
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyl scanning approach for enhancing the biological activity of the linear peptidic natural product, efrapeptin C†
Yuanqi
Lin
a,
Hiroaki
Itoh
 a,
Shingo
Dan
b and
Masayuki
Inoue
*a
a,
Shingo
Dan
b and
Masayuki
Inoue
*a
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp
bDivision of Molecular Pharmacology, Cancer Chemotherapy Center, Japanese Foundation for Cancer Research, 3-8-31 Ariake, Koto-ku, Tokyo 135-8550, Japan
First published on 19th October 2024
Abstract
Efrapeptin C (1a) is a large peptidic natural product comprising a 15-mer linear sequence and exerts potent anticancer activity by inhibiting mitochondrial FoF1-ATP synthase. Residues 1–6 and 9–15 of 1a fold into two 310-helical domains and interact with ATP synthase, while the central β-Ala-7–Gly-8 region functions as a flexible linker of the two domains. To enhance the function of 1a by minimally modifying its structure, we envisioned attaching one small methyl group to the β-Ala-7–Gly-8 and designed six methylated analogues 1b–1g, differing only in the position and configuration of the methyl group. We enabled the first solid-phase total synthesis of 1a and unified syntheses of 1b–1g. The growth inhibitory activities of 1a–1g against MCF-7 cells varied significantly: 1f with (S)-β3-hAla-7 and its epimer 1g with (R)-β3-hAla-7 were 4-fold more and 5-fold less potent, respectively, than 1a. Remarkably, the most potent 1f had the most stabilized 310-helical conformation and the highest hydrophobicity, which likely contributed to its effective transfer to the target protein within mitochondria. Moreover, 1f exhibited higher proteolytic stability than 1a. Therefore, the present methyl scanning approach provides a new strategy for changing the original properties of linear peptidic natural products to develop new pharmaceuticals.
Introduction
Numerous peptidic natural products have been characterized from bacteria, fungi, plants, and animals. These peptides have been evolutionarily optimized for a certain function and many of them display significant biological activities. Accordingly, these compounds often serve as drugs or as lead structures for the discovery of new pharmaceuticals.1–3 Potent and selective bioactivities originate from large linear or cyclic architectures comprising various unusual monomers, including non-proteinogenic amino acids. Among peptidic natural products, cyclic peptides are intensively investigated by medicinal chemists because the macrocycle generally preorganizes the peptide conformation and functional group orientations for target-binding interactions.4–8 Linear peptides, on the other hand, have been considered less suitable for drug development due to their higher conformational freedom than cyclic peptides and lability toward proteolytic enzymes.Efrapeptin C (1a, Fig. 1) is a linear peptidic natural product with a molecular weight of 1607.9,10 Compound 1a was isolated from cultures of an ascomycete fungus, Tolypocladium inflatum, and structurally determined to possess an N-terminal acetyl group (Ac-cap), 15 hydrophobic amino acid residues, and a C-terminal cationic heterocycle (C-cap) with the 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) structure. The sequence comprises seven α-aminoisobutyric acids (Aib-2, -4, -5, -9, -10, -12, and -15), three L-pipecolic acids (L-Pip-1, -3, and -11), two L-leucines (L-Leu-6 and -14), two glycines (Gly-8 and -13), and one β-alanine (β-Ala-7). Thus, 11 of the 15 amino acids are non-proteinogenic residues.
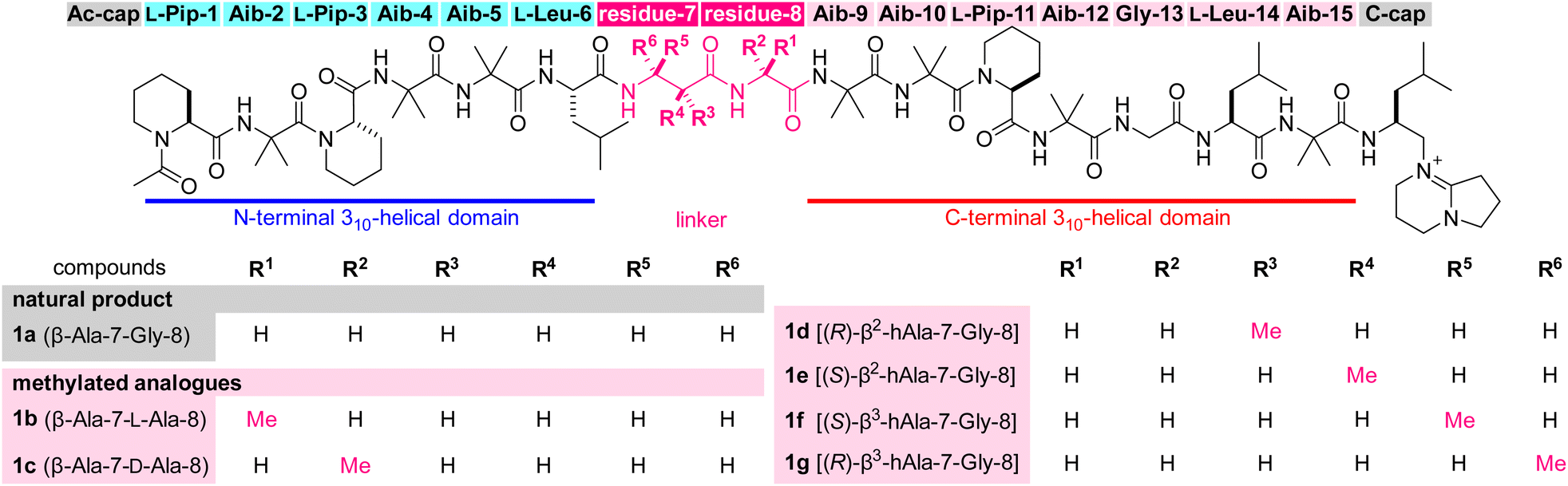 | ||
| Fig. 1 Structures of efrapeptin C (1a) and its mono-methylated analogues 1b–1g. The abbreviations for amino acids of residues 7–8 are also displayed with the compound numbers. Aib = 2-aminoisobutyric acid, hAla = homoalanine, Pip = pipecolic acid. | ||
Although 1a is toxic to various species, such as bacteria, fungi, and insects,11 potent anticancer activity is its most pharmacologically valuable feature. For example, a mixture of 1a and its natural congeners displays strong growth inhibitory activity against human breast cancer MCF-7 cells [50% inhibitory concentration (GI50) = 27 nM], and shows 60% growth inhibition at a dose of 0.15 mg kg−1 in mice xenografted with MCF-7 cells.12
Compound 1a exerts its activity by binding to FoF1-ATP synthase in mitochondria and inhibiting ATP synthesis in oxidative phosphorylation (OXPHOS).13–16 An X-ray analysis reported by Walker (Fig. 2a)17 demonstrated that 1a interacts with the F1 subunit of FoF1-ATP synthase and decreases nucleotide binding affinity for ATP synthesis. Upon binding, the Aib-rich residues 1–6 and 9–15 of 1a adopt 310-helical conformations by forming hydrogen bonding networks between the carbonyl oxygen of residue-(i) and the NH group of residue-(i + 3). In this bound form, the central β-Ala-7–Gly-8 with no branched alkyl group serves as a flexible linker and organizes the two helical domains to contact the target protein. Therefore, structural modifications in β-Ala-7–Gly-8 would potentially alter the entire three-dimensional (3D) structure, the bioactivity, and the proteolytic stability toward the development of new anticancer agents. As the precise functions of the flexible linker are unknown, however, finding analogues with improved properties is neither trivial nor obvious.
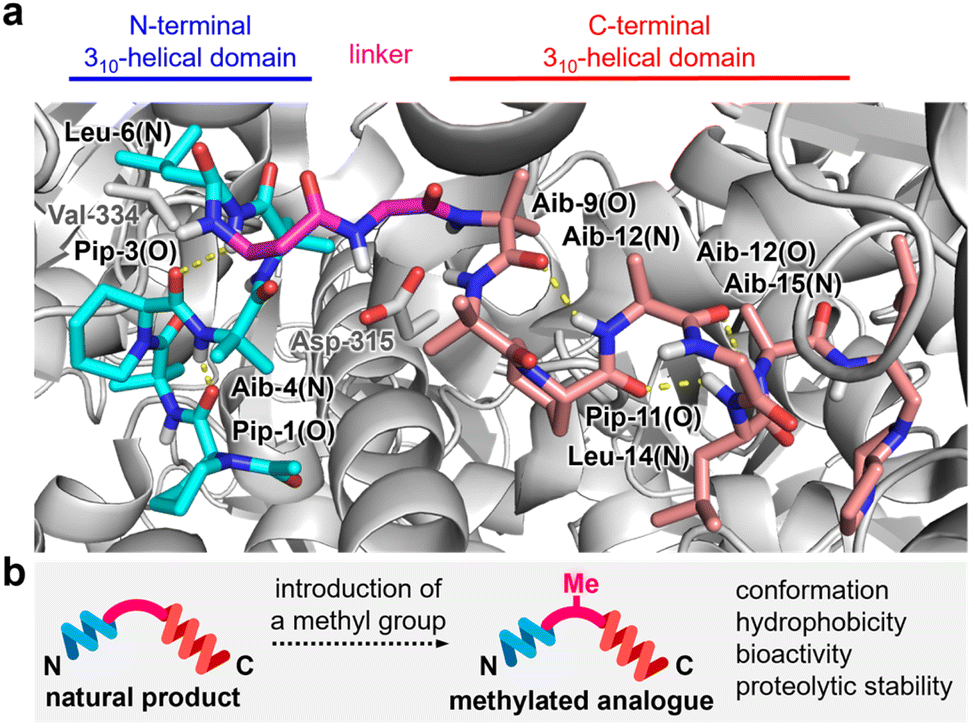 | ||
| Fig. 2 (a) Intramolecular hydrogen bonds (yellow dotted lines) of 1a in complex with the F1 domain of bovine FoF1-ATP synthase (PDB ID: 1EFR). N-terminal and C-terminal 310-helical domains are highlighted in cyan and pink, respectively. The flexible linker is highlighted in magenta. The F1 domain is displayed in gray. Val-334 of subunit α and Asp-315 of subunit β of the F1 domain form hydrogen bonds with 1a. (b) Plan for altering the properties of 1a by attaching one methyl group. | ||
The Sewald group reported the total synthesis of 1a and its analogues by a combination of solution- and solid-phase methods.18,19 A structure–activity relationship study revealed the significance of β-Ala-7–Gly-8 for the inhibitory activity of 1a against ATP hydrolysis by the F1-domain. Attaching a benzyl group to the C3 position of β-Ala-7 led to 10-fold weaker activity and exchanging β-Ala-7 with Aib-7 abolished the activity.20 Hence, subtle structural changes of the central flexible linker negatively influence the biological properties of 1a.
Our continued interest in discovering new natural product analogues with superior functions motivated us to identify potential structural modifications of β-Ala-7–Gly-8 of 1a to improve its original bioactivity and proteolytic stability (Fig. 2b).21–25 To minimally perturb the parent structure, we envisioned functionalizing the structure of 1a with the smallest aliphatic functionality, a methyl group, and designed six methylated analogues 1b–1g, differing only in the position and configuration of the methyl group. Here we disclose the first solid-phase total synthesis of efrapeptin C (1a), the syntheses of 1b–1g, and a comparison of the physicochemical and biological properties of 1a–1g. We found that 1f had a more stabilized 310-helical conformation, higher hydrophobicity, 4-fold more potent growth inhibition against MCF-7 cells, and higher proteolytic stability than parent 1a. The present methyl scanning approach led to the discovery of a fully synthetic efrapeptin analogue with superior properties, thereby providing a novel method for changing the intrinsic disadvantages of linear peptidic natural products as pharmaceuticals.26–31
Results and discussion
The profound influence of the methyl group on the physicochemical and biological behaviors has been utilized for developing drug candidates from synthetic small molecules for decades.32–34 On the other hand, the methyl effect is rarely applied to linear structures in comparison to their cyclic counterparts, presumably due to lower predictability of the appropriate position and orientation of the methyl group for enhancing the original activities. To screen the methyl effect on the conformation, hydrophobicity, bioactivity, and proteolytic stability (Fig. 2b), we aimed to comprehensively install one methyl group to the three methylene sites of β-Ala-7–Gly-8 of 1a in both (R)- and (S)-configurations, thereby designing six analogues 1b–1g (Fig. 1). Namely, 1b (β-Ala-7–L-Ala-8)/1c (β-Ala-7–D-Ala-8), 1d [(R)-β2-hAla-7–Gly-8]/1e [(S)-β2-hAla-7–Gly-8], and 1f [(S)-β3-hAla-7–Gly-8]/1g [(R)-β3-hAla-7–Gly-8] are epimeric pairs with one methyl group at the α position of Gly-8, the C2 position of β-Ala-7, and the C3 position of β-Ala-7, respectively.To investigate the methyl effect on β-Ala-7–Gly-8, we simulated the conformations of 1a–1g by molecular dynamics (MD). Two thousand structures were randomly generated for each peptide in n-octanol using an OPLS3e forcefield, starting from the initial structure that was built based on the protein-bound structure of 1a (Fig. 2a). To evaluate the impact of the methylated linker, the relative location of the two biologically important helical domains was analyzed (Fig. 3). Specifically, the distance and orientation of residues 1–6 and 9–15 were extracted as the length (r) between the carbonyl carbon atom (B) of L-Leu-6 and the nitrogen atom (C) of Aib-9, and the torsional angle (θ) defined by the α and carbonyl carbon atoms (A–B) of L-Leu-6 and the nitrogen and α carbon atoms (C–D) of Aib-9. The 2000 structures of each peptide were classified by the values of r and θ, plotted as a square dot, and color-coded from blue to red based on the frequency. The r and θ values of the protein-bound conformation of 1a are 7.7 and 162, respectively (indicated as a pink-colored cross mark. Fig. 3).
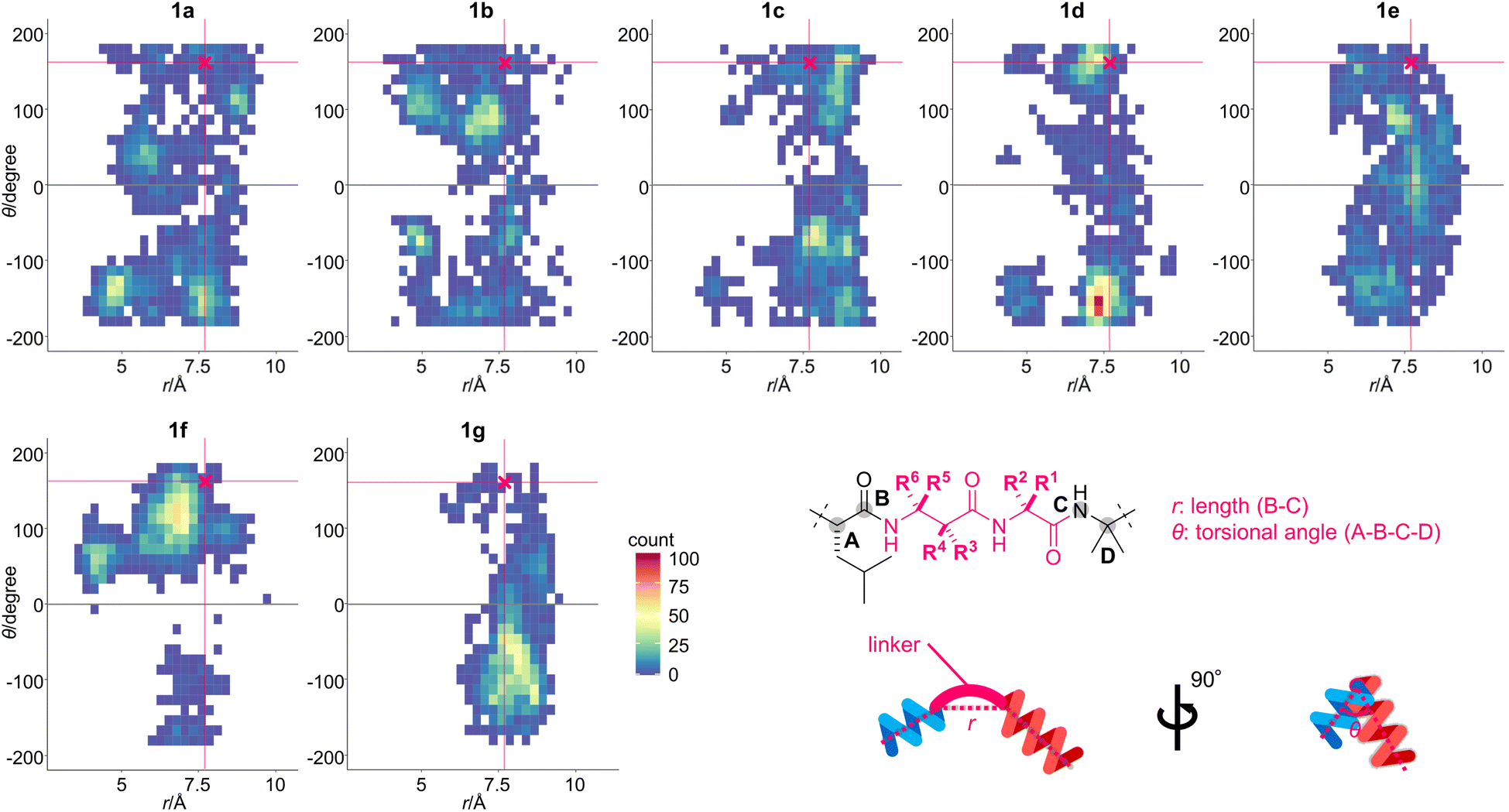 | ||
| Fig. 3 Two-dimensional histograms of the torsional angle (θ) against the length (r) of 1a–1g. The 2000 structures for each peptide were generated using a 40 ns trajectory of the replica exchange with solute tempering (REST) simulation in n-octanol (OPLS3e forcefield). The initial structure for each peptide was built based on the protein-bound structure of 1a (PDB ID: 1EFR). The specific conformation of β-Ala-7–Gly-8 was plotted according to r and θ, and the square dots are color-coded according to the obtained frequency. The conformation of 1a in complex with the F1-ATP synthase is indicated as a pink-colored cross mark [(r, θ) = (7.7, 162)]. Definitions of r and θ are also shown. | ||
According to the area of the colored square dots, the linker conformation of 1a varied significantly, indicating the unrestricted orientation of the two helical domains. In comparison, the methylated analogues 1b–1g displayed a narrower conformational distribution than 1a. The θ values of 1f with (S)-β3-hAla-7 and its epimer 1g with (R)-β3-hAla-7 were more localized than those of 1a or 1b–1e, indicating that the C3-methyl group of β-Ala-7 had the highest rigidifying effect. The r values of 1b–1f were mostly smaller than or equivalent to that of the cross mark (7.7 Å), while the r values of 1g were mostly larger. Thus, the linker of 1b–1f adopted a more bent conformation than that of 1g. In addition, the color of the square dots revealed that conformations close to the cross mark were frequently found for 1a–1f, but rarely found for 1g. It was thus anticipated that 1g would have weaker bioactivity compared with 1a–1f, due to a mismatch of its conformation with the protein-bound form. Together, the obtained results clarified that the position and configuration of the methyl group on the linear structure differentially influence the orientation of the two helical domains. Therefore, the linkers of 1b–1g were expected to effectively modify the original structure and functions of 1a.
Next, we focused on the syntheses of 1a and the six methylated analogues 1b–1g (Fig. 1). To chemically construct 1a–1g in a unified fashion, we selected a solid-phase strategy over a solution-phase counterpart because of the simplicity of the operations and ease in replacing the monomers for analogue preparations. The solid-phase total synthesis of 1a had not yet been achieved, and the multiple non-proteinogenic and sterically cumbersome components of 1a considerably amplify the synthetic challenge. In fact, we faced three major problems during the preliminary study.35 First, the bulky Aib residues impeded efficient amidation even with the highly reactive reagent combination of O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) and 1-hydroxy-7-azabenzotriazole (HOAt).36 Second, treatment with a strong acid such as trifluoroacetic acid cleaved the peptide sequence at the Pip residues, presumably through oxazolinium ring formation from the carbonyl groups of Pip and the preceding residue in a 1,4-relationship, followed by hydrolysis.37,38 Third, conjugation of the C-cap was decelerated due to the presence of the α,α-dimethyl group of Aib-15 and the sterically encumbered DBN of C-cap.
Retrosynthetically, 1a and the six methylated analogues 1b–1g were disassembled into 2–15 (Fig. 4). We planned to start the solid-phase synthesis from resin-linked Fmoc-protected Aib-15 8. The acid-labile trityl linker of 8 would allow for peptide release without damaging the acid-sensitive sequence, and the poly(ethylene glycol) (PEG)-based resin (ChemMatrix)39 of 8 would facilitate coupling of the bulky amino acids due to its high swelling properties in various solvents.40 Upon elongation, Fmoc-L-Leu-OH (2), Fmoc-Gly-OH (3), Fmoc-Aib-OH (4), Fmoc-L-Pip-OH (5), acetic anhydride (6), and C-Cap 7 would be utilized as the common structures of all the peptides. Alternatively, Fmoc-β-Ala-OH (9) of residue-7 or Fmoc-Gly-OH (3) of residue-8 for 1a was to be replaced with Fmoc-(R)-β2-hAla-OH (10)/Fmoc-(S)-β2-hAla-OH (11) for 1d/1e, with Fmoc-(S)-β3-hAla-OH (12)/Fmoc-(R)-β3-hAla-OH (13) for 1f/1g, and with Fmoc-L-Ala-OH (14)/Fmoc-D-Ala-OH (15) for 1b/1c. After the release of the sequence, C-cap 7 would be conjugated at the C-terminal Aib-15. To circumvent the unproductive amidation of the bulky Aib residues, the double coupling protocol was applied for attaching Fmoc-Aib-OH (4). Moreover, HATU/HOAt was replaced with (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholinocarbenium hexafluorophosphate (COMU)41 to form the amide bonds of Aib-2–L-Pip-3, Aib-4–Aib-5, Aib-9–Aib-10, Aib-10–L-Pip-11, and Aib-15–C-cap. The corresponding activated ester of ethyl 2-cyano-2-(hydroxyimino)acetate generated from COMU is less sterically shielded than that of HOAt and thus beneficial for facilitating the coupling of these sterically encumbered components.
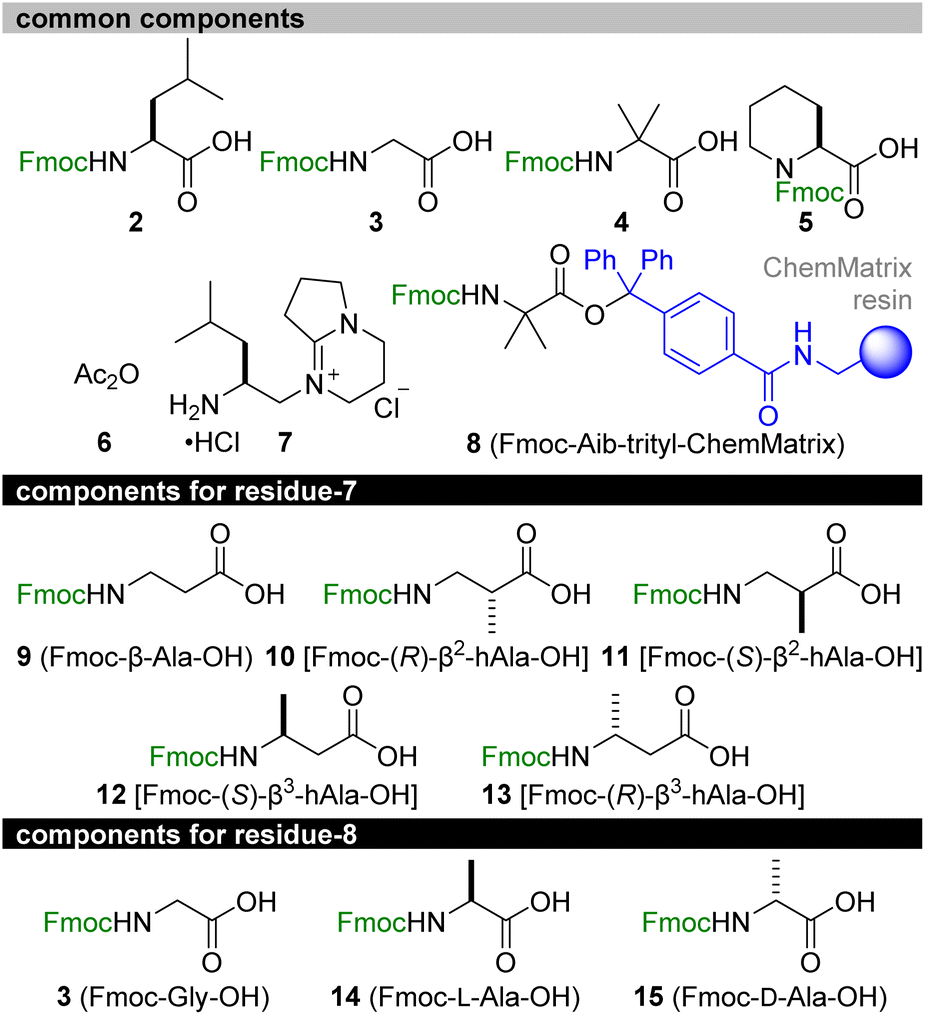 | ||
| Fig. 4 Synthetic components for total syntheses of 1a–1g. Fmoc = 9-fluorenylmethoxycarbonyl. | ||
The above considerations allowed for the first solid-phase total synthesis of 1a from Fmoc-protected Aib-15-loaded trityl-ChemMatrix resin 8 (Scheme 1). The first four Fmoc-protected amino acids (2, 3, 4, and 5) were condensed by repeating piperidine-mediated Fmoc removal in NMP at 40 °C and HATU/HOAt/i-Pr2NEt-promoted amidation in NMP at 60 °C under microwave irradiation,42 leading to residues 11–15 16. Subsequently, for the stepwise introduction of Fmoc-Aib-OH (4) to the secondary amine of Pip-11 and the primary amine of Aib-10, COMU was used as the condensation agent instead of HATU/HOAt. Thus, Fmoc removal and COMU/i-Pr2NEt-promoted elongation of 4 in NMP at 40 °C were performed to attach Aib-10 and Aib-9 to convert 16 to residues 9–15 18. Further chain extension from 18 was carried out using 3, 9, 2, and 4 by alternating applications of piperidine and HATU/HOAt/i-Pr2NEt, giving rise to residues 5–15 19a. Subsequently, Aib-5 of 19a was deprotected and reacted with 4 in the presence of COMU to 19a to provide 20a. After the Fmoc removal, Aib-4 of 20a was condensed with 5 using HATU/HOAt to afford 21a. The ensuing piperidine-treatment and COMU-promoted amidation of 4 converted 21a to residues 2–15 22a. Liberation of the N-terminal amine from 22a and the HATU/HOAt-mediated amidation with 5 were followed by exchange of the Fmoc group with the acetyl group via sequential treatment with piperidine and Ac2O (6), producing acetylated residues 1–15 23a. Finally, 23a was subjected to mildly acidic conditions using (CF3)2CHOH/CH2Cl2 to detach 24a from the trityl linker without degrading the 15-mer sequence.
 | ||
| Scheme 1 Solid-phase total syntheses of 1a and its methylated analogues 1b–1g. The positions and configurations of methyl groups in compounds 19a–19g, 20a–20g, 21a–21g, 22a–22g, 23a–23g, 24a–24g, and 25a–25g correspond to those of 1a–1g displayed in Fig. 1. COMU = (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate, HATU = O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, HOAt = 1-hydroxy-7-azabenzotriazole, NMP = N-methyl-2-pyrrolidone. | ||
The final incorporation of C-cap into the carboxylic acid of 24a turned out to be a difficult task. Prior to the coupling, amine hydrochloride 7 was neutralized with Et3N to generate the corresponding primary amine, and 10 equivalents of the amine were applied to 24a in the presence of COMU (10 equiv) and i-Pr2NEt (20 equiv).35 Whereas the stable azlactone of 25a was readily formed at the Aib-15 of 24a, only a minute amount of the desired 1a was detected after several hours. Hence, we utilized excess amounts of the reagents and extended the reaction time to realize this challenging coupling. Compound 24a was treated with the free amine of 7 (30 equiv), COMU (20 equiv), and i-Pr2NEt (30 equiv) at 30 °C for 3 d, generating 1a with the full consumption of azlactone 25a. Preparative reversed-phase HPLC led to the isolation of pure 1a in an exceptionally high overall yield (26% over 32 steps from resin 8). Remarkably, the average yield of each step was approximately 96%, demonstrating the high efficiency of the amidation reactions of the bulky components.
The route from 8 to 1a was then directly applied to devise the routes to the six methylated analogues 1b–1g (Scheme 1). Fmoc-(R)-β2-hAla-OH (10), Fmoc-(S)-β2-hAla-OH (11), Fmoc-(S)-β3-hAla-OH (12), and Fmoc-(R)-β3-hAla-OH (13) were employed instead of Fmoc-β-Ala-OH (9) of residue-7 of 1a to prepare 1d, 1e, 1f, and 1g, respectively. On the other hand, Fmoc-Gly-OH (3) of residue-8 of 1a was replaced with Fmoc-L-Ala-OH (14) and Fmoc-D-Ala-OH (15) to synthesize 1b and 1c, respectively. After HPLC purification, sufficient amounts of 1b–1g were isolated for further analyses of their physicochemical and biological properties (5.2% for 1b, 9.4% for 1c, 4.1% for 1d, 14% for 1e, 16% for 1f, 15% for 1g), corroborating the robustness of the established synthetic procedure.
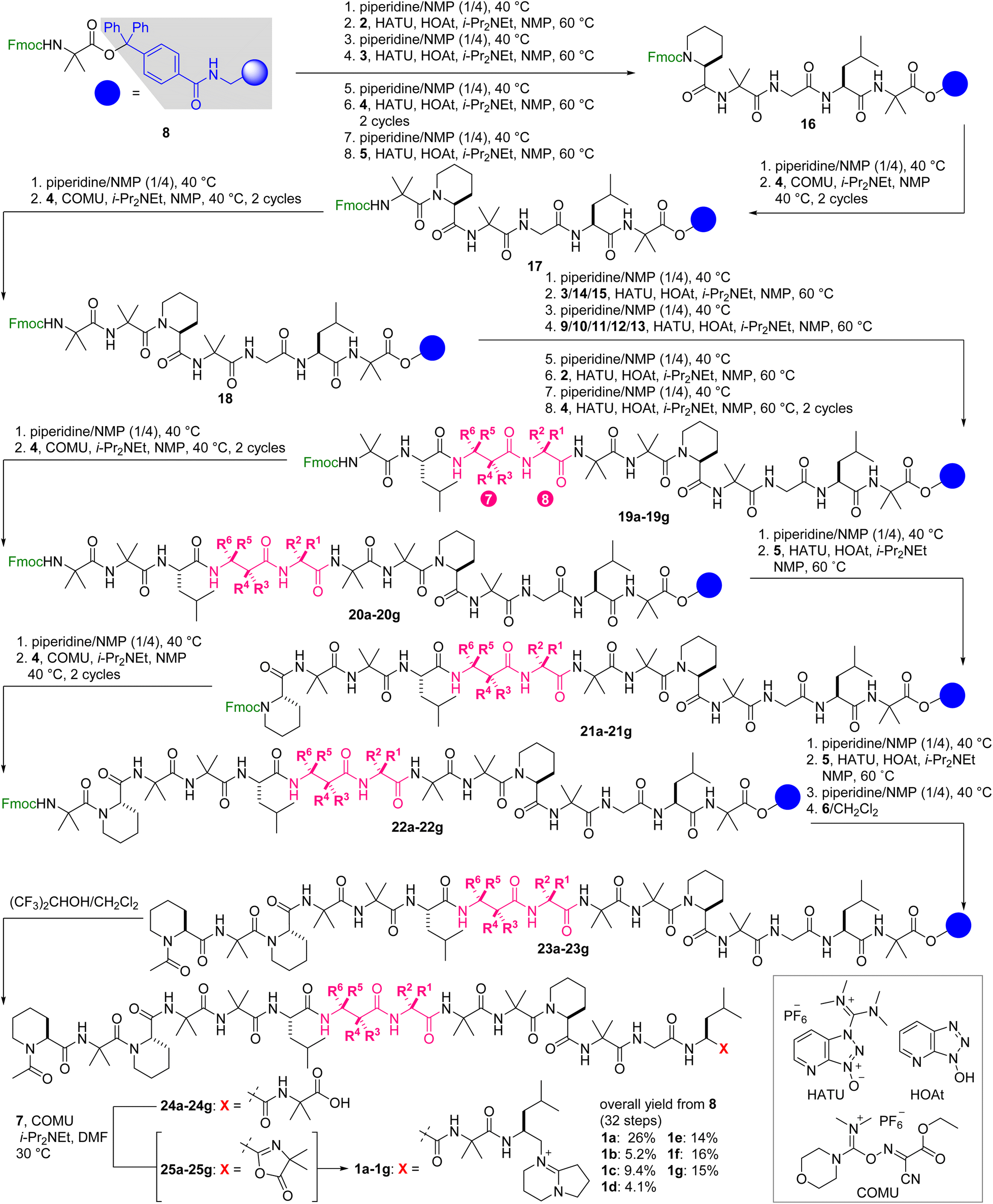
With the seven fully synthetic peptides in hand, we analyzed the conformation and hydrophobicity of 1a and six methylated analogues 1b–1g. To evaluate the folding structures of the seven peptides, electronic circular dichroism (ECD) spectra were measured in CF3CH2OH, which is known to mimic a hydrophobic environment of a lipid bilayer (Fig. 5). Natural product 1a exhibited a large band at 203–206 nm and a small band at 225–235 nm corresponding to the π → π* and n → π* transitions, respectively,43 thereby confirming the 310-helical conformations of residues 1–6 and 9–15. Two similar negative bands were observed in the spectra of all the methylated analogues 1b–1g. Even though the MD simulations indicated the disparate conformational characters of residues 7 and 8 of 1b–1g, installation of the small methyl group at residue 7 or 8 minimally disturbed the two 310-helices of 1b–1g. Interestingly, the intensities of the two bands of 1b, 1d, 1e, and 1f are higher than those of parent 1a, and those of 1c and 1g are comparable, clarifying the higher helical content of 1b/1d/1e/1f than 1c/1g. According to the spectra, 1f and its epimer 1g have the most and least stabilized 310-helical structures, respectively. Hence, the configurational switch at the C3-position of β-Ala-7 most significantly influenced the conformational equilibrium of the 310-helical domains, which should reflect the strongly rigidifying effect of the C3-methyl groups (Fig. 3).
 | ||
| Fig. 5 Electronic circular dichroism (ECD) spectra of 1a–1g. Data were recorded at a peptide concentration of 61 μM in CF3CH2OH. | ||
The hydrophobicities of 1a–1g were estimated by determining the n-octanol/water distribution coefficients (log D). To quantify the log D values, the retention times of the peptides and standard samples with known log D values were compared using ultra high-performance liquid chromatography (UHPLC) equipped with a C18 stationary phase.44,45 As a result, the log D values were calculated to be 2.03 (1a), 2.43 (1b), 2.04 (1c), 2.11 (1d), 2.35 (1e), 2.54 (1f), and 1.93 (1g) (Table 1). Thus, the positions and configurations of the methyl groups modulated the overall hydrophobicity of parent 1a. The more hydrophobic group, consisting of 1b, 1d, 1e, and 1f, corresponded to peptides with higher 310-helical contents. Alternatively, less hydrophobic peptides 1c and 1g possessed less stable helical structures. In general, the intramolecular hydrogen bonding associated with helix formation reduces the exposure of the polar amide backbone and increases the hydrophobicity. Thus, we deduced that the diverse hydrophobicity of 1a–1g originated from the different populations of the 310-helical conformations indicated by the ECD spectra (Fig. 5).
| a EC50 values were determined by evaluating the relative ATP production rate. ATP production rate was calculated from the concentration change within 1 h. b Mean values of three independent experiments are displayed with SD. c *p < 0.05 and **p < 0.01 determined by Dunnett's test. d GI50 values were determined by sulforhodamine B assay. The cells were incubated for 48 h. |
|---|

|
Next, various biological activities of the six methylated analogues 1b–1g were assessed and compared with those of 1a. We first evaluated the ability of 1a–1g to inhibit ATP synthesis (Table 1). In this assay, intact mitochondria are obtained by permeabilizing the plasma membrane of MCF-7 cells with digitonin and applied to an enzymatic ATP-detection system employing NADP.46 As the production of NADPH from NADP is coupled with ATP consumption, the amount of ATP synthesized by FoF1-ATP synthase can be evaluated from the UV absorbance of NADPH. To standardize the inhibitory activities of 1a–1g, we calculated the 50% effective concentrations (EC50). Parent 1a and the five analogues 1b–1f were similar in their activities, and the EC50 values were approximately 1 nM (1.2 nM for 1a, 1.2 nM for 1b, 0.86 nM for 1c, 1.1 nM for 1d, 0.95 nM for 1e, and 0.92 nM for 1f). Thus, the methylation patterns of 1b–1f had a negligible impact on the inhibition of FoF1-ATP synthase because they can adopt the appropriate binding conformation, as predicted by the MD calculations (Fig. 3). In contrast, the EC50 value of 1g (2.6 nM) was 2-fold larger than those of 1a–1f. The lower activity of 1g would be attributable to the inappropriate 3D orientation of the two helical domains and the lower content of the helical conformations, which were observed in the MD simulation and the ECD spectrum, respectively. Nevertheless, all six methylated analogues 1b–1g retained the ATP-production inhibitory activities of 1a.
A JFCR39 cancer panel screening was then conducted to examine the activity profiles of 1a–1g (Table 1).47,48 To evaluate the potency of 1a–1g, mean values of 50% growth inhibition concentrations (GI50) against 39 cancer cell lines were quantified. While the mean GI50 value of 1a was determined to be 646 nM, the strongest 1f (155 nM) and the weakest 1g (2950 nM) were 4-fold more and 5-fold less potent than parent 1a, respectively. Thus, the C3-position of β-Ala-7 was the most effective site for increasing and decreasing the potency of 1a. Despite the variable potency of 1a–1g, these seven peptides induced similar response patterns toward the 39 cell lines. For example, HBC-5/MCF-7 (breast), SF-295 (brain), and NCI-H522/DMS114 (lung) cell lines were more susceptible to 1a–1g than the other tested cell lines. The correlation coefficient values of the response patterns of 1b–1g turned out to be 0.85–0.90 compared with that of 1a.35 Taken together, the panel assays verified that 1b–1g shared the same mode of action as 1a.
To more precisely quantify the GI50 values, we individually evaluated the growth inhibitory activity of 1b–1g against 1a-sensitive MCF-7 cells using a sulforhodamine B assay49 with 3-fold serial dilutions of peptides (Table 1). The GI50 value of parent 1a was 37 nM in this assay. While all six methylated analogues displayed growth inhibitory activities, 1b–1g were categorized into the three classes according to their GI50 values: 1e (14 nM) and 1f (8.8 nM) were stronger analogues, 1b (27 nM) and 1d (30 nM) were comparable analogues, and 1c (104 nM) and 1g (173 nM) were weaker analogues. Analogue 1f and its epimer 1g exhibited 4-fold more and 5-fold less potent activities than 1a, respectively, and these data were consistent with those of the JFCR39 panel assays. Thus, the configurational switch of the C3-methyl groups of β3-hAla-7 of 1f and 1g resulted in a 20-fold difference in the activity. The observed most and least potent activities of 1f and 1g are in accordance with their behaviors presumed in Fig. 3, corroborating the predictive power of the MD calculation for the bioactivities. Most importantly, the discovery of the 4-fold more potent analogue 1f validated the utility of minimum structural modification of the natural product to enhance its bioactivity.
Moreover, we evaluated the cancer cell selectivity of 1a–1g using the MCF-10A immortalized human breast epithelial cell line as a model of normal cells (Table 1). The GI50 values of 1a–1g against MCF-10A cells were 6 to 11 times greater than those against MCF-7 cells, showing the prominent cancer cell selectivity of 1a–1g. In particular, the most potent 1f showed 6-fold more potent growth inhibitory activity against MCF-7 cancer cells (GI50 = 8.8 nM) compared with MCF-10A cells (GI50 = 57 nM), demonstrating its promising feature as a seed compound of anticancer agents.
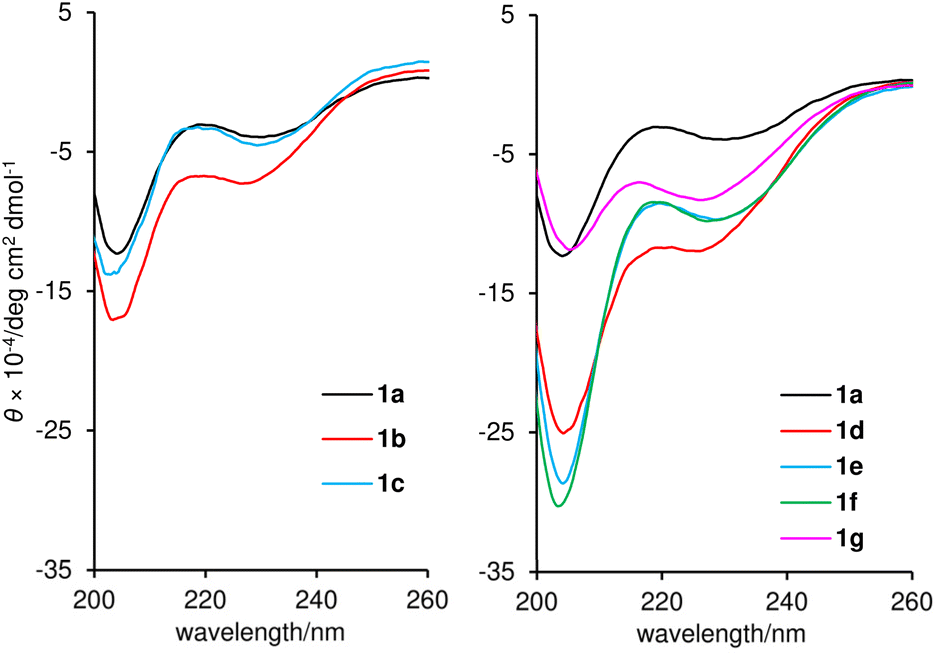
While the EC50 values of 1a–1g using mitochondrial FoF1-ATP synthase of plasma membrane-permeabilized MCF-7 cells ranged from 0.86 to 2.6 nM, the GI50 values obtained against intact MCF-7 cells ranged from 8.8 to 173 nM. Accordingly, the GI50 values not only varied more in terms of the concentration range, but were also at least 10 times higher than the EC50 values. We attributed the discrepancy in the two data sets to the distinct hydrophobicities of 1a–1g. When the log D numbers of 1a–1g were plotted against −log(GI50) numbers, the points of 1b–1g displayed a linear relationship (R2 = 0.823) with 1f and its epimer 1g being the high and low ends, respectively (Fig. 6). Thus, the more hydrophobic analogues exhibit higher growth inhibition activity. Because increased hydrophobicity generally reduces the barrier to membrane penetration, the analysis suggested the importance of the transportation of 1a–1g to FoF1-ATP synthase. The most potent and hydrophobic 1f would be most efficiently partitioned into plasma and mitochondrial membranes, and readily transferred to the inner mitochondrial membrane, thereby effectively inhibiting ATP production by FoF1-ATP synthase. On the other hand, the least hydrophobic 1g indeed exhibited the weakest activity, presumably due to the inefficient transportation to the target protein.50
 | ||
| Fig. 6 Plot of −log(GI50) values against log D values of 1a–1g. The red line represents the linear fit of analogues 1b–1g. The correlation coefficient (R2) and P-value are also displayed. | ||
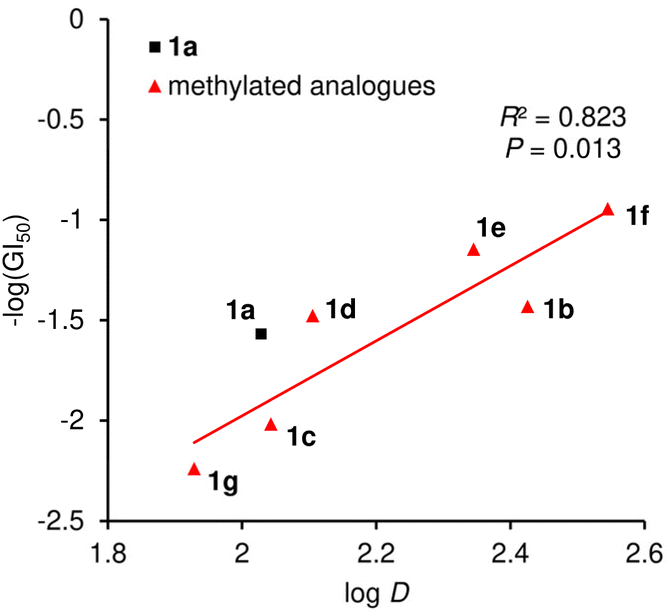
Lastly, the methyl effect on proteolytic stability was assessed using parent natural product 1a and the most potent analogue 1f to explore their bioavailability. Toward this aim, 1a and 1f were subjected to Pronase51 and papain.52 Pronase, a mixture of multiple proteases from Streptomyces griseus, failed to hydrolyze 1a or 1f at 37 °C even after 96 h, uncovering the unusual proteolytic stability of these linear peptides with multiple non-proteinogenic amino acid residues.35 When papain, a cysteine protease, was utilized, L-BAPA (benzoyl-L-arginine-p-nitroanilide)53 was completely degraded at 37 °C within 1 h (Fig. 7). Compounds 1a and 1f were significantly more stable than L-BAPA but underwent gradual hydrolysis. After 24 h incubation with papain, 41% of 1a and 61% of 1f remained in the reaction solution, which proved the improved protease resistance of 1f. Inspection of the hydrolyzed peptides of 1a revealed that hydrolysis occurred at the amide bonds at L-Leu-6–β-Ala-7, Gly-8–Aib-9, and Gly-13–L-Leu-14.35 Thus, the flexible linker at residues 7–8 was found to be the hot spot of proteolysis of 1a. Because proteases bind peptides in an extended rather than helical conformation and alkyl groups decelerate the hydrolysis rate, the higher content of the 310-helical structure and introduction of the C3-methyl group at β-Ala-7 would both contribute to the higher protease stability of 1f. Consequently, the data demonstrated the superiority of 1f to parent 1a as a potential anticancer agent.
 | ||
| Fig. 7 Proteolytic stability of 1a and 1f (71 μM) against papain (0.81 mM). The amount of remaining compound (%) was analyzed during 24 h incubation. L-BAPA = benzoyl-L-arginine-p-nitroanilide. | ||
Conclusions
In summary, we achieved the first solid-phase total synthesis of efrapeptin C (1a), syntheses of the six methylated analogues 1b–1g, and a detailed evaluation of the physicochemical and biological properties of 1a–1g. Natural product 1a possesses seven Aib, three L-Pip residues, and a C-terminal alkylated DBN as the sterically demanding components in its large 15-mer linear sequence. Upon binding to mitochondrial FoF1-ATP synthase, Aib-rich residues 1–6 and 9–15 fold into a 310-helical conformation and β-Ala-7–Gly-8 functions as a flexible linker of the two helical domains. Utilizing the trityl linker for the solid support and COMU as the powerful condensation reagent, we successfully constructed acid-sensitive 1a with multiple nonproteinogenic monomers in 26% overall yield. Analogues 1b–1g were then designed to have a single methyl group at β-Ala-7–Gly-8 to modify the original properties of 1a with minimum structural perturbation. Application of the route to 1a enabled us to prepare 1b–1g in a unified fashion, demonstrating the robustness of the devised strategy and tactics. Although 1b–1g only differ in the position and configuration of one methyl group, the methyl group considerably altered the flexibility of the linker, the stability of the 310-helix structures, and the hydrophobicities. Compounds 1a–1g were single-digit nanomolar inhibitors of the ATP production of mitochondrial FoF1-ATP synthase, and two- or three-digit nanomolar inhibitors of human breast cancer MCF-7 cell growth, indicating that the original bioactivities of 1a were maintained in 1b–1g. The most remarkable analogue was determined to be 1f with (S)-β3-hAla-7–Gly-8 because it has the most stable 310-helicity, the highest hydrophobicity, and the most potent activities against cancer cells. Analogue 1f displayed 4-fold more potent growth inhibitory activity against MCF-7 cells compared with parent 1a. The biological analyses suggested that the overall hydrophobicity of 1f originates from the largest population of the 310-helical conformation and contributes to the efficient membrane partition before reaching FoF1-ATP synthase inside the mitochondria. The cancer cell selectivity of 1f (MCF-7 vs. MCF-10A) and the superior proteolytic stability of 1f compared with 1a further supports the promising role of 1f as a lead structure for the development of new anticancer agents.The methyl effect has been used to optimize various properties of numerous drug candidates. It is rarely utilized for conformationally flexible molecules, however, because the effect is not easily predictable. Here we addressed this issue by comprehensively changing the position and configuration of the methyl group of the flexible linker, resulting in the discovery of 1f with better physicochemical, biological, and proteolytic profiles. The methyl scanning approach demonstrated here will be widely applicable for improving the pharmacologically unfavorable properties of linear peptidic natural products, allowing for the discovery of new drug candidates based on this underappreciated class of compounds.
Data availability
The experimental procedures and additional data can be found in the ESI.†Author contributions
H. I. and M. I. conceived and designed the study. Y. L. performed the synthesis of compounds and the assays. S. D. performed the cancer panel assay. H. I. and M. I. co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by Grants-in-Aid for Scientific Research (S) (JSPS, JP22H04970) to M. I., and for Scientific Research (C) (JSPS, JP21K05286) and Transformative Research Area (A) “Latent Chemical Space” (JSPS, JP23H04880 and JP23H04889) to H. I. A fellowship from JST SPRING (JPMJSP2108) to Y. L. is gratefully acknowledged. The JFCR39 cancer panel assay of 1a and synthetic analogues was supported by AdAMS (JSPS, JP22H04922). We thank Profs. Tomohiko Ohwada and Yuko Otani (The University of Tokyo) for supporting MD simulation.Notes and references
- J. Szychowski, J.-F. Truchon and Y. L. Bennani, J. Med. Chem., 2014, 57, 9292–9308 CrossRef PubMed.
- T. Dang and R. D. Süssmuth, Acc. Chem. Res., 2017, 50, 1566–1576 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef PubMed.
- E. Valeur, S. M. Guéret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef PubMed.
- H. Itoh and M. Inoue, Chem. Rev., 2019, 119, 10002–10031 CrossRef PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef PubMed.
- S. Gupta, S. B. Krasnoff, D. W. Roberts, J. A. A. Renwick, L. S. Brinen and J. Clardy, J. Am. Chem. Soc., 1991, 113, 707–709 CrossRef.
- S. Gupta, S. B. Krasnoff, D. W. Roberts, J. A. A. Renwick, L. S. Brinen and J. Clardy, J. Org. Chem., 1992, 57, 2306–2313 CrossRef.
- A. R. Bandani, B. P. S. Khambay, J. L. Faull, R. Newton, M. Deadman and T. M. Butt, Mycol. Res., 2000, 104, 537–544 CrossRef.
- A. E. Papathanassiu, N. J. MacDonald, D. R. Emlet and H. A. Vu, Cell Stress Chaperones, 2011, 16, 181–193 CrossRef PubMed.
- R. L. Cross and W. E. Kohlbrenner, J. Biol. Chem., 1978, 253, 4865–4873 CrossRef.
- For our previous study on yaku’amide B, a new inhibitor of FoF1-ATP synthase, see: K. Kitamura, H. Itoh, K. Sakurai, S. Dan and M. Inoue, J. Am. Chem. Soc., 2018, 140, 12189–12199 CrossRef PubMed.
- For a review of natural product-derived inhibitors of FoF1-ATP synthase, see: B. A. Patel, T. L. D'Amico and B. S. J. Blagg, Eur. J. Med. Chem., 2020, 207, 112779 CrossRef PubMed.
- For the potential utility of OXPHOS inhibitors as anticancer agents, see: Y. Xu, D. Xue, A. Bankhead, III and N. Neamati, J. Med. Chem., 2020, 63, 14276–14307 CrossRef.
- J. P. Abrahams, S. K. Buchanan, M. J. Van Raaij, I. M. Fearnley, A. G. Leslie and J. E. Walker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9420–9424 CrossRef PubMed.
- In the total synthesis of 1a by the Sewald group, the Aib unit was introduced as 2-azidoacetyl chloride to increase the reactivity for the amidation. In addition, the three synthetic fragments (Ac-L-Pip-1–Gly-8, Aib-9–Gly-13, and L-Leu-14–Aib-15–C-cap) were separately prepared both in solution and on solid phase, and subsequently assembled in solution (8.0% overall yield from the C-cap unit as a starting material). For details, see: M. Jost, J. C. Greie, N. Stemmer, S. D. Wilking, K. Altendorf and N. Sewald, Angew. Chem., Int. Ed., 2002, 41, 4267–4269 CrossRef.
- S. Weigelt, T. Huber, F. Hofmann, M. Jost, M. Ritzefeld, B. Luy, C. Freudenberger, Z. Majer, E. Vass, J.-C. Greie, L. Panella, B. Kaptein, Q. B. Broxterman, H. Kessler, K. Altendorf, M. Hollósi and N. Sewald, Chem.–Eur. J., 2012, 18, 478–487 CrossRef PubMed.
- M. Jost, S. Weigelt, T. Huber, Z. Majer, J. C. Greie, K. Altendorf and N. Sewald, Chem. Biodiversity, 2007, 4, 1170–1182 CrossRef PubMed.
- For our study on enhancing the original bioactivities of peptidic natural products, see ref. 21–23: H. Itoh, K. Tokumoto, T. Kaji, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Nat. Commun., 2019, 10, 2992 CrossRef PubMed.
- Y. Takada, H. Itoh, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Nat. Commun., 2020, 11, 4935 CrossRef.
- J. Fu, Y. Nakata, H. Itoh, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Chem.–Eur. J., 2023, 29, e202301224 CrossRef.
- For reviews of the structural modification of natural products, see ref. 24 and 25: A. M. Szpilman and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9592–9628 CrossRef.
- Z.-C. Wu and D. L. Boger, Nat. Prod. Rep., 2020, 37, 1511–1531 RSC.
- Numerous structurally related naturally occurring congeners of 1a have been reported. The strategy presented here can be directly applied to these compounds. For details, see ref. 26 and 27: T. Degenkolb, J. Kirschbaum and H. Brückner, Chem. Biodiversity, 2007, 4, 1052–1067 CrossRef.
- A. H. El-Desoky, Y. Hitora, Y. Nishime, Y. Sadahiro, T. Kawahara and S. Tsukamoto, J. Nat. Med., 2024, 78, 505–513 CrossRef.
- Since efrapeptins exhibit a wide range of pharmacologically useful functions other than anticancer activity for the development of new pharmaceuticals. For antimalarial activity of efrapeptins, see: G. Nagaraj, M. V. Uma, M. S. Shivayogi and H. Balaram, Antimicrob. Agents Chemother., 2001, 45, 145–149 CrossRef.
- For antiviral activity of efrapeptins, see: J. Eichberg, M. Oberpaul, C. Hartwig, A. H. Geißler, C. Culmsee, A. Vilcinskas, E. Böttcher-Friebertshäuser, H. Brückner, T. Degenkolb and K. Hardes, Arch. Pharm., 2024, 357, e2400384 CrossRef.
- The β-Ala-Gly linker is not only found in efrapeptins and congeners but also incorporated in PROTAC and radio-labeled molecules. See ref. 30 and 31: S. Wang, Z. Feng, C. Qu, S. Yu, H. Zhang, R. Deng, D. Luo, C. Pu, Y. Zhang and R. Li, J. Med. Chem., 2024, 67, 9842–9856 CrossRef PubMed.
- A. Ritler, M. S. Shoshan, X. Deupi, P. Wilhelm, R. Schibli, H. Wennemers and M. BéBé, Bioconjugate Chem., 2019, 30, 657–666 CrossRef PubMed.
- For reviews of the methyl effects in medicinal chemistry, see ref. 26–28: E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- P. S. M. Pinheiro, L. S. Franco and C. A. M. Fraga, Pharmaceuticals, 2013, 16, 1157 CrossRef.
- See the ESI† for details.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef.
- C. J. Creighton, T. T. Romoff, J. H. Bu and M. Goodman, J. Am. Chem. Soc., 1999, 121, 6786–6791 CrossRef.
- C. Rubini, A. Osler, A. Calderan, A. Guiotto and P. Ruzza, J. Pept. Sci., 2008, 14, 989–997 CrossRef PubMed.
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef.
- S. Lawrenson, M. North, F. Peigneguy and A. Routledge, Green Chem., 2017, 19, 952–962 RSC.
- A. El-Faham, R. S. Funosas, R. Prohens and F. Albericio, Chem.–Eur. J., 2009, 15, 9404–9416 CrossRef PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- C. Toniolo, A. Polese, F. Formaggio, M. Crisma and J. Kamphuis, J. Am. Chem. Soc., 1996, 118, 2744–2745 CrossRef.
- OECD, Test No. 117. Partition Coefficient (n-octanol/water), HPLC method, OECD Guidelines for the Testing of Chemicals, Section 1, OECD Publishing, Paris, 2004 Search PubMed.
- H. Mutoh, Y. Sesoko, T. Kuranaga, H. Itoh and M. Inoue, Org. Biomol. Chem., 2016, 14, 4199–4204 RSC.
- A. Signorile, L. Micelli, D. De Rasmo, A. Santeramo, F. Papa, R. Ficarella, G. Gattoni, S. Scacco and S. Papa, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 675–684 CrossRef.
- T. Yamori, Cancer Chemother. Pharmacol., 2003, 52, 74–79 CrossRef PubMed.
- S. Yaguchi, Y. Fukui, I. Koshimizu, H. Yoshimi, T. Matsuno, H. Gouda, S. Hirono, K. Yamazaki and T. Yamori, J. Natl. Cancer Inst., 2006, 98, 545–556 CrossRef.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef PubMed.
- The second least potent 1c exhibited the larger r value than that of the bioactive conformation in the MD simulation, which also supported that the calculation results were consistent with the hydrophobicity .
- L. L. Cline and M. L. Waters, Biopolymers, 2009, 92, 502–507 CrossRef.
- Structurally related neoefrapeptin A and N were hydrolyzed by papain. A. Fredenhagen, L.-P. Molleyres, B. Böhlendorf and G. Laue, J. Antibiot., 2006, 59, 267–280 CrossRef CAS.
- S. Tokura, N. Nishi and J. Noguchi, J. Biochem., 1971, 69, 599–600 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04384g |
| This journal is © The Royal Society of Chemistry 2024 |