
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2-actuated spin transition tuning in an interdigitated Hofmann-type coordination polymer†
Abhik
Paul
 a,
Wataru
Kosaka
bc,
Bhart
Kumar‡
a,
Dibya Jyoti
Mondal‡
a,
Hitoshi
Miyasaka
*bc and
Sanjit
Konar
*a
a,
Wataru
Kosaka
bc,
Bhart
Kumar‡
a,
Dibya Jyoti
Mondal‡
a,
Hitoshi
Miyasaka
*bc and
Sanjit
Konar
*a
aMolecular Magnetism Lab, Department of Chemistry, Indian Institute of Science Education and Research, Bhopal, Madhya Pradesh, India 462066. E-mail: skonar@iiserb.ac.in
bInstitute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: miyasaka@imr.tohoku.ac.jp
cDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aramaki-Aza-Aoba, Aoba-ku, Sendai 980-8578, Japan
First published on 30th August 2024
Abstract
The increased anthropogenic emission level of CO2 urges the development of CO2-responsive materials, but is it possible to regulate the inherent electronic properties through weak physisorption of a ubiquitous gas such as CO2? Herein, we intended to answer this imperative question by the first case of CO2-actuated variable spin-state stabilisation in an interdigitated Hofmann-type coordination polymer [FeIIPd(CN)4L2] (1, L = methyl isonicotinate), showing a wide shift in transition temperature (Teq) from 178 K at PCO2 = 0 kPa to 229 K at PCO2 = 100 kPa. Interestingly, the emergence of a stepped behaviour in the heating process below PCO2 = 10 kPa and overlapping magnetic susceptibility values above PCO2 = 10 kPa elucidate the selective LS state stabilisation solely correlated with the extent of CO2 accommodation. Based on the magnetic response and phase transition diagrams obtained under respective PCO2, a plausible scenario of the spin-state switching can be interpreted as (1ls + 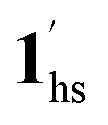 ) → (1hs +
) → (1hs + 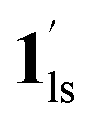 ) → 1hs at PCO2 ≤ 10 kPa,
) → 1hs at PCO2 ≤ 10 kPa,  → 1hs at 100 kPa < PCO2 ≥ 32 kPa and
→ 1hs at 100 kPa < PCO2 ≥ 32 kPa and 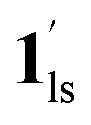 →
→ 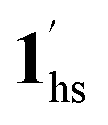 → 1hs at 100 kPa, where 1 and 1′ represent CO2-free and CO2-encapsulated states, respectively. The cooperative CO2 sorption with SCO based on the varied CO2 pressure corroborates a novel case for developing CO2-responsive magnetic materials henceforth.
→ 1hs at 100 kPa, where 1 and 1′ represent CO2-free and CO2-encapsulated states, respectively. The cooperative CO2 sorption with SCO based on the varied CO2 pressure corroborates a novel case for developing CO2-responsive magnetic materials henceforth.
Introduction
Switchable magnetic materials that can be reversibly interconverted between two stable states in response to an external stimulus are promising candidates for potential memory devices.1–5 Metal complexes, such as metal–organic frameworks (MOFs)6–8 or porous coordination polymers (PCPs),9,10 that are capable of accommodating guest molecules are of high interest in this regard as the inherent porous nature can be used to separate similar guest species11–13 or even exhibit guest-induced changes in their intrinsic properties,14 including emission,15 magnetic16,17 and conductive properties,17 amongst others.18–21Preceded by the remarkable example of guest-responsive bidirectional spin-state switching,22,23 the spin-crossover (SCO) research outcome investigated the influence of subtle interactions exerted by small guest molecules on effective spin-state stabilisation of PCPs.24–26 Nonetheless, the use of ubiquitous gas molecules (such as N2, O2 or CO2) to modulate SCO behaviour in PCPs is underdeveloped,27–29 as the significant variation of the crystal lattice volume resulting from the change in the spin-state with relatively weak physisorption of the gas remains highly unlikely,30–32 compared to the chemical pressure33 exerted by guests such as small organic molecules or solvents.34,35 Recent advances in the gas-responsive magnetic phase transitions revolve around utilising nanometric pores and36,37 open metal sites,38,39 the use of paramagnetic gases,40 or even interlayer charge transfer41–43 by manipulating the electronic states. However, obtaining a significant SCO response only through the physisorption of a gas such as CO2 can be narrowed down to a monomeric CoII complex44 and a 3D pseudo-Hofmann type framework.45 While the former case demonstrates the SCO induction in the CO2-admixed state, the latter forfeits the SCO activity. Herein, we bridge this leap in the literature by reporting the first case of fine-tuning the spin states of an interdigitated Hofmann-type PCP [FeIIPd(CN)4L2] (1, L = methyl isonicotinate), solely based on the selective physisorption of CO2 molecules in the framework.
Traditionally, for a flexible porous crystal, the “breathing effect” is demonstrated by a drastic expansion/contraction in the pore volume upon increasing the partial pressure of the adsorbent, usually leading to a two-step gas uptake, constituted of (i) a transition from the originally opened pore to a closed pore and (ii) opening of the closed pore (Scheme 1, left). On the other hand, the abrupt increase in the adsorption capacity due to the structural change or oscillations induced by the guest admission/release is referred to as the “gating effect” (Scheme 1, right), which is much more diverse and involves intricate structural flexibility than simple expansion/contraction.14,46
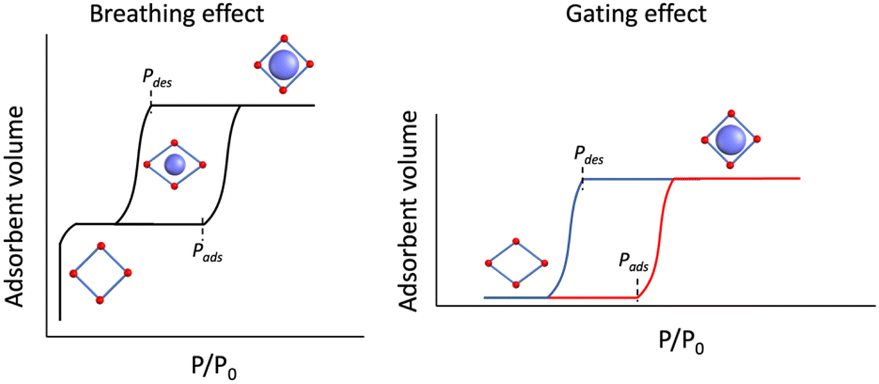 | ||
| Scheme 1 (Left) Breathing effect vs. (right) gating effect in gas adsorption of porous coordination polymers. | ||
Compared to the typically observed classical type I adsorption isotherms (Fig. 1a), a more energetically favourable cooperative transition constitutes adsorption over a narrow pressure range and engages high working capacities defined as the amount of gas adsorbed (Fig. 1b). On a temperature scale, the consecutive temperatures of such a bistable process can be defined as TGO and TGC (GO, gate opening and GC, gate closing, for adsorption and desorption, respectively), and the difference between them comprises the admixed state, based on the mode of temperature swing. Interestingly, such a “gating effect” can be correlated with the cooperative SCO dynamics where spin-state change defines the working capacity between the two states (Fig. 1c) using a thermal hysteresis loop (ΔT = Theat − Tcool).47 However, to successfully integrate the SCO behaviour under the particular effect of physisorption, i.e., without the coordination to open metal centres or any of the functional groups, the adsorbate must (i) possess a flexible and porous structural pattern to allow reversible insertion and removal of the adsorbent and (ii) the adsorbate–adsorbent interactions must be present as a subtle balance between the exerted chemical pressure in favour of the HS state and the electrostatic interactions in favour of the LS state, to regulate high cooperativity in the crystal lattice. In addition, the adsorbent must possess a necessary dipole or quadrupole moment that promotes insertion into the adsorbate framework.
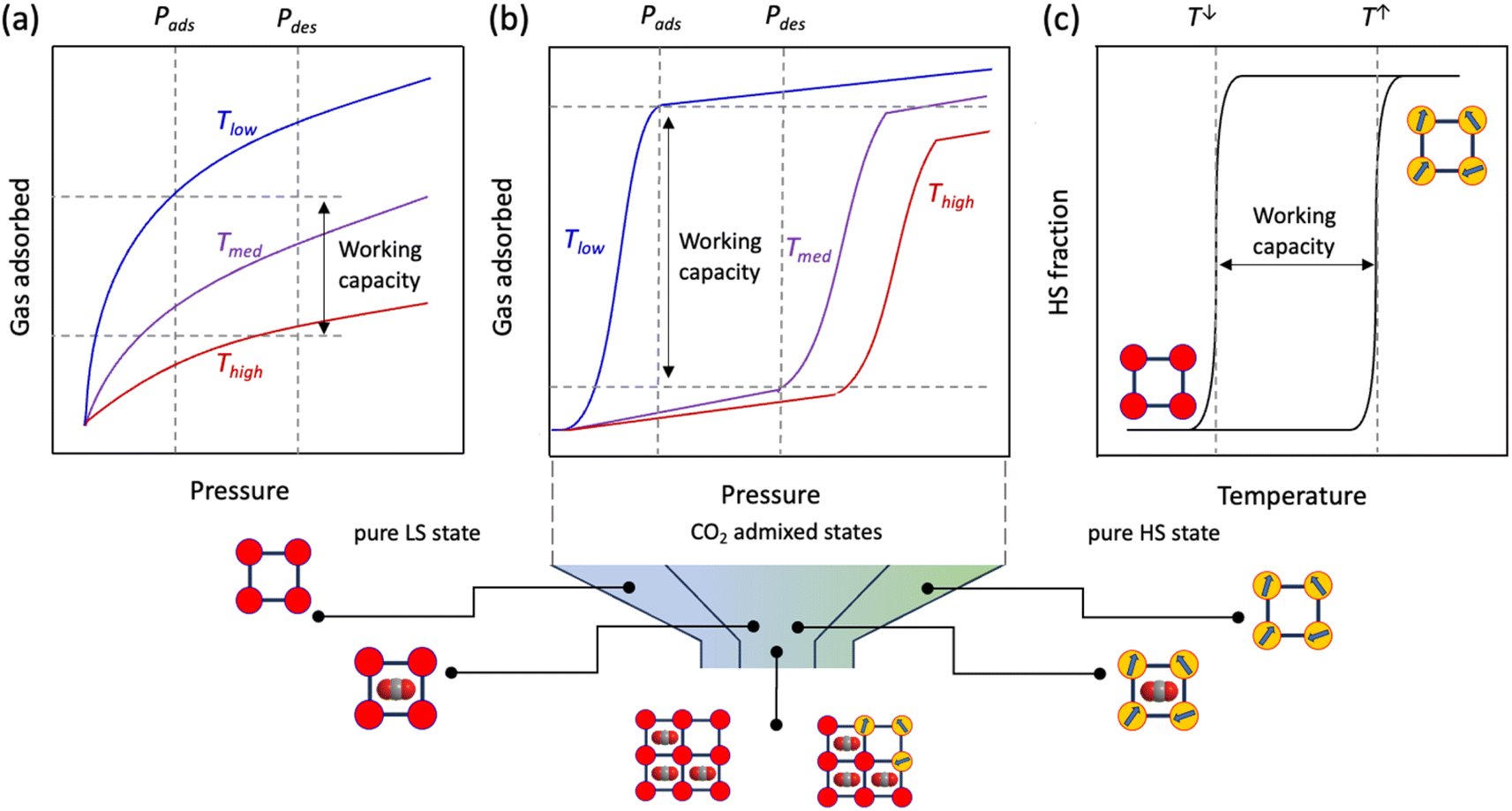 | ||
| Fig. 1 Idealised adsorption isotherms and the cooperative spin-transition mechanism. (a) and (b) Comparison of the working capacities for idealised gas adsorption isotherms of (a) classical and (b) cooperative adsorbents using a smaller temperature swing, and (c) temperature-dependent hysteretic spin transition. (Below) Schematic representation of the cooperative spin-transition under a CO2 atmosphere and pure LS states at low temperature before CO2 adsorption (1ls), followed by CO2 admixed states (1′), based on TGO and TST and pure HS states above TGC. TGO, TGC and TST correspond to gate opening, gate closing and spin transition temperatures, respectively. | ||
Inspired by our earlier demonstration of the modulation of cooperativity from a single-to-four-step transition by de/re-solvation of MeOH molecules in a 2D Hofmann-type PCP 1·1.3MeOH,48 we sought to rationalise the system under the effect of a supercritical gas, such as CO2, which could only impart weak dispersive interactions in the system. Notably, removal of the MeOH molecules from 1·1.3MeOH exerts stronger π-stacking interactions between the panelling ligands, leading to our assumption that the intriguing gate-opening/closing mechanism might be achieved under the influence of CO2. Herein, we report the first example of CO2-actuated spin transition tuning in 1, associated with a wide transition temperature shift from 171 K (cooling)/185 K (heating) at PCO2 = 0 kPa to 213 K (cooling)/244 K (heating) at PCO2 = 100 kPa with a maximum thermal hysteresis loop of ∼65 K at PCO2 = 10 kPa. The highly selective CO2 sorption and CO2-responsive spin-state stabilisation can be tuned by varying the amount of CO2 uptake, i.e., PCO2.
Results and discussion
Single crystal X-ray diffraction analysis of 1·1.3MeOH showed that each of the FeII atoms is coordinated axially by two methyl isonicotinate ligands and equatorially by the cyanide linkers of four [Pd(CN)4]2− units, forming a {Fe[Pd(CN)4]}∝ grid with Fe2Pd2 square windows. Two such grids are arranged by inter-ligand hydrogen bonding and lone pair⋯π interactions between the carboxylate groups and π e− cloud of the two adjacent pyridyl rings, resulting in an interdigitated (2D) framework (Fig. 2).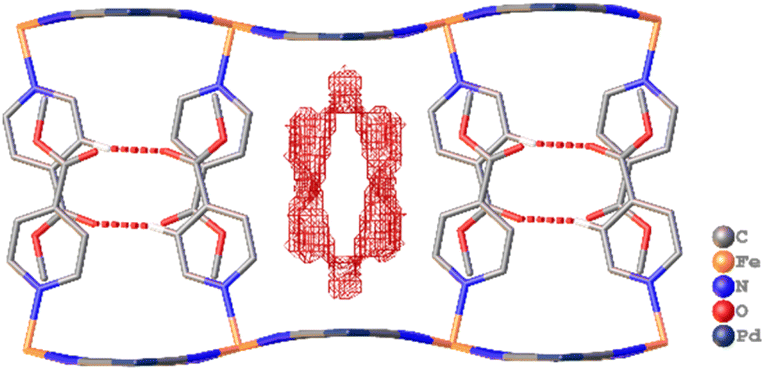 | ||
| Fig. 2 Interdigitated structure of framework 1·1.3MeOH, stabilised by inter-ligand hydrogen bonding and lone pair⋯π interactions, and adsorbent accessible voids are shown along crystallographic axis a. Solvent MeOH molecules and H atoms are omitted for visual clarity. | ||
As indicated by the thermogravimetric analysis, framework 1 possesses excellent thermal stability up to 230 °C (Fig. S1, ESI†). Comparison of the PXRD spectrum of 1 with that of the parent framework 1·1.3MeOH showed that the framework pattern in the desolvated phase is preserved (Fig. S2, ESI†), although a structural phase transition is evident from the shifting of peaks in the middle region (2θ = 15–25°). FTIR spectra of 1·1.3MeOH and 1 indicate that the bonding of 1·1.3MeOH is preserved in 1 (Fig. S3, ESI†). The variable temperature PXRD diffraction analyses of 1 showed a thermally induced-phase transition accompanied by SCO, as indicated by the peak shift near the SCO transition temperature in both cooling and heating processes (Fig. S4, ESI†). Structural flexibility deduced for 1 to house small guest molecules such as MeOH encouraged us to check its adsorption capability for any gas molecules. However, no adsorption for N2 (77 K) and O2 (90 K) was observed even at 120 K (for both N2 and O2, Fig. S5, ESI†). In contrast, above T ≥ 195 K, a sharp transition in the CO2 adsorption appeared, ∼15 kPa CO2 pressure (PCO2), which resulted in the gate opening (GO) transition, and on increasing PCO2, reached an adsorbed amount of ∼50 mlSTP g−1 (∼1 molecule per formula unit) at 100 kPa (Fig. 3a). The GO pressure (for adsorption) and GC pressure (for desorption) steadily increased with the temperature increase (up to 230 K). The temperature dependence of the GO and GC pressure obeyed the Clausius–Clapeyron relationship, d(ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PG)/d(lnTG−1) = ΔHTrans/Rg, where ΔHTrans is the variation in the transition enthalpy and Rg is the universal gas constant (Fig. 3c, S6 and S7, ESI†), with estimated enthalpy values of −27 kJ mol−1 and −52 kJ mol−1 for GO and GC, respectively.49 The gated behaviour was also characterised using the isobar plots (Fig. 3b), and for both cases, wide hysteresis was observed (Fig. S6, ESI†). Based on these data, the phase diagram of 1 under CO2 (T vs. PCO2 plot in Fig. 3c) was created for the GO and GC processes, revealing an apparent phase transition between 1 and 1·CO2, respectively.
PG)/d(lnTG−1) = ΔHTrans/Rg, where ΔHTrans is the variation in the transition enthalpy and Rg is the universal gas constant (Fig. 3c, S6 and S7, ESI†), with estimated enthalpy values of −27 kJ mol−1 and −52 kJ mol−1 for GO and GC, respectively.49 The gated behaviour was also characterised using the isobar plots (Fig. 3b), and for both cases, wide hysteresis was observed (Fig. S6, ESI†). Based on these data, the phase diagram of 1 under CO2 (T vs. PCO2 plot in Fig. 3c) was created for the GO and GC processes, revealing an apparent phase transition between 1 and 1·CO2, respectively.
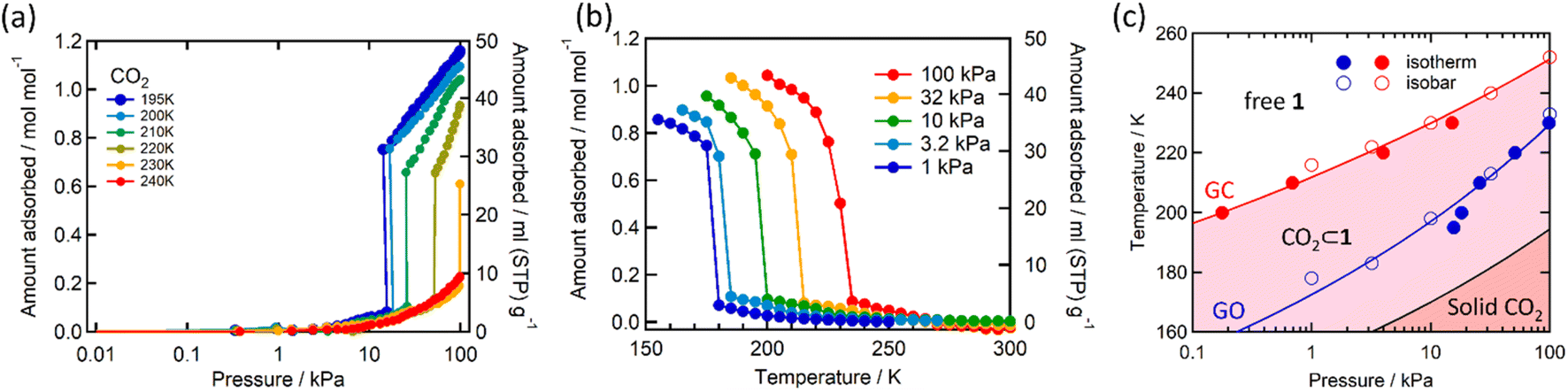 | ||
| Fig. 3 CO2 adsorption in 1 studied as (a) isotherms at different temperatures, (b) isobars at different CO2 pressures (solid lines are a guide for the eye), and (c) phase diagram for 1/1′/CO2 determined from these measurements. The red (GC) and blue (GO) lines represent the fitting curves based on the Clausius–Clapeyron equation (ESI†). The black solid line represents the saturated vapor pressure curve, distinguishing between the solid and gas phases of bulk CO2. | ||
Since 1 possesses a more restricted accessible pore, i.e., structural reorganisation accessible only under the influence of a small guest, we expected an interplay of the SCO response with CO2 adsorption. To check the consequence of the CO2-gated behaviour in the respective magnetic properties of 1, variable temperature magnetic susceptibility measurements (10–300 K) at 0.5 K min−1 under an external magnetic field (Hdc) of 1 kOe were conducted under varied CO2 pressure using a home-built gas cell (Experimental details, ESI†). First, after loading the sample in the magnetometer, the gas cell was filled with 100 kPa He (instead of vacuum) to enhance thermal conductivity. Upon cooling, a HS-to-LS transition was observed at 171 K (171 K in vacuo), while a complete LS-to-HS transition was observed upon heating at 185 K (186 K in vacuo) revealing a bistable region of 14 K (ΔT = TH − TC; Fig. 4a, closed circle), which perfectly reproduces the SCO behaviour (Teq, ∼178 K; Teq = (TH + TC)/2, where Teq, TH, and TC refer to the average temperature of ST and transition temperature in heating and cooling mode, respectively) of the guest-free framework 1 under vacuum (Fig. 4a, open circle). Notably, the coincidence of ST phenomena in these two cases dismissed the possibility of any additional LS state stabilisation under the effect of applied gas pressure. Although the slight changes in the transition temperatures (Tc and TH) and the thermal hysteresis (ΔT, 15 K in vacuo vs. 14 K under 100 kPa He) can be attributed to the modification in experimental setups.
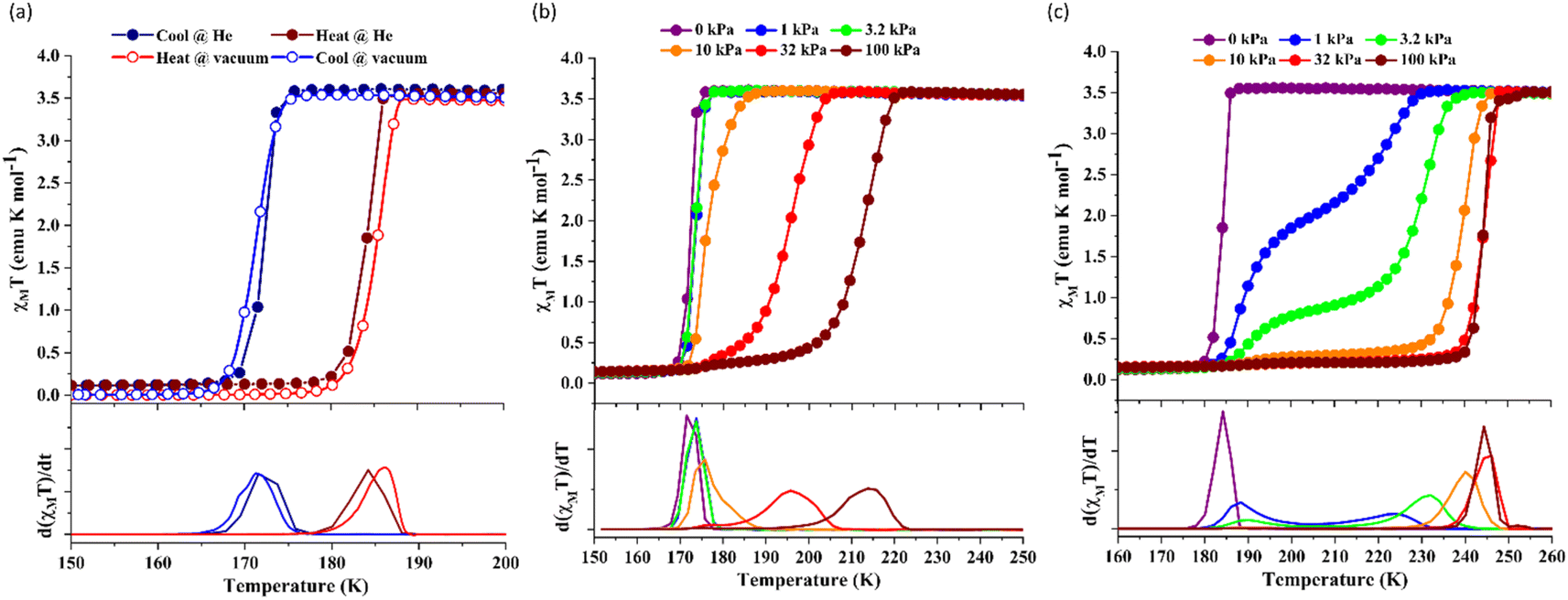 | ||
| Fig. 4 Variation of the magnetic properties of framework 1 under CO2 adsorption under Hdc = 1 kOe. (a) Magnetic susceptibility plots in the absence of any gas (open circles) and under 100 kPa He gas (filled circles). (b) The CO2 pressure dependence of the magnetic susceptibility measurements in cooling and (c) heating mode. First derivative plots are displayed at the bottom. | ||
Next, CO2 gas at PCO2 = 1, 3.2, 10, 32, and 100 kPa was introduced in the cell at 300 K, and the M–T measurements at Hdc = 1 kOe were performed consecutively in the cooling and heating mode (10–300 K). For the cooling mode shown in Fig. 4b, a one-step transition from HS-to-LS was observed with a gradual increase in the ST temperature (Tc; 173 K at PCO2 = 1.0 kPa to 213 K at PCO2 = 100 kPa) with increased CO2 pressure. Meanwhile, the LS-to-HS transition in the heating mode appeared in a stepwise fashion at PCO2 ≤ 10 kPa, in which TH(1) for the transition at lower temperature was approximately at 188 K in the all PCO2 range of 1–10 kPa, while TH(2) for the transition at higher temperature was increased from 224 K (PCO2 = 1.0 kPa) to 240 K (PCO2 = 10 kPa), elucidating that the physisorption of the CO2 molecules in the framework stabilise the LS state. Interestingly, the TC and TH(2), defined by the peak maxima of δχMT/δT vs. T plot (Fig. 4, bottom), differ significantly under the same PCO2, indicating the presence of thermal hysteresis (ΔT) with a temperature spread up to ∼65 K at PCO2 = 10 kPa (Fig. 4 and Table S1, ESI†). Similarly, the observed Teq variation correlates well with the result obtained from CO2 adsorption, which indicates that the abrupt SCO behaviour observed for 1 under the CO2 atmosphere is mainly associated with the gate-opening CO2 adsorption process.
Apparently, the emergence of the stepped behaviour in the heating process under low CO2 pressure (PCO2 < 10 kPa) intrigued us to investigate the plausible scenario that occurs during the transition under the CO2 atmosphere. The phase, as well as the spin state, for each process strongly depends on a relationship of three factors: (i) the CO2 occupation rate in the compound, (ii) GO and GC temperatures at each CO2 pressure applied, and (iii) intrinsic ST temperatures of pristine 1 and the CO2-accommodated phase 1′. Furthermore, the nature of each phase is basically determined by the cooling process under an applied CO2 pressure.
As evidenced from the phase diagram, the GO process operates in the temperature range of ∼160 to 230 K, while the GC process is valid in the temperature region of ∼190–250 K. Thus, based on the PCO2 applied, a variation of the LS state stabilisation for the CO2-admixed state (1′) is presumed eventually in the temperature range of ∼190–230 K (Fig. 5, ESI†), based on the amount of CO2 accommodated in the framework.
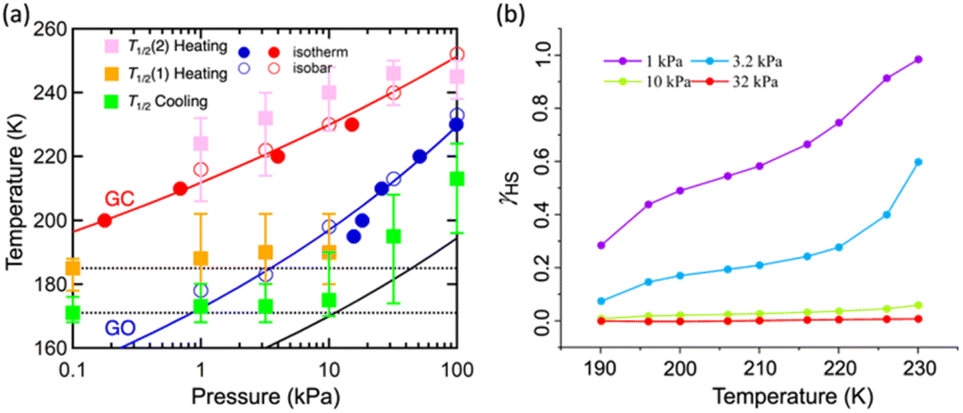 | ||
| Fig. 5 (a) Correlation of the gated CO2 sorption behaviour with the SCO transition temperatures of framework 1 under variable CO2 pressures in the temperature range of 160–260 K; (b) HS fraction population (γHS) of 1 under variable CO2 pressures (PCO2 = 1 to 32 kPa) in the temperature range of 190–230 K. | ||
To delineate the magnetic transition under CO2 sorption, the overall process can be described by a total of four states, i.e., without CO2 (1ls and 1hs) and with CO2 ( and
and 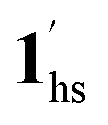 ). In the cooling process at PCO2 = 1 kPa, the ST at 173 K is owed to the intrinsic ST of pristine 1, exhibiting a change from 1hs → 1ls, because of the presence of CO2 adsorption GO at 170 K, namely, the 1ls phase adsorbed CO2. Notably, the magnetic susceptibility changes around this temperature (Fig. 6a, green asterisk) manifest that the transition from 1ls to
). In the cooling process at PCO2 = 1 kPa, the ST at 173 K is owed to the intrinsic ST of pristine 1, exhibiting a change from 1hs → 1ls, because of the presence of CO2 adsorption GO at 170 K, namely, the 1ls phase adsorbed CO2. Notably, the magnetic susceptibility changes around this temperature (Fig. 6a, green asterisk) manifest that the transition from 1ls to  might not be perfect due to the slow CO2 adsorption kinetics, and the two states are mixed at temperatures below 170 K (1hs → 1ls →
might not be perfect due to the slow CO2 adsorption kinetics, and the two states are mixed at temperatures below 170 K (1hs → 1ls → 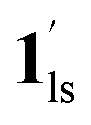 + 1ls, for PCO2 = 1 kPa, Fig. 6a), whereas for PCO2 > 10 kPa, the GO process occurs in the range of 200–230 K, and the gradual transition to the diamagnetic (LS) state occurs after or with the CO2 adsorption (1hs →
+ 1ls, for PCO2 = 1 kPa, Fig. 6a), whereas for PCO2 > 10 kPa, the GO process occurs in the range of 200–230 K, and the gradual transition to the diamagnetic (LS) state occurs after or with the CO2 adsorption (1hs → 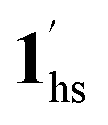 →
→ 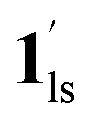 , for PCO2 = 100 kPa, Fig. 6b). Therefore, in the cooling cycle for PCO2 > 10 kPa, the CO2-accommodated phase prevailed because of TGO > TC; the 1hs phase adsorbed CO2, indicating 1hs →
, for PCO2 = 100 kPa, Fig. 6b). Therefore, in the cooling cycle for PCO2 > 10 kPa, the CO2-accommodated phase prevailed because of TGO > TC; the 1hs phase adsorbed CO2, indicating 1hs → 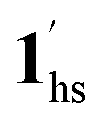 , followed by the ST at TC. However, the CO2 occupation in the material was still dependent on the CO2 pressure, so the phase at PCO2 = 10 kPa can be assigned to be a mixture of (1hs +
, followed by the ST at TC. However, the CO2 occupation in the material was still dependent on the CO2 pressure, so the phase at PCO2 = 10 kPa can be assigned to be a mixture of (1hs +  ) followed by the ST to the (1ls +
) followed by the ST to the (1ls + 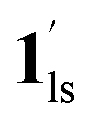 ) phase at 175 K, although the ratio of 1hs/1ls (i.e., 1·CO2) was much lower than that of
) phase at 175 K, although the ratio of 1hs/1ls (i.e., 1·CO2) was much lower than that of 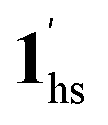 / (Fig. 5, right). The heating process well displays the nature of the CO2-accommodated mixture. Interestingly, at PCO2 < 10 kPa, the gated adsorption in the framework occurs in a stepwise fashion, with an increase in temperature, leading to two distinctive forms A and B (Fig. 6c shows an instance at PCO2 = 3.2 kPa). Below 173 K, a mixed state of
/ (Fig. 5, right). The heating process well displays the nature of the CO2-accommodated mixture. Interestingly, at PCO2 < 10 kPa, the gated adsorption in the framework occurs in a stepwise fashion, with an increase in temperature, leading to two distinctive forms A and B (Fig. 6c shows an instance at PCO2 = 3.2 kPa). Below 173 K, a mixed state of  + 1ls (form A) is active, and the SCO occurs from the non-CO2 adsorbed domain, i.e., 1ls → 1hs. The (
+ 1ls (form A) is active, and the SCO occurs from the non-CO2 adsorbed domain, i.e., 1ls → 1hs. The (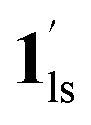 + 1hs) mixed state B prevails in a step-level in the temperature range of 173–220 K. The second form B undergoes a CO2 release at TGC = 220 K, accompanied by SCO transition
+ 1hs) mixed state B prevails in a step-level in the temperature range of 173–220 K. The second form B undergoes a CO2 release at TGC = 220 K, accompanied by SCO transition 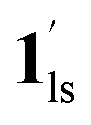 → 1hs. On the other hand, the transition at PCO2 = 100 K at TH = 245 K is due to the intrinsic SCO phenomenon for 1·CO2, i.e.,
→ 1hs. On the other hand, the transition at PCO2 = 100 K at TH = 245 K is due to the intrinsic SCO phenomenon for 1·CO2, i.e., 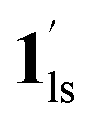 →
→ 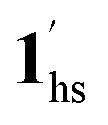 , because TGC = 253 K > TH (Fig. 6d), unlike the desorption-induced SCO observed at PCO2 < 10 kPa. For PCO2 = 32 kPa, the steep transition at ∼240 K just overlapped with the SCO transition of
, because TGC = 253 K > TH (Fig. 6d), unlike the desorption-induced SCO observed at PCO2 < 10 kPa. For PCO2 = 32 kPa, the steep transition at ∼240 K just overlapped with the SCO transition of 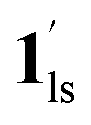 →
→ 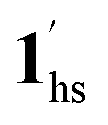 and CO2 release from
and CO2 release from 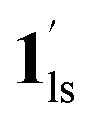 → 1hs. The materials at PCO2 < 10 kPa are mixtures of 1 and 1·CO2, whose ratios were determined to be ∼72%, ∼85% and ∼98% 1·CO2 states induced at 226 K by CO2 accommodation at PCO2 = 1, 3.2, and 10 kPa, respectively (Table S2 and Fig. S11, ESI†). We also carried out in situ PXRD measurements to check the structural changes during the adsorption and SCO process (Fig. S12, ESI†); four states (1ls, 1hs,
→ 1hs. The materials at PCO2 < 10 kPa are mixtures of 1 and 1·CO2, whose ratios were determined to be ∼72%, ∼85% and ∼98% 1·CO2 states induced at 226 K by CO2 accommodation at PCO2 = 1, 3.2, and 10 kPa, respectively (Table S2 and Fig. S11, ESI†). We also carried out in situ PXRD measurements to check the structural changes during the adsorption and SCO process (Fig. S12, ESI†); four states (1ls, 1hs, 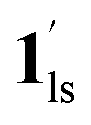 , and
, and 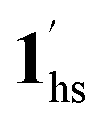 ) are distinguishable in PXRD patterns, providing further evidence of the SCO and gated adsorption process shown in Fig. 6.
) are distinguishable in PXRD patterns, providing further evidence of the SCO and gated adsorption process shown in Fig. 6.
 | ||
| Fig. 6 (Top) Comparative magnetic transition plots under the influence of a CO2 atmosphere in cooling ((a) 1.0 kPa; (b) 100 kPa) and heating ((c) 3.2 kPa; (d) 100 kPa) cycles; (bottom) the GO and GC temperatures denoted by green asterisk in the respective phase diagram, and a representative cartoon diagram of the SCO phenomenon occurring between different spin states of the framework 1. The red (GC) and blue (GO) lines in the phase diagram represent the fitting curves based on the Clausius–Clapeyron equation. | ||
Based on the above phenomenon of the CO2-adsorption actuated spin transition, we were intrigued to check the generality of this method for similar guest molecules such as CS2, which was reported to stabilise the LS states for 3D Hofmann-type framework {Fe(pyrazine)[Pt(CN)4]}.23,50,51 Thus, 1·CS2 was prepared by a liquid-to-liquid slow diffusion technique using a methanolic solution of CS2 as the buffer layer (Experimental section, ESI†). Single crystal X-ray diffraction analysis performed at 100 K showed that 1·CS2 crystallises in a monoclinic crystal system in space group I2/c, similar to the parent framework 1·1.3MeOH (monoclinic C2/c, at 90 K). While the interdigitated nature was retained, two of the CS2 molecules were found to lie in the middle of the Fe2Pd2 windows of two parallel {Fe[Pd(CN)4]}∝ grids with a short contact distance of 3.871 Å with the nearest Pd centres and an array of C–H⋯S interactions with the surrounding ligands (Fig. S13, ESI†). However, the Fe–N equatorial (2.041 and 2.030 Å with Pd(CN)4 units) and axial (2.117 Å with panelling ligands) bond lengths were found more towards the HS state, in stark contrast to 1·1.3MeOH. However, the instability of the sample at room temperature due to rapid guest loss puts a restriction on carrying out the dc magnetic susceptibility measurements of the CS2 analogue.
This structural anomaly for the CS2-encapsulated framework 1·CS2 can be attributed to the larger atomic size and low electronegativity of the S in CS2 exerting a significant chemical pressure to result in the HS state of the Fe atoms, diminishing the cooperative communication between the metal centres. On the other hand, the relatively smaller size and high electronegativity of O in CO2 facilitate the electrostatic interactions within framework 1, which is a prerequisite for gated sorption behaviour and predominantly stabilises the LS state with increased absorbate pressure.
Conclusions
Although the investigation of CO2 capture using MOFs has been quite explored lately, it is rare that they display CO2-indued changes in their structure and properties.52 The present study provides an efficient and straightforward strategy for the magnetic phase changes in situ by tuning the physisorption amount of CO2 without any change in the chemical properties of framework 1. Based on the magnetic response and the phase transition diagram obtained under the respective PCO2, the key factor of the overall spin-state switching can be interpreted as the stabilisation of LS states under the influence of CO2. We are hopeful that the insights gathered from this work into understanding the mechanism by which switchable properties can be controlled by gas adsorption will be helpful for the development of future CO2-responsive materials for practical applications.Data availability
Experimental and crystallographic details, additional structural views, PXRD patterns, FT-IR spectra, TGA data, and additional magnetic measurement information are provided in the ESI.†Author contributions
All authors have given approval to the final version of the manuscript. AP and SK conceptualised the project; BK and DJM prepared and performed physical characterisation of the frameworks; WK performed the adsorption studies and magnetic measurements under CO2; AP and WK interpreted the data and the working concept; AP, SK, and HM prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Engineering Research Board (SERB), India, under the project Core Research Grant (Project No. CRG/2022/001676), IISER Bhopal, a Grant-in-Aid for Scientific Research (no. 20H00381, 21H01900, and 21K18925) from MEXT, Japan, and the GIMRT and the E-IMR projects at the Institute for Materials Research, Tohoku University. AP, DJM and BK thank the Council of Scientific and Industrial Research (CSIR), India, for providing Senior Research Fellowships. SK acknowledges IISER Bhopal for instrumental support.Notes and references
- A. Paul, A. Gupta and S. Konar, Cryst. Growth Des., 2021, 21, 5473–5489 CrossRef CAS.
- A. Paul and S. Konar, J. Mater. Chem. C, 2022, 10, 4980–4984 RSC.
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS.
- A. Paul, R. Nasani, A. Mondal, S. Roy, S. Vela and S. Konar, Cryst. Growth Des., 2020, 20, 6296–6301 CrossRef CAS.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- O. I.-F. Chen, C.-H. Liu, K. Wang, E. Borrego-Marin, H. Li, A. H. Alawadhi, J. A. R. Navarro and O. M. Yaghi, J. Am. Chem. Soc., 2024, 146, 2835–2844 CrossRef CAS.
- E. Coronado and G. Mínguez Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539 RSC.
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- S. Li, W. Han, Q.-F. An, K.-T. Yong and M.-J. Yin, Adv. Funct. Mater., 2023, 2303447 CrossRef CAS.
- S.-M. Wang, M. Shivanna, S.-T. Zheng, T. Pham, K. A. Forrest, Q.-Y. Yang, Q. Guan, B. Space, S. Kitagawa and M. J. Zaworotko, J. Am. Chem. Soc., 2024, 146, 4153–4161 CrossRef CAS.
- M. Shivanna, K.-i. Otake, S. Hiraide, T. Fujikawa, P. Wang, Y. Gu, H. Ashitani, S. Kawaguchi, Y. Kubota, M. T. Miyahara and S. Kitagawa, Angew. Chem., Int. Ed., 2023, 62, e202308438 CrossRef CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- J. Zhang, W. Kosaka, K. Sugimoto and H. Miyasaka, J. Am. Chem. Soc., 2018, 140, 5644–5652 CrossRef CAS PubMed.
- J. Zhang, W. Kosaka, Y. Kitagawa and H. Miyasaka, Angew. Chem., Int. Ed., 2019, 58, 7351–7356 CrossRef CAS PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- S. Parshamoni, R. Nasani, A. Paul and S. Konar, Inorg. Chem. Front., 2021, 8, 693–699 RSC.
- S. Khatua, S. Goswami, S. Biswas, K. Tomar, H. S. Jena and S. Konar, Chem. Mater., 2015, 27, 5349–5360 CrossRef CAS.
- A. Paul, S. D. Adhikary, S. Kapurwan and S. Konar, J. Mater. Chem. A, 2022, 10, 13152–13169 RSC.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed.
- M. Ohba, K. Yoneda, G. Agustí, M. C. Muñoz, A. B. Gaspar, J. A. Real, M. Yamasaki, H. Ando, Y. Nakao, S. Sakaki and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 4767–4771 CrossRef CAS.
- B. Kumar, A. Paul, D. J. Mondal, P. Paliwal and S. Konar, Chem. Rec., 2022, 22, e202200135 CrossRef CAS PubMed.
- M. Feng, Z.-Y. Ruan, Y.-C. Chen and M.-L. Tong, Chem. Commun., 2020, 56, 13702–13718 RSC.
- S.-G. Wu, L.-F. Wang, Z.-Y. Ruan, S.-N. Du, S. Gómez-Coca, Z.-P. Ni, E. Ruiz, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2022, 144, 14888–14896 CrossRef CAS.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS.
- J. W. Shin, A. R. Jeong, S. Jeoung, H. R. Moon, Y. Komatsumaru, S. Hayami, D. Moon and K. S. Min, Chem. Commun., 2018, 54, 4262–4265 RSC.
- E. Coronado, M. Gimenez-Marques, G. Minguez Espallargas, F. Rey and I. J. Vitorica-Yrezabal, J. Am. Chem. Soc., 2013, 135, 15986–15989 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS.
- C. Mellot-Draznieks, C. Serre, S. Surblé, N. Audebrand and G. Férey, J. Am. Chem. Soc., 2005, 127, 16273–16278 CrossRef CAS.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS.
- K. Lin, Q. Li, R. Yu, J. Chen, J. P. Attfield and X. Xing, Chem. Soc. Rev., 2022, 51, 5351–5364 RSC.
- D. J. Mondal, B. Kumar, A. Paul and S. Konar, J. Mater. Chem. C, 2023, 11, 6750–6759 RSC.
- R. Turo-Cortés, C. Bartual-Murgui, J. Castells-Gil, M. C. Muñoz, C. Martí-Gastaldo and J. A. Real, Chem. Sci., 2020, 11, 11224–11234 RSC.
- W. Kosaka, H. Nemoto, K. Nagano, S. Kawaguchi, K. Sugimoto and H. Miyasaka, Chem. Sci., 2023, 14, 791–800 RSC.
- W. Kosaka, Y. Hiwatashi, N. Amamizu, Y. Kitagawa, J. Zhang and H. Miyasaka, Angew. Chem., Int. Ed., 2023, 62, e202312205 CrossRef CAS.
- E. Coronado, M. Giménez-Marqués, G. M. Espallargas and L. Brammer, Nat. Commun., 2012, 3, 828 CrossRef.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- W. Kosaka, Z. Liu, J. Zhang, Y. Sato, A. Hori, R. Matsuda, S. Kitagawa and H. Miyasaka, Nat. Commun., 2018, 9, 5420 CrossRef CAS.
- J. Zhang, W. Kosaka, Y. Kitagawa and H. Miyasaka, Nat. Chem., 2021, 13, 191–199 CrossRef CAS PubMed.
- J. Zhang, W. Kosaka, H. Sato and H. Miyasaka, J. Am. Chem. Soc., 2021, 143, 7021–7031 CrossRef CAS.
- J. Zhang, W. Kosaka, Q. Liu, N. Amamizu, Y. Kitagawa and H. Miyasaka, J. Am. Chem. Soc., 2023, 145, 26179–26189 CrossRef CAS.
- M. Nakaya, W. Kosaka, H. Miyasaka, Y. Komatsumaru, S. Kawaguchi, K. Sugimoto, Y. Zhang, M. Nakamura, L. F. Lindoy and S. Hayami, Angew. Chem., Int. Ed., 2020, 59, 10658–10665 CrossRef CAS.
- M. Magott, K. Płonka, B. Sieklucka, K. Dziedzic-Kocurek, W. Kosaka, H. Miyasaka and D. Pinkowicz, Chem. Sci., 2023, 14, 9651–9663 RSC.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Coord. Chem. Rev., 2014, 258, 119–136 CrossRef.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- D. J. Mondal, A. Mondal, A. Paul and S. Konar, Inorg. Chem., 2022, 61, 4572–4580 CrossRef CAS PubMed.
- K. Uemura, S. Kitagawa, K. Fukui and K. Saito, J. Am. Chem. Soc., 2004, 126, 3817–3828 CrossRef CAS.
- K. Scanda, Y. Avila, R. Mojica, R. Amaro, B. Portales-Martínez, M. González, M. Avila, B. D. Moreno and E. Reguera, Colloids Surf., A, 2023, 676, 132114 CrossRef CAS.
- H. Ando, Y. Nakao, H. Sato, M. Ohba, S. Kitagawa and S. Sakaki, Chem. Phys. Lett., 2011, 511, 399–404 CrossRef CAS.
- N. Yanai, K. Kitayama, Y. Hijikata, H. Sato, R. Matsuda, Y. Kubota, M. Takata, M. Mizuno, T. Uemura and S. Kitagawa, Nat. Mater., 2011, 10, 787–793 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details, additional structural views, PXRD patterns, FT-IR spectra, TGA data, and additional magnetic measurement information. CCDC 2336307. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04266b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |