
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-demand controlled bidirectional DNAzyme path for ultra-sensitive heavy metal ion detection†
Jing
Xu
a,
Yujin
Li
ab,
Futing
Wang
b,
Xinqi
Luo
a,
Wei
Zhang
f,
Yifan
Lyu
b,
Hongfen
Yang
e,
Ren
Cai
 *b and
Weihong
Tan
bcd
*b and
Weihong
Tan
bcd
aCollege of Chemistry and Chemical Engineering, Xinyang Normal University, Xinyang 464000, China
bMolecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio-Sensing and Chemometrics, College of Material Science and Engineering, College of Chemistry and Chemical Engineering, College of Biology, Hunan University, Changsha, Hunan 410082, China. E-mail: cairen@hnu.edu.cn
cThe Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, Zhejiang 310022, China
dInstitute of Molecular Medicine, Renji Hospital, Shanghai Jiao Tong University School of Medicine, College of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
eHunan Key Laboratory of Typical Environmental Pollution and Health Hazards, School of Public Health, Hengyang Medical School, University of South China, Hengyang 421001, China
fCollege of Life Science, Xinyang Normal University, Dabie Mountain Laboratory, Xinyang 464000, China
First published on 11th October 2024
Abstract
A bidirectional self-powered biosensor is constructed for the quasi-simultaneous detection of Pb2+ and Hg2+ based on MoS2@CuS heterostructures as an accelerator and hybridization chain reaction (HCR) as a signal amplification strategy. MoS2@CuS heterostructures significantly facilitate electron transfer between glucose and bioelectrodes, thereby greatly improving the detection signal of self-powered biosensors. This novel biosensor employs the unique sequences of DNAzymes to isolate Pb2+ and Hg2+ by the cleavage effect and thymine (T)–Hg2+–thymine (T) structures, respectively. In the process, Pb2+ cuts the sequence of DNAzyme at the bioanode to trigger glucose oxidation to monitor Pb2+. The as-formed T–Hg2+–T structures activate HCR to reduce [Ru(NH3)6]3+ to detect Hg2+ at the biocathode. It is noteworthy that this biosensor not only realizes Pb2+ or Hg2+ detection in a single-electrode, respectively, but also can quasi-simultaneously detect both Pb2+ and Hg2+ in the bioanode and the biocathode. The novel self-powered biosensor identifies Pb2+ in the range of 106 fM to 10 fM with a limit of detection (LOD) of 3.1 fM and Hg2+ in the range of 106 fM to 1 fM with an LOD of 0.33 fM.
Introduction
Heavy metal ion pollution is a severe hazard for human health because of the extremely low biodegradability of most heavy metals.1–4 At present, many novel detection methods are employed to monitor heavy metal ions, such as surface enhanced Raman spectroscopy (SERS),5 atomic absorption spectroscopy,6 inductively coupled plasma mass spectrometry,7 fluorescence spectroscopy,8–10 colorimetric method,11 and electrochemical methods.12,13 For example, an on-site visual approach was designed for toxic Cr(VI) detection based on an imidazolium-functionalized conjugated polymer;14 an aptamer-functionalized fluorescent DNA sensor was used to monitor Cu(II) in living tumor cells;15 an ultra-sensitive and selective electrochemical method was utilized for Cd(II) detection by the synergistic activation of phosphorus and the orbital coupling effect of Fe-doped CoP.16Compared with the above methods, self-powered biosensors are widely used in the detection of biomolecules. In addition to eliminating the need for external power, these sensors exhibit strong anti-interference ability, ultra-sensitive detection capability, and highly identifiable specificity.17–21 However, there has been no report of self-powered biosensors with the bidirectional detection to monitor heavy metal ions. The major reason is the difficulty in forming double bioelectrodes to simultaneously achieve linear detection.22–24 Therefore, a bidirectional self-powered biosensor toward multiple heavy metal ions detection is urgently needed.
This work reports a bidirectional self-powered biosensor for the quasi-simultaneous detection of Pb2+ and Hg2+ based on MoS2@CuS heterostructures as a co-accelerator and a hybridization chain reaction (HCR) as a signal amplification strategy (Scheme 1). Unique sequences of DNAzymes isolate Pb2+ and Hg2+ by the cleavage effect and thymine (T)–Hg2+–thymine (T) structures, respectively.25–29 In the process, Pb2+ cuts the sequence of the DNAzyme at the bioanode to trigger glucose oxidation to monitor Pb2+. The as-formed T–Hg2+–T structures activate HCR to reduce [Ru(NH3)6]3+ to detect Hg2+ at the biocathode. As a result, this biosensor not only monitors Pb2+ or Hg2+ through the current changes caused by the oxidation–reduction reactions in a single-electrode, respectively, but also could quasi-simultaneously detect both Pb2+ and Hg2+ by observing the open circuit voltage (Eocv) in the bioanode and the biocathode. The bidirectional biosensor also exhibits considerable practicability for detecting Pb2+ and Hg2+ in tap water.
 | ||
| Scheme 1 (a) The synthetic process of MoS2@CuS heterostructures. (b and c) The detection process of Pb2+ and Hg2+ of the bidirectional self-powered biosensor. | ||
Results and discussion
In the TEM images (Fig. 1a and S1†), uniform Cu2O templates, CuS nanocubes, and MoS2@CuS heterostructures (size of ∼500 nm) are observed, and there are many small nanosheets on the surface of MoS2@CuS heterostructures. The selected area electron diffraction pattern (SAED) confirms the polycrystalline feature (inset, Fig. 1a), which is consistent with the X-ray diffraction (XRD) pattern (Fig. 1b). All peaks are indexed to MoS2 (JCPDS: 37-1492) and CuS (JCPDS: 06-0462).30,31 After adding Au nanoparticles (AuNPs, ∼20 nm, 5 mM, Fig. S2†), many AuNPs are observed on the surface of MoS2@CuS heterostructures (Fig. 1c). The elemental mapping images and X-ray photoelectron spectra (XPS) confirm that Au, Cu, Mo, and S are present (Fig. 1d and S3†).32 In addition, UV-vis spectra and FTIR spectroscopy confirmed the successful recombination of heterostructures. For MoS2@CuS heterostructures, the FTIR absorption peaks at 1605 and 895 cm−1 were to the vibration of Mo–O bond and Mo–S bond, respectively (Fig. S4 and S5†).33 Finally, the Brunauer–Emmett–Teller (BET) specific surface of MoS2@CuS heterostructures is calculated to be 30.8 cm2 g−1 with aperture size of ∼5 nm (Fig. S6†). | ||
| Fig. 1 Characterization of MoS2@CuS heterostructures: (a) TEM image and SAED (insert); (b) XRD. (c) TEM image and (d) EDX images of AuNPs/MoS2@CuS heterostructures. | ||
The assembly process of the bioanode and biocathode was explored by cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Monitoring the maximum current value, the optimal recognition and cleavage time of Pb2+ on the DNAzyme was 60 min (Fig. 2a). After DNAzyme was cut by Pb2+, it was split into two sequences of single strand DNAs: S1 and S2, leaving as-split S2 on the surface of AuNPs/MoS2@CuS/CP by Au–S bonding. When S3-GOD is added, the as-split S2 hybridizes with S3-GOD by complementary base pairing to form the bioanode. The optimal hybridization time between S2 and S3-GOD is 50 min (Fig. 2b). In the biocathode, the optimal incubation time among Hg2+, S5, and S4 is 30 min to form S5–Hg2+/S4/AuNPs/MoS2@CuS/CP by the T–Hg2+–T structure (Fig. 2c). Finally, the as-formed T–Hg2+–T structure further combines with H1 and H2 from the HCR process to generate a multi-duplex structure of H1, H2, and S5 to form H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP.
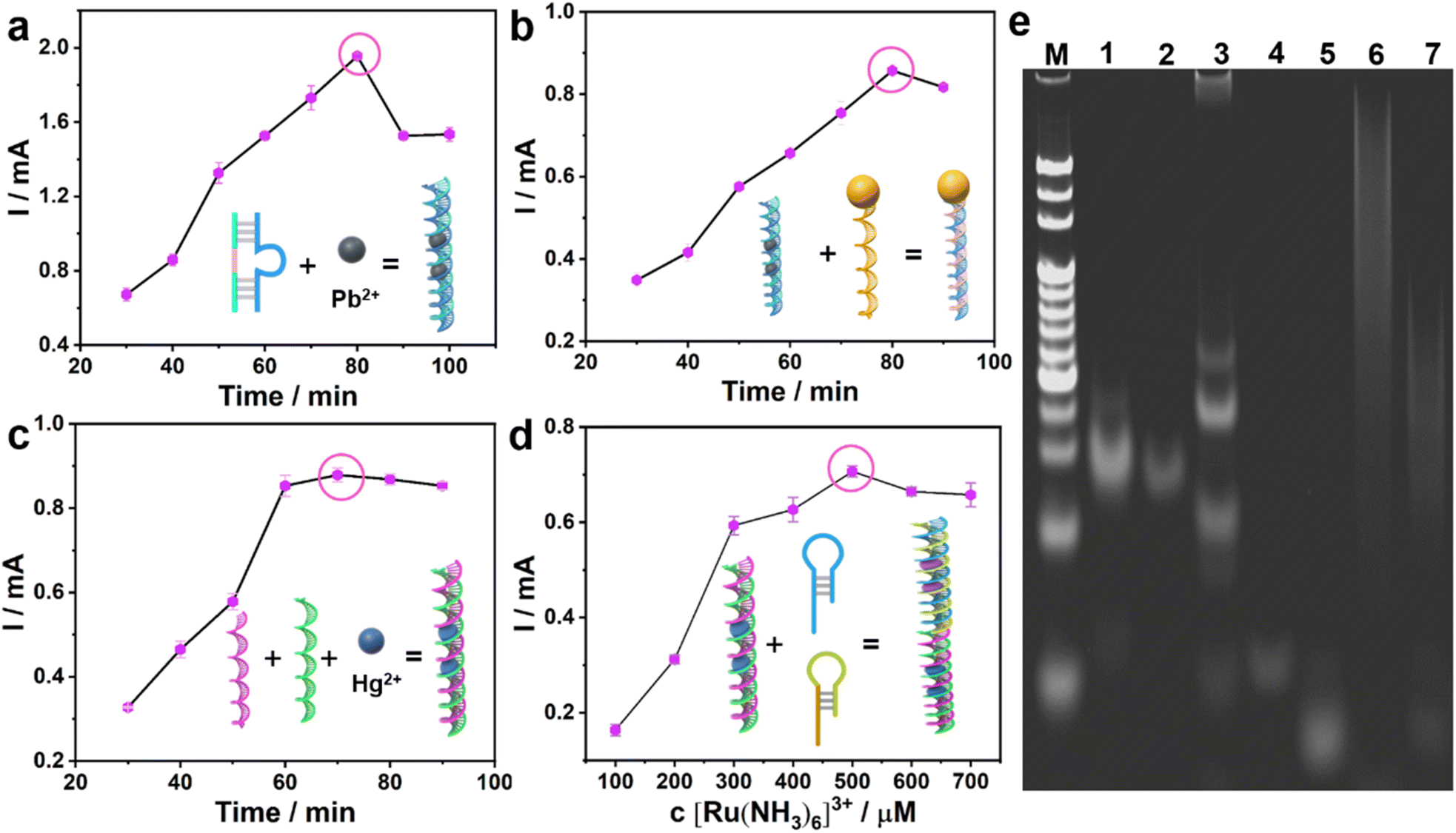 | ||
| Fig. 2 The optimization of the hybridization reaction times: (a) incubation between Pb2+ and DNAzyme; (b) incubation between S3-GOD and as-split S2; (c) incubation among S4, S5 and Hg2+; (d) the optimization of [Ru(NH3)6]3+ concentration. (e) Gel electrophoresis analysis. | ||
Next, [Ru(NH3)6]3+ inserts into the multi-duplex structure of H1, H2, and S5 on the surface of H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP (Fig. 2d) with optimal concentration of 500 μM [Ru(NH3)6]3+. These assembly processes of the bioanode and biocathode were further verified by gel electrophoresis (Fig. 2e). Line M represents molecular weight standards. For the bioanode, a fading, shallow band with low molecular weight appears when Pb2+ (lane 2) is incubated with DNAzyme (lane 1). After S3-GOD is added, S3-GOD hybridizes with as-split S2 to form many new bright bands (lane 3). For the biocathode, there is no visible band for S4 (lane 4) or S4 + S5 (lane 5), respectively. When the T–Hg2+–T (i.e., S4–Hg2+–S5) structure is formed, a higher bright band (lane 6) is observed. Finally, when the T–Hg2+–T structure triggers the HCR reaction, a new band is observed (lane 7). These results are consistent with circular dichroic chromatography (Fig. S7†). In the sensing mechanism, the Pb2+ could cleave the RNA sites and change the DNAzyme structure; the thymine–thymine mismatch base pair (T–T, H1–H2) can capture the Hg2+ to form a stable neutral T–Hg2+–T base pair.
Electrochemical impedance spectroscopy (EIS) was used to study the preparation process of the bioanode (Fig. 3a). The resistance (Ret) value of CP (curve A) is higher than that of AuNPs/MoS2@CuS/CP (curve B), confirming the good electrical conductivity of AuNPs/MoS2@CuS. After DNAzyme, Pb2+, and S3-GOD are sequentially incubated with AuNPs/MoS2@CuS/CP, the Ret increases from 14 Ω to 30 Ω (Fig. 3a). For the biocathode (Fig. 3b), H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP shows the highest Ret after S4, S5 and Hg2+, and H1, H2 are modified the biocathode. In addition, the ζ-potential measurements confirmed the successful assembly of the bioanode and biocathode (Fig. S8†). CV and linear scanning voltammetry (LSV) were performed to verify the catalytic reaction of GOD on the bioanode. In PBS with 5 mM glucose, a distinct oxidation peak at −0.48 V is observed (blue line), which corresponds to the splitting of DNAzyme to S1 and S2 by Pb2+, and the as-split S2 triggers the oxidation of glucose (Fig. 3c and d). The feasibility of the biocathode for Hg2+ detection was investigated by DPV. In PBS with 500 μM [Ru(NH3)6]3+, as-adsorbed [Ru(NH3)6]3+ acquires electrons and is reduced to [Ru(NH3)6]2+ at the biocathode. When AuNPs/MoS2@CuS (black line), S4 (blue line), S5, Hg2+ (green line), and H1, H2 (red line) are successively coated on the CP surface, H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP exhibits the largest peak current value (Fig. 3e). Finally, the current of DPV was increased when the biocathode combined with the HCR strategy (Fig. S9†), which confirmed the signal amplification effect of HCR. These results prove the efficient signal amplification strategy from HCR for the biocathode.
 | ||
| Fig. 3 (a) EIS of the bioanode: bare CP (A), AuNPs/MoS2@CuS/CP (B), DNAzyme/AuNPs/MoS2@CuS/CP (C), DNAzyme/AuNPs/MoS2@CuS/CP (D), Pb2+/BSA/DNAzyme/AuNPs/MoS2@CuS/CP (E), S3-GOD/Pb2+/DNAzyme/AuNPs/MoS2@CuS/CP (F) (inset: the circuit diagram). (b) EIS of the biocathode: bare CP (A), AuNPs/MoS2@CuS/CP (B), S4/AuNPs/MoS2@CuS/CP (C), BSA/S4/AuNPs/MoS2@CuS/CP (D), S5–Hg2+/S4/AuNPs/MoS2@CuS/CP (E), H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP (F) (inset: the circuit diagram). (c) CV and (d) LSV curves of the bioanode in PBS (pH 7.4) without glucose and with 5 mM glucose in the presence of Pb2+. (e) DPV curves of the assembly process of the biocathode in PBS (pH 7.4) with [Ru(NH3)6]3+. | ||
In Fig. 4a and b, there is a linear relationship between the peak current with increasing scan rate in the range of 60 to 200 mV s−1 (peak potential difference ≤ −20 mV), which proves that the as-prepared bioanode and biocathode exhibit accurate identification ability.34,35 Here, one biosensor was constructed based AuNPs/CP as the bioanode and the biocathode; the other one was fabricated based on AuNPs/MoS2@CuS/CP as the bioanode and the biocathode. The Eocv of the biosensor based on AuNPs/MoS2@CuS/CP is about 0.58 V, which is 0.13 V higher than that of the biosensor based on AuNPs/CP (Fig. 4c). Similarly, the maximum power output of the biosensors based on AuNPs/MoS2@CuS/CP and AuNPs/CP is 49.5 μW cm−2 and 33.2 μW cm−2, respectively (Fig. 4d), which proves high-efficiency conduction of MoS2@CuS heterostructures.
 | ||
| Fig. 4 (a and b) CV of S3-GOD/Pb2+/DNAzyme/AuNPs/MoS2@CuS/CP (i.e., bioanode) (yellow to red lines) and H1, H2/S5 Hg2+/S4/AuNPs/MoS2@CuS/CP (i.e., biocathode) (pink to blue lines) with different scan rates (ν): (a–h) 60, 80, 100, 120, 140, 160, 180, 200 mV s−1. (c) Eocv and (d) power output of the self-powered biosensor constructed of: (A) both S3-GOD/Pb2+/DNAzyme/AuNPs/MoS2@CuS/CP (the bioanode) and H1, H2/S5–Hg2+/S4/AuNPs/MoS2@CuS/CP (the biocathode), and (B) both S3-GOD/Pb2+/DNAzyme/CP (the bioanode) and H1, H2/S5–Hg2+/S4/CP (the biocathode). | ||
For the single detection of Pb2+, the peak intensity of glucose oxidation at −0.48 V increases with increasing Pb2+ concentration under optimal conditions in a CV test at the bioanode (Fig. S10†). Moreover, there is a good linear relationship between the logarithm of Pb2+ concentration and current intensity in the range of 10 fM ∼1 nM. The equation is: I = 0.01 × log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CPb2+ + 0.20 (correlation coefficient, R2 = 0.995), and the detection of limit (LOD) is calculated to be 3.1 fM (S/N = 3). Analogously, the current response at −0.18 V is attributed to the reduction of [Ru(NH3)6]3+ in the DPV test at the biocathode, which gradually increases with increasing Hg2+ concentration (Fig. S11†). A linear correlation between the logarithm of Hg2+ concentration and current intensity in the range of 1 fM to 1 nM was fitted by the regression equation: I = 0.1 × logCHg2+ + 2.4 (R2 = 0.995), and LOD is calculated to be 0.32 pM (S/N = 3).
CPb2+ + 0.20 (correlation coefficient, R2 = 0.995), and the detection of limit (LOD) is calculated to be 3.1 fM (S/N = 3). Analogously, the current response at −0.18 V is attributed to the reduction of [Ru(NH3)6]3+ in the DPV test at the biocathode, which gradually increases with increasing Hg2+ concentration (Fig. S11†). A linear correlation between the logarithm of Hg2+ concentration and current intensity in the range of 1 fM to 1 nM was fitted by the regression equation: I = 0.1 × logCHg2+ + 2.4 (R2 = 0.995), and LOD is calculated to be 0.32 pM (S/N = 3).
For the bidirectional detection of Pb2+ and Hg2+, Eocv of the novel biosensors was investigated at the double electrodes (Fig. 5). For instance, Hg2+ concentration is determined to be 1 nM at the biocathode in a tap water sample (Fig. 5a and b). A good linear relationship between Eocv and the logarithm of Pb2+ concentration is calculated: Eocv = 0.078 logCPb2+ + 1.21 (R2 = 0.994) with the LOD of 3.1 fM (S/N = 3) in the range of 10 fM to 106 fM. Here, the Eocv increases with the increment of Pb2+ concentration at the bioanode. In another example, Pb2+ concentration is determined to be 1 nM at the bioanode in the same tap water sample (Fig. 5c and d). There is a linear relationship between Eocv and the logarithm of Hg2+ concentration in the range of 1 fM to 106 fM: Eocv = 0.083 × logCHg2+ + 1.34 (R2 = 0.995), and the LOD is calculated to be 0.33 fM (S/N = 3). Here, the Eocv increases with the increment of Hg2+ concentration at the biocathode. Compared with previously reported sensors (Tables S2 and S3†), the bidirectional self-powered biosensor shows wider detection ranges and lower LODs for Pb2+ and Hg2+ detection. In order to further visualize the on-site detection of Pb2+ and Hg2+, our capacitor amplification strategy was explored by smart Bluetooth data transfer with a smartphone for real-time read-out of test results (Fig. S12 and see Movie S1†).36–38 In the test process, the mobile phone reads out the instantaneous current when different concentrations of Pb2+ and Hg2+ are in an unknown water sample, furthering realizing on-site and real-time detection (Fig. S13 and S14†).
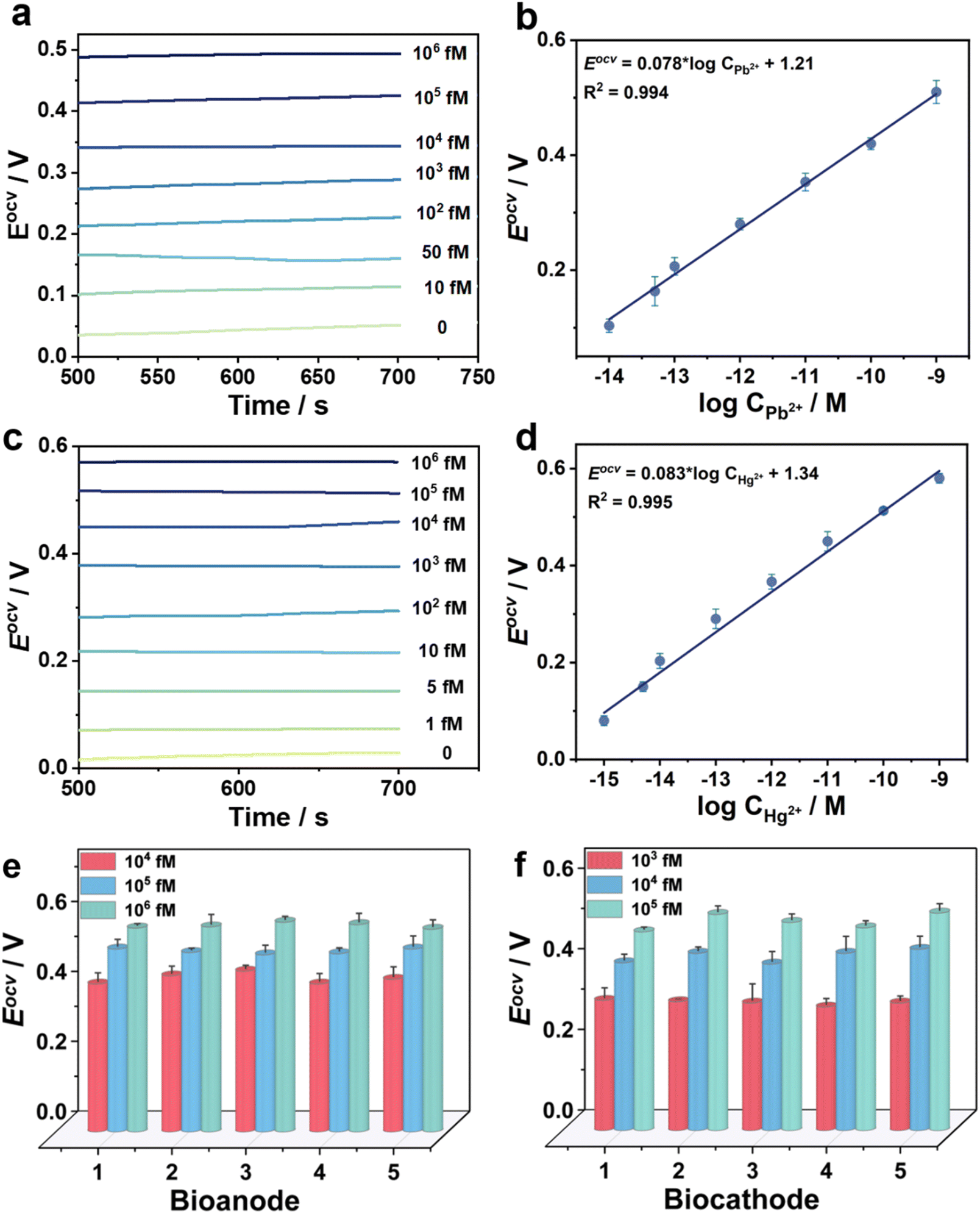 | ||
| Fig. 5 (a) Eocv of different concentrations of Pb2+ and (b) relationship between Eocv and Pb2+ concentration; (c) Eocv of different concentrations of Hg2+ and (d) relationship between Eocv and Hg2+ concentration. (e and f) The repeatability of the self-powered biosensor. | ||
The signal responses of the self-powered biosensor toward Pb2+ and Hg2+ detection remained at 83.5% and 82.1% of original signal after the biosensors were stored at 4 °C for 15 days, further confirming the long-time stability (Fig. S15†). The recyclability of one as-designed biosensor was tested by analyzing different concentrations of Pb2+ and Hg2+. All test results maintained within 5% of the error, which confirmed good recyclability for the biosensor (Fig. S16†). To confirm the specificity, there was no Eocv signal when six possible interferences, i.e., Na+, K+, Mg2+, Cu2+, Co2+, and blank water sample, were detected by the as-designed biosensor. The solution with Pb2+ or Hg2+ showed significantly high Eocv, thus verifying the good specificity of the bidirectional biosensor (Fig. S17†). In addition, the relative standard deviations (RSDs) were measured to be ≤5% in five parallel experiments, indicating the good repeatability of the novel biosensor (Fig. 5e and f). To further test the practical application, Pb2+ and Hg2+ with the concentrations of 1, 10, 50, and 100 nM in tap water and lake water were prepared to perform a labelled recovery experiment. The RSDs values were calculated to be the range from 3.7% to 5.3% for Pb2+ and 2.1% to 5.9% for Hg2+; and the recovery rates were in the range from 96.4% to 105.0% for Pb2+ and 97.0% to 104.8% for Hg2+ (Tables S4 and S5†). These results demonstrate that the bidirectional self-powered biosensor possesses good selectivity, stability, and reproducibility, and could be used for practical sample detection.
Conclusions
In summary, a bidirectional self-powered biosensor is designed to simultaneously monitor Pb2+ at the bioanode and Hg2+ at the biocathode. Here, the MoS2@CuS heterostructures are successfully prepared to accelerate the electron transfer between glucose and the bioelectrode to increase the power output. Moreover, the novel biosensor realizes Pb2+ detection by the cutting effect of Pb2+ toward DNAzyme to trigger glucose oxidation at the bioanode, and monitors Hg2+ by forming unique T–Hg2+–T structures to activate HCR for the reduction of [Ru(NH3)6]3+ at the biocathode. The self-powered biosensor could not only perform linear detection of Pb2+ at the bioanode or Hg2+ at the biocathode, but also quasi-simultaneously realizes linear detection for both Pb2+ and Hg2+ at the bioanode and the biocathode. The bidirectional biosensor exhibits wide detection ranges of 106 fM ∼10 fM toward Pb2+ and 106 fM ∼1 fM toward Hg2+, respectively; and the LODs of Pb2+ and Hg2+ are as low as 3.1 fM and 0.33 fM, respectively. This biosensor possesses highly efficient, ultra-sensitive, highly selective, and good reproducible detection of Pb2+ and Hg2+. Meanwhile, smart and portable mobile phones and capacitors are well used for on-site direct reading detection. This work opens a new direction for designing novel bidirectional biosensors to detect multiple heavy metal ions.Data availability
All underlying data are available in the published article itself and its ESI.†Author contributions
J. X., F. W., X. L., W. Z., and H. Y. directed the project. Y. L performed the main experimental work. F. L provided experimental equipment. R. C., and W. T. provided some constructive suggestions for the experiments. All the authors discussed the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Joint Fund Project for Technology Research and Development Plan of Henan Technology (Young Scientists, 235200810056), Hunan Provincial Natural Science Foundation of China (2024JJ5348 and 2021JJ20020), and National Natural Science Foundation of China (22004032). The authors also acknowledge the great support from the Dabie Mountain Laboratory, Nanhu Scholars Program for Young Scholars of XYNU and Analysis Testing Center of Xinyang Normal University for materials characterization.References
- S. H. Chen, J. J. Zhu, P. H. Li, Y. F. Sun, M. Yang and X. J. Huang, In-situ growth of zero-valent iron in FeOx/Mn3O4 to improve the surficial redox for high-efficient electrocatalysis of Pb(II), Chem. Eng. J., 2022, 430, 132959 CrossRef.
- A. Amalraj, R. Pavadai and P. Perumal, Recyclable target metal-enhanced fluorometric naked eye aptasensor for the detection of Pb2+ and Ag+ ions based on the structural change of CaSnO3@PDANS-constrained GC-rich ssDNA, ACS Omega, 2021, 6(45), 30580–30597 CrossRef.
- J. Xu, Y. J. Li, Y. B. Liu, X. Wu and K. J. Huang, Ultra-sensitive self-powered sensor based on the hollow Rubik's cube substrate and multiple signal amplification strategy for real-time heavy metal detection, Sens. Actuators, B, 2023, 394, 134377 CrossRef.
- M. Rana, M. Balcioglu, N. M. Robertson, M. S. Hizir, S. Yumak and M. V. Yigit, Low picomolar, instrument-free visual detection of mercury and silver ions using low-cost programmable nanoprobes, Chem. Sci., 2017, 8(2), 1200–1208 RSC.
- T. T. X. Ong, E. W. Blanch and O. A. H. Jones, Surface enhanced Raman spectroscopy in environmental analysis, monitoring and assessment, Sci. Total Environ., 2020, 720, 137601 CrossRef.
- İ. Narin, M. Soylak, L. Elçi and M. Doğan, Determination of trace metal ions by AAS in natural water samples after preconcentration of pyrocatechol violet complexes on an activated carbon column, Talanta, 2000, 52(6), 1041–1046 CrossRef PubMed.
- T. Van Acker, S. Theiner, E. Bolea-Fernandez, F. Vanhaecke and G. Koellensperger, Inductively coupled plasma mass spectrometry, Nat. Rev. Methods Primers, 2023, 3(1), 52 CrossRef.
- H. Yuan, T. Ren, Q. Luo, Y. Huang, Y. Huang, D. Xu, X. Guo, X. Li and Y. Wu, Fluorescent wood with non-cytotoxicity for effective adsorption and sensitive detection of heavy metals, J. Hazard. Mater., 2021, 416, 126166 CrossRef.
- J. Liu, L. Zhang, W. Zeng, L. Zhang, N. He and Z. Lu, High-throughput quantitative detection of triple-negative breast cancer-associated expressed miRNAs by rolling circle amplification on fluorescence-encoded microspheres, Chin. Chem. Lett., 2023, 34(9), 108141 Search PubMed.
- R. Pavadai, A. Amalraj, S. Subramanian and P. Perumal, High catalytic activity of fluorophore-labeled Y-shaped DNAzyme/3D MOF-MoS2NBs as a versatile biosensing platform for the simultaneous detection of Hg2+, Ni2+, and Ag+ ions, ACS Appl. Mater. Interfaces, 2021, 13(27), 31710–31724 CrossRef PubMed.
- Y. Liu, T. Li, G. Yang, Y. Deng, X. Mou and N. He, A simple AuNPs-based colorimetric aptasensor for chlorpyrifos detection, Chin. Chem. Lett., 2022, 33(4), 1913–1916 CrossRef CAS.
- M. Yang, P. Li, S. Chen, X. Xiao, X. Tang, C. Lin, X. Huang and W. Liu, Heavy metal detection: nanometal oxides with special surface physicochemical properties to promote electrochemical detection of heavy metal ions, Small, 2020, 16(25), 2070140 CrossRef.
- G. Yang, Z. Li, R. Usman, Z. Chen, Y. Liu, S. Li, H. Chen, Y. Deng, Y. Fang and N. He, DNA walker induced “signal on” fluorescence aptasensor strategy for rapid and sensitive detection of extracellular vesicles in gastric cancer, Chin. Chem. Lett., 2024, 109930 Search PubMed.
- Z. Li, S. Shen, S. Hussain, B. Cui, J. Yao, Y. Su, Y. Wang, Y. Hao and R. Gao, Imidazolium-functionalized conjugated polymer for on-site visual and ultrasensitive detection of toxic hexavalent chromium, Anal. Chem., 2024, 96(13), 5150–5159 CrossRef.
- Y. Mou, P. Yin, M. Chen, C. Wei, Y. Zhang, J. Zhang, Y. Zhao, X. Luo and Y. Wang, Engineering of an aptamer-functionalized fluorescent DNA sensor for Cu(II) responding in living tumor cells, Anal. Chem., 2023, 95(21), 8348–8356 CrossRef.
- Z. Gao, Y. Li, P. Li, Y. Yang, Y. Zhao, M. Yang, S. Chen, Z. Song and X. Huang, Synergistic activation of P and orbital coupling effect for ultra-sensitive and selective electrochemical detection of Cd(II) over Fe-doped CoP, J. Hazard. Mater., 2024, 463, 132842 CrossRef.
- Y. J. Li, J. Xu, Y. Y. Hou, J. Q. Lu, K. J. Huang and R. Cai, 3D hierarchically electrode combined with DNA circuit strategy powered highly sensitive sensing devices, Sens. Actuators, B, 2024, 401, 134963 CrossRef.
- C. Gu, L. Bai, L. Pu, P. Gai and F. Li, Highly sensitive and stable self-powered biosensing for exosomes based on dual metal-organic frameworks nanocarriers, Biosens. Bioelectron., 2021, 176, 112907 CrossRef PubMed.
- C. Gu, P. Gai, X. Kong, T. Hou and F. Li, Self-powered biosensing platform based on “Signal-On” enzymatic biofuel cell for DNA methyltransferase activity analysis and inhibitor screening, Anal. Chem., 2020, 92(7), 5426–5430 CrossRef CAS PubMed.
- J. Xu, Y. Liu, K. Huang, Y. Hou, X. Sun and J. Li, Real-time biosensor platform based on novel sandwich graphdiyne for ultrasensitive detection of tumor marker, Anal. Chem., 2022, 94(49), 16980–16986 CrossRef PubMed.
- Y. Hou, J. Xu, W. Xie, K. Huang, X. Tan, B. Zhao, S. Zhang and M. Gao, 3D DNA walker recognition-driven homogeneous dual-mode sensing strategy based on enzyme biofuel cell for ultrasensitive detection of HER2, Sens. Actuators, B, 2023, 376, 132998 Search PubMed.
- J. Fang, C. Yuan, J. Li, J. Li, T. Yang, Y. Guo, D. Wang, J. Xue, W. Fu and G. Xie, An enzyme-powered, three-dimensional lame DNA walker, Biosens. Bioelectron., 2021, 177, 112981 CrossRef PubMed.
- J. Xu, Y. Li, F. Wang, H. Yang, K. Huang, R. Cai and W. Tan, A smartphone-mediated “all-in-one” biosensing chip for visual and value-assisted detection, Anal. Chem., 2024, 96(39), 15780–15788 CrossRef PubMed.
- A. Arunjegan, P. Rajaji, S. Sivanesan and P. Panneerselvam, A Turn-ON fluorometric biosensor based on ssDNA immobilized with a metal phenolic nanomaterial for the sequential detection of Pb(II) and epirubicin cancer drug, RSC Adv., 2021, 11(20), 12361–12373 RSC.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, MercuryII-mediated formation of thymine−HgII−thymine base pairs in DNA duplexes, J. Am. Chem. Soc., 2006, 128(7), 2172–2173 CrossRef.
- A. Ono and H. Togashi, Highly selective oligonucleotide-based sensor for Mercury(II) in aqueous solutions, Angew. Chem., Int. Ed., 2004, 43(33), 4300–4302 CrossRef.
- S. W. Santoro and G. F. Joyce, A general purpose RNA-cleaving DNA enzyme, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(9), 4262–4266 CrossRef PubMed.
- Y. Fang, Y. Wang, L. Zhu, H. Liu, X. Su, Y. Liu, Z. Chen, H. Chen and N. He, A novel cartridge for nucleic acid extraction, amplification and detection of infectious disease pathogens with the help of magnetic nanoparticles, Chin. Chem. Lett., 2023, 34(8), 108092 CrossRef.
- R. Huang, L. He, L. Jin, Z. Li, N. He and W. Miao, Recent advancements in DNA nanotechnology-enabled extracellular vesicles detection and diagnosis: A mini review, Chin. Chem. Lett., 2023, 34(6), 107926 CrossRef.
- M. Li, L. Wang, Y. Qian and J. Du, Facile synthesis of MoS2/CuS nanoflakes as high performance electrocatalysts for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2022, 47(8), 5319–5325 CrossRef.
- H. Liu, Y. He, H. Zhang, S. Wang, K. Cao, Y. Jiang, X. Liu and Q. S. Jing, Heterostructure engineering of ultrathin SnS2/Ti3C2Tx nanosheets for high-performance potassium-ion batteries, J. Colloid Interface Sci., 2022, 606, 167–176 CrossRef.
- J. W. Stiles, A. L. Soltys, X. Song, S. H. Lapidus, C. B. Arnold and L. M. Schoop, Unlocking high capacity and fast Na+ diffusion of HxCrS2 by proton-exchange pretreatment, Adv. Mater., 2023, 35(10), 2209811 CrossRef PubMed.
- A. Amalraj, R. Pavadai, S. Subramanian and P. Perumal, Fabrication of multi-functional CuO@PDA-MoS2 mediated dual-functional fluorescence aptamer for the detection of Hg2+ ions and chloramphenicol through desulfurization cleavage reaction and exonuclease I activity, Appl. Surf. Sci., 2022, 602, 154222 CrossRef.
- H. Chen, X. Ma, X. Zhang, G. Hu, Y. Deng, S. Li, Z. Chen, N. He, Y. Wu and Z. Jiang, Novel aerosol detection platform for SARS-CoV-2: Based on specific magnetic nanoparticles adsorption sampling and digital droplet PCR detection, Chin. Chem. Lett., 2023, 34(1), 107701 CrossRef PubMed.
- Z. Guo, B. Jin, Y. Fang, L. Jin, S. Li, Y. Deng, Z. Chen, H. Chen, Y. Zhang, R. Usman and N. He, Automated screening of primary cell-based aptamers for targeting and therapy of pancreatic cancer, Chin. Chem. Lett., 2024, 35(2), 108528 CrossRef.
- F. Wang, P. Wang, H. Yang, R. Cai and W. Tan, Self-powered biosensing system with multivariate signal amplification for real-time amplified detection of PDGF-BB, Anal. Chem., 2023, 95(44), 16359–16365 CrossRef.
- Z. Guo, B. Jin, Y. Fang, Y. Deng, Z. Chen, H. Chen, S. Li, F. W. N. Chow, P. H. M. Leung, H. Wang, L. Cai and N. He, Selected aptamer specially combing 5-8F cells based on automatic screening instrument, Chin. Chem. Lett., 2022, 33(9), 4208–4212 CrossRef.
- F. Wang, H. Luo, Y. Hou, Y. Ya, K. J. Huang, S. Li, L. Wang, Y. Song and X. Tan, Dual microRNAs-driven enzyme biofuel cells: Visualization biosensing system with capacitor assistance output signal amplification, Sens. Actuators, B, 2022, 365, 131970 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04404e |
| This journal is © The Royal Society of Chemistry 2024 |