
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExclusive generation of a superoxide radical by a porous aromatic framework for fast photocatalytic decontamination of mustard gas simulant in room air†
Jian
Song‡
,
Hengtao
Lei‡
,
Yuhui
Zhai
,
Zilong
Dou
,
Yongyue
Ding
,
Xueyan
Han
,
Fengchao
Cui
 ,
Yuyang
Tian
* and
Guangshan
Zhu
*
,
Yuyang
Tian
* and
Guangshan
Zhu
*
Key Laboratory of Polyoxometalate and Reticular Material Chemistry of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun, Jilin 130024, China. E-mail: tianyy100@nenu.edu.cn; zhugs@nenu.edu.cn
First published on 27th August 2024
Abstract
Mustard gas and other chemical warfare agents (CWAs) are a global threat to public security, arising from unpredictable emergencies and chemical spill accidents. So far, photocatalysts such as metal clusters, polyoxometalates and porous solids have been exploited for oxidative degradation of mustard gas, commonly with 1O2 as reactive species. However, the production of 1O2 is oxygen-dependent and requires a high oxygen concentration to sustain the detoxication process. For safety and operation process considerations, it is always preferable to rapidly detoxify dangerous chemicals in the atmosphere of room air. In this work, a porous aromatic framework, PAF-68, was synthesized as a metal-free photocatalyst. In the presence of PAF-68, fast detoxication occurred in typical room air atmosphere. The half-life (t1/2) for the complete conversion of mustard gas simulant to nontoxic product in room air was only 1.7 min, which is comparable to the performance in pure oxygen, surpassing that of any other porous photocatalysts. It was found that ˙O2− rather than 1O2 is the predominant reactive species initiated by PAF-68 for mustard gas detoxication. Unlike the formation of 1O2 which prefers the environment of pure oxygen, generation of the ˙O2− is an oxygen-independent process. It is suggested that amorphous PAFs possess low exciton binding energy and long decay lifetime, which facilitate the generation of ˙O2−, and this offers a general design strategy to detoxifying chemical warfare agents under real-world conditions.
Introduction
CWAs are a global threat because of their strong lethality, ready availability, and diverse exposure pathways.1,2 Among them, mustard gas (HD) is a typical representative of corrosive poisons, and it is widely regarded as a historically significant chemical weapon, referred to as the “king of poisons” for its potent corrosiveness, chemical stability, long-lasting effectiveness, and especially difficult detoxication.3 Compared to other detoxifying approaches such as dehydrochlorination and hydrolysis, oxidation of HD is especially regarded as more promising for its fast conversion and innocuous products. Among the oxidation processes, photocatalytic oxidation driven by light, as the source of energy, is the most attractive due to its efficiency, sustainability and ease of operation.4 In pioneering research, Farha et al. used the metal organic framework (MOF) PCN-222 as a photocatalyst for mustard gas simulant detoxication. In an atmosphere of pure oxygen and under LED light irradiation, a mustard gas simulant, 2-chloroethyl ethyl sulfide (CEES), was selectively oxidized to its nontoxic product 2-chloroethyl ethyl sulfoxide (CEESO) with a half-life (t1/2) of 13 min.5 There has since been extensive work on the design of photocatalysts to faster convert mustard gas and its simulants.6–8 With regard to oxidation, selective conversion of mustard gas to its sulfoxide product is preferred because over-oxidation results in the production of a sulfone compound, which is as toxic as HD itself.9 Besides their demanding selectivity, catalysts and oxidants should be carefully selected to achieve fast detoxication. However, almost all currently reported photocatalytic reactions require pure oxygen, or even more dangerously, hydrogen peroxide as oxidants, because the reactive oxygen species (ROS) usually relies on the concentration of the oxidants.10,11 Despite singlet oxygen (1O2) being commonly used to selectively oxidize CEES to CEESO, a high concentration of oxygen molecules is needed to ensure a viable oxidation rate, due to the oxygen-dependent feature of 1O2 formation.12,13 Consequently, under an atmosphere of typical room air, with an oxygen partial pressure around 20%, the rate of CEES oxidation by 1O2 is significantly reduced.5,14 In real-world conditions, carrying pure oxygen or even explosive hydrogen peroxide as oxidants is difficult and risky. Therefore, the design and synthesis of photocatalysts for rapid degradation of CEES in an atmosphere of room air is an urgent goal of current research.Porous solids, including metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), have exhibited significant performance as photocatalysts owing to their massive accessible active sites and adjustable energy band structures.15–17 However, in practice, most of these materials serving as heterogeneous catalysts face the challenge of weak stability. In contrast, porous aromatic frameworks (PAFs) are a class of porous materials entirely constructed from organic building units, where the building units are linked via stable carbon–carbon covalent bonds.18,19 Due to their structural features, PAFs possess numerous accessible internal surfaces, a stable framework, and sub-nanometer-scaled pores, which are used as functional materials for adsorption,20 separation,20,21 catalysis,22 and as solid electrolytes,23,24 to name but a few. Apart from their exceptional stability, the physical and chemical properties of PAFs can be readily designed and modulated by the screening and modification of suitable building units.25 Therefore, the development of photoactive PAFs for ROS-related processes would be an alluring option.
Herein, a photoactive PAF (PAF-68) with high specific surface area and high stability was successfully synthesized, with tris(4-ethynylphenyl)amine (EPA) and meso-tetra(p-bromophenyl)porphyrin (TBPP) as building units (Fig. 1). PAF-68 showed good efficacy for the aerobic oxidation of mustard gas simulant. In humid O2, CEES was completely converted into the nontoxic CEESO with a half-life of 1.5 min. In an atmosphere of room air, regardless of the low oxygen partial pressure and the humidity, the half-life of the conversion of CEES to CEESO by PAF-68 reached an ultrafast pace of 1.7 min, which is quite close to that in pure oxygen. To the best of our knowledge, this is the fastest conversion among any reported photocatalysts in air. Both a reactive oxygen species (ROS) quenching experiment and electron paramagnetic resonance (EPR) spectroscopy suggested that PAF-68 rapidly initiated ˙O2− rather than 1O2. Compared to 1O2, the generation of ˙O2− is an oxygen concentration-independent process,26–28 thus allowing a fast conversion rate without needing an atmosphere of pure oxygen. Different from ordered porous solids such as MOFs and COFs, the amorphous PAFs are a metal-free photocatalyst that initiates ˙O2− as ROS. Mechanism investigation suggested that the porphyrin-based amorphous framework of PAF-68 is a feature of low exciton binding energy and long decay lifetime when excited by light, both of which would facilitate the generation of ˙O2−. The tough carbon–carbon linkages of the PAFs hindered photodegradation of the skeleton, resulting in durable photocatalytic degradation performance even after several cycles. This work not only provides a promising material for the fast detoxication of mustard gas simulant in general environments, but also presents an innovative but inspiring design principle for the detoxication of chemical warfare agents with other types of ROS.
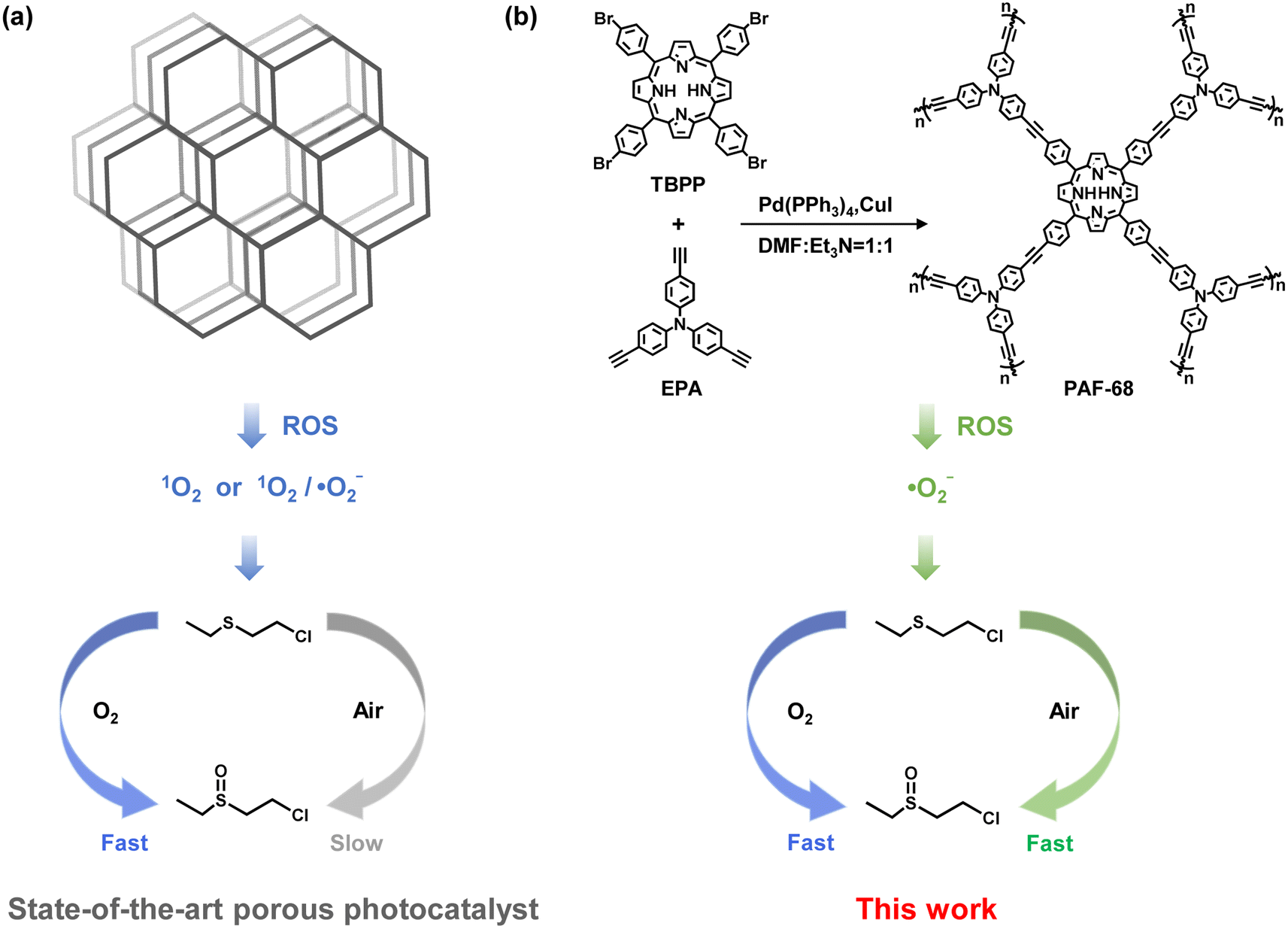 | ||
| Fig. 1 (a) Previously reported porous photocatalysts for detoxification of mustard gas and simulants can ensure rapid oxidative degradation under pure oxygen, while the catalytic rate is greatly reduced in room air. (b) In contrast, PAF-68 in this work degrades CEES in room air as fast as in pure oxygen. | ||
Results and discussion
Synthesis and characterization of PAF-68
Using photoactive TBPP and EPA as the building units, PAF-68 was obtained under solvothermal conditions in the form of a purple powder. The structural characteristics of PAF-68 were characterized by infrared spectroscopy (FT-IR), solid-state 13C CP/MAS NMR and X-ray photoelectron spectroscopy. The FT-IR spectrum is shown in Fig. 2a. The peaks at 564 and 515 cm−1 are related to the stretching vibration of C–Br on the monomer TBPP, while the peak at 3281 cm−1 was attributed to the characteristic vibration from the terminal alkynyl C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) H bond in EPA. The fact that these characteristic peaks vanished in the product indicated that the building units had fully reacted, and the new, low-intensity peak at 2204 cm−1 was assigned to the stretching vibration peak of the newly formed alkyne bond in PAF-68. In the solid-state 13C NMR, the chemical shift at around 90 ppm corresponds to the carbon–carbon triple bonds. Those at 118–152 ppm were assigned to aromatic carbons from the phenylporphine and phenylamine motifs in PAF-68 (Fig. 2b). X-ray photoelectron spectroscopy (XPS) proved that PAF-68 is metal-free and has two N 1s characteristic peaks of porphyrin and triphenylamine (Fig. S1†). Thus, the connection of the building units and the bonding mode of PAF-68 could be confirmed as expected. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were used to investigate the morphology of PAF-68. As can be observed in the SEM and TEM photographs (Fig. S2 and S3†), its morphology exhibited spherical nanoparticles with a size of 400–600 nm.
H bond in EPA. The fact that these characteristic peaks vanished in the product indicated that the building units had fully reacted, and the new, low-intensity peak at 2204 cm−1 was assigned to the stretching vibration peak of the newly formed alkyne bond in PAF-68. In the solid-state 13C NMR, the chemical shift at around 90 ppm corresponds to the carbon–carbon triple bonds. Those at 118–152 ppm were assigned to aromatic carbons from the phenylporphine and phenylamine motifs in PAF-68 (Fig. 2b). X-ray photoelectron spectroscopy (XPS) proved that PAF-68 is metal-free and has two N 1s characteristic peaks of porphyrin and triphenylamine (Fig. S1†). Thus, the connection of the building units and the bonding mode of PAF-68 could be confirmed as expected. Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were used to investigate the morphology of PAF-68. As can be observed in the SEM and TEM photographs (Fig. S2 and S3†), its morphology exhibited spherical nanoparticles with a size of 400–600 nm.
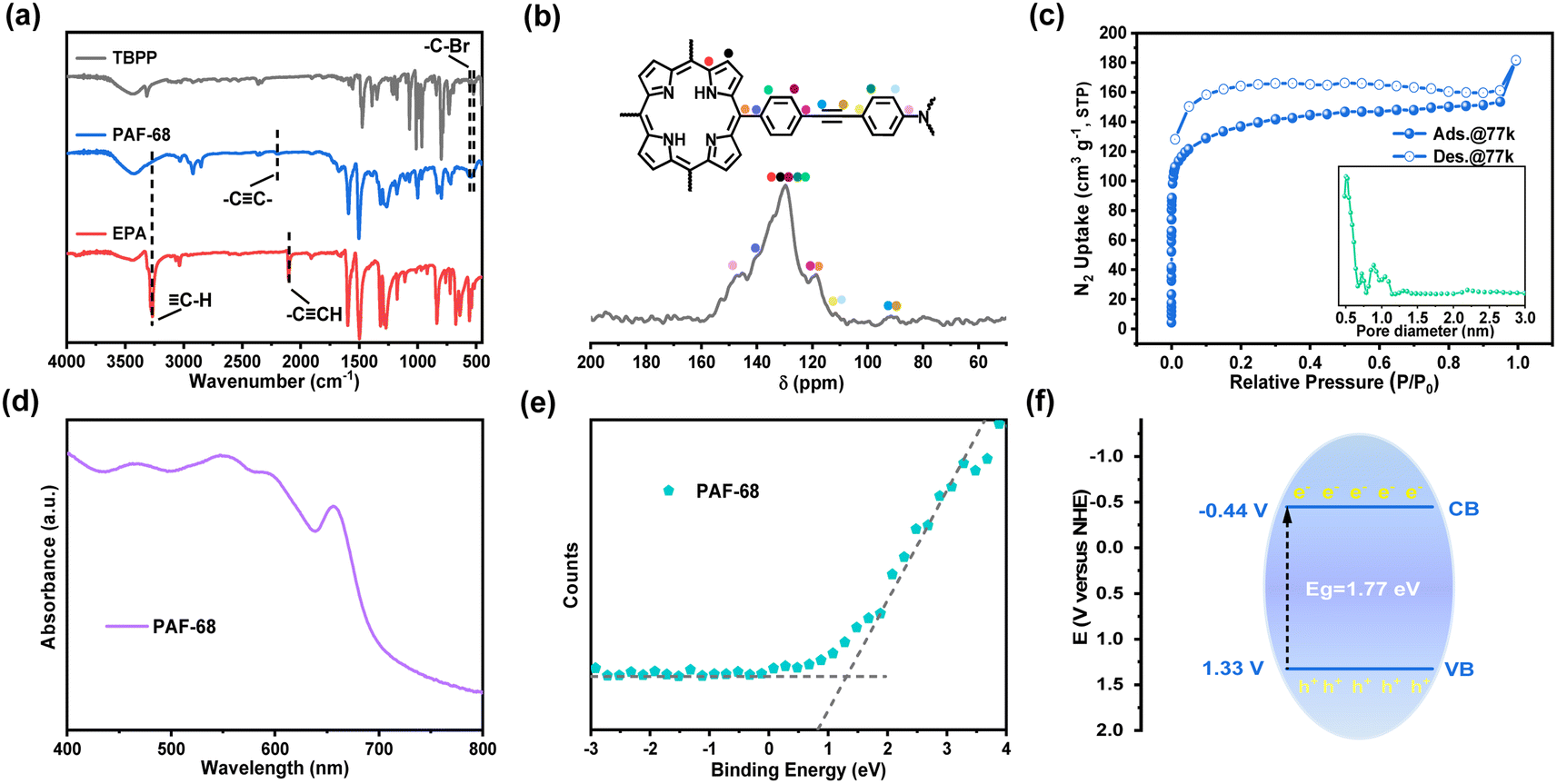 | ||
| Fig. 2 Structural and optical characterization of PAF-68. (a) FT-IR spectra of PAF-68, TBPP, and EPA for comparison. (b) Solid-state 13C CP/MAS NMR spectrum of PAF-68. (c) Nitrogen-sorption isotherm curves measured at 77 K and pore distribution of PAF-68. (d) UV-Vis DRS pattern of PAF-68. (e) VB-XPS spectrum of PAF-68 (the VB positions of PAF-68 are 1.33 eV below the Fermi level (EF)). (f) Band positions of PAF-68. | ||
The permanent porosity of PAF-68 was confirmed by N2 adsorption analysis at 77 K. The N2 adsorption–desorption isotherms of PAF-68 showed a typical type I curve as predicted (Fig. 2c), and the hysteresis in the isotherms was attributed to the swelling of the entirely organic framework with increased pressure.29 According to the Brunauer–Emmett–Teller model, the BET surface area of PAF-68 was estimated to be 509 m2 g−1. The total pore volume of PAF-68 was 0.28 cm3 g−1, with two primary pore diameters centered at 0.5 nm and 0.9 nm, respectively. The micropores facilitate substrate diffusion through the pores and provide accessibility to the reaction sites during catalytic conversion.
From the thermogravimetric analysis (TGA) curve (Fig. S4†) obtained under air flow, it is evident that the synthesized PAF-68 showed almost no weight loss up to 400 °C. As the temperature rose further, a sharp decrease in weight was observed, which was attributed to decomposition of the skeleton. A chemical stability assessment was performed by porosity measurements on the PAF-68 sample after stirring under harsh conditions and in organic solvents. The sample was either stirred in 3 M HCl (Fig. S5†) or 6 M KOH (Fig. S6†) for 48 h, or separately immersed in commonly used organic solvents, including tetrahydrofuran, methanol, dichloromethane, ethanol, and acetone for 24 h (Fig. S7†), then kept in ambient atmosphere for a whole year (Fig. S8†). The retained surface area indicated no obvious dissolution or degradation for PAF-68.
Light absorption measurements were applied to understand the electronic energy band levels of PAF-68. The UV-Vis diffuse reflectance spectrum of PAF-68 showed absorption bands in the wavelength range from 400 to 700 nm. The wide absorption band extends to the near-infrared region, which indicates its efficient light-harvesting ability due to the existence of the porphyrin macrocycle (Fig. 2d). A Kubelka–Munk-transformed reflectance spectrum based on UV-Vis DRS was recorded (Fig. S9†). According to the calculations derived from the Tauc plots, the corresponding optical bandgap (Eg) of PAF-68 was estimated to be 1.77 eV, indicating that PAF-68 is a semiconductor and possesses strong light-collecting capability. The valence band (VB) spectrum was also obtained using XPS for PAF-68 to evaluate the energy band, and the VB potential was found to be 1.33 eV (Fig. 2e). Deducing from the experimental energy levels of Eg and VB, the conduction band (CB) was calculated to be −0.44 eV (Fig. 2f). The CB, with enough negative potential, provides multiple pathways for oxygen activation.
Photocatalytic activity of PAF-68
The porous structure and light response of PAF-68 encouraged us to evaluate the catalytic oxidation of the toxic compounds, either in pure oxygen or in room air. CEES, a mustard gas simulant, was used as the modeling compound for safety reasons. The conversion and selectivity of the reaction were confirmed by 1H NMR and 13C NMR (Fig. 3a and S10†). In the 1H NMR results, the chemical shifts at 1.24, 2.58, 2.85, and 3.65 ppm were assigned to CEES, and those at 1.35, 2.82, 2.92, 3.17, and 3.95 ppm were attributed to the expected product CEESO. Under the condition of pure oxygen, the chemical shifts from CEES gradually vanished, and those signals corresponding to CEESO began to appear as soon as CEES was injected (Fig. S11†). Fig. 3b showed that with the aid of PAF-68, CEES can be completely converted into the nontoxic oxidation product CEESO in 3.7 min. The half-life (t1/2) for conversion of CEES to CEESO was evaluated to be 1.7 min, revealing the very fast degradation rate. It is noteworthy that although the detoxication process lasted for 2 h, no highly toxic 2-chloroethyl ethyl sulfone (CEESO2) was detected at all, suggesting that CEESO was the only product, with a selectivity of 100% (Fig. S12†). As PAFs are highly stable with carbon–carbon linkages, the humid environment would not affect the photocatalytic activity of PAF-68. Even more, when pure oxygen flux was bubbled in water, the trace humidity from the atmosphere accelerated the catalytic reaction, and the half-life in humid oxygen was further accelerated to 1.5 min (Fig. S13†).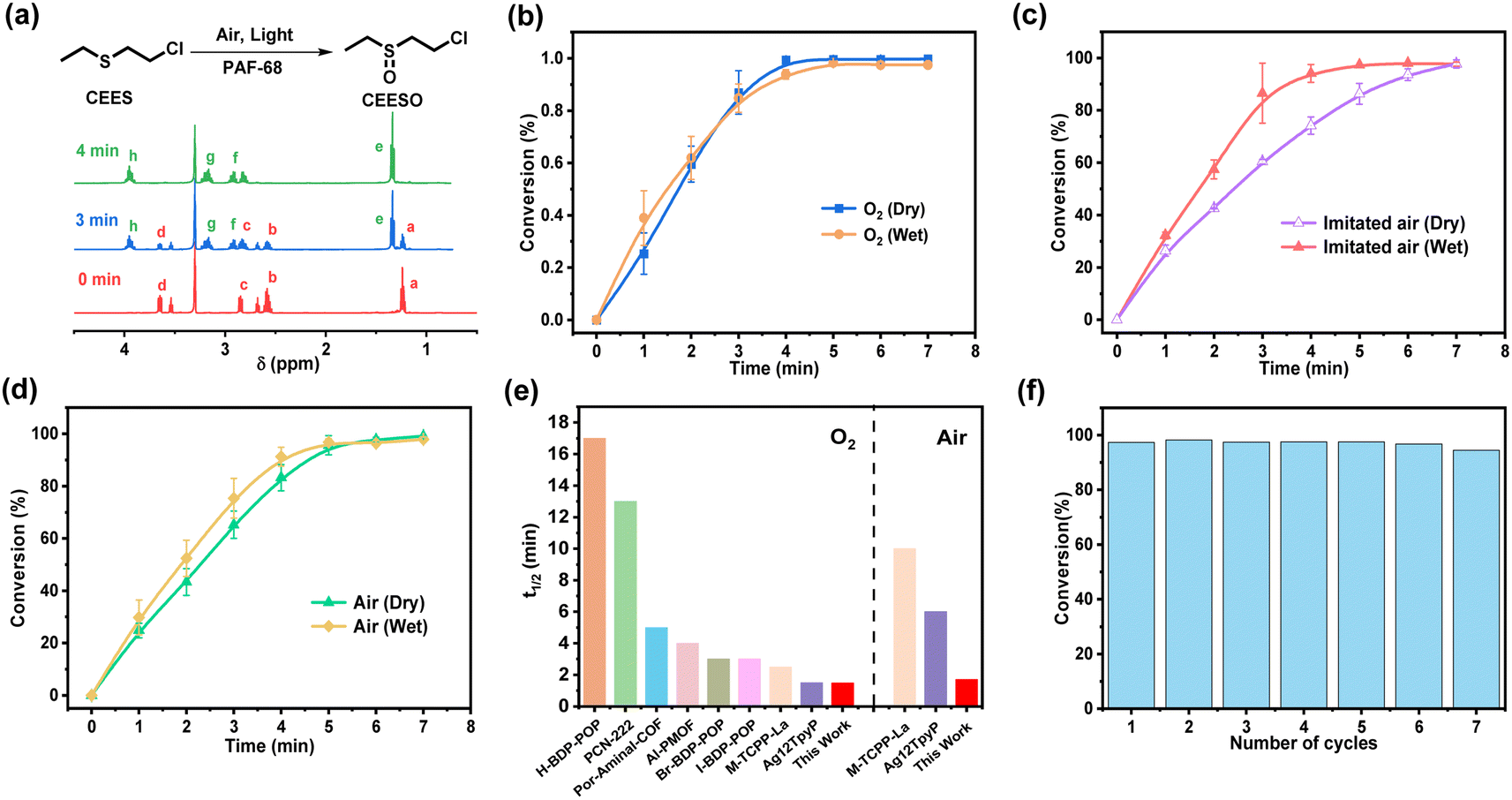 | ||
| Fig. 3 Detoxification of CEES catalyzed by PAF-68. (a) Photooxidation reaction of CEES by PAF-68 and 1H NMR spectra measured 0 min, 3 min and 5 min before and after photooxidation. (b) Conversion of CEES in the presence of PAF-68 under dry (blue) and wet (orange) pure O2. (c) Conversion of CEES in the presence of PAF-68 under dry (purple) and wet (red) simulated air. (d) Conversion of CEES in the presence of PAF-68 under dry (green) and wet (yellow) room air. (e) The half-life of CEES degradation using PAF-68 compared with other POP photosensitizers in pure oxygen and in air. (f) Recycling performance of PAF-68 towards photooxidation of CEES. | ||
The oxygen content in room air environment is only 21% (i.e., un-enriched in terms of oxygen). Under such a low oxygen partial pressure, the reported photocatalysts for CEES detoxification universally showed a significantly prolonged conversion half-life compared with oxidation in pure oxygen. For example, a porphyritic ligand-coordinated silver cluster exhibited a CEES degradation half-life of 1.5 min in pure oxygen, but the half-life of the same material in air was 6 min.30 Another porphyrin-based MOF, Fe–TCPP–La, selectively converted CEES to CEESO with a half-life of 2.5 min in oxygen atmosphere, but in air the half-life was about 10 min.14 Air atmosphere was also applied to PAF-68 to further investigate the impact of oxygen concentrations on the conversion of CEES. Surprisingly, PAF-68 exhibited selective conversion from CEES to CEESO in air almost as fast as in pure oxygen. Under an atmosphere of dry imitated air (N2/O2, v/v = 80, 20), CEES was fully converted into nontoxic CEESO within 7 min, with a t1/2 of 2.4 min (Fig. S14†). Trace humidity from the atmosphere by gas bubbling also expedited the catalytic reaction, with the half-life reduced to 1.7 min (Fig. 3c and S15†). In a real room air atmosphere, PAF-68 showed its wide suitability (Fig. S16 and S17†). Complete conversion from CEES to CEESO under humid air took 5.1 min, with a half-life of only 1.9 min (Fig. 3d). This conversion rate was comparable to that in imitated humid air (t1/2 = 1.7 min) and humid oxygen (t1/2 = 1.5 min). Even under an extremely low-oxygen condition (N2/O2, v/v = 98/2), CEES could still be completely converted to CEESO with a t1/2 of 2.7 min (Fig. S18–S21†), further demonstrating that the catalytic effect of PAF-68 is oxygen concentration-independent. Compared with other reported materials with greatly reduced conversion in air, the detoxification rate of PAF-68 to CEES in air atmosphere is very close to that in pure oxygen (Fig. 3e and Table S1†).
The covalent carbon–carbon linkages also render their excellent chemical stability in PAFs over other MOF-based catalysts. PAF-68 was used as a catalyst in humid air and recovered after the photocatalytic degradation. The results indicated that PAF-68 retained similar conversion rate and selectivity after 7 cycles, equivalent to those of the original catalyst (Fig. 3f). The FT-IR, solid-state 13C NMR, N2 adsorption and XPS spectra indicated that cycled PAFs have unaltered structure and permanent porosity, and no CEES and CEESO residues (Fig. S22†). At the same time, after keeping PAF-68 at room temperature in room air for a year, it still exhibited the same catalytic activity (Fig. S23†), indicating its structural stability and availability for long-term storage as well. For comparison, CEES detoxification was carried out without light and without PAF-68, respectively. The results showed that CEES did not convert within 15 min (Fig. S24†). When the reaction is carried out in aprotic solvents such as deuterated chloroform, CEESO2 and the oxidation elimination product ethyl vinyl sulfoxide (EVSO) signals are detected, consistent with previous reports (Fig. S25†).31 In order to prove the rapid detoxification caused by the superiority of PAF-68 rather than the experimental conditions, a typical PCN-222 was synthesized and compared (Fig. S26–S28†). The results showed that under the same conditions, the half-life of CEES degradation by PCN-222 in pure oxygen was 27.2 min, and the half-life in air was 33.6 min, which was close to the previously reported white light conditions.
Mechanism investigation
As a metal-free organic photocatalyst, PAF-68 generates ROS for fast decontamination of mustard gas simulant. It is rewarding to understand the generation of specific ROS for the corresponding photocatalytic process and shed light on the design of photocatalysts for CWA detoxification. In general, photocatalysts initiate the generation of different kinds of ROS such as ˙O2−, ˙OH, H2O2, and 1O2 from molecular oxygen. To identify the type of ROS involved in this process, 1,4-benzoquinone, tert-butanol, catalase, and carotene were employed as quenching agents to selectively scavenge ˙O2−, ˙OH, H2O2, and 1O2, respectively. Oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) was selected as an indicative reaction because colorless TMB is converted into the blue oxidized state (oxTMB) when exposed to light. As shown in Fig. 4a, the degree of oxidation was monitored by the characteristic UV-Vis absorption at 370 and 652 nm. The results were recorded and summarized in Fig. 4b. When 1,4-benzoquinone was used as the quenching agent for ˙O2−, the oxidation of TMB was completely prevented, leaving no color change of the TMB solution and no absorbance signal in UV-Vis. With regards to other trapping agents for ˙OH, H2O2, and 1O2, quenching was not observed, indicating the absence of these ROS. Therefore, it is believed that ˙O2− is the predominant ROS in the photocatalytic process initiated by PAF-68. Further convincing evidence for the identification of ROS was given by electron paramagnetic resonance spectroscopy (EPR). As a spin-trapping agent, 5,5-dimethyl-1-pyrroline-oxide (DMPO) was added to the photocatalysis reaction. Under light irradiation, four peaks were observed, and their appearance was attributed to the existence of DMPO*˙O2−. The peaks were significantly weakened in the dark, indicating the initiation of ˙O2− was driven by light irradiation (Fig. 4c). In contrast, there was no spin signal for ˙OH or 1O2 observed either under light irradiation or in darkness. (Fig. S29†). Together with the results of the quenching experiments, both of them showed that ˙O2− was the predominant ROS in the photoinduced process involving PAF-68. Compared with other reported HD/CEES detoxication photocatalysts such as MOFs and COFs,30,32 where 1O2 is usually present as the main ROS, PAF materials exclusively generating ˙O2− as ROS would be an exciting result for photocatalyst design and photodynamic therapy, as ˙O2− is independent of the oxygen concentration. For this reason, there is merit in unveiling the reason for ROS generation by metal-free or entirely organic compounds.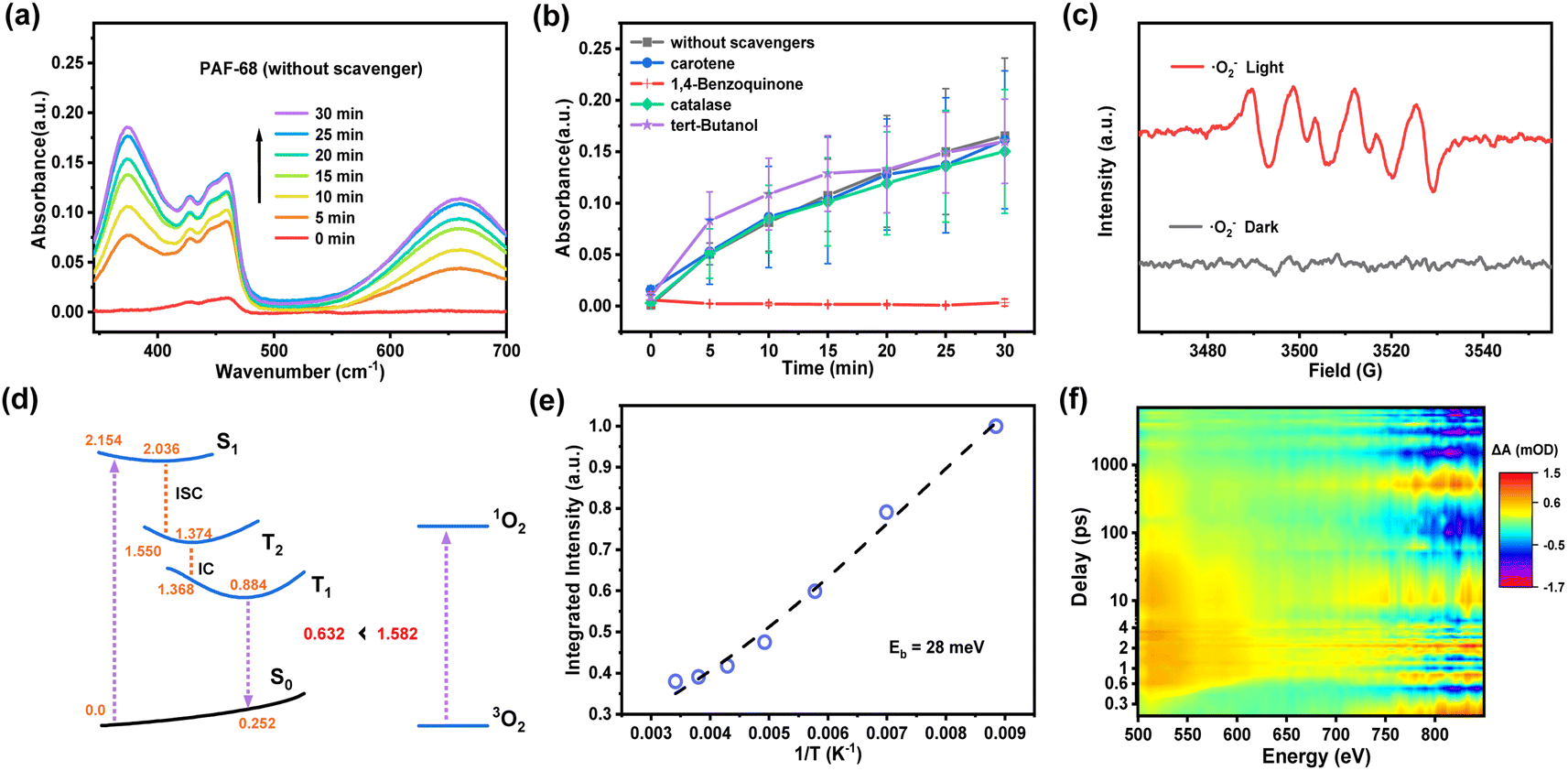 | ||
| Fig. 4 Practicability and carrier dynamics of PAF-68. (a) Time-dependent UV-Vis absorption spectra of TMB oxidation over PAF-68. (b) PAF-68 in the presence of different scavengers under visible light irradiation. (c) EPR spectra of PAF-68 in the presence of DMPO. (d) Suggested excited-state relaxation pathway from the initially populated S1 to T1 states of PAF-68 mediated by an intermediate T2 state, and energy profile for the type-II energy transfer from the T1PAF-68 to 3O2. (e) Integrated PL intensity of PAF-68 as a function of temperature from 83 K to 293 K. (f) fs-TA spectra of PAF-68 following excitation at 400 nm. | ||
Herein, we proposed a plausible reaction mechanism of the photocatalytic detoxification of CEES by PAF-68 as shown in Fig. S30.† Under visible light excitation, PAF-68 changes from the ground state to the excited state, producing photoinduced electrons and photoinduced holes. Subsequently, O2 is activated by electron transfer to generate O2−. On the other hand, the hole combines with CEES to form a positively charged intermediate A. Intermediate A combines with O2 to form peroxide B. Finally, peroxide B reacts with CEES to form the product CEESO. Quantum chemical calculations have been further conducted to explore the excited-state properties of PAF-68 and get in-depth insights into the underlying mechanism of ˙O2−. Our calculations suggest (Fig. S31†) that the first electronically excited singlet state S1 of PAF-68 is of local π–π* character, and thus is the vertical transition to the Franck–Condon point with the S0 stable structure. S1 state corresponds to the excitation of electrons from the two highest occupied molecular orbitals (HOMOs) to the two lowest unoccupied molecular orbitals (LUMOs), which can be explained by the Gouterman model (so-called four-orbital model).33 The orbital contributions of HOMO to LUMO and HOMO−1 to LUMO+1 for S0 to S1 excitation account for 62.9% and 35.0%, respectively. We also revealed the excited-state relaxation process from the S0 to T1 states by optimizing the minima of the corresponding states and computing linearly interpolated internal coordinate paths. The corresponding relaxation pathway is depicted in Fig. 4d. Upon photoexcitation, PAF-68 is vertically excited to the Franck–Condon point, and then relaxes rapidly to the S1 minimum with the S0–S1 energy gap of 2.036 eV. Subsequently, PAF68 further decays to the T2 intermediate state through the intersystem crossing process, with the S1–T2 energy gap of 0.486 eV. In T2 state, PAF68 relaxes rapidly to its minimum, with the S0–T2 energy gap of 1.374 eV, and then goes on decaying to the T1 state followed by rapid internal conversion process from T2 to T1, with the T2–T1 energy gap of 0.006 eV. In the T1 state, PAF68 relaxes to its minimum. The vertical T1–S0 emission energy was calculated to be 0.632 eV, which is significantly lower than the energy gap of 1.582 eV that is required to excite 3O2 to its singlet excited state to produce 1O2, indicating that this process could be endothermic. Therefore, in thermodynamics, the excitation energy transfer from the T1 state of PAF-68 to 3O2 is not beneficial.
As the excitation energy transfer process is unfavorable, the other pathway to ROS would be a charge transfer process that generates ˙O2−. To understand the photoinduced electron–hole pair recombination and separation in PAF-68, the exciton-binding energy (Eb) was determined by temperature-dependent photoluminescence (PL) measurement. The PL intensity of PAF-68 decreases rapidly with the increase of temperature from 83 K to 293 K (Fig. S32†). Arrhenius equation fitting of the integral PL intensity as a function of temperature indicated the Eb of PAF-68 was about 28 meV (Fig. 4e), which is much lower than the values for typical polymers at about 100 meV. The low Eb of PAF-68 is in favor of the formation of free electrons and thus facilitates the generation of ˙O2−. The thermal dissociation ratio was also calculated (Fig. S33†). It was recently reported that exciton dissociation at the order–disorder interfaces could be boosted by the simultaneous electron injection and hole blocking.34 As PAFs are usually considered long-range disordered structures with local regularity, the unique structural feature of PAFs would be a possible reason for the low binding energy.
To further explore the excitonic processes involved in PAF-68, the photoexcited carrier kinetics in PAF-68 was studied by femtosecond time-resolved transient absorption (fs-TA) spectroscopy in air at 298 K.35,36 A 400 nm pump pulse was used to excite PAF-68. The two-dimensional pseudo-color (ΔA) plot in Fig. 4f was generated by fitting the transient absorption spectrum of PAF-68 with the change of delay time and probe wavelength. As the global fitting analysis has been completed, the steady-state absorption spectrum was then obtained. A broad excited-state absorption over the range of 500–650 nm was attributed to the photoinduced absorption (PA) of electrons in PAF-68 (Fig. S34†), which relates to the formation of free carriers.37 The positive signal of PAF-68 increases continuously at 522 nm before 0.6 ps, and the continuous absorption of PAF-68 at 0.8 ps to 7.5 ns at 522 nm reflects the intramolecular charge transfer. The excited state decay after 516 nm optical excitation was plotted to further describe the lifetime of the photogenerated charge carriers. The fitting results showed that the average lifetime of the photogenerated charge of PAF-68 was about 1017 ps (Fig. S35†). The long decay lifetime, which is in favor of effective electron–hole separation, is feasible in PAF-68. The delayed excited state decay results are consistent with low exciton binding energy, which excellently supports that PAF-68 has a long-term carrier lifetime and effective exciton dissociation, and both facilitate the efficient generation of ˙O2−. These findings together support that the amorphous PAFs could affect ROS production, and eventually leading to their rapid catalytic detoxification of CEES in room air.
Conclusions
In summary, a porphyrin-based PAF was synthesized, and its photocatalytic decontamination of a mustard gas simulant was studied. Compared to other leading CEES detoxication catalysts, PAF-68 rapidly degraded CEES to nontoxic CEESO in room air, with a t1/2 of 1.7 min and a selectivity of 100%. The performance in air not only surpasses that of many photocatalysts under pure oxygen; it is also much desired due to the availability and safety of the oxidant supply. Mechanism investigation revealed that ˙O2− was initiated as a ROS by photoactivated PAF-68 and quickly detoxicated mustard gas simulant in room air. PAF-68 has low exciton binding energy and a long decay lifetime, which are in favor of the generation of ˙O2−. Their stability and applicability in typical room air support PAFs as promising photocatalysts for CEES degradation in real-world environments, offering an original foundation for the future development of effective and widely applicable photocatalysts.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. S. and H. L. contributed equally to this work. G. Z. conceived the project. Y. T. and J. S. supervised the project. J. S., H. L. and Y. T. designed the experiments, analyzed the data, and wrote the manuscript. H. L. carried out most of the experiments. Y. Z., Y. D. and X. H. participated in photocatalysis tests. Z. D. participated in the TMB quenching experiment. F. C. provided the theoretical support. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the Science and Technology Development Plan Project of Jilin Province, China (20240602105RC), China Postdoctoral Science Foundation (2024T170120, 2023M740577), National Natural Science Foundation of China (22131004, U21A20330, 22075040), the “111” project (B18012), Fundamental Research Funds for the Central Universities (2412023QD013), Postdoctoral Funding Project of Jilin Province, China.References
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Coord. Chem. Rev., 2017, 346, 101–111 CrossRef CAS.
- J. Dong, J. Hu, Y. Chi, Z. Lin, B. Zou, S. Yang, C. L. Hill and C. Hu, Angew. Chem., Int. Ed., 2017, 56, 4473–4477 CrossRef CAS PubMed.
- Y. Li, D. Zhang, W. Qiao, H. Xiang, F. Besenbacher, Y. Li and R. Su, Chem. Synth., 2022, 2, 9 CrossRef CAS.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS.
- C. T. Buru, M. C. Wasson and O. K. Farha, ACS Appl. Nano Mater., 2020, 3, 658–664 CrossRef CAS.
- A. Atilgan, M. M. Cetin, J. Yu, Y. Beldjoudi, J. Liu, C. L. Stern, F. M. Cetin, T. Islamoglu, O. K. Farha, P. Deria, J. F. Stoddart and J. T. Hupp, J. Am. Chem. Soc., 2020, 142, 18554–18564 CrossRef CAS.
- Q.-Y. Wang, J. Liu, M. Cao, J.-H. Hu, R. Pang, S. Wang, M. Asad, Y.-L. Wei and S.-Q. Zang, Angew. Chem., Int. Ed., 2022, 61, e202207130 CrossRef CAS.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- Y. Hou, H. An, Y. Zhang, T. Hu, W. Yang and S. Chang, ACS Catal., 2018, 8, 6062–6069 CrossRef CAS.
- L. Ma, Y. Liu, Y. Liu, S. Jiang, P. Li, Y. Hao, P. Shao, A. Yin, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2019, 58, 4221–4226 CrossRef CAS PubMed.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS.
- L. Jiang, H. Bai, L. Liu, F. Lv, X. Ren and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 10660–10665 CrossRef CAS PubMed.
- Z.-H. Long, D. Luo, K. Wu, Z.-Y. Chen, M.-M. Wu, X.-P. Zhou and D. Li, ACS Appl. Mater. Interfaces, 2021, 13, 37102–37110 CrossRef CAS.
- Z.-X. Sun, K. Sun, M.-L. Gao, Ö. Metin and H.-L. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202206108 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- Y. Peng, Q. Tan, H. Huang, Q. Zhu, X. Kang, C. Zhong and B. Han, Chem. Synth., 2022, 2, 15 CrossRef CAS.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- A. A. Uliana, N. T. Bui, J. Kamcev, M. K. Taylor, J. J. Urban and J. R. Long, Science, 2021, 372, 296–299 CrossRef CAS PubMed.
- Y. Ma, F. Cui, H. Rong, J. Song, X. Jing, Y. Tian and G. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202113682 CrossRef CAS PubMed.
- J. Song, Y. Li, P. Cao, X. Jing, M. Faheem, Y. Matsuo, Y. Zhu, Y. Tian, X. Wang and G. Zhu, Adv. Mater., 2019, 31, 1902444 CrossRef CAS PubMed.
- J. Zou, A. Trewin, T. Ben and S. Qiu, Angew. Chem., Int. Ed., 2020, 59, 769–774 CrossRef CAS PubMed.
- Z. Li, L. Wang, M. Yu, Y. Liu, B. Liu, Z. Sun, W. Hu and G. Zhu, ACS Appl. Mater. Interfaces, 2022, 14, 53798–53807 CrossRef CAS PubMed.
- Z. Zhang, Y. Zheng, Z. Dou, M. Gu, M. Sun, J. Song, N. Gao, F. Cui, Y. Tian and G. Zhu, ACS Cent. Sci., 2023, 9, 488–493 CrossRef CAS PubMed.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- J. An, S. Tang, G. Hong, W. Chen, M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, Nat. Commun., 2022, 13, 2225 CrossRef CAS.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, J. Am. Chem. Soc., 2023, 145, 8130–8140 CrossRef CAS PubMed.
- J. Jeromenok and J. Weber, Langmuir, 2013, 29, 12982–12989 CrossRef CAS PubMed.
- M. Cao, R. Pang, Q.-Y. Wang, Z. Han, Z.-Y. Wang, X.-Y. Dong, S.-F. Li, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2019, 141, 14505–14509 CrossRef CAS PubMed.
- Y. Hao, E. K. Papazyan, Y. Ba and Y. Liu, ACS Catal., 2022, 12, 363–371 CrossRef CAS.
- S. Wang, Q. Sun, W. Chen, Y. Tang, B. Aguila, Y. Pan, A. Zheng, Z. Yang, L. Wojtas, S. Ma and F.-S. Xiao, Matter, 2020, 2, 416–427 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- H. Wang, X. Sun, D. Li, X. Zhang, S. Chen, W. Shao, Y. Tian and Y. Xie, J. Am. Chem. Soc., 2017, 139, 2468–2473 CrossRef CAS.
- J. Hu, K. Xu, L. Shen, Q. Wu, G. He, J.-Y. Wang, J. Pei, J. Xia and M. Y. Sfeir, Nat. Commun., 2018, 9, 2999 CrossRef PubMed.
- X. Zhang, K. Geng, D. Jiang and G. D. Scholes, J. Am. Chem. Soc., 2022, 144, 16423–16432 CrossRef CAS PubMed.
- Z. Chen, Q. Zhang and Y. Luo, Angew. Chem., Int. Ed., 2018, 57, 5320–5324 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04551c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |