′,3′:4,5]phenaleno[1,2-d]furan (DTPF, highlighted in cyan in Fig. 1A). Intriguingly, DTPF, when adorned with four hexylphenyl substituents at two sp3 carbon atoms, abbreviated as DTPF4, demonstrates a considerable solubility of 679 mg mL−1 in chloroform. The homopolymer derived from DTPF4 (p-DTPF4, Fig. 1A) exhibits notably superior hole mobility compared to p-O5H-E-POZ-E at a given hole density, emphasizing the importance of high-level polycyclic heteroaromatic monomers in conjugated polymer synthesis. However, density functional theory calculations with periodic boundary conditions reveal that the HOMO energy level of p-DTPF4 is −4.67 eV, 180 meV deeper than that of spiro-OMeTAD (−4.49 eV).
Considering the theoretical HOMO energy level of 5,5′-(2,5-bis(hexyloxy)-1,4-phenylene)bis(3,4-ethylenedioxythiophene) (EBEH) as −4.80 eV, shallower than DTPF4 (−5.05 eV), we alternatively copolymerized DTPF4 with EBEH, yielding the copolymer p-DTPF4-EBEH (Fig. 1A), with a theoretical HOMO energy level of −4.33 eV. To systematically elucidate the structure–property relationship, we also synthesized the homopolymer p-EBEH (as depicted in Fig. 1A), with a notably high theoretical HOMO energy level of −3.89 eV. Employing p-DTPF4-EBEH as the primary constituent of the hole transport layer, we fabricated PSCs with an average PCE reaching 25.8%, significantly outperforming those prepared with p-DTPF4 (17.8%), p-EBEH (10.5%), and spiro-OMeTAD (24.3%). Notably, PSCs based on p-DTPF4-EBEH also demonstrated excellent thermal storage stability at 85 °C and operational stability at 45 °C. Degradation analysis of the devices underscored the pivotal role of the hole transport layer—ensuring uniform morphology, high elastic modulus, excellent waterproofing, and slow species diffusivities.
2 Results and discussion
2.1 Monomer and polymer synthesis
For details on the synthesis and structural characterization of monomers and conjugated polymers, please refer to the Experimental Section in ESI.† As delineated in Scheme S1 of ESI,† the synthesis of the monomer DTPF4 commenced from the readily accessible and cost-effective 5,9-dibromodinaphtho[2,1-b:1′,2′-d]furan. Sequential steps, including Miyaura borylation, Suzuki–Miyaura cross-coupling, carbonyl addition, and intramolecular Friedel–Crafts cyclization, were employed, resulting in DTPF4 in four steps with an overall yield of 50%. The monomer EBEH was synthesized following established procedures.38 DTPF4 and EBEH were dibrominated using N-bromosuccinimide respectively, yielding monomers DTPF4-2Br and EBEH-2Br.As illustrated in Scheme S2 of ESI,† palladium-catalyzed direct arylation39,40 was employed to synthesize homopolymers p-DTPF4 and p-EBEH, as well as the alternating copolymer p-DTPF4-EBEH, typically by polycondensation of a thiophene derivative with a dibrominated compound. High-temperature gel permeation chromatography analysis using polystyrene as a standard revealed that p-DTPF4 had a number-average molecular weight (Mn) of 11 kDa, with a polydispersity index (PDI) of 1.59; p-DTPF4-EBEH showed an Mn of 12 kDa, with a PDI of 2.50; p-EBEH exhibited an Mn of 4.2 kDa, with a PDI of 2.06. It is noteworthy that, in this study, we employed a catalyst system with moderate activity to prevent carbon–carbon coupling side reactions at the beta position of thienyl.41 The lower Mn of p-EBEH may be attributed to its limited solubility in the reaction solvent or the likelihood of intermediates undergoing debromination.42 Attempts were made to synthesize p-EBEH via ferric chloride-based oxidative polymerization;26 however, the resulting polymer exhibited extremely low solubility (rendering it unsuitable for PSC fabrication), likely due to its high molecular weight and strong intermolecular π–π interactions.
2.2 Polymer glass transition
Differential scanning calorimetry (DSC) analyses revealed the Tg values of spiro-OMeTAD, p-DTPF4, p-DTPF4-EBEH, and p-EBEH as 122 °C, 199 °C, 164 °C, and 62 °C, respectively (Fig. S2, ESI†). We observed that the planar and rigid conjugated core led to a weaker main chain relaxation signal in p-DTPF4.43 Interestingly, the Tg value of the alternating copolymer p-DTPF4-EBEH falls within the range observed for the homopolymers p-DTPF4 and p-EBEH, providing valuable insights for tailoring copolymers with suitable Tg values.Additionally, molecular dynamics simulations were conducted to determine the specific volume values of the amorphous solid across various temperatures (Fig. S3A–D, ESI†). Linear fittings were applied to the specific volume data in both low-temperature and high-temperature regions, with the intersection of the fitted lines providing the theoretical Tg value. It is noteworthy that the Tg values obtained from molecular dynamics simulations were higher due to the rapid cooling rate during the simulation.43 Furthermore, the molecular weight of all three polymers was presumed to be around 10 kDa in the simulations. Nevertheless, a strong correlation was observed between the Tg values obtained from DSC measurements and those from molecular dynamics simulations (Fig. S3E, ESI†).
2.3 Energy levels and hole extraction
Fig. S4A in ESI† depicts the cyclic voltammograms of the three conjugated polymers. Analyses revealed HOMO energy levels of −5.34 eV for p-DTPF4, −5.17 eV for p-DTPF4-EBEH, and −4.94 eV for p-EBEH. Additionally, HOMO energy levels were determined via ultraviolet photoelectron spectroscopy after depositing the conjugated polymers onto ITO substrates coated with PEDOT:PSS, yielding values of −5.22 eV for p-DTPF4, −4.96 eV for p-DTPF4-EBEH, and −4.65 eV for p-EBEH (Fig. S5, ESI†). While discrepancies exist in the absolute values of the HOMO energy levels obtained from ultraviolet photoelectron spectroscopy and cyclic voltammogram for each conjugated polymer, these experimental values align well with the theoretically calculated HOMO energy levels (Fig. 1B and S4B, ESI†).Fig. 1C illustrates the HOMO contour plots of the conjugated polymers. In p-DTPF4, the HOMO predominantly distributes over the thiophene ring and the dibenzofuran fragment, with minimal presence on the terminal benzene ring of oxa[5]helicene and virtually none on the hexylphenyl moiety. In p-EBEH, the HOMO primarily distributes across the thiophene ring, benzene ring, and oxygen atoms, with limited presence on the 1,2-ethylene and hexyl groups. Contrasting with homopolymers, the alternating copolymer p-DTPF4-EBEH displays HOMO distribution over the thiophene ring, the benzene ring directly linked to 3,4-ethylenedioxythiophene, oxygen atoms, and the dibenzofuran fragment, with reduced presence on the terminal benzene ring of oxa[5]helicene and virtually none on the 1,2-ethylene, hexyl, and hexylphenyl groups.
Subsequently, the UV-vis absorption spectra of these organic semiconductor films were recorded, and the optical bandgaps were determined via Tauc plots (Fig. S6, ESI†). These data were integrated with the HOMO energy levels acquired from ultraviolet photoelectron spectroscopy to generate the lowest unoccupied molecular orbital (LUMO) energy levels. The energy level diagram for PSCs is depicted in Fig. 1D. Thermodynamic analysis underscored that, similar to spiro-OMeTAD, these conjugated polymers effectively impeded the injection of electrons from the photoexcited FAPbI3 perovskite to the gold electrode, but ensured the driving force of hole extraction.
The hole extraction capacity of these organic semiconductors was assessed via time-resolved photoluminescence decay measurements (Fig. S7, ESI†). Detailed information on sample preparation and measurements can be found in the Experimental Section of ESI.† Upon excitation with picosecond pulsed laser, the amplitude-weighted average lifetime of photoluminescence at 810 nm for the FAPbI3 thin film coated with polystyrene was determined to be 5.4 μs (Table S1, ESI†). Substituting polystyrene with organic semiconductors led to a substantial reduction in photoluminescence lifetime, by over two orders of magnitude. Subsequent calculation of hole extraction rate constants (Table S1, ESI†) revealed a relationship between the rate constant and the driving force for hole extraction, which can be described by the Marcus electron transfer theory (Fig. 1E). Further computations suggested that these organic semiconductors all achieve hole extraction yields approaching 100% (Table S1, ESI†).
2.4 Hole density, conductivity, and mobility
In intrinsic organic semiconductors, a limited number of charge carriers are generated through thermally activated disproportionation mechanisms. For materials with optical bandgaps exceeding 1.5 eV, the charge carrier density produced by this mechanism at room temperature is below 104 cm−3. Metal–insulator–semiconductor (MIS) devices were constructed, and their impedance spectra were recorded to determine the capacitances of organic semiconductors at varying bias potentials. The hole density (p) in pristine organic semiconductor film was determined using the Mott–Schottky relationship. For details, see the Experimental Section in ESI.† The results indicated that the p values for spiro-OMeTAD was 1.3 × 1017 cm−3, for p-DTPF4 was 5.3 × 1016 cm−3, for p-DTPF4-EBEH was 3.0 × 1017 cm−3, and for p-EBEH was 2.8 × 1018 cm−3. Notably, these p values notably surpass those generated by thermally activated disproportionation. Intriguingly, organic semiconductors with high HOMO energy levels appear to have inadvertently experienced doping by oxygen from the atmosphere, even without deliberate introduction of additional dopants. At this stage, oxygen is converted to superoxide anions, establishing electrostatic equilibrium with the cationic free radicals generated through the oxidation of the organic semiconductor. Analysis reveals a strong linear correlation between the logarithm of p and the HOMO energy level obtained from ultraviolet photoelectron spectroscopy (Fig. 2A). This observation implies that the equilibrium constant for air doping in organic semiconductors relies on the Gibbs free energy of the reaction. Consequently, the shallower the HOMO energy level of the organic semiconductor, the higher the Gibbs free energy of the chemical reaction with oxygen in the air, leading to a greater equilibrium constant and, consequently, a higher p value produced.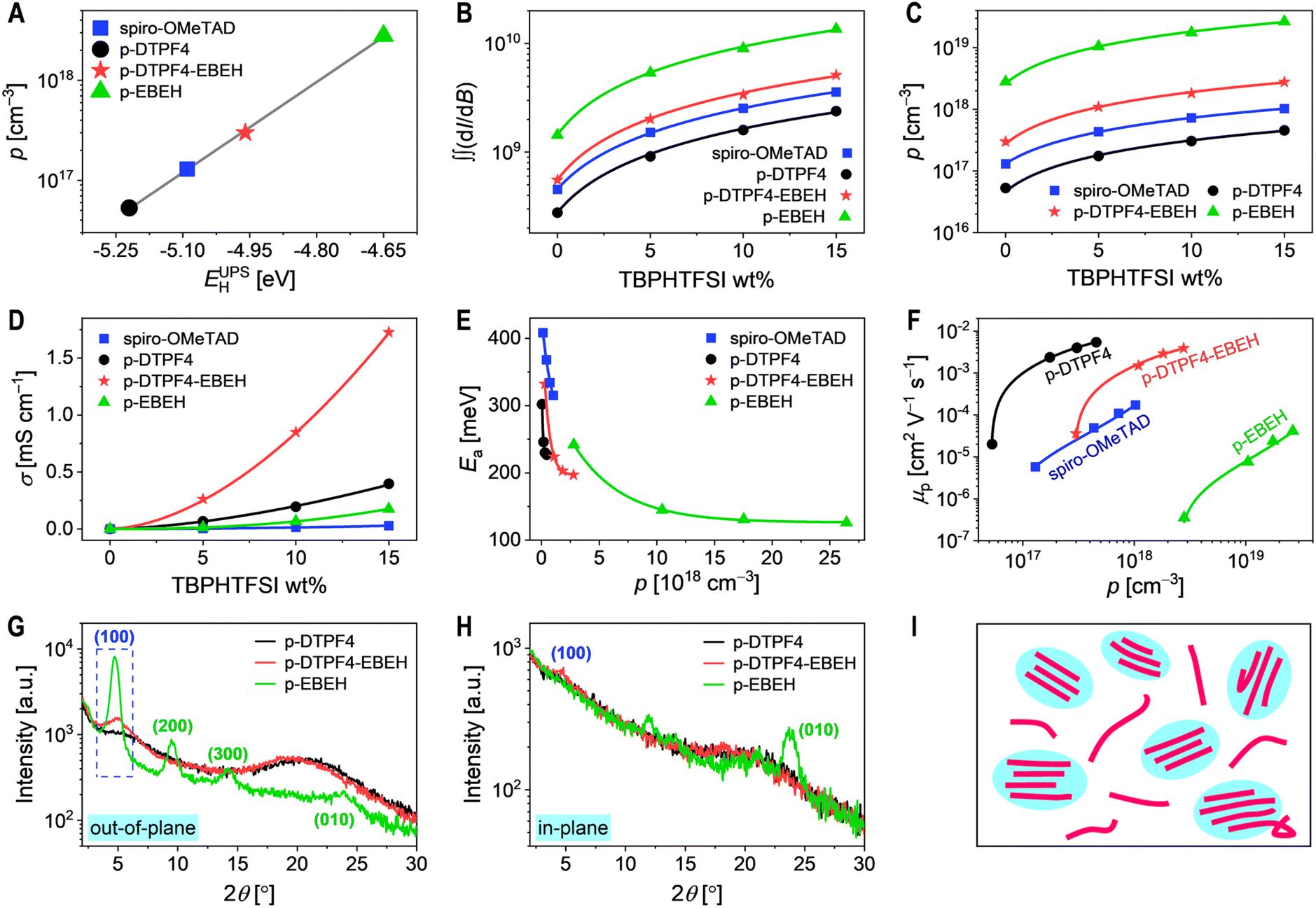 | ||
| Fig. 2 (A) Semi-logarithmic plot depicting the relationship between hole density (p) and the energy levels of HOMO, acquired through ultraviolet photoelectron spectroscopy measurements (EUPSH). The solid gray line represents a linear fit. (B) Semi-logarithmic plots illustrating the quadratic integral of the electron paramagnetic resonance signal (∬(dI/dB)) as a function of TBPHTFSI weight percentage. Linear fits in conventional coordinates are denoted by solid lines. (C) Semi-logarithmic plots displaying the variation of hole density (p) with respect to TBPHTFSI weight percentage. Linear fits in conventional coordinates are delineated by solid lines. (D) Plots depicting the conductivity (σ) as a function of TBPHTFSI weight percentage. Solid lines represent power-law fits. (E) Plot of activation energy (Ea) versus hole density (p). Solid lines depict mono-exponential decay fits. (F) Logarithmic plots illustrating the dependence of hole mobility (μp) on hole density (p). Solid lines serve as visual guide lines. (G and H) Out-of-plane and in-plane grazing-incidence X-ray diffraction patterns of conjugated polymer thin films on monocrystalline silicon substrates. (I) Schematic representation elucidating the microstructure of p-EBEH thin film, where crystalline regions are discontinuous. | ||
To enhance the p generated by air doping, we blended a small quantity of 4-tert-butylpyridinium bis(trifluoromethanesulfonyl)imide (TBPHTFSI) with organic semiconductor.44 As depicted in Fig. 2B, the quadratic integral intensity (∬(dI/dB)) of the electron paramagnetic resonance signal (Fig. S8, ESI†) exhibited a linear increase with the rise in TBPHTFSI's weight percentage. By comparing the magnitude of ∬(dI/dB) with and without TBPHTFSI, we estimated the p values in the organic semiconductor composite films. As illustrated in Fig. S9 of ESI,† the p values in the organic semiconductor composite films increased linearly with the increasing weight percentage of TBPHTFSI, confirming that TBPHTFSI facilitates air oxidation doping in these organic semiconductors, with a reaction order close to 1. At a TBPHTFSI weight percentage of 15%, the p values in the composite films based on spiro-OMeTAD, p-DTPF4, p-DTPF4-EBEH, and p-EBEH were determined to be 1.0 × 1018 cm−3, 4.6 × 1017 cm−3, 2.8 × 1018 cm−3, and 2.6 × 1019 cm−3, respectively (Fig. 2C). Furthermore, a correlation was observed between the HOMO energy level of the organic semiconductor and the p value, suggesting that higher HOMO energy levels correspond to higher p values.
Next, we evaluated the direct-current conductivities (σ) of both pristine and TBPHTFSI-containing organic semiconductor films using interdigital gold electrodes, unveiling a power-law augmentation of σ as the weight percentage of TBPHTFSI increased (Fig. 2D). Pristine organic semiconductor films displayed σ values of 0.12 μS cm−1 for spiro-OMeTAD, 0.17 μS cm−1 for p-DTPF4, 1.7 μS cm−1 for p-DTPF4-EBEH, and 0.16 μS cm−1 for p-EBEH. Conversely, in organic semiconductor composite films containing 15% weight percentage of TBPHTFSI, σ values were substantially elevated: 28 μS cm−1 for spiro-OMeTAD, 396 μS cm−1 for p-DTPF4, 1727 μS cm−1 for p-DTPF4-EBEH, and 175 μS cm−1 for p-EBEH. Additionally, we conducted temperature-dependent conductivity measurements (Fig. S10, ESI†). Utilizing the Arrhenius equation for analysis enabled us to determine the activation energy for hole conduction. As illustrated in Fig. 2E, the activation energy displayed a single-exponential decay with increasing p.
Based on p and σ, the hole mobility (μp) can be determined using the formula μp = σ/qp, with q representing the elementary charge. As depicted in Fig. 2F, μp shows a positive correlation with p. Organic semiconductors are characterized by energy disorder and may contain trace impurities.45 When the p is low, more holes tend to be trapped by deep-level traps, hindering their thermal activation and participation in transport, resulting in reduced μp. Conversely, as the p increases, more holes occupy shallow traps, increasing the chances of thermal activation, thereby promoting faster transport and higher μp. At a given p value, the μp ranking among these four organic semiconductors is as follows: p-DTPF4 > p-DTPF4-EBEH > spiro-OMeTAD > p-EBEH. Notably, the p-EBEH film exhibits the highest crystallinity as revealed by the grazing incidence X-ray diffraction measurements (Fig. 2G and H). However, due to the low molecular weight and thus the short single-chain length, the connectivity between crystalline regions is poor, thereby impeding hole transport (Fig. 2I). This highlights the critical role of molecular weight in the hole transport of conjugated polymers.46,47
2.5 Photovoltaic performance
The organic semiconductor was blended with TBPHTFSI at an 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 weight ratio and dissolved in chlorobenzene solution containing 4-tert-butylpyridine. This solution was then spin-coated to form a hole transport layer for constructing PSCs with an ITO/SnO2/FAPbI3/hole transport layer/Au architecture. Detailed fabrication procedures and measurement methods are outlined in the Experimental Section of ESI.† Photocurrent density–voltage (J–V) curves were recorded under simulated AM1.5G irradiation at 100 mW cm−2. Statistical analysis of photovoltaic parameters, including short-circuit current density (JSC), open-circuit voltage (VOC), fill factor (FF), and power conversion efficiency (PCE), was conducted on ten cells of each type (Fig. 3A–D), suggesting a good reproducibility. Representative J–V curves are shown in Fig. 3E, with corresponding photovoltaic parameters listed in Table 1. The spiro-OMeTAD-based cell exhibited a JSC of 26.18 mA cm−2, a VOC of 1.165 V, an FF of 79.6%, and a PCE of 24.3%. In contrast, the p-DTPF4-based cell showed reduced JSC (25.49 mA cm−2), VOC (1.120 V), FF (62.4%), and consequently, a lower PCE of 17.8%. Similarly, the p-EBEH-based cell demonstrated even lower JSC (23.02 mA cm−2), VOC (0.925 V), and FF (49.1%), resulting in the lowest PCE of 10.5%. Encouragingly, the p-DTPF4-EBEH cell displayed a JSC (26.19 mA cm−2) comparable to that of the spiro-OMeTAD-based cell, a higher VOC (1.190 V), a higher FF (82.8%), and ultimately achieved a superior PCE of 25.8%. Comparison of forward and reverse scan J–V curves revealed minimal hysteresis in the p-DTPF4-EBEH-based cell (Fig. S11, ESI†). Furthermore, when the p-DTPF4-EBEH-based cell was switched from open circuit to a bias voltage of 1.035 V, steady-state photocurrent density (24.85 mA cm−2) was rapidly attained, with a steady-state PCE output of 25.72% (Fig. S12, ESI†), consistent with J–V measurements.
:15 weight ratio and dissolved in chlorobenzene solution containing 4-tert-butylpyridine. This solution was then spin-coated to form a hole transport layer for constructing PSCs with an ITO/SnO2/FAPbI3/hole transport layer/Au architecture. Detailed fabrication procedures and measurement methods are outlined in the Experimental Section of ESI.† Photocurrent density–voltage (J–V) curves were recorded under simulated AM1.5G irradiation at 100 mW cm−2. Statistical analysis of photovoltaic parameters, including short-circuit current density (JSC), open-circuit voltage (VOC), fill factor (FF), and power conversion efficiency (PCE), was conducted on ten cells of each type (Fig. 3A–D), suggesting a good reproducibility. Representative J–V curves are shown in Fig. 3E, with corresponding photovoltaic parameters listed in Table 1. The spiro-OMeTAD-based cell exhibited a JSC of 26.18 mA cm−2, a VOC of 1.165 V, an FF of 79.6%, and a PCE of 24.3%. In contrast, the p-DTPF4-based cell showed reduced JSC (25.49 mA cm−2), VOC (1.120 V), FF (62.4%), and consequently, a lower PCE of 17.8%. Similarly, the p-EBEH-based cell demonstrated even lower JSC (23.02 mA cm−2), VOC (0.925 V), and FF (49.1%), resulting in the lowest PCE of 10.5%. Encouragingly, the p-DTPF4-EBEH cell displayed a JSC (26.19 mA cm−2) comparable to that of the spiro-OMeTAD-based cell, a higher VOC (1.190 V), a higher FF (82.8%), and ultimately achieved a superior PCE of 25.8%. Comparison of forward and reverse scan J–V curves revealed minimal hysteresis in the p-DTPF4-EBEH-based cell (Fig. S11, ESI†). Furthermore, when the p-DTPF4-EBEH-based cell was switched from open circuit to a bias voltage of 1.035 V, steady-state photocurrent density (24.85 mA cm−2) was rapidly attained, with a steady-state PCE output of 25.72% (Fig. S12, ESI†), consistent with J–V measurements.
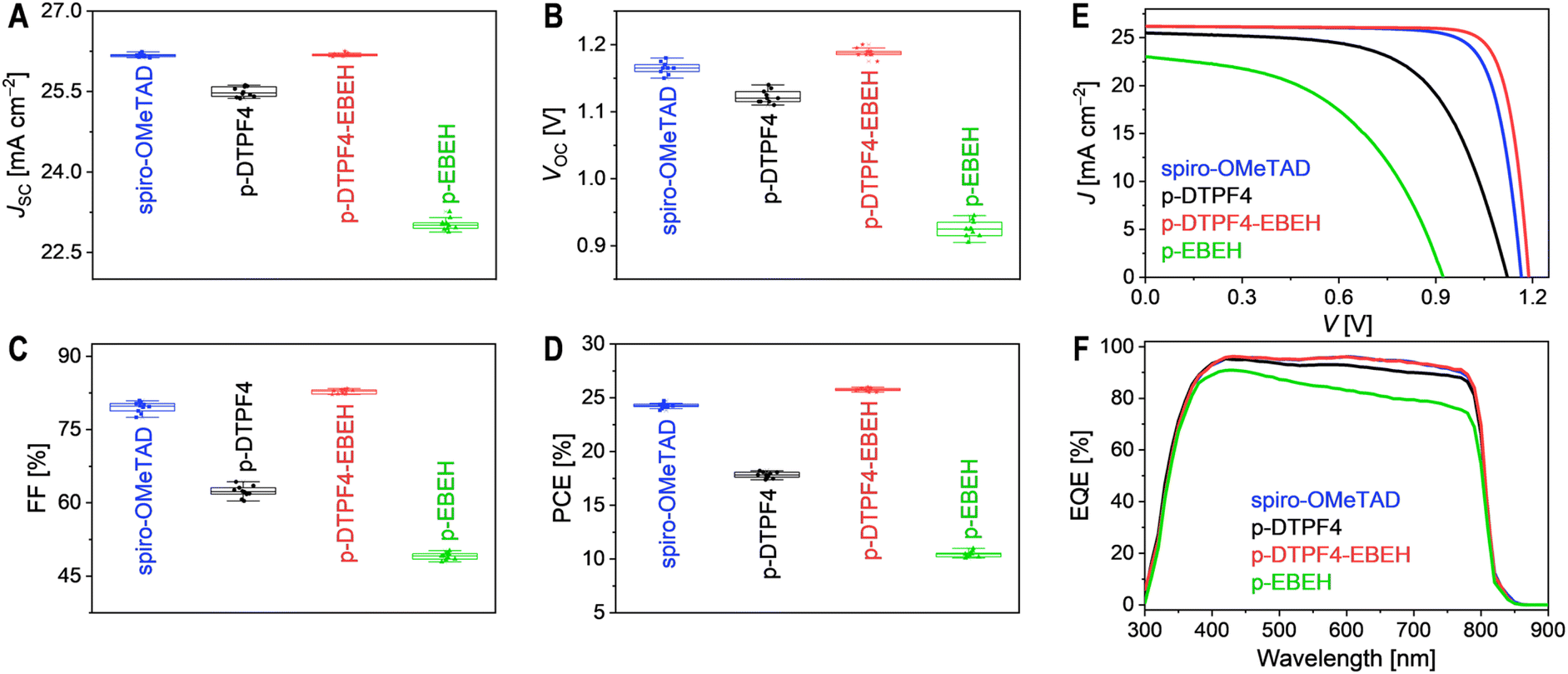 | ||
| Fig. 3 (A–D) Statistical analysis of photovoltaic parameters of PSCs fabricated with varying hole transport layers under simulated AM1.5G irradiation at 100 mW cm−2: (A) short-circuit photocurrent density (JSC); (B) open-circuit voltage (VOC); (C) fill factor (FF); (D) power conversion efficiency (PCE). (E) Representative photocurrent density–voltage (J–V) curves. (F) External quantum efficiency (EQE) spectra. | ||
| Cell | J SC [mA cm−2] | V OC [V] | FF [%] | PCE [%] | J EQESC [mA cm−2] | R sh [kΩ] | R s [Ω] | I s [pA] |
|---|---|---|---|---|---|---|---|---|
| a J SC: short-circuit current density; VOC: open-circuit voltage; FF: fill factor; PCE: power conversion efficiency; JEQESC: short-circuit current density estimated by external quantum efficiency (EQE) spectrum; Rsh: shunt resistance; Rs: series resistance; Is: reverse saturation current. | ||||||||
| Spiro-OMeTAD | 26.18 | 1.165 | 79.6 | 24.3 | 25.93 | 42 | 19 | 0.004 |
| p-DTPF4 | 25.49 | 1.120 | 62.4 | 17.8 | 25.24 | 20 | 12 | 228 |
| p-DTPF4-EBEH | 26.19 | 1.190 | 82.8 | 25.8 | 25.94 | 76 | 9 | 0.002 |
| p-EBEH | 23.02 | 0.925 | 49.1 | 10.5 | 22.78 | 2.3 | 14 | 2608 |
Subsequently, we conducted external quantum efficiency (EQE) measurements for the PSCs under various monochromatic light irradiations (Fig. 3F). These EQE spectra were then combined with the standard AM1.5G solar spectrum (ASTM G173-03, Fig. S13A, ESI†) to estimate the short-circuit photocurrent density of the cells under AM1.5G irradiation (JEQESC, Table 1). The results reveal a strong linear correlation between JEQESC and JSC (Fig. S13B, ESI†). Specifically, PSCs employing spiro-OMeTAD and p-DTPF4-EBEH exhibited similar EQE spectra, with maximal EQE values reaching 96%; accordingly, their JEQESC values were nearly identical, at 25.93 mA cm−2 and 25.94 mA cm−2, respectively. Conversely, as the incident monochromatic light wavelength gradually shifted from 420 nm to 800 nm, the EQE of the p-DTPF4-based cell displayed a gradual decline, accompanied by a corresponding decrease in JEQESC to 25.24 mA cm−2. Furthermore, the EQE reduction observed in the p-EBEH-based cell was more pronounced, with JEQESC registering the lowest value at 22.78 mA cm−2. The decrease in EQE in the longer wavelength region is attributed to diminished electron collection efficiency, reflecting variations in charge recombination at the interface between the hole transport layers and the perovskite layer. In other words, faster charge recombination leads to more significant EQE decay with wavelength redshift.
To understand the differences in VOC and FF resulting from distinct hole transport layers, we applied the Shockley diode equation to fit the J–V curves,48 unveiling crucial parameters like shunt resistance (Rsh), series resistance (Rs), and reverse saturation current (Is), collected in Table 1. Broadly, elevated Rsh and diminished Is contribute to heightened VOC, while reduced Rs, augmented Rsh, and diminished Is foster enhanced FF. Noteworthy is the progressive decline in Rs across cells utilizing spiro-OMeTAD, p-EBEH, p-DTPF4, and p-DTPF4-EBEH, attributed to the incremental conductivity of the hole transport layer (refer to Fig. 2D). The FF of cells employing p-EBEH, p-DTPF4, and p-DTPF4-EBEH exhibited a consecutive rise, primarily owing to the escalating Rsh and dwindling Is. For spiro-OMeTAD-based cell, the second-highest Rsh and second-smallest Is assuaged the adverse effects of the largest Rs, resulting in the second-highest FF. In p-EBEH-based cell, the lowest Rsh might correlate with its highest hole density due to the highest HOMO energy level, leading to rapid charge recombination at the interface between the hole transport layer and the perovskite layer; the highest Is could stem from the incomplete morphology of the hole transport layer. The confluence of these factors led to the lowest VOC, FF, and JSC. Despite possessing the deepest HOMO energy level and lowest hole density, p-DTPF4 failed to manifest the highest VOC, attributed to its lower Rsh and larger Is, indicative of rapid charge recombination at the hole transport layer/perovskite layer interface, possibly due to incomplete morphology of the hole transport layer.
Upon removal of the encapsulation materials and gold electrodes from the PSCs, we scrutinized the morphology of the hole transport layers using scanning electron microscopy. On the surface of the perovskite polycrystalline thin film composed of micrometer-sized grains (Fig. S14, ESI†), the TBPHTFSI-containing composite material derived from spiro-OMeTAD formed a relatively uniform film with a few pinholes (Fig. 4A and an enlarged view in Fig. S15, ESI†). In contrast, the surface of the p-DTPF4 composite film exhibited densely scattered pinholes (an enlarged view in Fig. S15, ESI†), accompanied by several penetrating circular pores measuring several hundred nanometers in diameter (Fig. 4B). Moreover, the p-EBEH composite film displayed a rough morphology with numerous worm-like protrusions and minor localized fractures distributed on the surface (Fig. 4D). Remarkably, the composite film based on p-DTPF4-EBEH appeared smooth, dense, and intact (Fig. 4C).
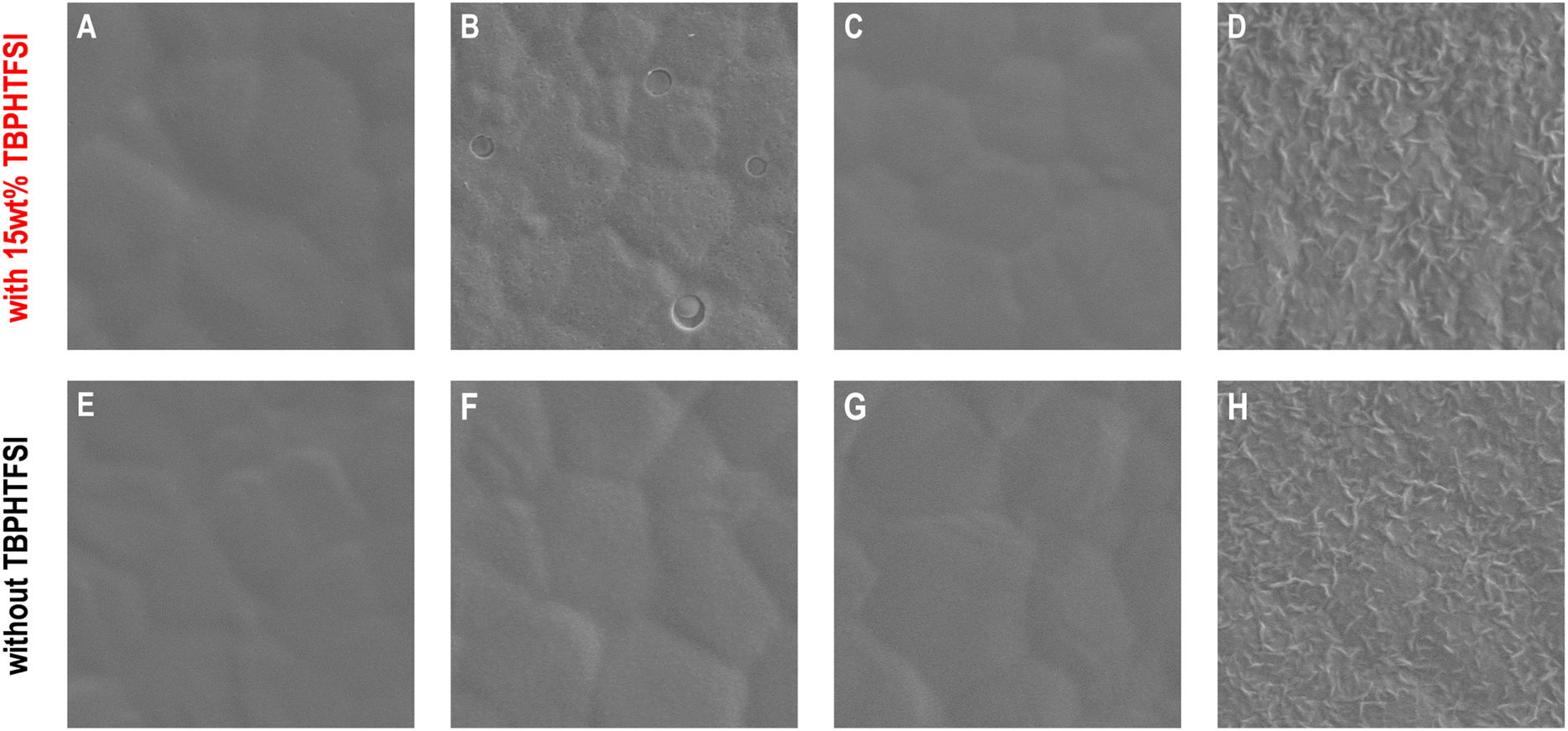 | ||
| Fig. 4 Top-view scanning electron microscopy images of the organic semiconductor–TBPHTFSI composite layers deposited on the surface of FAPbI3 perovskite polycrystalline thin films and the corresponding pristine organic semiconductor layers: (A and E) spiro-OMeTAD, (B and F) p-DTPF4, (C and G) p-DTPF4-EBEH, (D and H) p-EBEH. Image size: 5 μm × 5 μm. Encapsulation materials and gold electrodes were removed before imaging. | ||
Some composite films display suboptimal morphology—could this stem from inadequate compatibility between the organic semiconductor and TBPHTFSI due to the large polarity difference? To investigate this possibility, we conducted control experiments by depositing pristine organic semiconductor films onto the surface of the perovskite polycrystalline thin film. As depicted in Fig. 4E–G, the pristine films of spiro-OMeTAD, p-DTPF4, and p-DTPF4-EBEH all exhibit uniformity, smoothness, and integrity. Obviously, the compatibility of organic semiconductor and ionic doping promoter plays a crucial role in controlling the morphology of hole transport layer and thus photovoltaic performance. However, the pristine film of p-EBEH remains notably rough (Fig. 4H). Furthermore, when deposited on monocrystalline silicon substrates, p-EBEH demonstrates the highest degree of crystallinity (as shown in Fig. 2G and H). Typically, crystalline or semi-crystalline polymer films are less smooth than amorphous ones.
2.6 Thermal stability at 85 °C
In light of the observed low PCEs (<20%) in PSCs utilizing p-DTPF4 and p-EBEH, our subsequent investigation delved into the thermal stability at 85 °C, comparing cells employing p-DTPF4-EBEH and spiro-OMeTAD. Six cells of each type underwent aging in an 85 °C oven (ambient relative humidity: 45–90%; with oven humidity at 5% when ambient humidity was 61%). Periodically removed from the aging process, the cells underwent J–V measurements under AM1.5G irradiation at room temperature. The temporal evolution of PCE is illustrated in Fig. 5A. After 1000 hours, the p-DTPF4-EBEH-based cell exhibited a decrease in PCE from 25.7% to 23.9%, retaining 93% of its initial value. In contrast, the spiro-OMeTAD-based cell retained only 63% of its initial PCE under identical conditions. Statistical analysis of photovoltaic parameters for aged cells is presented in Fig. S16, ESI.† For easy comparison, J–V curves of representative cells before and after aging are plotted in Fig. 5B, with corresponding photovoltaic parameters listed in Table S2, ESI.† The decline in PCE of the p-DTPF4-EBEH-based cell primarily stemmed from a 3.3% decrease in FF, a 2.5% decrease in VOC, and a 1.1% decrease in JSC. In contrast, the spiro-OMeTAD-based cell experienced more substantial declines, with FF decreasing by 19%, VOC by 9%, and JSC by 14%. The EQE spectra of cells based on both organic semiconductors before and after aging are depicted in Fig. 5C. Aging led to a reduction in the EQE maximum. Specifically, the maximal EQE value decreased from 96% to 82% for the spiro-OMeTAD-based cell, whereas for the p-DTPF4-EBEH-based cell, the decrease was only 1.4%. Notably, a strong linear correlation between JEQESC and JSC was observed for both aged and unaged cells (Fig. S17, ESI†).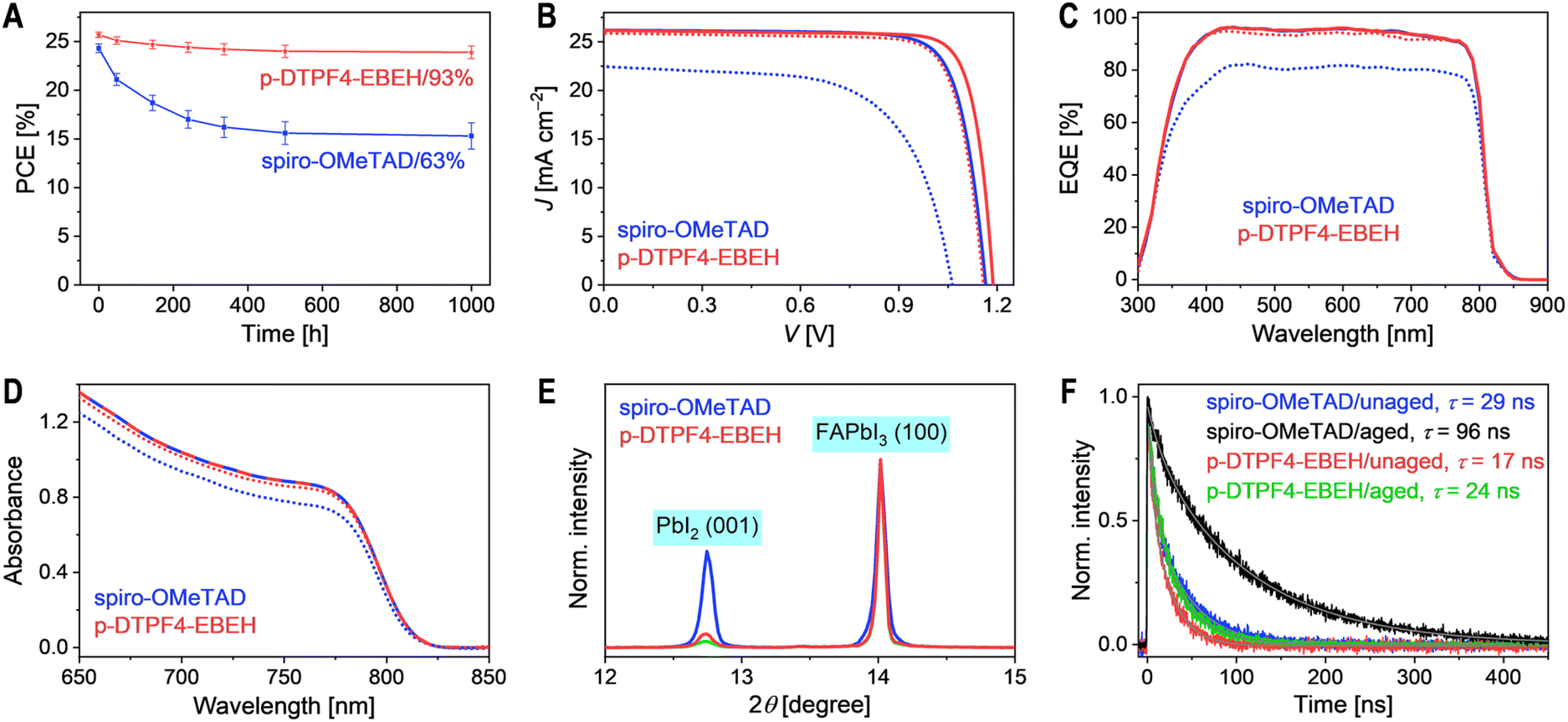 | ||
| Fig. 5 (A) Temporal changes in power conversion efficiency (PCE) over a 1000 hour aging period at 85 °C (ISOS-D-2). The PCE data from six cells of each type are averaged and subjected to error analysis. (B) Representative photocurrent density–voltage (J–V) curves of unaged and aged cells under simulated AM1.5G irradiation at 100 mW cm−2. In panels (B)–(D), data from unaged and 1000 hour 85 °C aged cells are represented by solid and dashed lines, respectively. (C) External quantum efficiency (EQE) spectra before and after aging. (D) UV-vis absorption spectra before and after aging. (E) X-ray diffraction patterns before (green) and after (blue and red) aging. The patterns from unaged cells are nearly identical and are indicated by green solid lines. (F) Time-resolved photoluminescence decays at 810 nm before and after aging. Bi-exponential decay fits are shown by gray solid lines; amplitude-weighted average photoluminescence lifetimes (τ) are calculated using fit parameters. Excitation wavelength: 670 nm. | ||
UV-vis absorption measurements (Fig. 5D) revealed a reduction in optical density within the 650–850 nm wavelength range, indicating perovskite layer degradation. Remarkably, degradation of the perovskite layer in the p-DTPF4-EBEH-based cell was notably less severe compared to the spiro-OMeTAD-based counterpart. Additionally, X-ray diffraction analysis (Fig. 5E) illustrated an increase in the height of the PbI2 (001) diffraction peak after aging at 85 °C. The area of this diffraction peak in the spiro-OMeTAD-based cell expanded to 10.0 times its initial value, 5.6 times greater than observed in the p-DTPF4-EBEH-based cell (1.8 times). Fig. 5F demonstrated that after thermal aging, the photoluminescence lifetime of the p-DTPF4-EBEH-based cell extended from 17 ns to 24 ns, while that of the spiro-OMeTAD-based cell elongated from 29 ns to 96 ns. Notably, the photoluminescence lifetime of the SnO2-free dummy cell with polystyrene decreased from 5425 ns to 2626 ns after aging (Fig. S18, ESI†), suggesting that thermal aging induces the generation of more defects in the perovskite layer and accelerates defect-assisted non-radiative recombination. Therefore, the prolonged photoluminescence lifetime of aged cells may be linked to the formation of wide-bandgap species at the perovskite layer/hole transport layer interface. Moreover, this scenario might also be associated with the fracture of the hole transport layer, potentially impeding hole extraction from the photoexcited perovskite.
Following aging, the hole transport layer employing spiro-OMeTAD displayed numerous nanopores (Fig. 6A). This pronounced morphological degradation likely impedes hole extraction and transport, and leads to the formation of microcontacts between the perovskite layer and the gold electrode. In contrast, the morphology of the hole transport layer utilizing p-DTPF4-EBEH remained nearly unchanged, preserving relatively good integrity (Fig. 6B). Moreover, fluorescence optical microscopy imaging revealed no discernible microstructures in the unaged cells (Fig. S19, ESI†). However, post-thermal aging, the perovskite layer with spiro-OMeTAD displayed many green luminescent spots (Fig. 6C), whereas the layer with p-DTPF4-EBEH exhibited only a few such spots (Fig. 6D). UV-vis absorption, X-ray diffraction, photoluminescence decay, scanning electron microscopy, and fluorescence optical microscopy measurements collectively affirm that p-DTPF4-EBEH more effectively suppresses the thermal decomposition of the perovskite layer compared to spiro-OMeTAD.
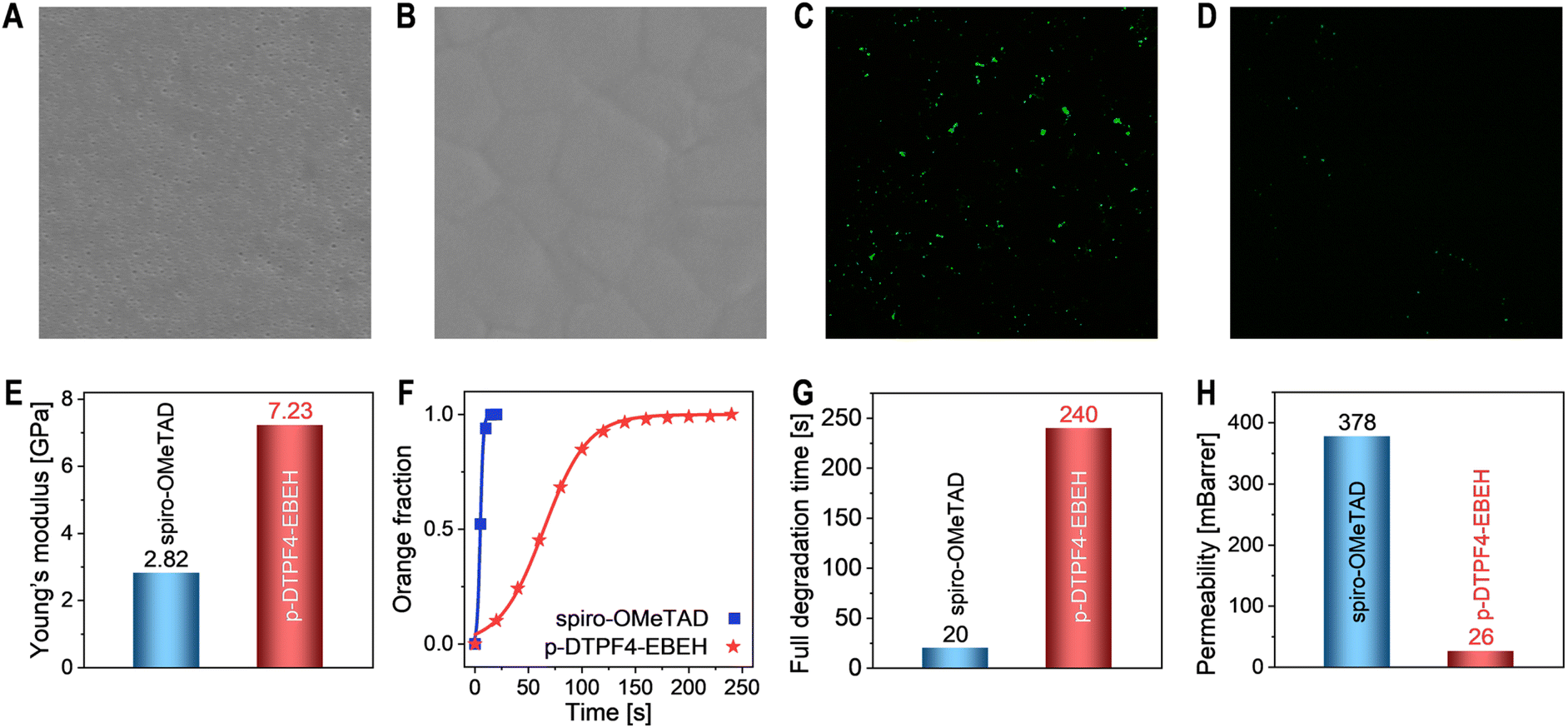 | ||
| Fig. 6 (A and B) Top-view scanning electron microscopy images of hole transport layers in aged PSCs: (A) spiro-OMeTAD; (B) p-DTPF4-EBEH. Image size: 5 μm × 5 μm. (C and D) Fluorescence optical microscopy images of aged cells: (C) spiro-OMeTAD; (D) p-DTPF4-EBEH. Image size: 25 μm × 25 μm. (E) Young's moduli of organic semiconductor composite films. (F) Plots of the proportion of orange-yellow areas in polarized microscopy images over time subsequent to the application of a water droplet. (G) Full degradation time of FAPbI3 films coated with different organic semiconductor composite films. (H) Permeabilities of water molecules in different hole transport layers as obtained from molecular dynamics simulations. | ||
To unravel the origins of the morphological evolution of the hole transport layer, we conducted nanoindentation tests (Fig. S20A and B, ESI†) to evaluate the Young's moduli of the composite films, yielding values of 2.82 GPa for spiro-OMeTAD and 7.23 GPa for p-DTPF4-EBEH, respectively (Fig. 6E). Notably, p-DTPF4-EBEH exhibited a significantly higher Young's modulus, indicating its enhanced resistance to deformation within the hole transport layer. Supplementary molecular dynamics simulations, investigating temperature-dependent variations in the Young's modulus, supported these results (see Fig. S20C in ESI†). Despite a reduction observed in the Young's modulus with increasing temperature, p-DTPF4-EBEH consistently demonstrated superior Young's moduli compared to spiro-OMeTAD across all temperature regimes. It is essential to emphasize the crucial role of cohesive energy density in elucidating intermolecular interactions, wherein heightened cohesive energy density aligns with elevated Young's modulus, as demonstrated in Fig. S20D, ESI.†
Organic–inorganic hybrid perovskites, such as FAPbI3, display remarkable sensitivity to humidity. Here, we evaluated the waterproofing capability of the hole transport layer through an accelerated experiment. In a control scenario, a water droplet applied to FAPbI3 film maintained an almost constant contact angle of 55° within 10 seconds, while the polarized optical microscopy window rapidly transitioned from black to orange-yellow (see Fig. S21A in ESI†), indicating the emergence of birefringent PbI2 microcrystals. Upon spin-coating a hole transport layer (∼80 nm) based on spiro-OMeTAD onto the FAPbI3 film surface, the water contact angle increased to 78° (Fig. S21B, ESI†), while a larger angle of 93° was observed with a hole transport layer (∼80 nm) based on p-DTPF4-EBEH (Fig. S21C, ESI†). Importantly, these contact angles remained stable within 20 seconds. However, polarized optical microscopy measurements revealed the effective mitigation of perovskite film degradation by the hole transport layers. Using Spyder software, we quantified the proportion of orange-yellow regions in polarized micrographs. Subsequent fitting of the relationship between orange-yellow regions and time with the Bohart–Adams model (Fig. 6F) revealed a complete degradation time of 240 seconds for the FAPbI3 film with p-DTPF4-EBEH and 20 seconds for the one with spiro-OMeTAD (Fig. 6G), underscoring the better waterproofing ability of p-DTPF4-EBEH. Furthermore, molecular dynamics and grand canonical Monte Carlo simulations were employed to determine the permeability of water molecules in the hole transport layers, yielding values of 26 mBarrer for the p-DTPF4-EBEH-based composite film and 378 mBarrer for the spiro-OMeTAD-based composite film (Fig. 6H).
The comprehensive analyses of device degradation elucidated that, in comparison to the reference material spiro-OMeTAD, p-DTPF4-EBEH displayed an increased Young's modulus and an enhanced waterproofing capability. This augmentation plays a pivotal role in maintaining the integrity of the hole transport layer under 85 °C thermal stress and effectively retards the degradation of the perovskite layer under heat and humidity conditions.
2.7 Operational stability
Utilizing perturb and observe method, we scrutinized the operational stability of PSCs within a nitrogen environment at 45 °C, subjected to simulated AM1.5G irradiation. The temporal evolution of crucial parameters, encompassing photocurrent density (JMPP), photovoltage (VMPP), and power conversion efficiency (PMPP) at the maximum power point (MPP), are shown in Fig. 7A–C. Within a mere 112 hour span, the PMPP of spiro-OMeTAD-based cell underwent a notable deterioration of 27%, chiefly attributed to a 11% reduction in JMPP and a 19% decline in VMPP. In contrast, across an extended period of 500 hours, the PMPP of p-DTPF4-EBEH-based cell displayed only a marginal decrease, diminishing from 25.0% to 23.8%. This result underscored a good retention rate of 95%, primarily stemming from a 2.6% decrease in JMPP and a 2.3% decline in VMPP.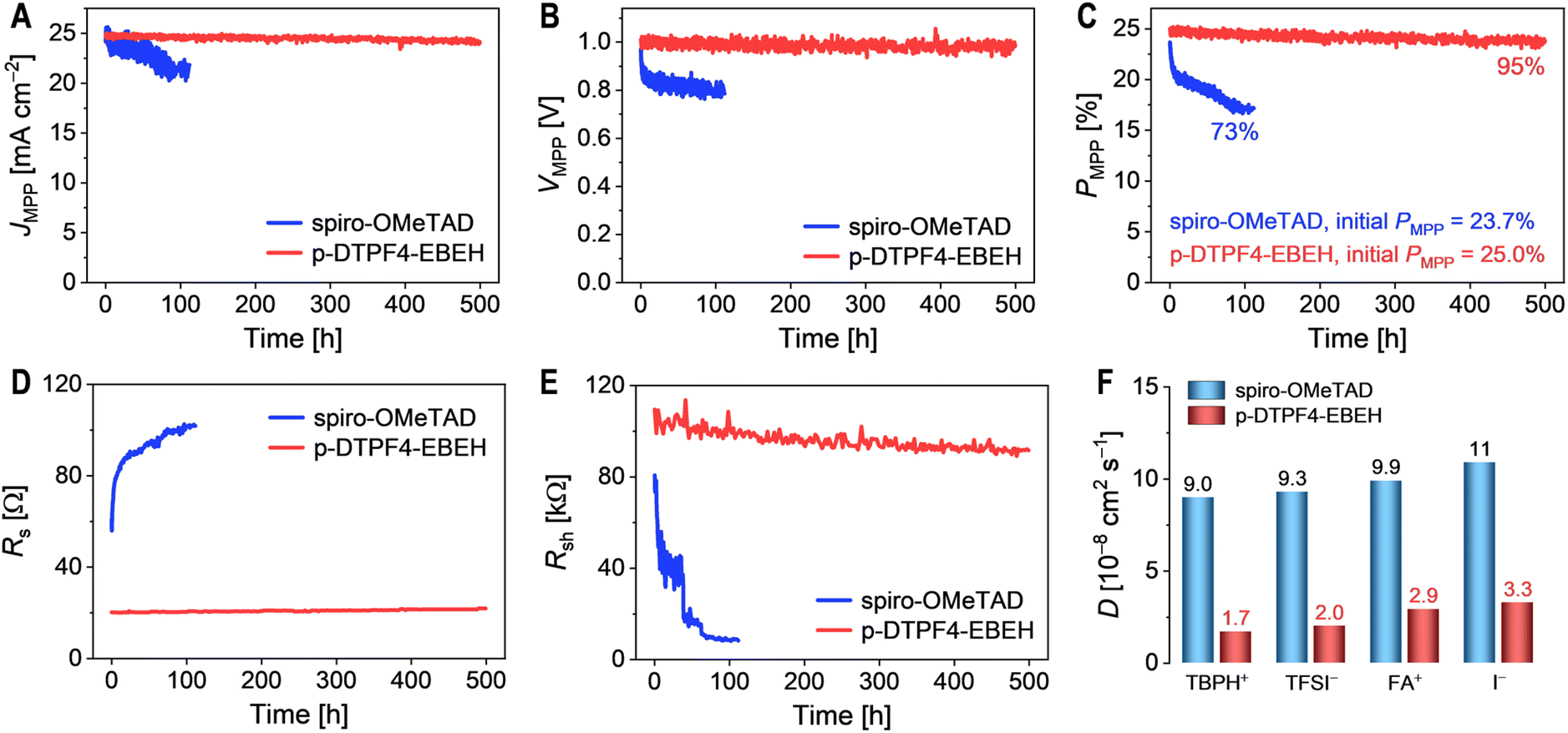 | ||
| Fig. 7 (A–C) Photovoltaic parameters obtained from maximum power point (MPP) tracking under simulated AM1.5G irradiation at 100 mW cm−2: (A) photocurrent density (JMPP); (B) photovoltage (VMPP); (C) power conversion efficiency (PMPP). (D and E) Evolution of series resistance (Rs) and shunt resistance (Rsh) over time during MPP tracking. (F) Diffusivities of ions within the hole transport layer at 45 °C. | ||
Throughout the MPP tracking process, J–V curves were intermittently measured and fitted using the Shockley diode equation. Fig. 7D and E depict the temporal evolution of the Rs and Rsh of the cells. As the cell operated, Rs exhibited a gradual increase, while Rsh showed a decrease. The decline in Rsh suggests an acceleration in charge recombination, likely associated with an increase in defects within the perovskite layer. The increase in Rs could be attributed to the rise in defects and scattering centers within the perovskite layer. Importantly, compared to the spiro-OMeTAD-based cell, the p-DTPF4-EBEH-based cell demonstrated a significantly reduced magnitude of variation in both Rsh and Rs.
Subsequently, employing molecular dynamics simulations, we delved into the ion diffusivities within the hole transport layer, encompassing 4-tert-butylpyridinium cation (TBPH+), bis(trifluoromethanesulfonyl)imide anion (TFSI−), formamidinium cation (FA+), and iodide anion (I−). At 45 °C, the diffusivities of each ion within the p-DTPF4-EBEH-based hole transport layer were notably lower compared to those within the spiro-OMeTAD-based counterpart (Fig. 7F). The magnitudes of diffusivities may correlate with the cohesive energy density within the hole transport layer. It is evident that the judicious selection of an appropriate hole transport layer to mitigate ion migration under the electric field is pivotal for enhancing the operational stability of PSCs.
3 Conclusions
In summary, we have synthesized a high-solubility heteroaromatic, DTPF4, characterized by a helical structure comprising nine fused rings and four hexylphenyl moieties. Leveraging palladium-catalyzed direct arylation polymerization, we have crafted the homopolymer p-DTPF4 with remarkable hole mobility. Through a strategic integration of the more electron-rich EBEH in an alternating copolymerization with DTPF4, we have engineered p-DTPF4-EBEH, which not only demonstrates appropriate HOMO energy level but also exhibits enhanced conductivity. Notably, compared to the conventional material spiro-OMeTAD, the hole transport layer founded on p-DTPF4-EBEH displays a plethora of superior attributes, including a more uniform film morphology, elevated Young's modulus, heightened waterproofing capability, and reduced diffusivities of external species. Harnessing the potential of p-DTPF4-EBEH, perovskite solar cells have achieved an impressive average efficiency of 25.8% alongside commendable stability. This exploration elucidating the intricate interplay among chemical structure, physical properties, and photovoltaic performance of conjugated polymers has furnished instrumental insights for the prospective design of hole-transporting materials, thereby fostering the continuous advancement of perovskite solar cell technologies.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Conceptualization: P. W. Data curation: Y. C.; Y. Z. Funding acquisition: Y. Y.; P. W. Investigation: Y. C.; Y. Z.; L. F.; Y. R.; J. Z.; J. Z. Methodology: Y. C.; J. Z. Project administration: J. Z.; Y. Y. Supervision: P. W. Validation: J. Z. Visualization: Y. C.; Y. Z.; J. Z. Writing – original draft: Y. C.; Y. Z. Writing – review & editing: P. W.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22275160 and 52073250) and the National Key Research and Development Program of China (2022YFA1204800).References
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS PubMed.
- NREL Best Research-Cell Efficiencies, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.pdf, accessed: 2024/06/26 Search PubMed.
- L. Calió, S. Kazim, M. Grätzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522 CrossRef PubMed.
- C. Altinkaya, E. Aydin, E. Ugur, F. H. Isikgor, A. S. Subbiah, M. De Bastiani, J. Liu, A. Babayigit, T. G. Allen, F. Laquai, A. Yildiz and S. De Wolf, Adv. Mater., 2021, 33, 2005504 CrossRef CAS PubMed.
- M. Kim, J. Jeong, H. Lu, T. K. Lee, F. T. Eickemeyer, Y. Liu, I. W. Choi, S. J. Choi, Y. Jo, H.-B. Kim, S.-I. Mo, Y.-K. Kim, H. Lee, N. G. An, S. Cho, W. R. Tress, S. M. Zakeeruddin, A. Hagfeldt, J. Y. Kim, M. Grätzel and D. S. Kim, Science, 2022, 375, 302 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531 CrossRef CAS PubMed.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724 CrossRef CAS PubMed.
- H. J. Snaith and M. Grätzel, Appl. Phys. Lett., 2006, 89, 262114 CrossRef.
- X. Zhao, H.-S. Kim, J.-Y. Seo and N.-G. Park, ACS Appl. Mater. Interfaces, 2017, 9, 7148 CrossRef CAS PubMed.
- A. K. Jena, M. Ikegami and T. Miyasaka, ACS Energy Lett., 2017, 2, 1760 CrossRef CAS.
- Y. Ren, Y. Wei, T. Li, Y. Mu, M. Zhang, Y. Yuan, J. Zhang and P. Wang, Energy Environ. Sci., 2023, 16, 3534 RSC.
- J. H. Heo, S. H. Im, J. H. Noh, T. N. Mandal, C.-S. Lim, J. A. Chang, Y. H. Lee, H.-j. Kim, A. Sarkar, M. K. Nazeeruddin, M. Grätzel and S. I. Seok, Nat. Photonics, 2013, 7, 486 CrossRef CAS.
- B. Cai, Y. Xing, Z. Yang, W.-H. Zhang and J. Qiu, Energy Environ. Sci., 2013, 6, 1480 RSC.
- J. Lee, M. M. Byranvand, G. Kang, S. Y. Son, S. Song, G.-W. Kim and T. Park, J. Am. Chem. Soc., 2017, 139, 12175 CrossRef CAS PubMed.
- L. Zhang, C. Liu, J. Zhang, X. Li, C. Cheng, Y. Tian, A. K.-Y. Jen and B. Xu, Adv. Mater., 2018, 30, 1804028 CrossRef PubMed.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T.-Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511 CrossRef CAS PubMed.
- Q. Fu, Z. Xu, X. Tang, T. Liu, X. Dong, X. Zhang, N. Zheng, Z. Xie and Y. Liu, ACS Energy Lett., 2021, 6, 1521 CrossRef CAS.
- H. Opoku, J. H. Lee, J.-J. Lee, H. Ahn and J. W. Jo, ACS Mater. Lett., 2022, 4, 2515 CrossRef CAS.
- Y. Bai, Z. Zhou, Q. Xue, C. Liu, N. Li, H. Tang, J. Zhang, X. Xia, J. Zhang, X. Lu, C. J. Brabec and F. Huang, Adv. Mater., 2022, 34, 2110587 CrossRef CAS PubMed.
- H.-S. Lin, T. Doba, W. Sato, Y. Matsuo, R. Shang and E. Nakamura, Angew. Chem., Int. Ed., 2022, 61, e202203949 CrossRef CAS PubMed.
- Q. Fu, H. Liu, S. Li, T. Zhou, M. Chen, Y. Yang, J. Wang, R. Wang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210356 CrossRef CAS PubMed.
- Q. Fu, X. Tang, H. Liu, R. Wang, T. Liu, Z. Wu, H. Y. Woo, T. Zhou, X. Wan, Y. Chen and Y. Liu, J. Am. Chem. Soc., 2022, 144, 9500 CrossRef CAS PubMed.
- H. Zhang, X. Yu, M. Li, Z. Zhang, Z. Song, X. Zong, G. Duan, W. Zhang, C. Chen, W.-H. Zhang, Y. Liu and M. Liang, Angew. Chem., Int. Ed., 2023, 62, e2023142 Search PubMed.
- Y. Cai, Y. Zhang, J. Zhang, X. Pan, M. R. Andersson and P. Wang, Angew. Chem., Int. Ed., 2024, 63, e20231581 Search PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268 CrossRef CAS PubMed.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem, 2020, 4, 66 CrossRef CAS PubMed.
- C. Graebe, W. Knecht and J. Unzeitig, Liebigs Ann. Chem., 1881, 209, 134 CrossRef.
- A. Arienti, F. Bigi, R. Maggi, P. Moggi, M. Rastelli, G. Sartori and A. Trerè, J. Chem. Soc., Perkin Trans. 1, 1997, 1391 RSC.
- K. Nakahara, C. Mitsui, T. Okamoto, M. Yamagishi, K. Miwa, H. Sato, A. Yamano, T. Uemura and J. Takeya, Chem. Lett., 2013, 42, 654 CrossRef CAS.
- N. Xu, Y. Li, D. Ricciarelli, J. Wang, E. Mosconi, Y. Yuan, F. De Angelis, S. M. Zakeeruddin, M. Grätzel and P. Wang, iScience, 2019, 15, 234 CrossRef CAS PubMed.
- Y. Wei, A. Zhang, X. Xie, J. Zhang, L. He and P. Wang, ACS Mater. Lett., 2021, 3, 947 CrossRef CAS.
- L. He, Y. Zhang, Y. Wei, Y. Cai, J. Zhang and P. Wang, Matter, 2023, 6, 4013 CrossRef CAS.
- S. Ming, K. Lin, H. Zhang, F. Jiang, P. Liu, J. Xu, G. Nie and X. Duan, Chem. Commun., 2020, 56, 5275 RSC.
- T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056 CrossRef CAS PubMed.
- A. L. Mayhugh, P. Yadav and C. K. Luscombe, J. Am. Chem. Soc., 2022, 144, 6123 CrossRef CAS PubMed.
- M. Wakioka, H. Morita, N. Ichihara, M. Saito, I. Osaka and F. Ozawa, Macromolecules, 2020, 53, 158 CrossRef CAS.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27 RSC.
- Z. Qian, Z. Cao, L. Galuska, S. Zhang, J. Xu and X. Gu, Macromol. Chem. Phys., 2019, 220, 1900062 CrossRef.
- Y. Ren, M. Ren, X. Xie, J. Wang, Y. Cai, Y. Yuan, J. Zhang and P. Wang, Nano Energy, 2021, 81, 105655 CrossRef CAS.
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759 RSC.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Fréchet, Adv. Mater., 2003, 15, 1519 CrossRef CAS.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699 CrossRef CAS PubMed.
- S. Yoo, B. Domercq and B. Kippelen, J. Appl. Phys., 2005, 97, 103706 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04244a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |