
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting lysosomes by design: novel N-acridine thiosemicarbazones that enable direct detection of intracellular drug localization and overcome P-glycoprotein (Pgp)-mediated resistance†
Busra
Kaya
a,
Henry
Smith
a,
Yanbing
Chen
a,
Mahan Gholam
Azad
ab,
Tiffany
M. Russell
a,
Vera
Richardson
ab,
Paul V.
Bernhardt
 c,
Mahendiran
Dharmasivam
*ab and
Des R.
Richardson
*abd
c,
Mahendiran
Dharmasivam
*ab and
Des R.
Richardson
*abd
aCentre for Cancer Cell Biology and Drug Discovery, Griffith University, Nathan, Brisbane, 4111, Queensland, Australia. E-mail: d.richardson@griffith.edu.au; m.dharmasivam@griffith.edu.au
bDepartment of Pathology and Bosch Institute, Molecular Pharmacology and Pathology Program, University of Sydney, Sydney, New South Wales, Australia
cSchool of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, 4072, Australia
dDepartment of Pathology and Biological Responses, Nagoya University Graduate School of Medicine, Nagoya 466-8550, Japan
First published on 15th August 2024
Abstract
Innovative N-acridine thiosemicarbazones (NATs) were designed along with their iron(III), copper(II), and zinc(II) complexes. Lysosomal targeting was promoted by specifically incorporating the lysosomotropic Pgp substrate, acridine, into the thiosemicarbazone scaffold to maintain the tridentate N, N, S-donor system. The acridine moiety enables a significant advance in thiosemicarbazone design, since: (1) it enables tracking of the drugs by confocal microscopy using its inherent fluorescence; (2) it is lysosomotropic enabling lysosomal targeting; and (3) as acridine is a P-glycoprotein (Pgp) substrate, it facilitates lysosomal targeting, resulting in the drug overcoming Pgp-mediated resistance. These new N-acridine analogues are novel, and this is the first time that acridine has been specifically added to the thiosemicarbazone framework to achieve the three important properties above. These new agents displayed markedly greater anti-proliferative activity against resistant Pgp-expressing cells than very low Pgp-expressing cells. The anti-proliferative activity of NATs against multiple Pgp-positive cancer cell-types (colon, lung, and cervical carcinoma) was abrogated by the third generation Pgp inhibitor, Elacridar, and also Pgp siRNA that down-regulated Pgp. Confocal microscopy demonstrated that low Pgp in KB31 (–Pgp) cells resulted in acridine's proclivity for DNA intercalation promoting NAT nuclear-targeting. In contrast, high Pgp in KBV1 (+Pgp) cells led to NAT lysosomal sequestration, preventing its nuclear localisation. High Pgp expression in KBV1 (+Pgp) cells resulted in co-localization of NATs with the lysosomal marker, LysoTracker™, that was significantly (p < 0.001) greater than the positive control, the di-2-pyridylketone-4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) Zn(II) complex, [Zn(DpC)2]. Incorporation of acridine into the thiosemicarbazone scaffold led to Pgp-mediated transport into lysosomes to overcome Pgp-resistance.
Introduction
Thiosemicarbazones of the di-2-pyridylketone thiosemicarbazone (DpT) class have been demonstrated to possess potent anti-cancer efficacy in vitro and in vivo in a range of tumor models.1–25 The first generation DpT agent, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT; Fig. 1A(I)), potently and selectively inhibited the growth of a variety of tumor xenografts1,6,12,18 and also prevented tumor spread.15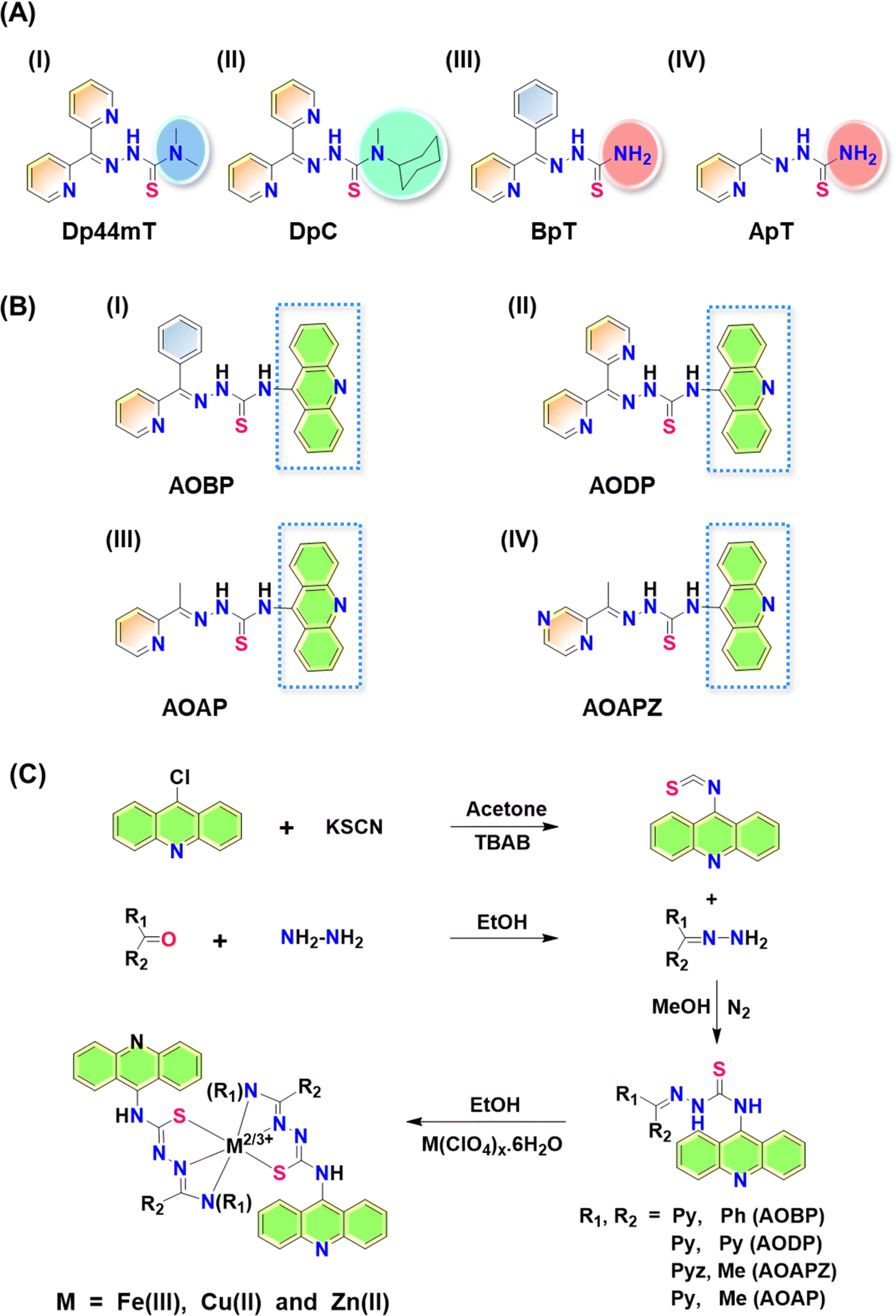 | ||
| Fig. 1 (A) Line drawings of the structures of: (I) Dp44mT; (II) DpC; (III) BpT; and (IV) ApT; (B) line drawings of the structures of the N-acridine thiosemicarbazones (NATs): (I) AOBP; (II) AODP; (III) AOAP; and (IV) AOAPZ. (C) Scheme for the synthesis of NATs and their metal ion complexes. | ||
Unfortunately, Dp44mT caused cardiac fibrosis in mice at high, non-optimal doses.6 This led to the second-generation DpT analogue, di-2-pyridylketone-4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) (Fig. 1A(II)), that markedly suppressed tumor xenograft growth and metastasis, showed good tolerability, potent anti-cancer activity, and no cardiotoxicity.9–14,16,17 Other related thiosemicarbazones were also prepared and characterized by our laboratory, including those of the 2-benzoylpyridine thiosemicarbazone (BpT) series (Fig. 1A(III)), and 2-acetylpyridine thiosemicarbazone (ApT) series (Fig. 1A(IV)).1,5,19
Despite their ability to bind iron, at the low doses used, Dp44mT or DpC had no appreciable effect on whole animal hematology.6,12,16,18 Other laboratories subsequently confirmed the marked anti-tumor activity of these agents in vitro and in vivo in terms of their selective inhibition of tumor growth and spread.15,20,23,25 Another important aspect of the activity of these thiosemicarbazones was their ability to broadly and effectively synergize with a variety of standard chemotherapies.9,22 Due to its excellent safety, anti-tumor activity,10–14,16,17 and pharmacokinetics,24 DpC entered multi-centre clinical trials in 2016.7,21
Multidrug resistance is a critical challenge for cancer patients and is defined as the resistance of cancer cells to multiple chemotherapeutic drugs with different structures and mechanisms of action.26 One of the best characterized and common resistance mechanisms in cancer involves the cellular efflux of chemotherapeutic drugs through drug “pumps” such as ATP-dependent translocase, ABCB1, which is also commonly known as P-glycoprotein (Pgp).1,27 The presence of Pgp on the plasma membrane leads to the efflux of a range of anti-cancer chemotherapeutics (e.g., Doxorubicin; DOX) that are Pgp substrates out of the cell, effectively decreasing their intracellular concentration and preventing their cytotoxicity (Fig. 2A).37
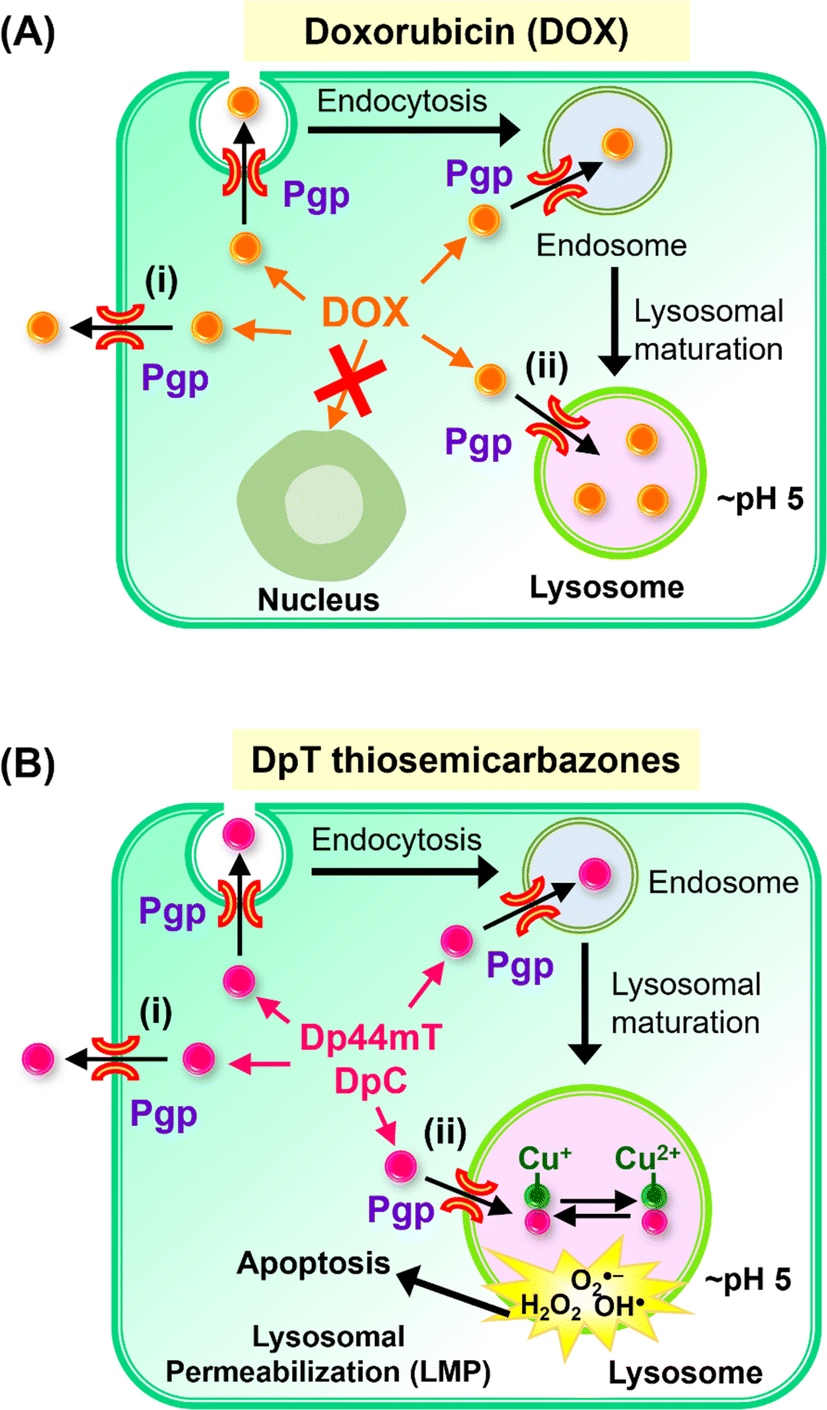 | ||
| Fig. 2 (A) Schematic demonstrating that P-glycoprotein (Pgp) is found in cells at the plasma membrane and also intracellularly in endosomes and lysosomes after endocytosis.2,28–34 Schematic demonstrating that Pgp induces resistance to Doxorubicin (DOX) through two mechanisms: (i) as an efflux pump at the plasma membrane that induces the release of DOX from tumor cells;28 and (ii) through the ability of lysosomal Pgp to sequester DOX in the lysosomal lumen, preventing its access to a key pharmacological target, the nucleus.5,28 The sequestration of DOX in lysosomes leads to resistance as it does not induce lysosomal membrane permeabilization.5,28 (B) Schematic showing that: (i) Pgp at the plasma membrane effluxes DpT thiosemicarbazones such as Dp44mT and DpC from cells;1 and (ii) lysosomal Pgp results in thiosemicarbazone sequestration in lysosomes that results in lysosomal membrane permeabilization1–4,35,36 due to the potent redox activity of their Cu(II) complexes. Lysosomal membrane permeabilization is a catastrophic process leading to apoptosis.18,36 | ||
More recently, investigations by our laboratory1,28 and others29–34 demonstrated that Pgp is not only expressed on the plasma membrane, but is also found intracellularly and is functional in lysosomes28 (Fig. 2A and B). In fact, Pgp acts to not only efflux drugs out of cells via its localization on the plasma membrane, but is also involved in sequestering anti-cancer drugs such as DOX and thiosemicarbazones into lysosomes (Fig. 2A and B).1–4,28 The quarantining of DOX into lysosomes prevents its access to sensitive targets (e.g., in the nucleus), leading to resistance (Fig. 2A).2,3,5,28 As such, the lysosome acts as a “waste bag” to segregate DOX, preventing its access to the nucleus.28
Previous studies have comprehensively demonstrated that the localization of Pgp in lysosomes occurs via endocytosis from the plasma membrane, leading to Pgp-containing endosomes and lysosomes (Fig. 2A and B).1,5,28–34 Notably, the Pgp transporter on the plasma membrane pumps drugs out of cells, while its internalization into endosomes and lysosomes leads to Pgp transporting drugs into the endosomal and lysosomal lumen1–5,28
Our laboratory showed that Dp44mT and DpC could overcome Pgp-mediated resistance in multiple tumor cell-types,1,2,4,5,19 with Dp44mT being significantly more effective against Pgp-expressing tumors in vivo than their non-Pgp-expressing counterparts.1 Further, our studies showed these agents are Pgp substrates and are pumped out of cells, while Pgp in the lysosomal membrane transports Dp44mT and DpC into lysosomes (Fig. 2B).1,2,4,5,19
In contrast to the anthracycline, DOX (Fig. 2A), transport of Dp44mT or DpC by Pgp into lysosomes results in a marked increase in lysosomal membrane permeabilization and cytotoxicity (Fig. 2B), selectively killing Pgp-expressing tumor cells.1,2,5,19 This effect is mediated, in part, by the lysosomotropic character of the thiosemicarbazones,36 with these compounds becoming positively charged and trapped in the organelle after transport into the lysosomal lumen by Pgp.1
Once in the lysosomal lumen, the thiosemicarbazones bind intra-lysosomal Cu released by the degradation of proteins, resulting in redox-active Cu(II) complexes, which generate reactive oxygen species (ROS) that induce lysosomal membrane permeabilization (Fig. 2B).1,4,35,36,38 Thus, Dp44mT and DpC use an innovative mechanism to overcome Pgp-resistance in cancer cells.1,2,5,19 In contrast, while DOX is a Pgp substrate that is also transported into lysosomes, it does not generate potently cytotoxic Cu complexes, and does not induce lysosomal membrane permeabilization5,28 (Fig. 2A).
Neither Dp44mT nor DpC were specifically designed to become trapped in the lysosome and to take advantage of the lysosomal Pgp transport process (Fig. 2B). The authors only deciphered this mechanism after extensive investigation of the medicinal chemistry and pharmacology of the DpT class of thiosemicarbazones.1,6,19,36,38 To improve their efficacy, one specific design strategy could be to incorporate well-characterized lysosomotropic moieties such as the acridine into the thiosemicarbazone framework to maintain the tridentate N, N, S donor system. The acridine moiety enables a significant advance in thiosemicarbazone design, because acridine: (1) is a Pgp substrate,39–43 it facilitates lysosomal targeting, resulting in the drug overcoming Pgp-mediated resistance; (2) it is lysosomotropic44–46 enabling effective targeting and trapping in lysosomes;47–51 and (3) another advantage is that acridine can be directly imaged due to its inherent fluorescence, enabling tracking of its intracellular location.52,53 These new N-acridine analogues are novel, with this being the first time that acridine has been added to the thiosemicarbazone framework to achieve the three important properties above.
Considering the many interesting properties of the acridine moiety, we report herein the synthesis and characterization of four novel N-acridine thiosemicarbazones (NATs) and their Cu(II), Fe(III), and Zn(II) complexes. These include ligands with acridine at the N4 position and a variety of aromatic substituents at the N1 position, including: 2-benzoyl ((E)-N-(acridin-9-yl)-2-(phenyl(pyridin-2-yl)methylene)hydrazine-1-carbothioamide; AOBP; Fig. 1B(I)); 2-dipyridyl ((E)-N-(acridin-9-yl)-2-(di(pyridin-2-yl)methylene)hydrazine-1-carbothioamide; AODP; Fig. 1B(II)); 2-acetylpyridine ((2E)-N-(acridin-9-yl)-2-[1-(pyridin-2-yl)ethylidene]hydrazine-1-carbothioamide; AOAP; Fig. 1B(III)); and 2-acetylpyrazine ((2E)-N-(acridin-9-yl)-2-[1-(pyrazin-2-yl)ethylidene]hydrazine-1-carbothioamide; AOAPZ; Fig. 1B(IV)).
Since acridine is a Pgp substrate,39–43 these novel NAT ligands demonstrate selectivity for cancer cells with high Pgp expression (KBV1; +Pgp) relative to their counterpart cells with very low Pgp levels (KB31; −Pgp). This effect was not only observed with these latter cell-types, but also in other tumor cells with substantially lower Pgp expression. Confocal microscopy demonstrated lysosomal targeting of the NATs, especially AOBP, that was markedly enhanced by Pgp expression. These results indicated our targeting strategy of including the lysosomotropic Pgp substrate, acridine, into the thiosemicarbazone structural framework was successful, leading to their ability to overcome Pgp-mediated resistance.
Results and discussion
Synthesis of the NATs was accomplished by adding aryl and diaryl hydrazones to isothiocyanatoacridine (Fig. 1C). Benzoylpyridine, pyridyl, and di-2-pyridyl systems were selected because they resulted in potent anti-cancer activity when incorporated into the thiosemicarbazones of the DpT, BpT, and ApT series (Fig. 1A(I–IV)), which are known Pgp substrates.1,5,9,19 Substitution with nitrogen heterocycles like pyridine and pyrazine was also critical for preserving the tridentate N, N, S donor atom ligand system employed by α-N-heterocyclic thiosemicarbazones.54 Analogues bearing aryl substituents other than benzoylpyridine and di-2-pyridyl were synthesized to dissect structure–activity relationships of the NATs and their Fe(III), Cu(II), and Zn(II) complexes.The X-ray crystallography of NAT ligands AOBP, AODP, AOAPZ and the [Zn(AOAPZ)2] complex, along with their analysis, are provided in ESI (see ESI Results and discussion; Fig. S1†). Packing diagrams are presented in Fig. S2.† The purification of the AOAPZ Fe(III) complex resulted in an unstable product due to frequent desulfurization and cyclization. A similar outcome was observed during the crystallization of [Fe(AOBP)2]+, leading to an unusual bicyclic structure in its perchlorate salt form, with H2S being eliminated from the thiosemicarbazone (Fig. S3†). The exact mechanistic details of this conversion remain unclear and were not the focus of this investigation. However, the synthesized Fe(III) complexes are stable prior to recrystallization.
The NAT ligands demonstrate selective anti-proliferative activity against Pgp-expressing KBV1 (+Pgp) cells versus KB31 (−Pgp) cells
Our previous investigations implemented a range of Pgp-expressing cell-types to demonstrate that Dp44mT and DpC overcome Pgp-mediated drug resistance.1,2,4,5,19 As such, studies were designed to assess whether the inclusion of a well-known Pgp substrate (acridine)39–41,43 into the thiosemicarbazone framework conferred selective anti-proliferative activity against Pgp-expressing tumor cells. In initial experiments, the novel NAT ligands and their Fe(III), Cu(II), and Zn(II) complexes were assayed against a well-characterized pair of cell lines: KBV1 (+Pgp) cells that express very high levels of functional Pgp, and KB31 (−Pgp) cells, which do not express significant Pgp levels.1,19,28The expression of Pgp in these cell-types was confirmed by western analysis prior to use, with KBV1 (+Pgp) cells expressing markedly and significantly (p < 0.0001) greater Pgp protein levels than KB31 (−Pgp) cells, where expression was extremely low (Fig. 3A). The anti-proliferative efficacy (i.e., IC50) of the novel NATs over incubations of 24, 48, and 72 h was compared to the positive control ligands, Dp44mT and DpC (Table 1), which are known Pgp substrates and selectively target Pgp-expressing cells.1,2,4,5,19
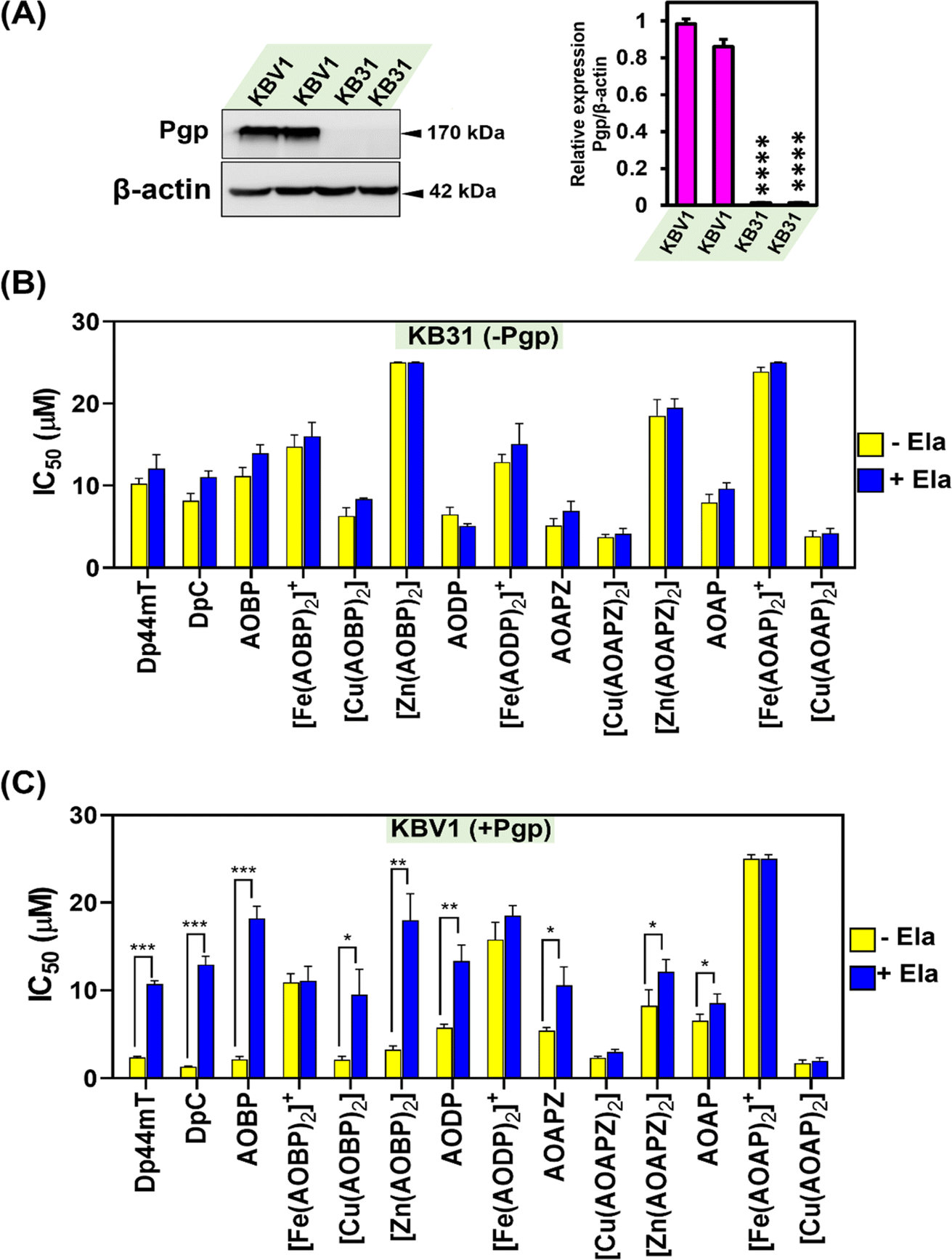 | ||
| Fig. 3 (A) Pgp is expressed at high levels in KBV1 (+Pgp) cells, while KB31 (−Pgp) cells do not express significant Pgp levels. Results are relative to the protein loading control, β-actin. The western blot shown is typical of 3 experiments, while the densitometry represents the mean ± SD (3 experiments). ****p < 0.0001 vs. KBV1 (+Pgp) cells. (B) Incubation of KB31 (−Pgp) cells with the Pgp inhibitor, Elacridar (Ela), has no significant (p > 0.05) effect on the anti-proliferative activity (IC50) of Dp44mT, DpC, and the NATs and their Fe(III), Cu(II), and Zn(II) complexes. (C) Incubation of KBV1 (+Pgp) cells with the Pgp inhibitor, Ela, significantly inhibits the anti-proliferative activity (IC50) of the positive controls, Dp44mT, DpC, and also AOBP, [Cu(AOBP)2], [Zn(AOBP)2], AODP, AOAPZ, [Zn(AOAPZ)2], and AOAP. In (B) and (C), KB31 (−Pgp) and KBV1 (+Pgp) cells were pre-incubated in the presence and absence of Ela (0.2 μM) for 1 h/37 °C. The cells were then incubated for 24 h/37 °C with the ligands and their complexes in the presence and absence of Ela (0.2 μM). Cellular proliferation was then examined to calculate the IC50. Results are mean ± SD (3 experiments). *p < 0.05; **p < 0.01; ***p < 0.001 as shown on the graph. | ||
| Compounds | IC50 values (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 24 h | Fold dif. | 48 h | Fold dif. | 72 h | Fold dif. | ||||
| KBV1 | KB31 | KBV1 | KB31 | KBV1 | KB31 | ||||
| Dp44mT | 2.39 ± 0.12 | 8.16 ± 0.88 | 3.42 | 0.23 ± 0.01 | 2.45 ± 0.24 | 10.54 | 0.01 ± 0.002 | 0.13 ± 0.02 | 10.68 |
| DpC | 1.31 ± 0.09 | 10.23 ± 0.7 | 7.82 | 0.16 ± 0.01 | 1.0 ± 0.05 | 6.71 | 0.19 ± 0.02 | 0.29 ± 0.07 | 1.53 |
| AOBP | 2.12 ± 0.30 | 11.16 ± 1.04 | 5.12 | 0.74 ± 0.02 | 4.56 ± 0.32 | 6.19 | 0.61 ± 0.06 | 0.97 ± 0.22 | 1.60 |
| [Fe(AOBP)2]+ | 10.93 ± 0.98 | 14.75 ± 1.98 | 1.35 | 0.40 ± 0.02 | 3.78 ± 0.75 | 9.42 | 0.25 ± 0.02 | 1.94 ± 0.24 | 7.68 |
| [Cu(AOBP)2] | 2.13 ± 0.37 | 8.39 ± 0.98 | 3.95 | 0.26 ± 0.03 | 10.39 ± 0.52 | 39.72 | 0.14 ± 0.02 | 1.63 ± 0.34 | 11.66 |
| [Zn(AOBP)2] | 3.25 ± 0.42 | >25 | >7.7 | 0.59 ± 0.01 | 4.74 ± 0.54 | 7.96 | 0.37 ± 0.08 | 2.78 ± 0.16 | 7.43 |
| AODP | 5.75 ± 0.42 | 6.49 ± 0.86 | 1.13 | 0.55 ± 0.01 | 2.27 ± 0.31 | 4.09 | 0.46 ± 0.04 | 2.13 ± 0.22 | 4.61 |
| [Fe(AODP)2]+ | 15.78 ± 1.97 | 17.66 ± 0.96 | 1.12 | 0.22 ± 0.03 | 5.23 ± 0.41 | 23.59 | 0.30 ± 0.010 | 2.40 ± 0.41 | 7.95 |
| AOAPZ | 5.43 ± 0.33 | 5.14 ± 0.86 | 0.95 | 1.32 ± 0.06 | 4.43 ± 0.39 | 3.36 | 1.42 ± 0.26 | 3.55 ± 0.51 | 2.49 |
| [Cu(AOAPZ)2] | 3.89 ± 0.18 | 3.73 ± 0.32 | 0.96 | 2.49 ± 0.05 | 3.64 ± 0.13 | 1.46 | 1.50 ± 0.19 | 2.84 ± 0.71 | 1.89 |
| [Zn(AOAPZ)2] | 12.13 ± 1.84 | 18.17 ± 2.46 | 1.49 | 0.63 ± 0.04 | 4.33 ± 0.45 | 6.85 | 0.69 ± 0.12 | 1.72 ± 0.18 | 2.47 |
| AOAP | 6.56 ± 0.74 | 7.95 ± 0.99 | 1.21 | 2.29 ± 0.18 | 4.62 ± 0.31 | 2.01 | 1.69 ± 0.33 | 2.12 ± 0.38 | 1.25 |
| [Fe(AOAP)2]+ | >25 | >25 | — | 1.21 ± 0.22 | 19.58 ± 2.03 | 7.96 | 2.09 ± 0.39 | >5 | >2.38 |
| [Cu(AOAP)2] | 2.96 ± 0.38 | 4.21 ± 0.68 | 1.42 | 2.46 ± 0.06 | 3.23 ± 0.26 | 1.31 | 0.87 ± 0.161 | 2.56 ± 0.61 | 2.97 |
Assessing Dp44mT and DpC after all incubation times (Table 1) demonstrated they showed significantly (p < 0.001–0.05) greater anti-proliferative activity (i.e., lower IC50) against KBV1 (+Pgp) cells than KB31 (−Pgp) cells, with a 1.53 to 10.68-fold decrease of the IC50 being evident in KBV1 (+Pgp) cells versus KB31 (−Pgp) cells. The greatest differential in anti-proliferative activity between KBV1 (+Pgp) and KB31 (−Pgp) cells was observed for Dp44mT after incubation periods of 48 and 72 h (10.54 to 10.68-fold), while for DpC the greatest differential occurred at 24 and 48 h (7.82 and 6.71-fold, respectively; Table 1). The mean differential anti-proliferative activity between KBV1 (+Pgp) and KB31 (−Pgp) cells over 24, 48, and 72 h for Dp44mT (3.42, 10.54, and 10.68-fold, respectively; mean: 8.21-fold) was on average greater than that observed for DpC (7.82, 6.71, and 1.53-fold, respectively; mean: 5.35-fold).
Examining the four new NAT ligands, AOBP resulted in on average the most pronounced selective anti-proliferative efficacy against Pgp-expressing cells (Table 1). In fact, AOBP demonstrated significantly (p < 0.01) greater activity (1.60–6.19-fold; average: 4.30-fold) against KBV1 (+Pgp) cells than KB31 (−Pgp) cells over all incubation times (Table 1). Similar, although less pronounced efficacy was observed for AODP. In this case, AODP demonstrated greater 1.13–4.61-fold (average 3.28-fold) activity against KBV1 (+Pgp) cells versus KB31 (−Pgp) cells after all incubation times, with it having significantly (p < 0.01) greater efficacy after 48 and 72 h (Table 1). AOAPZ, and particularly AOAP, demonstrated the least anti-proliferative efficacy of the NAT series (Table 1). In fact, the mean differential anti-proliferative activity between KBV1 (+Pgp) and KB31 (−Pgp) cells over 24, 48, and 72 h for AOAPZ (0.95, 3.36, and 2.49-fold, respectively; mean: 2.27-fold) was greater than AOAP (1.21, 2.01, and 1.25-fold, respectively; mean: 1.49-fold).
In summary, all NAT ligands demonstrate selective anti-proliferative activity against KBV1 (+Pgp) cells vs. KB31 (−Pgp) cells, with AOBP and AODP demonstrating the greatest efficacy and AOAP the least.
Effect of complexation on selective anti-proliferative activity against KBV1 (+Pgp) cells: identification of pronounced selectivity upon complexation of AOBP with Cu(II)
Complexation of the NAT ligands with Fe(III), Cu(II), and Zn(II) was then examined in terms of their selective anti-proliferative activity against KBV1 (+Pgp) cells versus their KB31 (−Pgp) counterparts (Table 1). This was done to assess the potential roles of these complexes in the selective anti-proliferative activity of the ligands against Pgp-expressing cells. Generally, the Cu(II) complexes demonstrated the greatest anti-proliferative activity versus the other complexes in both cell-types (Table 1). This observation correlated strongly with previous studies on other thiosemicarbazones and the known role of their Cu(II) complexes in redox activity and lysosomal membrane permeabilization.4,36,55,56Of all the agents examined in Table 1, [Cu(AOBP)2] demonstrated the greatest mean differential in anti-proliferative activity between KBV1 (+Pgp) cells and KB31 (−Pgp) cells over 24 h, 48 h, and 72 h (i.e., 3.95, 39.72, and 11.66-fold, respectively; mean: 18.44-fold; Table 1). Both the Zn(II) and Fe(III) complexes of AOBP demonstrated less, but still pronounced differential activity between KBV1 (+Pgp) and KB31 (−Pgp) cells over 24, 48, and 72 h. That is, for [Zn(AOBP)2] >7.70, 7.96, and 7.43-fold, respectively (mean: 7.70-fold), and for [Fe(AOBP)2]+, 1.35, 9.42, and 7.68-fold, respectively (mean: 6.15-fold; Table 1). Considering the known lability of Zn(II) thiosemicarbazone complexes,57 it cannot be excluded that transchelation of the NAT Zn(II) could occur with other cytosolic ligands. There are a plethora of labilizing ligands in cells that the Zn(II) complexes can be exposed to under physiological conditions. These ligands can potentially include glutathione (GSH) and cysteine, etc.58,59 and also 3000 proteins that bind Zn(II), which include 10% of the human proteome, such as the metallothioneins.60,61
Despite many attempts, the Cu(II) and Zn(II) complexes of AODP, as well as the Zn(II) complex of AOAP, always led to desulfurization and cyclization of the ligand during purification and/or recrystallization, similar to the Fe(III) complex of AOAPZ (see X-ray Crystallography section – ESI data†). However, relative to [Fe(AOBP)2]+, the AODP Fe(III) complex displayed greater mean differential activity between KBV1 (+Pgp) and KB31 (−Pgp) cells over 24, 48, and 72 h (1.12, 23.59, and 7.95-fold, respectively; mean: 10.89-fold; Table 1).
The Cu(II) and Zn(II) complexes of AOAPZ, and the Fe(III) and Cu(II) complexes of AOAP demonstrated lower mean differential activity between KBV1 (+Pgp) and KB31 (−Pgp) cells over 24, 48, and 72 h than the respective AOBP and AODP complexes (Table 1). It can be speculated this observation may relate to the differential ability of these complexes to act as Pgp substrates. However, further studies are required to validate this suggestion. The most active of the AOAPZ and AOAP complexes were [Zn(AOAPZ)2] and [Fe(AOAP)2]+ after a 48 h incubation, where the differential anti-proliferative activity between KBV1 (+Pgp) and KB31 (−Pgp) cells was 6.85-fold and 7.96-fold, respectively (Table 1).
Overall, of all the agents assessed herein, [Cu(AOBP)2] demonstrated the greatest mean differential activity against KBV1 (+Pgp) cells versus KB31 (−Pgp) cells. The Fe(III) and Zn(II) complexes of AOBP and the AODP Fe(III) complex also demonstrated marked efficacy against KBV1 (+Pgp) cells.
Pgp inhibition with Ela inhibits anti-proliferative activity against KBV1 (+Pgp) cells for multiple NATs including the potent complex, [Cu(AOBP)2]
Considering the results in Table 1, where Dp44mT, DpC, and several NAT ligands and their complexes demonstrated selective anti-proliferative activity against KBV1 (+Pgp) cells, studies then examined the ability of the well-characterized, third-generation Pgp inhibitor, Ela,1,19,28,62 to prevent this effect (Fig. 3B and C). In these studies, KBV1 (+Pgp) and KB31 (−Pgp) cells were pre-incubated in the presence or absence of Ela (0.2 μM) for 1 h/37 °C prior to treatment for 24 h/37 °C with the ligands or their complexes (0.012–25 μM) in the presence and absence of Ela (0.2 μM). The IC50 values were then calculated.First, as a relevant negative control, the anti-proliferative activity of the ligands and complexes was examined using KB31 (−Pgp) cells (Fig. 3B), where no significant (p > 0.05) difference in anti-proliferative activity was observed for all agents in the presence or absence of Ela. These results demonstrate that Ela has no non-specific effect on the anti-proliferative activity of these compounds in the absence of Pgp expression, as we showed previously for Dp44mT, DpC, and other thiosemicarbazones.19 These data validate the high specificity of Ela as a third-generation Pgp inhibitor.1,19,28,62
As a positive control using KBV1 (+Pgp) cells, inhibition of Pgp with Ela resulted in a marked and significant (p < 0.001) decrease in the anti-proliferative efficacy of Dp44mT and DpC (Fig. 3C), which our laboratory has previously demonstrated.1,19 Similarly, significant (p < 0.001–0.05) inhibition of anti-proliferative activity by Ela in KBV1 (+Pgp) cells was observed for all four NAT ligands (i.e., AOBP, AODP, AOAPZ and AOAP), and also [Cu(AOBP)2], [Zn(AOBP)2], and [Zn(AOAPZ)2] (Fig. 3C). In contrast, Ela had no significant (p > 0.05) effect on the anti-proliferative activity of [Fe(AOBP)2]+, [Fe(AODP)2]+, [Cu(AOAPZ)2], [Fe(AOAP)2]+, or [Cu(AOAP)2] in KBV1 (+Pgp) cells (Fig. 3C). This observation suggested the anti-proliferative activity of these latter complexes in KBV1 (+Pgp) cells was Pgp-independent.
In summary, the specific Pgp inhibitor, Ela, suppressed the anti-proliferative activity of multiple NATs and their complexes in KBV1 (+Pgp) cells, including AOBP and [Cu(AOBP)2] that displayed a marked differential in anti-proliferative efficacy between KBV1 (+Pgp) and KB31 (−Pgp) cells.
Inhibition of Pgp activity by Ela or Pgp silencing inhibits the anti-proliferative efficacy of AOBP in multiple Pgp-expressing tumor cell-types, but not KB31 (−Pgp) cells that express very low Pgp levels
The studies above demonstrated that of the NAT ligands, AOBP demonstrated the greatest differential anti-proliferative activity between KBV1 (+Pgp) and KB31 (−Pgp) cells (Table 1). Furthermore, Ela was demonstrated to inhibit the efficacy of AOBP in KBV1 (+Pgp) cells, but not KB31 (−Pgp) cells (Fig. 3B and C), with these results being consistent with Pgp playing a role in enhancing the anti-proliferative activity of this agent. Considering this, investigations then examined the anti-proliferative activity of AOBP using multiple Pgp-expressing cancer cell-types, namely KBV1 (+Pgp) cells, DMS-53 small cell lung carcinoma cells, and HCT-15 colon adenocarcinoma cells (Table 2(A and B)). The study also assessed the ability of either Ela (using the protocol above in Fig. 3), or Pgp siRNA versus a non-targeting negative control siRNA (NC siRNA), to inhibit the anti-proliferative efficacy of the thiosemicarbazones. The KB31 (−Pgp) cells were used as a relative negative control for the Pgp-expressing cell-types and were treated using the same protocol. In these silencing studies, Pgp siRNA or NC siRNA was incubated with the cells for 72 h/37 °C and then incubated for 24 h/37 °C in the presence or absence of DOX, Dp44mT, DpC, or AOBP (Table 2(B)).| (A) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Agents | IC50 (μM) | |||||||
| KBV1 | KBV1 + Ela | KB31 | KB31 + Ela | DMS-53 | DMS-53 + Ela | HCT-15 | HCT-15 + Ela | |
| DOX | >100 | 1.37 ± 0.10 | 1.36 ± 0.48 | 1.16 ± 0.24 | 17.96 ± 5.94 | 2.08 ± 0.89** | 33.15 ± 5.42 | 10.35 ± 2.54** |
| Dp44mT | 2.39 ± 0.12 | 10.70 ± 0.34*** | 8.16 ± 0.88 | 12.06 ± 1.34 | 1.49 ± 0.12 | 2.96 ± 0.16*** | 0.69 ± 0.08 | 5.88 ± 0.19**** |
| DpC | 1.31 ± 0.09 | 12.93 ± 0.95*** | 10.23 ± 0.66 | 11.04 ± 0.76 | 0.66 ± 0.03 | 3.51 ± 0.98** | 0.28 ± 0.11 | 2.01 ± 0.29** |
| AOBP | 2.12 ± 0.30 | 18.21 ± 1.38*** | 11.16 ± 1.04 | 13.94 ± 1.04 | 1.95 ± 0.53 | 4.07 ± 1.63* | 1.52 ± 0.05 | 9.95 ± 0.74**** |
| (B) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Agents | IC50 (μM) | |||||||
| KBV1 + NC siRNA | KBV1 + Pgp siRNA | KB31 + NC siRNA | KB31 + Pgp siRNA | DMS-53 + NC siRNA | DMS-53 + Pgp siRNA | HCT-15 + NC siRNA | HCT-15 + Pgp siRNA | |
| DOX | 55.01 ± 2.96 | 2.43 ± 0.15**** | 5.75 ± 0.66 | 5.48 ± 0.95 | 38.47 ± 5.30 | 2.57 ± 0.32*** | 58.29 ± 6.50 | 31.25 ± 4.15** |
| Dp44mT | 1.57 ± 0.66 | 8.34 ± 2.04** | >25 | >25 | 5.44 ± 0.80 | 19.37 ± 0.59**** | 5.49 ± 2.33 | 14.83 ± 2.44** |
| DpC | 2.45 ± 0.68 | 19.01 ± 5.69** | >25 | >25 | 5.57 ± 0.28 | 8.99 ± 0.88** | 4.90 ± 0.29 | 9.14 ± 0.27*** |
| AOBP | 2.79 ± 0.34 | 18.98 ± 5.62** | >25 | >25 | 22.87 ± 1.05 | >25 | 12.56 ± 1.17 | 21.42 ± 2.41** |
The DMS-53 and HCT-15 cell-types demonstrated significantly (p < 0.001) lower Pgp levels than those in KBV1 (+Pgp) cells, and markedly and significantly (p < 0.001) greater expression than KB31 (−Pgp) cells, where Pgp levels were exceptionally low (Fig. S4A†). As a relevant control, the effect of Pgp siRNA was examined on Pgp protein levels and was demonstrated to markedly and significantly (p < 0.0001) decrease its expression in KBV1 (+Pgp), DMS-53, and HCT-15 cells versus the NC siRNA (Fig. S4B†).
In these studies with Ela or Pgp siRNA (Table 2(A and B)), the activity of AOBP was compared to several well-characterized controls, namely: (1) DOX, the efficacy of which is promoted by inhibition of Pgp activity or Pgp silencing;28 or (2) Dp44mT or DpC whose anti-proliferative activity is known to be abrogated by Pgp inhibition or silencing.1,19 DOX is a classical Pgp substrate and is effectively effluxed by Pgp from cancer cells,28,63 or transported into lysosomes by Pgp where it is sequestered, without inducing lysosomal membrane permeabilization (Fig. 2A), leading to resistance.5,28 In all cell-types expressing Pgp (KBV1, DMS-53, and HCT-15; Fig. S4A†), incubation with Ela or Pgp siRNA resulted in a pronounced and/or significant (p < 0.0001–0.01) increase in anti-proliferative activity (i.e., decrease of the IC50) of DOX (Table 2(A and B)). As a pertinent example, Ela markedly enhanced the anti-proliferative activity of DOX in the three Pgp-expressing cell-types, but had no significant (p > 0.05) effect on the IC50 of DOX in KB31 (−Pgp) cells in the presence or absence of Ela (Table 2(A)). These data using KB31 (−Pgp) cells again confirmed the selectivity of Ela against Pgp-expressing cell-types observed above in Fig. 3B.
In terms of the effect of Pgp siRNA on anti-proliferative activity, assessing KBV1 (+Pgp) cells, the IC50 of DOX was 55.01 ± 2.96 μM for the NC siRNA-treated cells, while it was significantly (p < 0.0001) lower after incubation with the Pgp siRNA (2.43 ± 0.15 μM; Table 2(B)). It is notable that incubation of KB31 (−Pgp) cells with Pgp siRNA resulted in no significant (p > 0.05) change in the IC50 relative to NC siRNA (Table 2(A and B)), demonstrating this treatment did not non-specifically alter proliferation. Collectively, these results in Table 2(A and B) using Ela or Pgp siRNA are consistent with the role of Pgp in preventing the anti-proliferative activity of DOX, inducing resistance.
Opposite to their effect on enhancing the anti-proliferative efficacy of DOX, either Ela or Pgp siRNA decreased and/or significantly (p < 0.0001–0.05) decreased the anti-proliferative activity (i.e., increased the IC50) of Dp44mT, DpC, and AOBP in all three Pgp-expressing tumor cell-types versus control media alone or the NC siRNA, respectively (Table 2(A and B)). For example, examining KBV1 (+Pgp) cells after treatment with AOBP, an IC50 of 2.79 ± 0.34 μM was observed after incubation with NC siRNA, with this being significantly (p < 0.01) increased by Pgp siRNA to 18.98 ± 5.62 μM (Table 2(B)). Thus, these results suggested that Pgp plays a role in promoting the anti-proliferative efficacy of these thiosemicarbazones.
These results above assessing KBV1 (+Pgp) cells were in contrast to those obtained for KB31 (−Pgp) cells that demonstrated greater resistance to Dp44mT, DpC, and AOBP, having IC50 values of 8.16 to >25 μM in the presence or absence of Ela or Pgp siRNA (Table 2(A and B)). This resistance to the anti-proliferative activity of these thiosemicarbazones is consistent with KB31 (−Pgp) cells not expressing substantial Pgp (Fig. 3A and S4A†). In fact, incubation of KB31 (−Pgp) cells with Ela had no significant (p > 0.05) effect on the IC50 of AOBP (>25 μM) versus control media alone (Table 2(A and B)). Further, for all three thiosemicarbazones, treatment of KB31 (−Pgp) cells with Pgp siRNA had no significant (p > 0.05) effect on the IC50 (>25 μM) versus the NC siRNA.
In summary, the results in Table 2(A and B) using Ela and Pgp siRNA in multiple cell-types indicate that Pgp activity and expression play a role in enhancing the anti-proliferative activity of AOBP, Dp44mT, and DpC, while inhibiting the efficacy of DOX.
Cellular Pgp expression is increased by DOX, but decreased by Dp44mT, DpC, and the NAT ligands
The investigations above in Fig. 3 and Tables 1 and 2 indicate Pgp plays a key role in the anti-proliferative activity of the NAT ligands. To further investigate the role of Pgp in their activity, experiments investigated the effects of the agents on Pgp expression in cells (Fig. S5†). Our previous studies demonstrated a marked differential between the activity of DOX that increased Pgp protein expression in KBV1 (+Pgp) cells, while DpC and Dp44mT decreased its levels.5 These effects were rationalized by DOX selectively killing non-Pgp-expressing KBV1 (+Pgp) cells, leading to cultures enriched in Pgp-expressing cells.5 In contrast, DpC and Dp44mT were selectively cytotoxic to Pgp-expressing KBV1 (+Pgp) cells, effectively decreasing Pgp protein levels.5In these studies, KBV1 (+Pgp) cells (Fig. S5A†) or KB31 (−Pgp) cells (Fig. S5B†) were incubated for 48 h/37 °C with DOX (100–400 μM), Dp44mT (10–40 μM), DpC (1–20 μM), AOBP (10–40 μM), and AODP (10–40 μM). Our previous studies5 guided preliminary experiments to optimize the concentrations to ensure cellular selection without extreme toxicity that would prevent harvesting sufficient protein. As demonstrated previously,5 incubation of KBV1 (+Pgp) cells with increasing DOX concentrations caused a marked and significant (p < 0.0001) increase in Pgp expression versus the control. In contrast, Dp44mT, DpC, AOBP, and AODP all significantly (p < 0.0001–0.001) decreased Pgp expression, with this effect being more pronounced as the concentration increased (Fig. S5A†). Parallel control studies with KB31 (−Pgp) cells demonstrated no impact of DOX or these thiosemicarbazones on Pgp expression, which was at very low levels in these cells (Fig. S5B†).
Collectively, these results indicate that AOBP and AODP act similarly to Dp44mT and DpC and selectively target Pgp-expressing cells, demonstrating a distinct advantage over DOX, which promotes the selection of KBV1 (+Pgp) cells expressing high Pgp levels.
Pgp expression in KBV1 (+Pgp) cells prevents nuclear accumulation of AOBP and AOAP and their localization in a cytoplasmic compartment
The studies above in Tables 1 and 2 and Fig. 4 and S4† demonstrate that multiple NATs act analogously to Dp44mT and DpC and possess selective anti-proliferative efficacy against Pgp expressing cells, with this activity being mediated by Pgp. As our previous studies indicated that lysosomal Pgp was important for overcoming Pgp-mediated resistance by Dp44mT and DpC,1–3 investigations then examined intracellular localization of several NATs (Fig. 5). To assess if the inherent fluorescence of NATs and their Fe(III), Cu(II), and Zn(II) complexes were suitable for cellular imaging, fluorescence spectroscopy was initially performed to calculate quantum yield (Φ; Table S2†). Studies were performed using the compounds (10 μM) dissolved in DMSO implementing an excitation at 400 nm and a 10 nm slit width (Fig. S6†). The quantum yield of the agents was 0.15–0.92 × 10−3 (Table S2†), which was similar to, or greater, than fluorescent [Zn(DpC)2] (0.36 × 10−3), which targets lysosomes, and that we previously used in confocal microscopy studies.4,55,57 As such, the NATs were considered suitable for confocal microscopy analysis.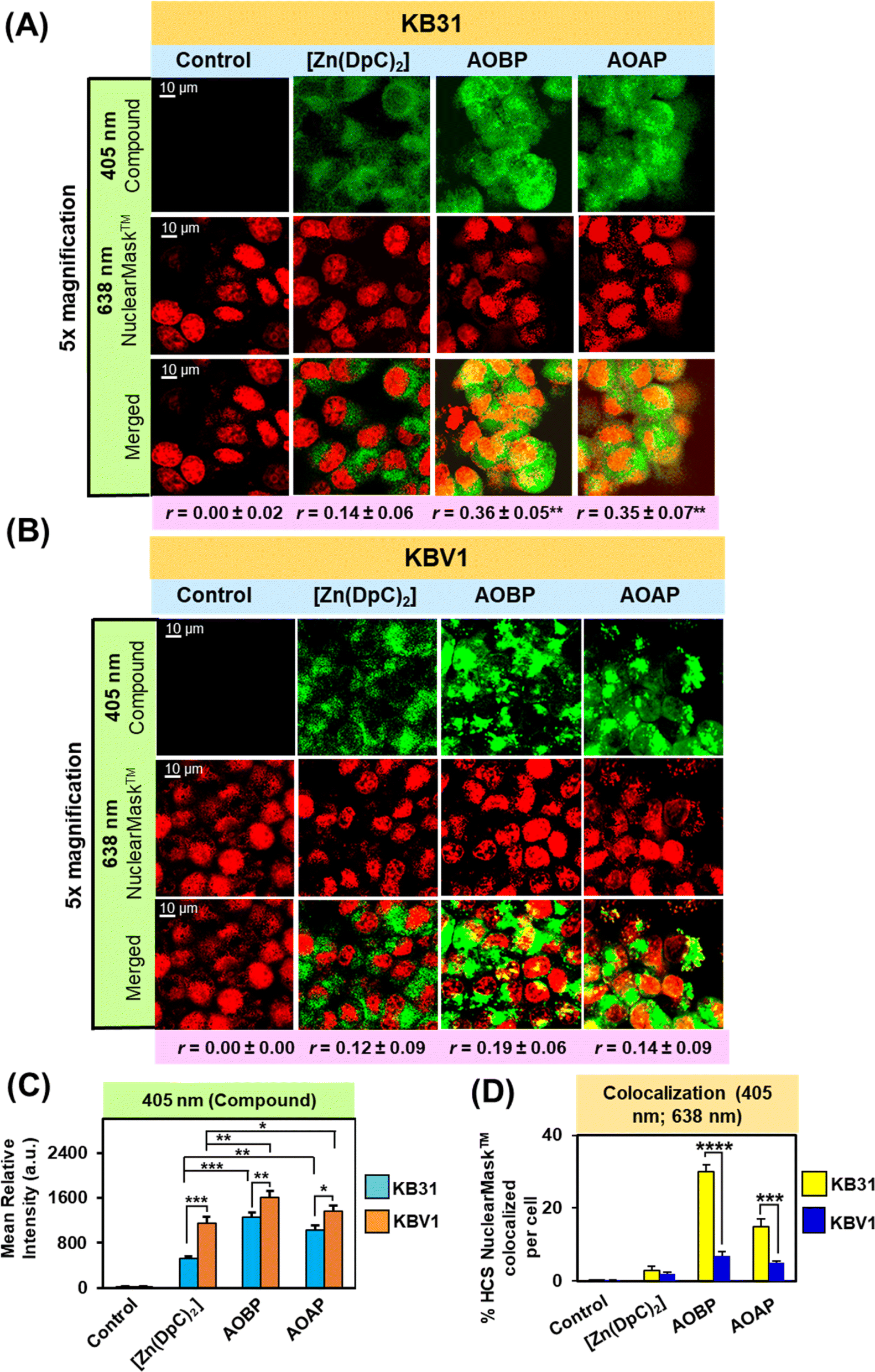 | ||
| Fig. 4 Confocal microscopy demonstrates AOBP and AOAP co-localize with the nuclear probe, HCS NuclearMask™ Deep Red, in KB31 (−Pgp) cells, while only cytosolic localization of AOBP and AOAP is observed in KBV1 (+Pgp) cells. (A) The KB31(−Pgp) and (B) KBV1 (+Pgp) cell-types were incubated with [Zn(DpC)2], AOBP, and AOAP (25 μM) for 2 h/37 °C and then examined using confocal microscopy. The images are typical from 3 experiments. (C) Quantification of the pixel intensity of [Zn(DpC)2], AOBP, and AOAP at 405 nm was performed with ImageJ. (D) Quantitation of the co-localization of HCS NuclearMask™ Deep Red (638 nm) with [Zn(DpC)2], AOBP, and AOAP (405 nm) was performed with ImageJ. Studies were performed using a 40× objective using a constant acquisition setting with Olympus Fluoview FV3000 software. Images were digitally magnified (5×) for demonstration of the co-localization. Results are mean ± SD (3 experiments). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 as shown on the graph. Scale bar: 10 μm. | ||
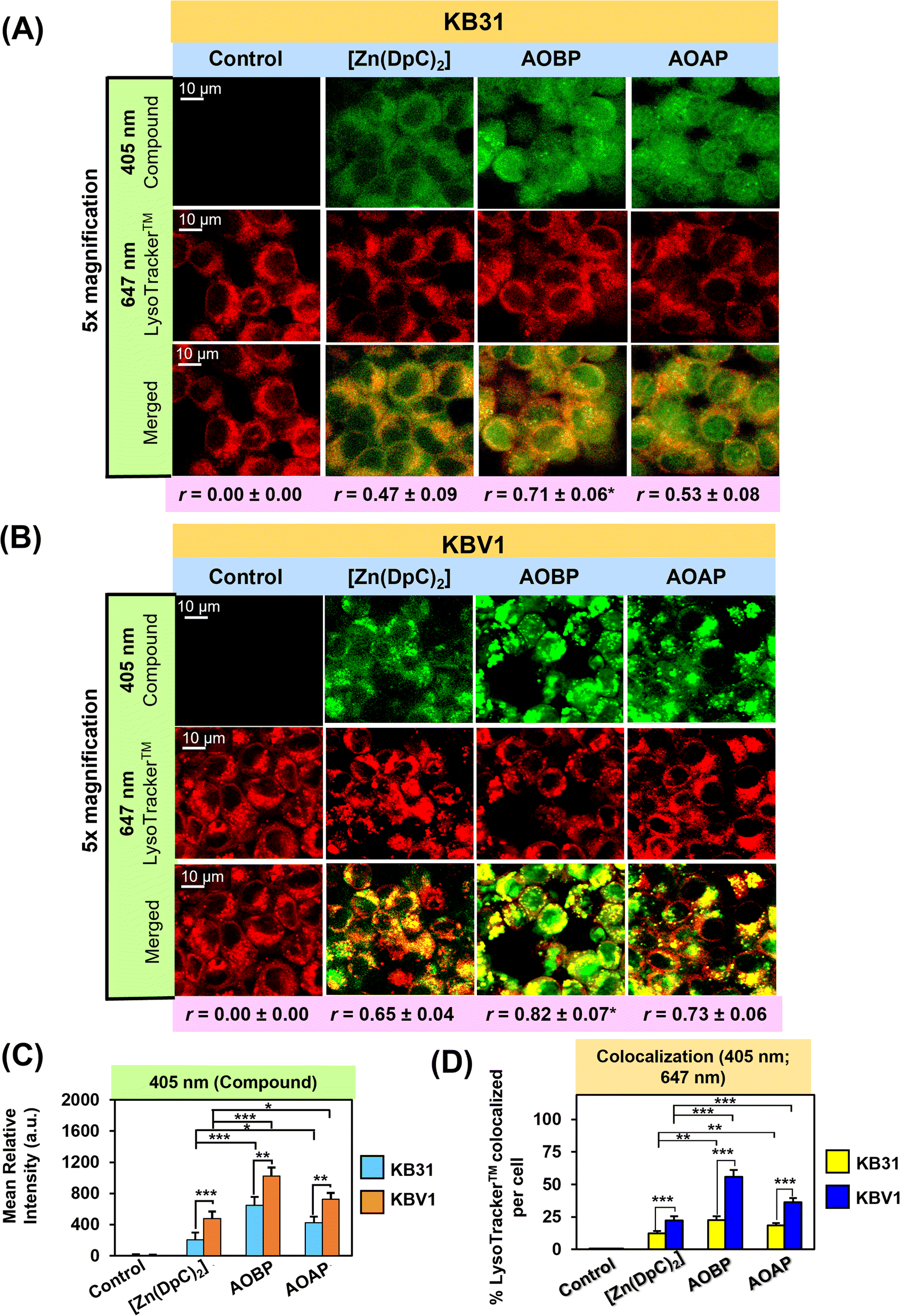 | ||
| Fig. 5 (A and B) Confocal microscopy demonstrates that AOBP and AOAP co-localize to a significantly greater extent with LysoTracker™ than [Zn(DpC)2] in (A) KB31 (−Pgp) cells and especially (B) KBV1 (+Pgp) cells. Both cell-types were incubated with inherently fluorescent, [Zn(DpC)2], AOBP, and AOAP (25 μM) for 2 h/37 °C resulting in a green (405 nm), punctate pattern. Incubation with LysoTracker™ (red) resulted in a typical, punctated pattern of red (647 nm) fluorescence. (C) Quantification of the pixel intensity of [Zn(DpC)2], AOBP, and AOAP at 405 nm was performed with ImageJ. (D) Quantification of the co-localization of [Zn(DpC)2], AOBP, and AOAP (405 nm) with LysoTracker™ (647 nm) was estimated with ImageJ. Co-localization of AOBP and LysoTracker™ was confirmed by the Pearson's correlation coefficients. The images are typical from three experiments. Studies were performed using a 40× objective using a constant acquisition setting with Olympus Fluoview FV3000 software. Images were digitally magnified (5×) for demonstration of the co-localization. Results are mean ± SD (3 experiments). *p < 0.05; **p < 0.01; ***p < 0.001 as shown on the graph. Scale bar = 10 μm. | ||
The observed lower quantum yield of the NATs free ligands (Table S2†) may be due to the occurrence of a photo-induced electron transfer (PET) process, which is caused by the presence of a lone pair of electrons on the nitrogen atoms. This PET process is prevented after complexation, resulting in an increase in fluorescence intensity.64–66 Interestingly, only the [Cu(AOAP)2] complex demonstrated markedly enhanced fluorescence intensity compared to the other complexes and their free ligands (Fig. S6†). This can be attributed to the separation of the excited states of the ligand and Cu(II) caused by the distortion in the geometry of the [Cu(AOAP)2] complex in the excited state.67,68 Similar observations have been noted in other thiosemicarbazone Cu(II) complexes as well.67–69 Unexpectedly, the paramagnetic NAT Fe(III) complexes slightly increased the fluorescence intensity relative to their free ligand, this may be due to the chelation-enhanced fluorescence and suppression of PET.70 As expected, enhancement of fluorescence intensity and quantum yield of the Zn(II) complexes (Table S2†) is probably due to the chelation of the ligand to the Zn(II) centre. Chelation enhances the rigidity of the ligand, thereby reducing the loss of energy by non-radiative decay of the intra-ligand emission excited state.71,72
Of the NAT ligands and complexes, AOBP and AOAP, were initially utilized in confocal microscopy studies as they demonstrated the strongest and weakest mean differential anti-proliferative activity, respectively, against KBV1 (+Pgp) versus KB31 (−Pgp) cells (Table 1). Furthermore, Pgp was involved in their anti-proliferative activity, as the Pgp inhibitor, Ela, was shown to significantly (p < 0.001–0.05) inhibit the anti-proliferative activity of AOBP and AOAP against KBV1 (+Pgp) cells, but not KB31 (−Pgp) cells (Fig. 3B, C and Table 2).
As part of our dissection of the intracellular localization of AOBP and AOAP, we first examined using confocal microscopy if AOBP and AOAP demonstrated nuclear co-localization (Fig. 4A and B). This assessment was performed considering the reported proclivity of acridine-substituted compounds to intercalate into DNA47,73,74 and accumulate in the nucleus.41,73,75–77 To demonstrate the nucleus, KB31 (−Pgp) and KBV1 (+Pgp) cells were probed with the well-established nuclear probe, HCS NuclearMask™ Deep Red.78,79 This latter probe was used for confocal microscopy rather than Hoechst 33342 to prevent interference with the fluorescence spectra of AOBP, AOAP, and [Zn(DpC)2]. Further, the inherently fluorescent, [Zn(DpC)2],4,55,57 was implemented as a relative control, as our laboratory demonstrated it did not localize to the nucleus, but was mainly co-localized in the cytoplasm with the classical lysosomal marker, LysoTracker™.4,55,57
After a 2 h/37 °C incubation with KB31 (−Pgp) cells (Fig. 4A) and KBV1 (+Pgp) cells (Fig. 4B), [Zn(DpC)2], AOBP, and AOAP (25 μM) exhibited a punctate distribution at 405 nm due to their own inherent fluorescence (Fig. 4A). The distribution of [Zn(DpC)2] was confined mainly to the outer (extra-nuclear) cytosol of KB31 (−Pgp) cells, while in contrast, AOBP and AOAP resulted in near homogeneous staining of the entire cell (Fig. 4A). Quantitation of compound fluorescence demonstrated significantly (p < 0.01) higher cellular levels of AOBP and to a lesser extent AOAP versus [Zn(DpC)2] (Fig. 4C). The HCS NuclearMask™ Deep Red probe assessed at 638 nm produced well-defined staining of the nucleus.
Following co-localization analysis of HCS NuclearMask™ (red channel) with [Zn(DpC)2] (green channel), no significant merge (yellow fluorescence) was apparent indicating the two probes were in different cellular compartments (Fig. 4A). In contrast, for both AOBP and AOAP, the green fluorescence of AOBP and AOAP substantially merged with red fluorescence of HCS NuclearMask™, leading to a pronounced and significant (p < 0.0001–0.001) increase in the intensity of yellow fluorescence and the Pearson's correlation coefficient (r = 0.36 ± 0.05 (3) and r = 0.35 ± 0.07 (3) for AOBP and AOAP, respectively) versus the control (r = 0.00 ± 0.02). These results indicated AOBP and AOAP co-localized with HCS NuclearMask™ in KB31 (−Pgp) cells, and this occurred to a significantly (p < 0.01) greater extent than that observed for [Zn(DpC)2], where the Pearson's correlation coefficient was r = 0.14 ± 0.06 (3) (Fig. 4A).
Examining KBV1 (+Pgp) cells, the fluorescence of [Zn(DpC)2] was similar although more defined and punctate relative to that observed in KB31 (−Pgp) cells (cf.Fig. 4A and B), being mainly confined to the extranuclear region (Fig. 4B). However, in contrast to the near homogenous cellular distribution (including nucleus) of AOBP and AOAP in KB31 (−Pgp) cells (Fig. 4A), in KBV1 (+Pgp) cells, the distribution of both ligands was intensively punctate and confined predominantly to the cytosolic extra-nuclear region (Fig. 4B). Quantitation of fluorescence at 405 nm demonstrated significantly (p < 0.001–0.05) higher levels of all 3 compounds in KBV1 (+Pgp) cells versus KB31 (−Pgp) cells, with AOAP and especially AOBP demonstrating significantly (p < 0.01–0.05) higher levels than [Zn(DpC)2] in KBV1 (+Pgp) cells (Fig. 4C). In fact, for [Zn(DpC)2], AOBP and AOAP, upon co-localization, there was no appreciable merge of their inherent fluorescence (green) with the red fluorescence of the HCS NuclearMask™ in KBV1 (+Pgp) cells (Pearson's correlation coefficients of r = 0.12 ± 0.09 (3) for [Zn(DpC)2]; r = 0.19 ± 0.06 (3) for AOBP; and r = 0.14 ± 0.09 (3) for AOAP; Fig. 4B). In fact, assessing quantitation, co-localization of AOBP and AOAP with HCS NuclearMask™ was markedly and significantly (p < 0.001) greater in KB31 (−Pgp) cells than KBV1 (+Pgp) cells (Fig. 4D).
Together, these data above demonstrate that Pgp expression in KBV1 (+Pgp) cells prevents nuclear accumulation of AOBP and AOAP (Fig. 4B), which was pronounced in KB31 (−Pgp) cells (Fig. 4A), probably due to the proclivity of acridine to intercalate into DNA.47,73,74 Instead, in KBV1 (+Pgp) cells, AOBP and AOAP (as well as [Zn(DpC)2]), accumulated in a cytoplasmic compartment, which considering our previous data examining Dp44mT, DpC, [Zn(DpC)2], and DOX,1,2,4,5,19,28 was hypothesized to be lysosomes. In fact, we previously reported analogous results with the fluorescent Pgp substrate, DOX, where in KBV1 (+Pgp) cells this drug was observed in the lysosomal compartment.5 In contrast, in KB31 (−Pgp) cells, DOX co-localized with the nucleus.5 This difference in drug localization between the two cell-types indicated the functional role of Pgp in KBV1 (+Pgp) cells to sequester DOX into lysosomes, preventing its entrance into the nucleus.5
In conclusion, Pgp expression in KBV1 (+Pgp) cells (Fig. 4B) prevented the strong nuclear co-localization of AOBP and AOAP observed in KB31 (−Pgp) cells (Fig. 4A), and instead resulted in their accumulation in an extranuclear cytoplasmic compartment.
AOBP and AOAP avidly co-localize with LysoTracker™-stained lysosomes in KB31 (−Pgp) and particularly KBV1 (+Pgp) cells to a greater extent than [Zn(DpC)2]
Previous studies from our laboratory indicated that Dp44mT, DpC, and [Zn(DpC)2] targeted the lysosome,1,4,5,19,36 and further, were markedly sequestered in lysosomes of cells with high Pgp levels.1,3–5 To directly visualize if AOBP and AOAP in KB31 (−Pgp) cells and KBV1 (+Pgp) were localized in lysosomes, live cell confocal microscopy imaging was performed using the well-characterized LysoTracker™ Deep Red (LysoTracker™) probe that resulted in the classical punctate cytoplasmic distribution of these organelles (Fig. 5A–D).80,81 In both cell-types, incubation with the inherently fluorescent, [Zn(DpC)2],4,55,57 AOBP, and AOAP, resulted in a punctate pattern of green fluorescence at 405 nm (Fig. 5A and B).As observed in Fig. 4A, in contrast to the extranuclear cytoplasmic distribution of [Zn(DpC)2], a distinctly different pattern of AOBP and AOAP fluorescence was observed in KB31 (−Pgp) cells, where almost homogenous cellular distribution was observed (Fig. 5A). Indeed, significantly (p < 0.001–0.05) higher levels of AOBP and AOAP relative to [Zn(DpC)2] were observed in KB31 (−Pgp) cells (Fig. 5C). Examining KB31 (−Pgp) cells, upon the merge of the inherent fluorescence of [Zn(DpC)2], AOBP, or AOAP with LysoTracker™, there was a significant (p < 0.001) increase in co-localization (yellow fluorescence) versus the control (Fig. 5D). Relative to [Zn(DpC)2], the co-localization intensity was significantly (p < 0.01) greater for AOAP and especially AOBP in KB31 (−Pgp) cells (Fig. 5D). This co-localization in KB31 (−Pgp) cells for AOBP and AOAP led to Pearson's correlation coefficients of r = 0.71 ± 0.06 (3) and r = 0.53 ± 0.08 (3), respectively. Notably, the AOBP correlation coefficient was significantly greater (p < 0.05) than that of [Zn(DpC)2] = 0.47 ± 0.09 (3) (Fig. 5A). These results indicated increased lysosomal targeting of AOAP and especially AOBP in KB31 (−Pgp) cells, indicating our targeting strategy of incorporating the lysosomotropic agent, acridine,44–46 into the thiosemicarbazone framework was successful.
Examining KBV1 (+Pgp) cells, the cellular distribution of the fluorescence of [Zn(DpC)2], AOBP, and AOAP was extranuclear in puncta and similar to the distribution of LysoTracker™ (Fig. 5B). In fact, the appearance of the puncta for [Zn(DpC)2], AOBP, and AOAP in KBV1 (+Pgp) cells was “tighter” and more punctate in KBV1 (+Pgp) cells than KB31 (−Pgp) cells (cf.Fig. 5A and B). In particular, the distribution of AOBP and AOAP was distinctly particulate and extranuclear in KBV1 (+Pgp) cells (Fig. 5B), which was clearly different to their homogenous cellular distribution that included the nucleus in KB31 (−Pgp) cells (Fig. 5A). Furthermore, significantly (p < 0.001–0.01) greater intracellular levels of all 3 compounds were observed in KBV1 (+Pgp) versus KB31 (−Pgp) cells, with the levels of AOBP and AOAP being significantly (p < 0.001–0.05) greater than [Zn(DpC)2] in both cell-types (Fig. 5C).
Co-localization of [Zn(DpC)2], AOBP, and AOAP (green) with LysoTracker™ (red) in KBV1 (+Pgp) cells resulted in a punctate pattern of yellow fluorescence that appeared more intense and distinct than that in KB31 (−Pgp) cells (cf.Fig. 5A and B). The co-localization intensity of [Zn(DpC)2], AOAP, and particularly AOBP with LysoTracker™, was significantly (p < 0.001) higher in KBV1 (+Pgp) cells than KB31 (−Pgp) cells (Fig. 5D).
This co-localization in KBV1 (+Pgp) cells was significantly (p < 0.001) increased for AOAP and especially AOBP relative to [Zn(DpC)2], again suggesting the greater lysosomal targeting of both NAT's (Fig. 5A and D). Hence, AOBP and AOAP demonstrated greater lysosomal targeting than the positive control, [Zn(DpC)2], in cells highly expressing Pgp. The Pearson's correlation coefficients of r = 0.82 ± 0.07 (3) and r = 0.73 ± 0.06 (3) for AOBP and AOAP, respectively, relative to r = 0.65 ± 0.04 (3) for [Zn(DpC)2] in KBV1 (+Pgp) cells (Fig. 5B), additionally supported lysosomal localization for all these compounds. Further, the more marked and significant (p < 0.01) lysosomal co-localization of AOBP compared to AOAP in KBV1 (+Pgp) cells (Fig. 5D) correlates with the greater Pgp-dependent anti-proliferative activity of AOBP versus AOAP (Table 1, Fig. 3B and C).
Collectively, the observations in Fig. 5A–D demonstrate our specific design strategy of including the acridine moiety that is lysosomotropic,44–46 but also a Pgp substrate,39–43 has successfully led to pronounced lysosomal targeting of AOAP and especially AOBP.
Conclusions
Dp44mT and DpC are well-characterized substrates of the drug transporter, Pgp, and are actively transported into lysosomes by utilizing Pgp on the lysosomal membrane (Fig. 2B).1–4,35 However, neither of these ligands were ever specifically designed to be Pgp substrates or to have lysosomotropic properties. Only after dissection of the mechanism of their anti-proliferative activity was the role of lysosomal targeting1,36 and Pgp1,2 realized as critical determinants of their efficacy and ability to overcome drug resistance.6Furthermore, precise determination of the subcellular distribution of Dp44mT and DpC were limited by the lack of inherent fluorescence exhibited by the ligands themselves. To overcome these limitations, we designed, synthesized, and characterized for the first time a new series of NAT ligands and their Fe(III), Cu(II), and Zn(II) complexes. An important aspect of their design was the incorporation of the fluorescent acridine moiety that imparts both lysosomotropic character and is a Pgp substrate.44–46,82
The NATs and their Fe(III), Cu(II), and Zn(II) complexes demonstrated higher anti-proliferative activity against KBV1 (+Pgp) versus KB31 (−Pgp) cells (Table 1). Noteworthy was the marked anti-proliferative selectivity of [Cu(AOBP)2] in KBV1 (+Pgp) versus KB31 (−Pgp) cells (Table 1). The potent anti-proliferative activity of AOBP against KBV1 (+Pgp) cells and Pgp-expressing DMS-53 lung cancer and HCT-15 colon carcinoma cells was abrogated by the specific Pgp inhibitor, Ela, or Pgp silencing (Fig. 3C, Table 2(A and B)). These observations further substantiated the role of Pgp transport activity in the selectivity of these agents (Fig. 6A). In marked contrast, the anti-proliferative efficacy of the anthracycline, DOX, was increased by Ela and Pgp silencing, which agrees with the well-known role of Pgp in decreasing the activity of this drug.28
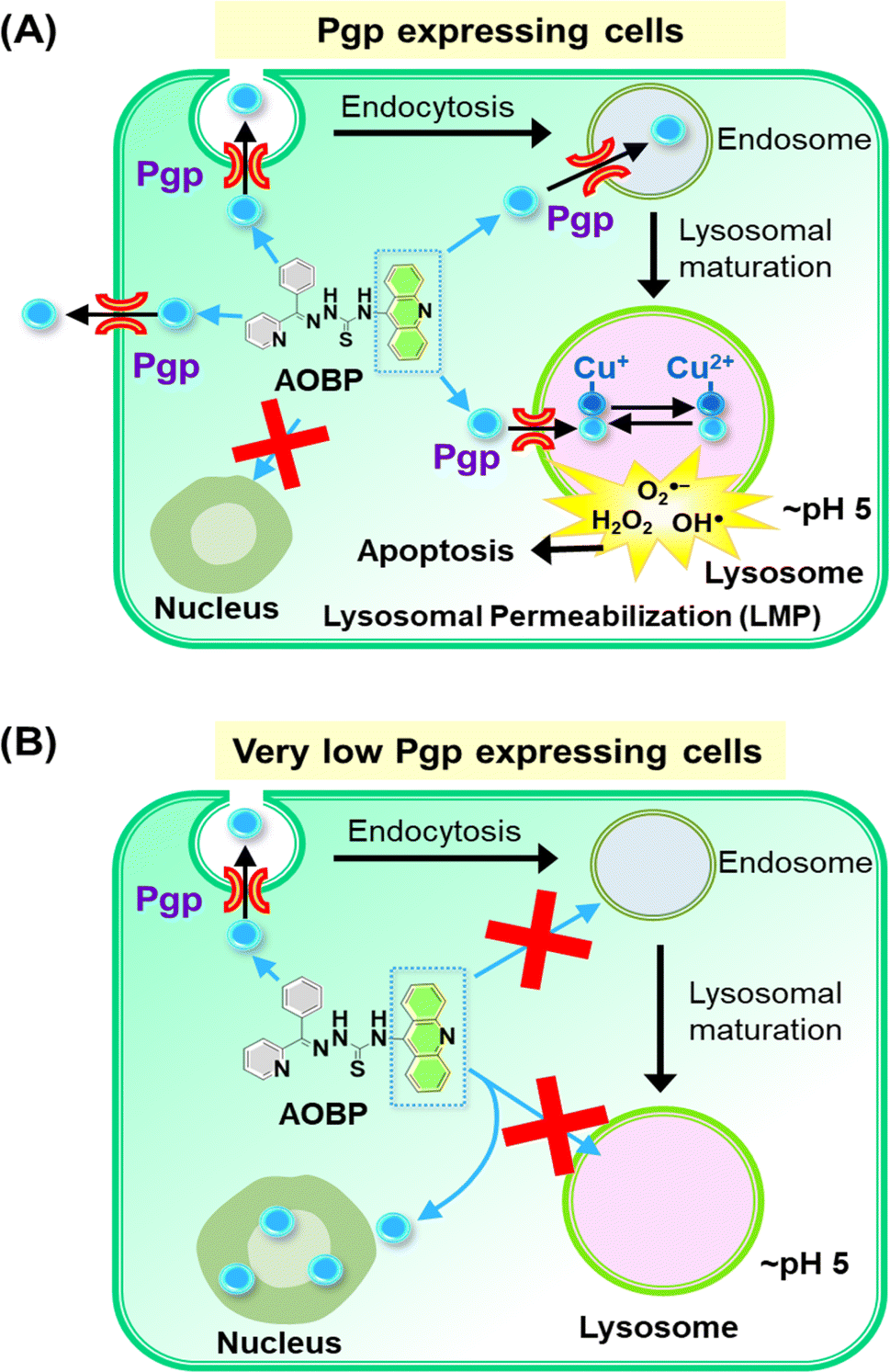 | ||
| Fig. 6 Schematic illustrating the mechanism of action of AOBP in overcoming Pgp-mediated drug resistance by targeting lysosomes. (A) In Pgp-expressing cells, Pgp on the plasma membrane becomes endocytosed into endosomes,1,28 with a proportion of endosomes maturing into lysosomes. The acridine moiety is lysosomotropic, and acts as a Pgp substrate, conferring these properties on AOBP. The AOBP can be transported by Pgp into the lysosomal lumen, where it becomes positively charged and trapped at lysosomal pH. This lysosomal sequestration prevents AOBP from entering the nucleus where it can target DNA. Once in the lysosome, AOBP then binds Cu liberated after lysosomal degradation of proteins to form potent, redox-active Cu complexes that generate ROS and causes lysosomal membrane permeabilization and apoptosis. In addition to entrance of AOBP into the lysosome via lysosomal Pgp, AOBP can theoretically also be effluxed out of the cell by Pgp at the plasma membrane. (B) In cells with very low Pgp expression, the acridine moiety of AOBP can result in strong nuclear association. This latter effect is presumably due to the known ability of acridine to intercalate into DNA,47,73,74 but Pgp overexpression prevents this effect due to lysosomal sequestration of the drug (see (A)). | ||
Confocal microscopy demonstrated that inherently fluorescent AOBP and AOAP co-localized with the nucleus in KB31 (−Pgp) cells, but not KBV1 (+Pgp) cells (Fig. 4A and B). These results suggested Pgp expression prevented nuclear accumulation of AOBP and AOAP, leading to cytosolic accumulation in the lysosomal compartment of KBV1 (+Pgp) cells as demonstrated by the well-characterized LysoTracker™ probe (Fig. 5B and D). The nuclear accumulation of AOBP and AOAP in KB31 (−Pgp) cells was probably mediated by the acridine moiety that has proclivity for the nucleus due to its ability to intercalate into DNA (Fig. 5A and 6B).47,73,74 In KBV1 (+Pgp) cells expressing high Pgp levels, nuclear localization of the acridine-substituted thiosemicarbazones was prevented due to their sequestration into lysosomes by Pgp (Fig. 6A). Our laboratory reported similar observations with DOX,28 which like the DNA intercalating agent, acridine,47,73,74 associates with the nucleus in KB31 (−Pgp) cells, but instead is sequestered by Pgp into lysosomes in KBV1 (+Pgp) cells.
Examining co-localization of AOBP and AOAP with lysosomes, it was demonstrated this was greater than the lysosomal targeting agent, [Zn(DpC)2],4,55,57 particularly in KBV1 (+Pgp) cells (Fig. 5B and D). These results indicate the specific design strategy of adding acridine to promote both lysosomotropic character and lysosomal Pgp transport has been successful. These results agree with the pronounced Pgp-dependent anti-proliferative activity of AOBP and AOAP in KBV1 (+Pgp) versus KB31 (−Pgp) cells (Table 1).
The significantly greater co-localization in lysosomes of AOBP relative to AOAP (Fig. 5A, B and D) was also consistent with the markedly greater anti-proliferative activity of AOBP relative to AOAP against KBV1 (+Pgp) cells (Table 1). Acridine is a well-known lysosomotropic agent44–46 with a pKa of 5.6 (ref. 83) with an assessment of its ionization as a function of pH (Marvin Sketch, ChemAxon software)84 indicating that ∼89% is charged at lysosomal pH (pH 5), while 93% is neutral at physiological pH (pH 7.4). Similar analysis of the four NATs demonstrated that 94% of the agent is charged at pH 5, while 93% is neutral. These properties would facilitate the entrance of the neutral NAT ligand from the extracellular milieu (pH 7.4) across the plasma membrane and into the cell. The neutral ligand would then transverse the cytosol and enter lysosomes and become charged and trapped in these organelles. This analysis demonstrates a marked improvement of the NATs relative to Dp44mT or DpC, where only ∼16% of the ligand was charged at lysosomal pH,36 and explains the pronounced lysosomal accumulation of the NATs (Fig. 5A–D).
The addition of the lipophilic acridine moiety to the thiosemicarbazone structure definitely has marked effects on their biological activity, as described above. While an optimum lipophilicity is an important consideration for effective anti-proliferative activity of thiosemicarbazone ligands,16 the current study did not determine its influence on the cellular uptake or efflux of the NATs or their ability to overcome Pgp-mediated resistance. Further investigation is required with a broader range of NATs to examine specific structure–activity relationships with regards to the role of varying lipophilicity on their uptake and ability to overcome Pgp resistance.
In conclusion, the addition of the acridine moiety into the thiosemicarbazone scaffold increases its lysosomotropic character and the targeting of Pgp in lysosomes that could be useful in overcoming deadly Pgp-mediated resistance, which is a major killer in advanced and resistant cancer. As such, this article substantially advances our understanding of targeting lysosomes by specifically designed anti-cancer therapeutics to overcome Pgp-mediated resistance.
Data availability
All data are available from the corresponding authors by request. The electronic ESI† contains detailed descriptions of the following general materials and methods: materials; general methods; X-ray crystallography; cell culture; a cellular proliferation assay; confocal microscopy analysis; western blot analysis; and statistical methods. The 1H NMR and 13C NMR spectra for the ligands are presented in Fig. S7–S14,† with the Zn(II) complexes of AOBP and AOAPZ being shown in Fig. S15–S18.† The HR-MS spectra of AOBP, AODP, AOAPZ, and AOAP are demonstrated in Fig. S19–S2.†Author contributions
D. R. R., M. D., and B. K. designed studies, supervised students and staff on the project, analyzed data, wrote the manuscript, and obtained grant funding. B. K. H. S. and M. D. synthesized and characterized compounds, analyzed data, conducted cellular or molecular studies, and/or wrote parts of the manuscript. B. K., M. D., Y. C., T. M. R., V. R., and M. G. A. edited the text, performed cellular or molecular experiments, and prepared figures. P. V. B. performed data analysis, conducted X-ray diffraction, and contributed to writing sections of the text.Conflicts of interest
The authors declare no competing conflict of interest.Acknowledgements
D. R. R thanks the National Health and Medical Research Council (NHMRC) of Australia for Senior Principal Research Fellowships (APP1062607 and 1159596), Australian Research Council Discovery Grant (DP200103530), NHMRC Ideas Grant (2010632), and NHMRC Project Grants (APP1144829; APP1128152; and APP1144456). D. R. R., M. D., and P. V. B thank the NHMRC for an Ideas Grant (2019160) and National Breast Cancer Foundation of Australia (NBCF) for an Investigator Initiated Research Scheme Grant (IIRS-23-004). M. D. and D. R. R. appreciate support from the Thrasher Research Fund USA Early Career Award. M. D. appreciates a Griffith University Postdoctoral Fellowship from Griffith University and an Elaine Henry Postdoctoral Fellowship from the NBCF. M. D, B. K and D. R. R thank the NHMRC MRFF for an Early to Mid-Career Researcher Grant (2027365). B. K. kindly acknowledges support from The Scientific and Technological Research Council of Turkey (TUBITAK) for a 2219-International Postdoctoral Research Fellowship (App. # 1059B192000031) and Griffith University Postdoctoral Fellowship. M. G. A and D. R. R. acknowledge the award of a Tour de Cure Postgraduate Research Student PhD Scholarship. M. G. A. and T. M. R. appreciate a Griffith University Postgraduate Research Scholarship (GUPRS).References
- P. J. Jansson, T. Yamagishi, A. Arvind, N. Seebacher, E. Gutierrez, A. Stacy, S. Maleki, D. Sharp, S. Sahni and D. R. Richardson, J. Biol. Chem., 2015, 290, 9588–9603 CrossRef CAS PubMed.
- N. A. Seebacher, D. J. Lane, P. J. Jansson and D. R. Richardson, J. Biol. Chem., 2016, 291, 3796–3820 CrossRef CAS PubMed.
- L. Al-Akra, D.-H. Bae, S. Sahni, M. L. Huang, K. C. Park, D. J. Lane, P. J. Jansson and D. R. Richardson, J. Biol. Chem., 2018, 293, 3562–3587 CrossRef CAS PubMed.
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef CAS PubMed.
- N. A. Seebacher, D. R. Richardson and P. J. Jansson, Cell Death Dis., 2016, 7, e2510 CrossRef CAS PubMed.
- M. Whitnall, J. Howard, P. Ponka and D. R. Richardson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14901–14906 CrossRef CAS PubMed.
- T. P. Wijesinghe, M. Dharmasivam, C. C. Dai and D. R. Richardson, Pharmacol. Res., 2021, 173, 105889 CrossRef CAS PubMed.
- Y. Yu, Y. S. Rahmanto and D. Richardson, Br. J. Pharmacol., 2012, 165, 148–166 CrossRef CAS PubMed.
- M. Dharmasivam, M. G. Azad, R. Afroz, V. Richardson, P. J. Jansson and D. R. Richardson, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130152 CrossRef CAS PubMed.
- B. Geleta, K. C. Park, P. J. Jansson, S. Sahni, S. Maleki, Z. Xu, T. Murakami, M. Pajic, M. V. Apte, D. R. Richardson and Z. Kovacevic, FASEB J., 2021, 35, e21347 CrossRef CAS PubMed.
- Z.-L. Guo, D. R. Richardson, D. S. Kalinowski, Z. Kovacevic, K. C. Tan-Un and G. C.-F. Chan, J. Hematol. Oncol., 2016, 9, 1–16 CrossRef PubMed.
- Z. Kovacevic, S. Chikhani, D. B. Lovejoy and D. R. Richardson, Mol. Pharmacol., 2011, 80, 598–609 CrossRef CAS PubMed.
- Z. Kovacevic, S. V. Menezes, S. Sahni, D. S. Kalinowski, D.-H. Bae, D. J. Lane and D. R. Richardson, J. Biol. Chem., 2016, 291, 1029–1052 CrossRef CAS PubMed.
- S. C. Lim, P. J. Jansson, S. J. Assinder, S. Maleki, D. R. Richardson and Z. Kovacevic, FASEB J., 2020, 34, 11511–11528 CrossRef CAS PubMed.
- W. Liu, F. Xing, M. Iiizumi-Gairani, H. Okuda, M. Watabe, S. K. Pai, P. R. Pandey, S. Hirota, A. Kobayashi and Y. Y. Mo, EMBO Mol. Med., 2012, 4, 93–108 CrossRef CAS PubMed.
- D. B. Lovejoy, D. M. Sharp, N. Seebacher, P. Obeidy, T. Prichard, C. Stefani, M. T. Basha, P. C. Sharpe, P. J. Jansson and D. S. Kalinowski, J. Med. Chem., 2012, 55, 7230–7244 CrossRef CAS PubMed.
- F. Shehadeh-Tout, H. H. Milioli, S. Roslan, P. J. Jansson, M. Dharmasivam, D. Graham, R. Anderson, T. Wijesinghe, M. G. Azad and D. R. Richardson, Pharmacol. Res., 2023, 193, 106806 CrossRef CAS PubMed.
- J. Yuan, D. B. Lovejoy and D. R. Richardson, Blood, 2004, 104, 1450–1458 CrossRef CAS PubMed.
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 8601–8620 CrossRef CAS PubMed.
- K. Ishiguro, Z. P. Lin, P. G. Penketh, K. Shyam, R. Zhu, R. P. Baumann, Y.-L. Zhu, A. C. Sartorelli, T. J. Rutherford and E. S. Ratner, Biochem. Pharmacol., 2014, 91, 312–322 CrossRef CAS PubMed.
- P. J. Jansson, D. S. Kalinowski, D. J. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed.
- E. Potuckova, H. Jansova, M. Machacek, A. Vavrova, P. Haskova, L. Tichotova, V. Richardson, D. S. Kalinowski, D. R. Richardson and T. Simunek, PLoS One, 2014, 9, e88754 CrossRef PubMed.
- V. A. Rao, S. R. Klein, K. K. Agama, E. Toyoda, N. Adachi, Y. Pommier and E. B. Shacter, Cancer Res., 2009, 69, 948–957 CrossRef CAS PubMed.
- V. Sestak, J. Stariat, J. Cermanova, E. Potuckova, J. Chladek, J. Roh, J. Bures, H. Jansova, P. Prusa and M. Sterba, Oncotarget, 2015, 6, 42411 CrossRef PubMed.
- J. Wang, D. Yin, C. Xie, T. Zheng, Y. Liang, X. Hong, Z. Lu, X. Song, R. Song and H. Yang, Oncotarget, 2014, 5, 8478–8491 CrossRef PubMed.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- J. A. Endicott and V. Ling, Annu. Rev. Biochem., 1989, 58, 137–171 CrossRef CAS PubMed.
- T. Yamagishi, S. Sahni, D. M. Sharp, A. Arvind, P. J. Jansson and D. R. Richardson, J. Biol. Chem., 2013, 288, 31761–31771 CrossRef CAS PubMed.
- P. Ferrao, P. Sincock, S. Cole and L. Ashman, Leuk. Res., 2001, 25, 395–405 CrossRef CAS PubMed.
- A. Molinari, A. Calcabrini, S. Meschini, A. Stringaro, P. Crateri, L. Toccacieli, M. Marra, M. Colone, M. Cianfriglia and G. Arancia, Curr. Protein Pept. Sci., 2002, 3, 653–670 CrossRef CAS PubMed.
- A. Rajagopal and S. M. Simon, Mol. Biol. Cell, 2003, 14, 3389–3399 CrossRef CAS PubMed.
- K. J. Gotink, H. J. Broxterman, M. Labots, R. R. De Haas, H. Dekker, R. J. Honeywell, M. A. Rudek, L. V. Beerepoot, R. J. Musters and G. Jansen, Clin. Cancer Res., 2011, 17, 7337–7346 CrossRef CAS PubMed.
- F. Colombo, E. Trombetta, P. Cetrangolo, M. Maggioni, P. Razini, F. De Santis, Y. Torrente, D. Prati, E. Torresani and L. Porretti, PLoS One, 2014, 9, e114787 CrossRef PubMed.
- A. Noack, B. Gericke, M. von Kockritz-Blickwede, A. Menze, S. Noack, I. Gerhauser, F. Osten, H. Y. Naim and W. Loscher, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9590–E9599 CrossRef CAS PubMed.
- E. Gutierrez, D. R. Richardson and P. J. Jansson, J. Biol. Chem., 2014, 289, 33568–33589 CrossRef CAS PubMed.
- D. B. Lovejoy, P. J. Jansson, U. T. Brunk, J. Wong, P. Ponka and D. R. Richardson, Cancer Res., 2011, 71, 5871–5880 CrossRef CAS PubMed.
- P. Gros and C. Shustik, Cancer Invest., 1991, 9, 563–569 CrossRef CAS PubMed.
- E. M. Gutierrez, N. A. Seebacher, L. Arzuman, Z. Kovacevic, D. J. Lane, V. Richardson, A. M. Merlot, H. Lok, D. S. Kalinowski, S. Sahni, P. J. Jansson and D. R. Richardson, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 1665–1681 CrossRef CAS PubMed.
- A. M. Teitelbaum, J. L. Gallardo, J. Bedi, R. Giri, A. R. Benoit, M. R. Olin, K. M. Morizio, J. R. Ohlfest, R. P. Remmel and D. M. Ferguson, Cancer Chemother. Pharmacol., 2012, 69, 1519–1527 CrossRef CAS PubMed.
- K. W. Ward and L. M. Azzarano, J. Pharmacol. Exp. Ther., 2004, 310, 703–709 CrossRef CAS PubMed.
- P. Prasher and M. Sharma, MedChemComm, 2018, 9, 1589–1618 RSC.
- S. Dallavalle, V. Dobricic, L. Lazzarato, E. Gazzano, M. Machuqueiro, I. Pajeva, I. Tsakovska, N. Zidar and R. Fruttero, Drug Resistance Updates, 2020, 50, 100682 CrossRef PubMed.
- J. Wang, T. Luo, S. Li, Y. Zhhang, C. Wang and J. Zhao, Curr. Med. Chem., 2013, 20, 4070–4079 CrossRef CAS PubMed.
- Z. Zawada, J. Šebestík, M. Šafařík and P. Bouř, Eur. J. Org Chem., 2011, 6989–6997 CrossRef CAS.
- Y. Geng, L. Kohli, B. J. Klocke and K. A. Roth, Neuro-Oncology, 2010, 12, 473–481 CAS.
- Y. Kubo, M. Yamada, S. Konakawa, S.-i. Akanuma and K.-i. Hosoya, Pharmaceutics, 2020, 12, 747 CrossRef CAS PubMed.
- A. J. Pickard, F. Liu, T. F. Bartenstein, L. G. Haines, K. E. Levine, G. L. Kucera and U. Bierbach, Chem.–Eur. J., 2014, 20, 16174–16187 CrossRef CAS PubMed.
- P. Boya and G. Kroemer, Oncogene, 2008, 27, 6434–6451 CrossRef CAS PubMed.
- W. Fu, X. Li, X. Lu, L. Zhang, R. Li, N. Zhang, S. Liu, X. Yang, Y. Wang and Y. Zhao, Cell Death Dis., 2017, 8, e3086 CrossRef PubMed.
- L. Čižeková, A. Grolmusová, Z. Ipothova, Z. Barbieriková, V. Brezová, J. Imrich, L. Janovec, I. Dovinová and H. Paulíková, Bioorg. Med. Chem., 2014, 22, 4684–4693 CrossRef PubMed.
- N. Seebacher, D. J. Lane, D. R. Richardson and P. J. Jansson, Free Radical Biol. Med., 2016, 96, 432–445 CrossRef CAS PubMed.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef PubMed.
- H. N. Lee, H. N. Kim, K. Swamy, M. S. Park, J. Kim, H. Lee, K.-H. Lee, S. Park and J. Yoon, Tetrahedron Lett., 2008, 49, 1261–1265 CrossRef CAS.
- D. R. Richardson, P. C. Sharpe, D. B. Lovejoy, D. Senaratne, D. S. Kalinowski, M. Islam and P. V. Bernhardt, J. Med. Chem., 2006, 49, 6510–6521 CrossRef CAS PubMed.
- M. Dharmasivam, B. Kaya, T. Wijesinghe, M. Gholam Azad, M. A. Gonzálvez, M. Hussaini, J. Chekmarev, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2023, 66, 1426–1453 CrossRef CAS PubMed.
- M. Dharmasivam, B. Kaya, T. P. Wijesinghe, V. Richardson, J. R. Harmer, M. A. Gonzalvez, W. Lewis, M. G. Azad, P. V. Bernhardt and D. R. Richardson, Chem. Sci., 2024, 15, 974–990 RSC.
- B. Kaya, M. Gholam Azad, M. Suleymanoglu, J. R. Harmer, T. P. Wijesinghe, V. Richardson, X. Zhao, P. V. Bernhardt, M. Dharmasivam and D. R. Richardson, J. Med. Chem., 2024, 67, 12155–12183 CrossRef CAS PubMed.
- A. Santoro, B. Vileno, Ò. Palacios, M. D. Peris-Díaz, G. Riegel, C. Gaiddon, A. Krężel and P. Faller, Metallomics, 2019, 11, 994–1004 CrossRef CAS PubMed.
- Y. Higashi, T. Aratake, T. Shimizu, S. Shimizu and M. Saito, Int. J. Mol. Sci., 2021, 22, 7765 CrossRef CAS PubMed.
- C. Andreini, I. Bertini and A. Rosato, Acc. Chem. Res., 2009, 42, 1471–1479 CrossRef CAS PubMed.
- B. Chen, P. Yu, W. N. Chan, F. Xie, Y. Zhang, L. Liang, K. T. Leung, K. W. Lo, J. Yu and G. M. Tse, Signal Transduction Targeted Ther., 2024, 9, 6 CrossRef CAS PubMed.
- R. P. Dash, R. Jayachandra Babu and N. R. Srinivas, Eur. J. Drug Metab. Pharmacokinet., 2017, 42, 915–933 CrossRef CAS PubMed.
- A. L. Harris and D. Hochhauser, Acta Oncol., 1992, 31, 205–213 CrossRef CAS PubMed.
- D. Das, B. Chand, K. Sarker, J. Dinda and C. Sinha, Polyhedron, 2006, 25, 2333–2340 CrossRef CAS.
- A. Majumder, G. M. Rosair, A. Mallick, N. Chattopadhyay and S. Mitra, Polyhedron, 2006, 25, 1753–1762 CrossRef CAS.
- N. Gondia and S. Sharma, Opt. Quantum Electron., 2017, 49, 1–12 CrossRef CAS.
- S. A. Ingle, A. N. Kate, A. A. Kumbhar, A. A. Khan, S. S. Rao and S. P. Gejji, RSC Adv., 2015, 5, 47476–47487 RSC.
- S. S. Kadam, R. P. Gotarne, M. N. Shinde, V. S. Mane, A. A. Khan and A. A. Kumbhar, Inorg. Chim. Acta, 2022, 536, 120887 CrossRef CAS.
- A. N. Kate, A. A. Kumbhar, A. A. Khan, P. V. Joshi and V. G. Puranik, Bioconjugate Chem., 2014, 25, 102–114 CrossRef CAS PubMed.
- M. Gao, P. Xie, L. Wang, X. Miao and F. Guo, Res. Chem. Intermed., 2015, 41, 9673–9685 CrossRef CAS.
- S.-L. Zheng, J.-H. Yang, X.-L. Yu, X.-M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830–838 CrossRef CAS PubMed.
- L. Y. Zhang, G. F. Liu, S. L. Zheng, B. H. Ye, X. M. Zhang and X. M. Chen, Eur. J. Inorg. Chem., 2003, 2003, 2965–2971 CrossRef.
- S. A. Perez, C. de Haro, C. Vicente, A. Donaire, A. Zamora, J. Zajac, H. Kostrhunova, V. Brabec, D. Bautista and J. Ruiz, ACS Chem. Biol., 2017, 12, 1524–1537 CrossRef CAS PubMed.
- S. Ding, X. Qiao, J. Suryadi, G. S. Marrs, G. L. Kucera and U. Bierbach, Angew. Chem., 2013, 52 Search PubMed.
- T. Esteves, C. Xavier, S. Gama, F. Mendes, P. D. Raposinho, F. Marques, A. Paulo, J. C. Pessoa, J. Rino and G. Viola, Org. Biomol. Chem., 2010, 8, 4104–4116 RSC.
- M. K. Gatasheh, S. Kannan, K. Hemalatha and N. Imrana, Karbala Int. J. Mod. Sci., 2017, 3, 272–278 CrossRef.
- W. D. Wilson and R. L. Jones, Adv. Pharmacol. Chemother., 1981, 18, 177–222 CAS.
- B. S. Mandavilli and M. S. Janes, Curr. Protoc. Cytom., 2010, 53, 9–35 Search PubMed.
- G. V. Orsinger, J. M. Watson, M. Gordon, A. C. Nymeyer, E. E. de Leon, J. W. Brownlee, K. D. Hatch, S. K. Chambers, J. K. Barton and R. K. Kostuk, J. Biomed. Opt., 2014, 19, 036020 CrossRef PubMed.
- D. I. Mundy, W. P. Li, K. Luby-Phelps and R. G. Anderson, Mol. Biol. Cell, 2012, 23, 864–880 CrossRef CAS PubMed.
- M. W. Robinson, R. Alvarado, J. To, A. T. Hutchinson, S. N. Dowdell, M. Lund, L. Turnbull, C. B. Whitchurch, B. A. O'Brien and J. P. Dalton, FASEB J., 2012, 26, 4614–4627 CrossRef CAS PubMed.
- T. Thalhammer, V. Stapf, L. Gajdzik and J. Graf, Environ. Toxicol. Pharmacol., 1994, 270, 213–220 CrossRef CAS.
- S. G. Schulman and A. C. Capomacchia, J. Am. Chem. Soc., 1973, 95, 2763–2766 CrossRef CAS.
- T. Németh, G. Dargó, J. L. Petró, Z. Petrik, S. Lévai, B. Krámos, Z. Béni, J. Nagy, G. T. Balogh and P. Huszthy, Chirality, 2017, 29, 522–535 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC AOBP: 2355274, AODP·MeCN: 2355275, AOAPZ: 2355276, [Zn(AOAPZ)2]·CH3OH·C2H5OH: 2359500 and [AOBP-S](ClO4)·CH2Cl2: 2355277. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04339a |
| This journal is © The Royal Society of Chemistry 2024 |