
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCascade-responsive size/charge bidirectional-tunable nanodelivery penetrates pancreatic tumor barriers†
Yiqi
Shi
a,
Jinghan
Liao
b,
Cuiyun
Zhang
a,
Qi
Wu
a,
Shanshan
Hu
a,
Ting
Yang
c,
Jihong
Liu
a,
Zhirong
Zhu
a,
Wei-Hong
Zhu
 a and
Qi
Wang
*a
a and
Qi
Wang
*a
aShanghai Key Laboratory of Functional Materials Chemistry, Key Laboratory for Advanced Materials and Institute of Fine Chemicals, Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: wangqi@ecust.edu.cn
bState Key Laboratory of Systems Medicine for Cancer, Shanghai Cancer Institute, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 2200/25 Xietu Road, Shanghai, 200032, China
cGuangdong Key Laboratory of Nanomedicine, CAS-HK Joint Lab for Biomaterials, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China
First published on 29th August 2024
Abstract
The pancreatic tumor microenvironment presents multiple obstacles for polymer-based drug delivery systems, limiting tumor penetration and treatment efficacy. Here, we engineer a hyaluronidase/reactive oxygen species cascade-responsive size/charge bidirectional-tunable nanodelivery (btND, G/R@TKP/HA) for co-delivery of gemcitabine and KRAS siRNA, capable of navigating through tumor barriers and augmenting anticancer efficiency. When penetrating the tumor stroma barrier, the hyaluronic acid shell of the nanodelivery undergoes degradation by hyaluronidase in an extracellular matrix, triggering size tuning from large to small and charge tuning from negative to positive, thereby facilitating deeper penetration and cellular internalization. After endocytosis, the nanodelivery protonizes in the endo/lysosome, prompting rapid endo/lysosomal escape, effectively overcoming the lysosome barrier. Intracellular ROS further disrupt the nanodelivery, inducing its size tuning again from small to large and a positive charge decrease for high tumor retention and controlled drug release. The btND shows remarkable antitumor activity in pancreatic cancer mouse models, highlighting the efficacy of this approach in penetrating tumor barriers and enhancing anticancer outcomes.
Introduction
Polymer-based drug delivery systems are increasingly recognized for their potential in pancreatic cancer therapy.1,2 However, the distinctive anatomical attributes of pancreatic cancer—its deep-seated location, restricted blood flow, and dense stroma—present formidable challenges to achieving effective tissue penetration and intratumoral retention, ultimately compromising therapeutic outcomes.3,4 To overcome these hurdles, an ideal drug delivery system should combine prolonged systemic circulation, deep stromal penetration, robust tumor accumulation, efficient tumor cell uptake, lysosomal escape, and controlled drug release.5In the realm of tumor therapeutics, the performance of delivery systems is closely tied to their size and surface charge.6,7 Larger systems offer extended circulation and increased tumor accumulation but struggle with deep penetration.8 Conversely, smaller systems penetrate well but often lack sustained intratumoral retention.9 While efforts have been made to develop size-tunable systems, unidirectional size adjustments present trade-offs: shrinking systems sacrifice retention, while expanding ones compromise penetration. Consequently, such unidirectional adjustments fail to concurrently optimize both the penetration depth and retention duration requisite for effective tumor targeting. Beyond size considerations, the surface charge of delivery systems serves as a pivotal determinant of their intratumoral behavior.10,11 Negatively charged systems extend circulation and reduce off-target effects, while positively charged ones enhance cellular uptake and cytotoxicity against tumor cells. In response, innovative carrier systems have been devised with a charge transition from negative to positive;12,13 however, the initial negative charge poses challenges for the encapsulation of gene-based therapeutics. Given these complexities, an integrated approach considering both size and charge, alongside tailored optimization strategies, is essential for next-generation delivery systems.
In response to the intricate challenges presented by size and charge across diverse biological barriers, we have developed a tumor microenvironment (TME)-responsive size and charge bidirectionally tunable nanodelivery (btND). This innovative btND is equipped with dual-cascade responsiveness to both hyaluronidase (HAase) and reactive oxygen species (ROS). It is fabricated by encapsulating the chemotherapeutic gemcitabine (Gem)14 and KRAS siRNA15 within a ROS-responsive polymer (TCM-TK-PEI), which is bridged by using an aggregation-induced emission (AIE) photosensitizer (TCM), and subsequently coated with hyaluronic acid (HA) (Fig. 1A). In the circulatory system, the btND adopts a large size and negative charge. This enlarged size facilitates prolonged circulation,16 while the negative charge effectively minimizes off-target toxicities. Upon infiltration into the pancreatic tumor extracellular matrix (ECM), the HA coating undergoes degradation by HAase,17 leading to a size reduction that enhances tissue penetration. Concurrently, the transition from a negative to positive charge profile not only augments cellular uptake efficiency but also invokes the “proton sponge” effect,18 thereby facilitating lysosomal escape. Upon irradiation with white light, the AIE photosensitizer TCM produces a significant amount of ROS, leading to photodynamic therapeutic effects and initiating the cleavage of thioketal (TK) bonds19 within the carrier material. This process prompts polymer degradation followed by TCM reaggregation. The resulting increase in size promotes enhanced tumor retention, while the decreased charge distribution aids in the release of gene drugs. Crucially, TCM's near-infrared fluorescence capability allows for precise tracking of drug delivery behavior (Fig. 1B). Overall, this btND exhibits a dynamic size modulation—from large to small and back to large—coupled with a charge profile that shifts from negative to positive and then decreases. This design holds substantial promise for surmounting the six pivotal barriers inherent to pancreatic cancer therapy.
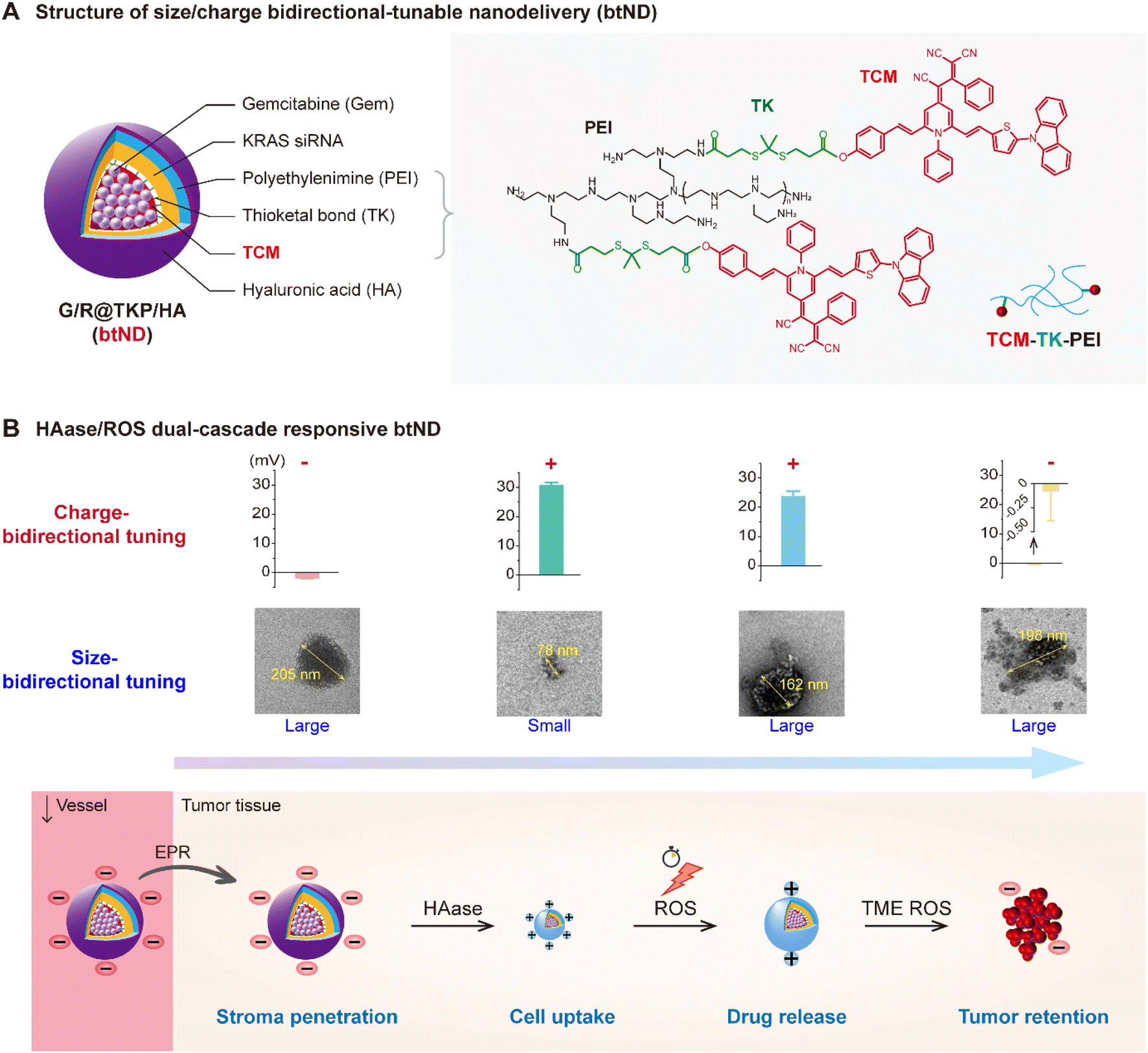 | ||
| Fig. 1 TME-cascade responsive size/charge bidirectional-tunable nanodelivery (btND). (A) Structure of the size/charge bidirectional-tunable nanodelivery Gem/KRAS siRNA/TCM-TK-PEI@HA (G/R@TKP/HA). TCM-TK-PEI is synthesized by conjugating an AIE photosensitizer (TCM) containing a ROS-responsive thioketal (TK) bond to polyethylenimine (PEI) via an amidation reaction. TCM-TK-PEI self-assembles into stable nanoparticles to encapsulate gemcitabine (Gem) (Gem@TCM-TK-PEI, G@TKP). Subsequently, negatively charged KRAS siRNA is complexed via electrostatic interaction (Gem/KRAS siRNA@TCM-TK-PEI, G/R@TKP). Finally, the negatively charged hyaluronic acid (HA) is modified in the outermost layer by electrostatic interaction to obtain G/R@TKP/HA. (B) Schematic illustration of TME-cascade responsive btND G/R@TKP/HA surmounting tumor barriers. (I) G/R@TKP/HA initially possesses a large size and negative charge, facilitating extended circulation time and reduced off-target effects. (II) Upon reaching tumor tissue via the enhanced permeability and retention (EPR) effect, the external hyaluronic acid coating of G/R@TKP/HA is degraded by the overexpressed hyaluronidase (HAase) in the tumor tissue. This degradation triggers size reduction and charge reversal, promoting deep tissue penetration and enhancing uptake by tumor cells. (III) Utilizing the reactive oxygen species (ROS) generated by TCM-mediated photodynamic therapy (PDT) to destroy the structure of G/R@TKP/HA, results in a size increase and positive charge decrease, facilitating the release of loaded drugs and siRNA. (IV) Subsequently, TME ROS cleaves the thioketal (TK) bonds within the G/R@TKP/HA's polymer skeleton, leading to further size enlargement and a decrease in positive charge, enhancing tumor retention and sufficient drug release. | ||
Results and discussion
Construction of a smart btND with size/charge bidirectional-tunability
In this study, we devised a HAase/ROS cascade-responsive, size- and charge-bidirectional tunable nanodelivery (btND) to overcome challenges associated with conventional drug administration. We synthesized a ROS-responsive polymer, termed TCM-TK-PEI, by covalently linking the aggregation-induced-emission (AIE) photosensitizer TCM to polyethyleneimine (PEI) with a 10 kDa molecular weight, utilizing a ROS-cleavable TK bond. This multifunctional polymer facilitates both photodynamic therapy (PDT) and fluorescence bioimaging, while also serving as an efficient carrier for gemcitabine (Gem) encapsulation within its hydrophobic TCM segment and for loading KRAS siRNA through electrostatic interactions with the PEI moiety. To further augment its biocompatibility and targeting specificity, we coated the TCM-TK-PEI nanoplatform with HA via electrostatic interactions. This advanced btND, denoted as Gem/KRAS siRNA@TCM-TK-PEI/HA (G/R@TKP/HA), exhibits dual responsiveness to HAase and ROS, thereby offering a promising approach for synergistic drug and gene co-delivery applications.The initial large size and negative charge of G/R@TKP/HA facilitate extended blood circulation and tumor accumulation. As G/R@TKP/HA penetrates the pancreatic tumor stroma barrier, its HA coating undergoes degradation in response to HAase,17 leading to a size reduction from large to small and a charge transition from negative to positive, thereby enabling deep tissue penetration. The cell membrane barrier is overcome through electrostatic interactions between the oppositely charged G/R@TKP/HA and cell membranes, further aided by residual HA on the G/R@TKP/HA surface that targets the overexpressed CD44 receptors on tumor cell membranes.20,21 Upon endocytosis, the endo/lysosomal barrier is breached through a combination of the “proton sponge” effect22 resulting from increased positive charge due to PEI protonation and ROS-induced lysosomal membrane disruption23,24 triggered by G/R@TKP/HA upon white light irradiation. Intracellular ROS induce polymer degradation and TCM reaggregation, leading to a size increase from small to large and a decrease in positive charge, facilitating high tumor retention and site-specific drug release. This synergistic approach combines chemotherapy, siRNA therapy, and PDT to enhance anticancer efficacy. The synthesis route and characterization of TCM-TK-PEI and its intermediates are detailed in the ESI.†
Cascade-response behavior of the btND with size/charge bidirectional-tunability
The synthesis pathway for the ROS-responsive polymer TCM-TK-PEI is extensively detailed in the ESI (Fig. S1†). Initially, we synthesized the AIE photosensitizer TCM, which underwent amidation to conjugate with PEI (10 kDa), yielding TCM-TK-PEI. We then meticulously compared the photophysical attributes of the AIE photosensitizer TCM with those of the resultant polymer TCM-TK-PEI. TCM exhibited minimal fluorescence in MeOH, EtOH, and DMSO with favorable dispersibility, whereas its aqueous solubility was limited, with absorption and fluorescence peaks at 512 nm and 636 nm (in 99% water), respectively, exhibiting AIE characteristics in DMSO-water mixtures (Fig. S2†). The polymer TCM-TK-PEI displayed solubility in alcoholic solvents and DMSO, with narrow absorption peaks and minimal fluorescence, yet in aqueous environments, it exhibited broadened absorption peaks and pronounced AIE fluorescence centered at 461 nm and 594 nm, indicating nanoparticle self-assembly (Fig. 2A and B). The self-assembly of TCM-TK-PEI in aqueous solution was also confirmed by DLS and TEM, showing a particle size of (76.95 ± 0.80) nm, with a spherical morphology (Fig. S3†). Crucially, both the absorption and emission spectra of TCM-TK-PEI displayed a blue shift relative to those of TCM, attributed to the influence of positive charges on PEI, with shifts of 51 nm, 39 nm, 42 nm, and 40 nm for absorption in water, MeOH, EtOH, and DMSO, respectively, and a 42 nm shift for emission in water (Fig. 2C, D and S4†). Following this, we assessed the ROS generation of TCM-TK-PEI using dichlorofluorescein diacetate (DCFH-DA),25 with TCM serving as the control, revealing a ROS generation trend similar to that of TCM, underscoring its potential for photodynamic therapy (PDT) (Fig. 2E and S5†). Moreover, further evaluation via1H NMR spectroscopy confirmed the ROS responsiveness of TCM-TK-PEI. Upon H2O2 addition, cleavage of the disulfide (TK) bond occurred, forming thiol groups and leading to the disappearance of the characteristic peak of the original methyl (Hα) at 1.60 ppm, affirming the ROS-responsive nature of TCM-TK-PEI (Fig. 2F). Upon ROS stimulation, the TEM image of the nanoparticles self-assembled by TCM-TK-PEI also validated the ROS response properties of TCM-TK-PEI, showing the adequate disruption of TCM-TK-PEI and the reaggregation of TCM residues (Fig S6†).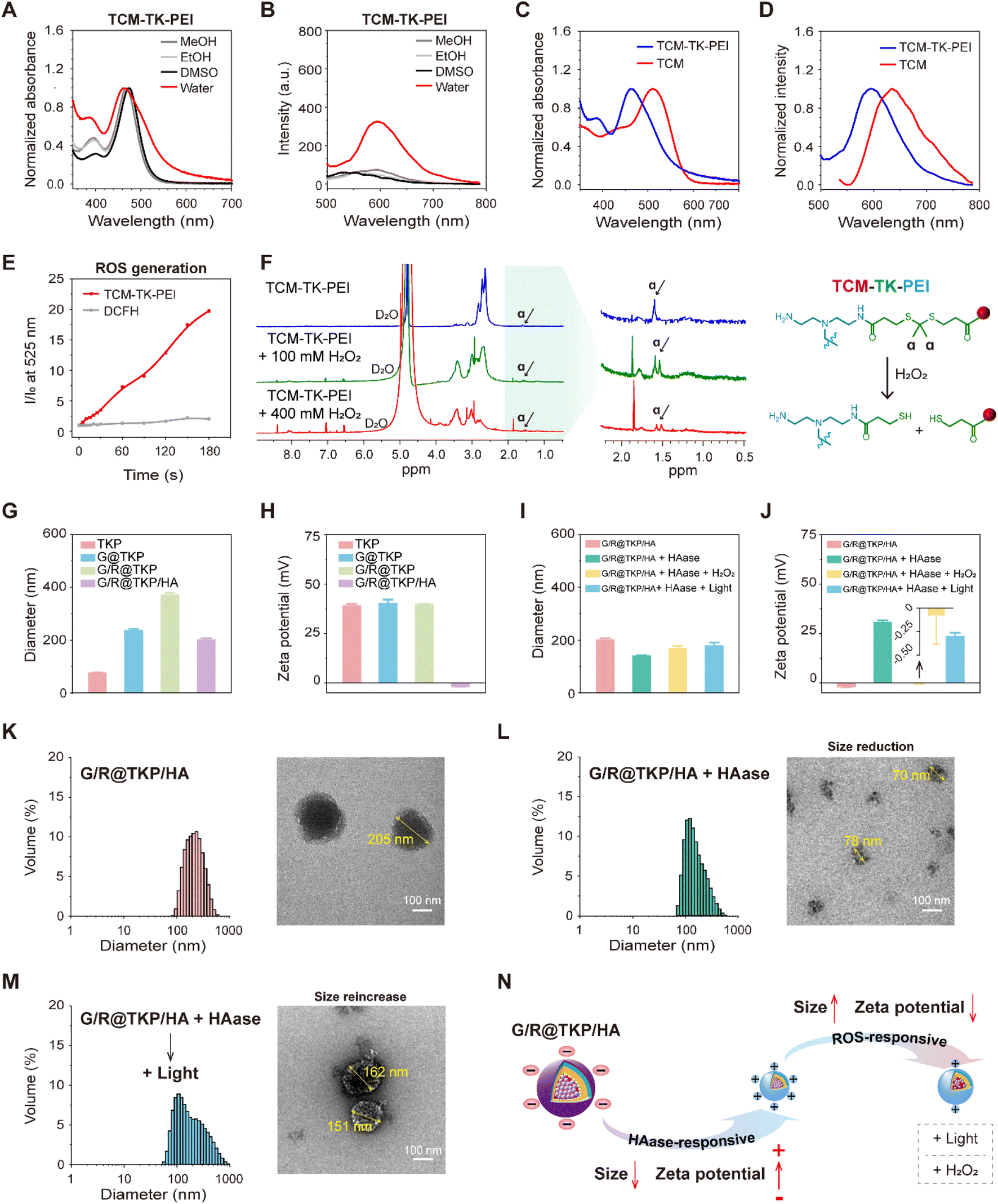 | ||
| Fig. 2 HAase/ROS dual-cascade responsive size/charge bidirectional-tunability of the btND. (A) Normalized absorption spectra of TCM-TK-PEI (0.15 mg mL−1) in different solvents (MeOH, EtOH, DMSO, and water). (B) Normalized fluorescence spectra of TCM-TK-PEI (0.15 mg mL−1) in different solvents (MeOH, EtOH, DMSO, and water); λex = λabs. (C) Normalized absorption and (D) normalized fluorescence spectra of TCM and TCM-TK-PEI in water; λex = λabs. (E) Total ROS generation by TCM-TK-PEI (0.15 mg mL−1) upon white light irradiation (40 mW cm−2) using DCFH-DA (40 μm) as an indicator; λex = 488 nm. (F) 1H NMR spectrum of TCM-TK-PEI before and after incubation with 100 mM and 400 mM H2O2 at 37 °C (left), and the mechanism of ROS-responsive cleavage of the TK bond (right). (G) Particle size of TKP (TCM-TK-PEI), G@TKP (TCM-TK-PEI@Gem), G/R@TKP (TCM-TK-PEI@Gem/siRNA) and G/R@TKP/HA (TCM-TK-PEI@Gem/siRNA/HA) (n = 3). (H) Zeta potential of T, T@G, T@G/si and G/R@TKP/HA (n = 3). (I) Particle size changes of G/R@TKP/HA before and after HAase/ROS-cascade stimuli (n = 3). (J) Zeta potential changes of G/R@TKP/HA before and after HAase/ROS-cascade stimuli (n = 3). (K) Size distribution (left) and TEM image (right) of G/R@TKP/HA. (L) Size distribution (left) and TEM image (right) of G/R@TKP/HA treated with HAase. (M) Size distribution (left) and TEM image (right) of the HAase/ROS-cascade response of G/R@TKP/HA after HAase treatment followed by white light irradiation (40 mW cm−2, 8 min). (N) Schematic illustration of HAase/ROS dual-cascade responsive size and charge bidirectional tunability of G/R@TKP/HA. | ||
The HAase/ROS dual-cascade btND, G/R@TKP/HA, was synthesized with bidirectional tunability in size and charge. Gem, encapsulated with high efficiency (97.17 ± 0.019%) and loaded effectively (6.81 ± 0.001%), was housed within the hydrophobic TCM segment (Fig. S7†). We further screened the siRNA load quantity via agarose gel electrophoresis assay and the TCM-TK-PEI/siRNA weight ratio (2.5/1) with efficient siRNA complexation was selected for subsequent experiments (Fig. S8A and B†). Sequentially, KRAS siRNA and HA were modified onto the outer hydrophilic PEI section, with a TCM-TK-PEI/KRAS siRNA/HA weight ratio of 2.5/1/1, via electrostatic interaction. Specifically, TCM-TK-PEI self-assembled into nanoparticles in water, denoted as TKP, measuring 77.0 ± 0.80 nm in diameter with a positive potential of 39.2 ± 0.73 mV. Loading Gem and KRAS siRNA increased nanoparticle size (G@TKP of 237.5 ± 4.98 nm and G/R@TKP of 371.4 ± 7.31 nm) while maintaining a positive zeta potential (G@TKP of 40.3 ± 1.85 mV and G/R@TKP of 39.9 ± 0.22 mV). Notably, coating with HA reduced the size of G/R@TKP/HA nanoparticles to 202.4 ± 5.77 nm and reversed the zeta potential to −1.90 ± 0.12 mV. This modification compacted the loose nanoparticles and shielded the positively charged PEI, fostering good biocompatibility and prolonged blood circulation (Fig. 2G and H). Agarose gel electrophoresis confirmed G/R@TKP/HA's complete siRNA retention, while dynamic light scattering revealed uniform particle size maintenance for 10 days, highlighting its exceptional stability for biological applications (Fig. S8B and C†).
Due to the abundant presence of HAase in the pancreatic tumor stroma17 and high levels of ROS in tumor cells,26 we investigated the responsive behavior of size/charge tunable G/R@TKP/HA in the presence of HAase and ROS. After HAase treatment, the particle size of G/R@TKP/HA decreased from 202.4 ± 5.77 nm to 141.7 ± 0.83 nm (Fig. 2I), and the charge reversed from −1.90 ± 0.12 mV to 30.73 ± 0.95 mV (Fig. 2J). This indicated that HAase degraded the HA coating of G/R@TKP/HA, leading to particle size reduction and charge reversal from negative to positive. The decreased particle size facilitated penetration through the tumor stroma barrier, while the charge reversal promoted cellular uptake. Continued white light irradiation (40 mW cm−2, 8 min) to induce ROS production resulted in the particle size increasing to 179.3 ± 11.81 nm (Fig. 2I), and the charge decreasing to 23.73 ± 1.71 mV (Fig. 2J). Similar results were obtained with additional ROS (H2O2), where the nanoparticle size increased to 168.2 ± 10.13 nm (Fig. 2I), and the potential significantly decreased to −0.08 ± 0.31 mV (Fig. 2J). This suggested that the combined use of HAase and ROS could cause particle enlargement and charge reduction. Transmission electron microscopy confirmed these findings, showing that the initial nanoparticles G/R@TKP/HA were uniformly spherical (Fig. 2K), while after treatment with HAase, the nanoparticles were degraded into surface-roughened particles of various sizes (Fig. 2L). Continued ROS production under light exposure or additional H2O2 led to the swelling of nanoparticles into loose spherical structures (Fig. 2M and S9†), primarily attributed to the detachment of the PEI portion due to the breakage of TK groups in the nanoparticles.
Moreover, agarose gel electrophoresis experiments demonstrated that the combined use of HAase and ROS could induce the migration of siRNA on the gel, indicating that the change in potential caused by the breakage of TK bonds might weaken the interaction between nanoparticles and siRNA, thereby promoting siRNA release. In summary, HAase stimulation triggered the reduction in size and reversal of charge of G/R@TKP/HA, facilitating deep penetration into tumor tissues and promoting cellular uptake. After the cascade ROS response, the particle size increased, and the positive charge reduced, ensuring high retention in tumor tissues and gene drug release (Fig. 2N). Therefore, this TME-cascade-responsive G/R@TKP/HA with size and charge bidirectional tunability can effectively traverse multiple tumor barriers, achieve deep delivery, and further enhance therapeutic efficacy.
Penetration of tumor stroma, cell membrane and lysosome barriers
Adhering to our design strategy, the size/charge bidirectional-tunable btND, G/R@TKP/HA, responsive to the pancreatic tumor microenvironment can effectively navigate through the tumor stroma, cell membrane and lysosomal barriers. Herein, we validated its penetration through these barriers using three-dimensional (3D) PANC1 multicellular tumor spheroids (MTSs), cellular uptake, and endo/lysosomal escape experiments. Initially, we evaluated the permeability of G/R@TKP/HA in PANC1 MTS, employing uncoated G/R@TKP as a control. Fluorescence imaging with AIE fluorescence of TCM was conducted by scanning along the z-axis, capturing images at 5 μm intervals throughout the MTS. The results showed that tumor spheroids treated with uncoated G/R@TKP exhibited reduced uptake and minimal fluorescence in deeper regions, while those treated with HA-coated G/R@TKP/HA displayed stronger fluorescence and deeper penetration, visible up to 55 μm from the surface (Fig. 3A and S10†). Additionally, HAase-responsive size reduction resulted in brighter fluorescence and enhanced overall fluorescence signal in G/R@TKP/HA-treated tumor spheroids compared to those treated with uncoated G/R@TKP, facilitating deeper tissue penetration (Fig. 3B, C, S10, Movie S1 and S2†).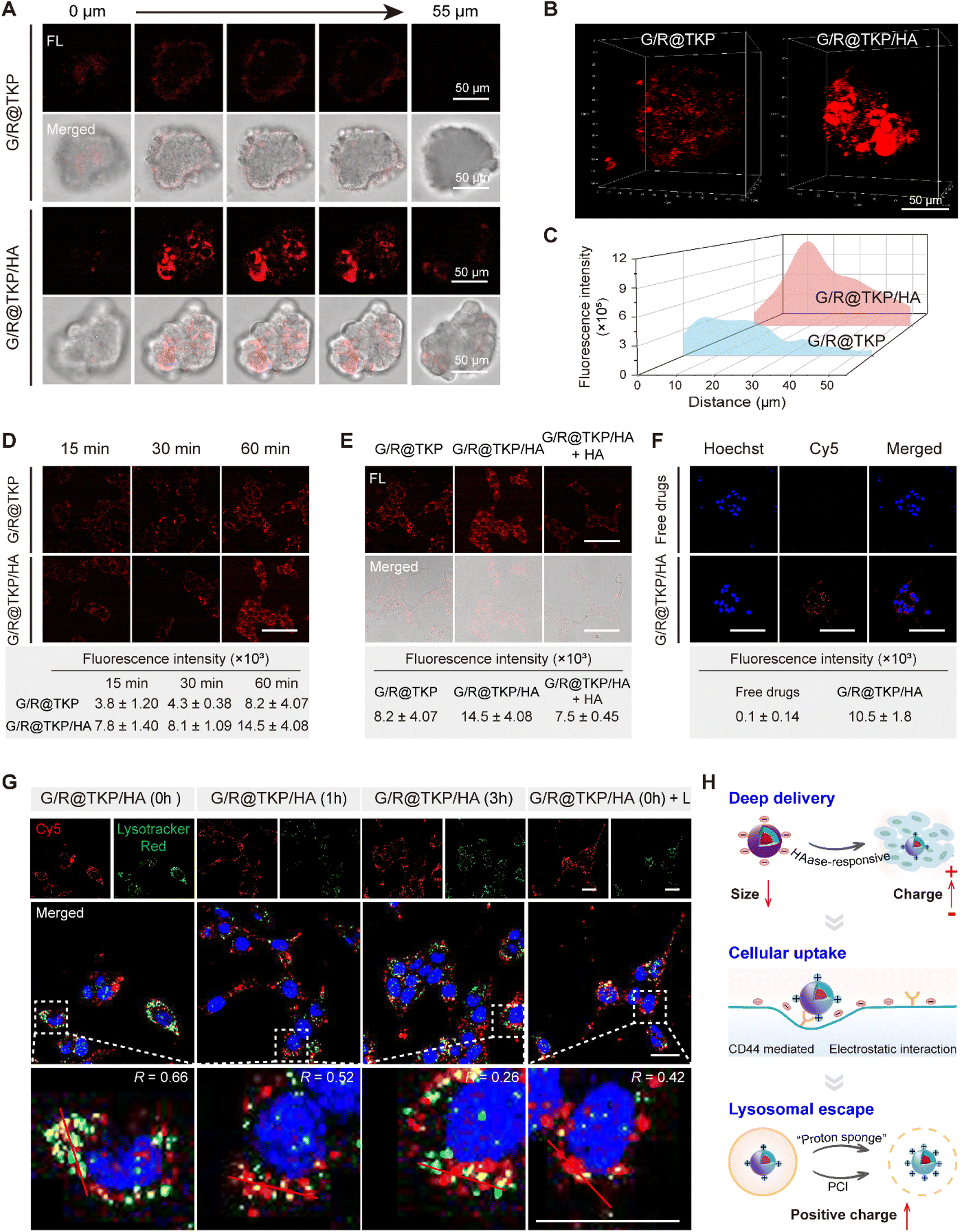 | ||
| Fig. 3 Tumor barriers penetration of the TME-cascade responsive btND with size/charge bidirectional-tunability. (A) Three-dimensional PANC1 MTS images obtained along the z-axis at different depths after treating with G/R@TKP or G/R@TKP/HA for 4 h. All images share the same scale bar of 50 μm. (B) Three-dimensional reconstruction of PANC1 MTSs in (A). (C) Quantitative analysis of fluorescence intensity in (A). (D) Confocal images of cells incubated with G/R@TKP or G/R@TKP/HA for different times. All images share the same scale bar of 75 μm. Quantitative analysis of TCM fluorescence intensity is shown in the table below. (E) Cellular uptake of G/R@TKP, G/R@TKP/HA and G/R@TKP/HA + HA by CLSM. G/R@TKP/HA + HA: cells pretreated with HA and then incubated with G/R@TKP/HA. All images share the same scale bar of 75 μm. Quantitative analysis of TCM fluorescence intensity is shown in the table below. (F) Cellular uptake of free drugs (Gem + Cy5siRNA) and G/R@TKP/HA in PANC1 cells by CLSM. Cell nuclei stained with Hoechst 33342 are shown in blue, and Cy5-labeled siRNA are shown in red. All images share the same scale bar of 100 μm. Quantitative analysis of Cy5 fluorescence intensity is shown in the table below. (G) Endo/lysosomal escape of G/R@TKP/HA by CLSM. After 1 h incubation with G/R@TKP/HA, the G/R@TKP/HA medium was replaced with a fresh medium and the cells were incubated for an additional 0 h, 1 h, and 3 h, respectively. For the light group, the cells were exposed to white light (40 mW cm−2, 8 min) after the replacement of the fresh medium. Cell nuclei stained with Hoechst 33342 are shown in blue, endo/lysosomes stained with LysoTracker Red are shown in green and Cy5-labeled siRNA are shown in red. Pearson's correlation coefficients of Cy5 and LysoTracker Red along the selected line across the cell (indicated by a red line in the zoomed-in image) are shown in the upper left corner of the zoomed-in image. All images share the same scale bar of 25 μm. (H) Schematic illustration of tumor stroma, cell membrane and lysosome barrier penetration of G/R@TKP/HA. | ||
Next, we studied the tumor cell membrane penetration ability of G/R@TKP/HA by tracking the fluorescence of the AIE photosensitizer TCM, and used G/R@TKP without HA coating as a control. As co-incubation time with PANC1 cells increased, intracellular fluorescence grew stronger, with G/R@TKP/HA showing a more robust signal than G/R@TKP, underscoring the enhanced cellular uptake due to the HA coating (Fig. 3D and S11†). To further explore this, we pre-incubated cells with free HA to block CD44 receptors on the cell membrane, assessing subsequent changes in cellular uptake. The data revealed a marked reduction in G/R@TKP/HA uptake post HA pretreatment, confirming G/R@TKP/HA's active uptake mediated by CD44 recognition of HA (Fig. 3E and S12†). Additionally, using Cy5-labeled siRNA to formulate G/R@TKP/HA, we co-incubated them with tumor cells to gauge the dual-responsive platform's gene drug delivery efficiency. The Cy5 fluorescence in pancreatic cancer cells treated with the G/R@TKP/HA group was significantly stronger than that in cells treated with free genes, highlighting the btND's capability to traverse the tumor cell barrier and amplify drug delivery efficacy (Fig. 3F and S13†).
Subsequently, we further examined the lysosomal escape capacity of G/R@TKP/HA. As shown in Fig. 3G and S14,† after a 1 hour co-incubation of cells with G/R@TKP/HA carrying Cy5-labeled siRNA without medium replacement, the Cy5 red fluorescence significantly overlapped with the LysoTracker green fluorescence, resulting in yellow dots with an overlap coefficient of 0.66. This suggests rapid G/R@TKP/HA uptake and lysosomal sequestration. With continued incubation in a fresh medium for 1 hour, the overlap coefficient decreased to 0.52 and further to 0.26 after 3 hours, indicating gradual G/R@TKP/HA escape from lysosomes. This escape is primarily driven by the protonation of PEI in the acidic lysosomal milieu, enhancing the positive charge density of G/R@TKP/HA and triggering the “proton sponge” effect, facilitating their release from lysosomes. Under an alternate condition, following a 1 hour co-incubation with G/R@TKP/HA, cells exposed to white light (40 mW cm−2) for 8 minutes showed a decrease in the overlap coefficient from 0.66 to 0.42. This reduction indicates PCI-induced lysosomal membrane disruption, promoting rapid escape of G/R@TKP/HA. The decrease in red fluorescence of acridine orange (AO)25 in endo/lysosomes indicates that the endo/lysosomes were damaged (Fig. S15†).
In summary, the TME-responsive size/charge bidirectional-tunable G/R@TKP/HA can effectively penetrate stroma barriers via HAase-responsive size reduction. Additionally, HAase-responsive charge reversal of G/R@TKP/HA promotes cell endocytosis through electrostatic interactions, while HA-mediated active targeting overcomes cell barriers. Moreover, G/R@TKP/HA can breach the endo/lysosome barrier utilizing the “proton sponge” effect and PCI-triggered lysosomal membrane disruption. This multifaceted approach enables deep tissue penetration and enhances therapeutic efficacy (Fig. 3H).
In vitro antitumor efficacy
According to our design, the TME-cascade responsive btND, G/R@TKP/HA, containing the AIE photosensitizer (TCM), Gem and KRAS siRNA, could achieve three-in-one synergistic treatment encompassing PDT, chemotherapy and siRNA therapy (Fig. 4A). First, the intracellular ROS production of TCM-TK-PEI/HA (TKP/HA) upon white light irradiation was confirmed using DCFH-DA as the indicator. As shown in Fig. 4B, in the TKP/HA + L group, non-fluorescent DCFH-DA was oxidized by ROS to produce fluorescent dichlorofluorescein (DCFH), indicating the effective intracellular ROS generation of TKP upon white light irradiation, which provides assurance for subsequent photodynamic therapy, TK bond cleavage, and PCI.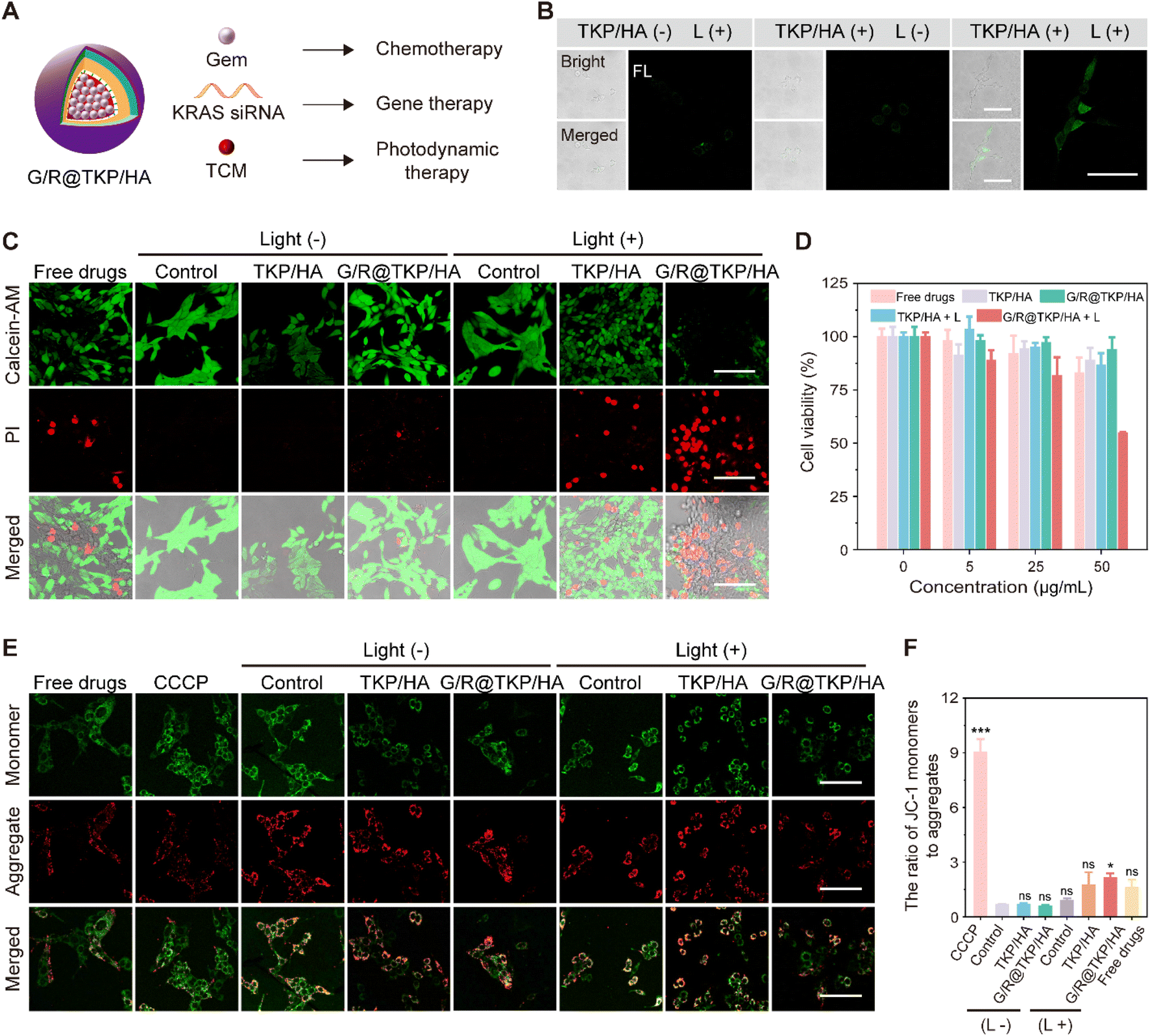 | ||
| Fig. 4 In vitro antitumor efficacy of the TME-cascade responsive btND with size/charge bidirectional-tunability. (A) Schematic illustration of a three-in-one synergistic therapy of G/R@TKP/HA for treating PANC1 cells. (B) Intracellular ROS generation of G/R@TKP/HA in PANC1 cells, detected by DCFH-DA. White light irradiation: 40 mW cm−2, 5 min. All images share the same scale bar of 75 μm. (C) Live/dead staining images of PANC1 cells following different treatments, assessed by using calcein-AM for live cells and PI for dead cells. White light irradiation: 40 mW cm−2, 15 min. All images share the same scale bar of 100 μm. (D) Cell viability of PANC1 cells following different treatments. White light irradiation: 40 mW cm−2, 15 min. (E) JC-1 assay of cell apoptosis in PANC1 cells following different treatments. The monomeric JC-1 form is shown in green for apoptotic cells, and the aggregated JC-1 form is shown in red for live cells. White light irradiation: 40 mW cm−2, 15 min. All images share the same scale bar of 100 μm. (F) The ratio of green/red fluorescence in (E) (n = 3). ns: P > 0.05, 0.01 < *P < 0.05, 0.001 < **P < 0.01, 0.0001 < ***P < 0.001, 0.00001 < ****P < 0.0001, calculated with the T-test. | ||
We further assessed the in vitro anti-tumor efficacy of different treatment groups, including Gem + KRAS siRNA (Free drugs), TKP/HA polymer material, G/R@TKP/HA, TKP/HA with light (TKP/HA + L), and G/R@TKP/HA with light (G/R@TKP/HA + L), using live/dead cell staining and MTT assay. We chose calcein-AM and propidium iodide (PI) to stain live (green fluorescence) and dead (red fluorescence) cells, respectively.27,28 As depicted in Fig. 4C, under non-light irradiation conditions, the blank control group and TKP/HA polymer material group showed strong green fluorescence and barely visible red fluorescence, indicating high cell viability and excellent biocompatibility of the delivery vector. In the Free drugs group, red fluorescence appeared, suggesting cell death. However, the red fluorescence signal in the G/R@TKP/HA group was significantly lower than that in the Free drugs group, indicating improved biocompatibility and reduced cell damage with drug encapsulation. When cells were exposed to light, the control group still exhibited obvious green fluorescence and negligible red fluorescence, indicating the minimal effect of white light irradiation on cell growth. However, in the TKP/HA + L group undergoing photodynamic therapy, red fluorescence emerged, indicating that ROS produced by TKP/HA under light irradiation killed cells. Particularly, the G/R@TKP/HA + L group, the three-in-one synergistic treatment group, exhibited the most significant red fluorescence and the weakest green fluorescence among all groups, demonstrating the most potent cytotoxic effect. MTT assay also confirmed that the combined treatment of G/R@TKP/HA + L showed the most effective treatment efficacy (Fig. 4D). It's worth noting that the negatively charged HA coating could shield the positive charge of PEI, thereby reducing its cytotoxicity (Fig. S16†). These results demonstrate the effective killing of PANC1 cells by the TME-cascade responsive size/charge bidirectional-tunable G/R@TKP/HA, indicating its potential for pancreatic cancer therapy.
Next, we investigated cell apoptosis using JC-1 as an indicator and carbonyl cyanide 3-chlorophenylhydrazone (CCCP) as the positive control.29,30 In normal cells, JC-1 exists in the aggregate form emitting red fluorescence at around 590 nm, while during cell apoptosis, it switches to the monomer form emitting green fluorescence at 529 nm. The ratio of green to red fluorescence can reflect cell apoptosis. As shown in Fig. 4E and F, in non-light groups, the green to red fluorescence ratio of the control and TKP/HA was ∼0.68 and ∼0.70, respectively, indicating the absence of cell apoptosis. The ratio of free drugs was ∼1.62, suggestive of cell apoptosis, while drug encapsulation can prevent cell damage (the ratio of G/R@TKP/HA: ∼0.59). When exposed to light, the control group showed a ratio of ∼0.89, confirming that white light irradiation did not induce cell apoptosis. Conversely, TKP/HA + L exhibited a ratio of ∼1.77, signifying that ROS produced by TKP/HA upon light irradiation could result in cell apoptosis. Remarkably, the G/R@TKP/HA + L group showed the most excellent cell apoptosis among all treatment groups (the ratio: ∼2.16), indicating that combining chemotherapy, gene therapy and PDT can effectively lead to cell death.
In vivo tumor targetability, penetrability and retention
Previous in vitro experiments have revealed the exceptional pancreatic tumor penetration capabilities of HAase/ROS-cascade responsive btND, G/R@TKP/HA, which can be attributed to its bidirectional tunability in size and charge during cascade stimulation (Fig. 5A). Building upon this observation, we conducted further assessments to evaluate the tumor targetability and penetrability of G/R@TKP or G/R@TKP/HA following intravenous (i.v.) injection into PANC1 tumor-bearing mice, with subsequent collection of in vivo fluorescence images at various time points using an IVIS spectrum imaging system. For mice injected with HA-uncoated G/R@TKP, fluorescence signals at the tumor site were scarcely detected between 0 to 5 hours, with only faint fluorescence observed at the 8-hour mark. Conversely, mice injected with HA-coated G/R@TKP/HA exhibited initially weak fluorescence at the tumor site, which gradually intensified from 0 to 5 hours before diminishing by the 8-hour time point (Fig. 5B, C and S17†). Tumor tissue sections were further analyzed to investigate the stroma penetrability of G/R@TKP and G/R@TKP/HA groups. The fluorescence signals in the G/R@TKP group were primarily concentrated in the superficial layers of the tumor tissue, with weaker signals observed beyond 30 μm depth, while the G/R@TKP/HA group exhibited strong fluorescence in both superficial and deeper regions, extending up to 200 μm depth, highlighting its superior tumor targetability and penetrability (Fig. 5D and E). This enhanced performance can be attributed to the negatively charged HA coating, which prolongs blood circulation, thereby enhancing tumor accumulation. Moreover, degradation of the HAase-responsive HA coating triggers size reduction of G/R@TKP/HA, facilitating deep tumor penetration, followed by a cascade-ROS response leading to size tuning from small to large, further enhancing tumor retention. Conversely, positively charged G/R@TKP may face clearance by the reticuloendothelial system, with its constant particle size limiting tumor stroma penetration, resulting in weak in vivo imaging fluorescence.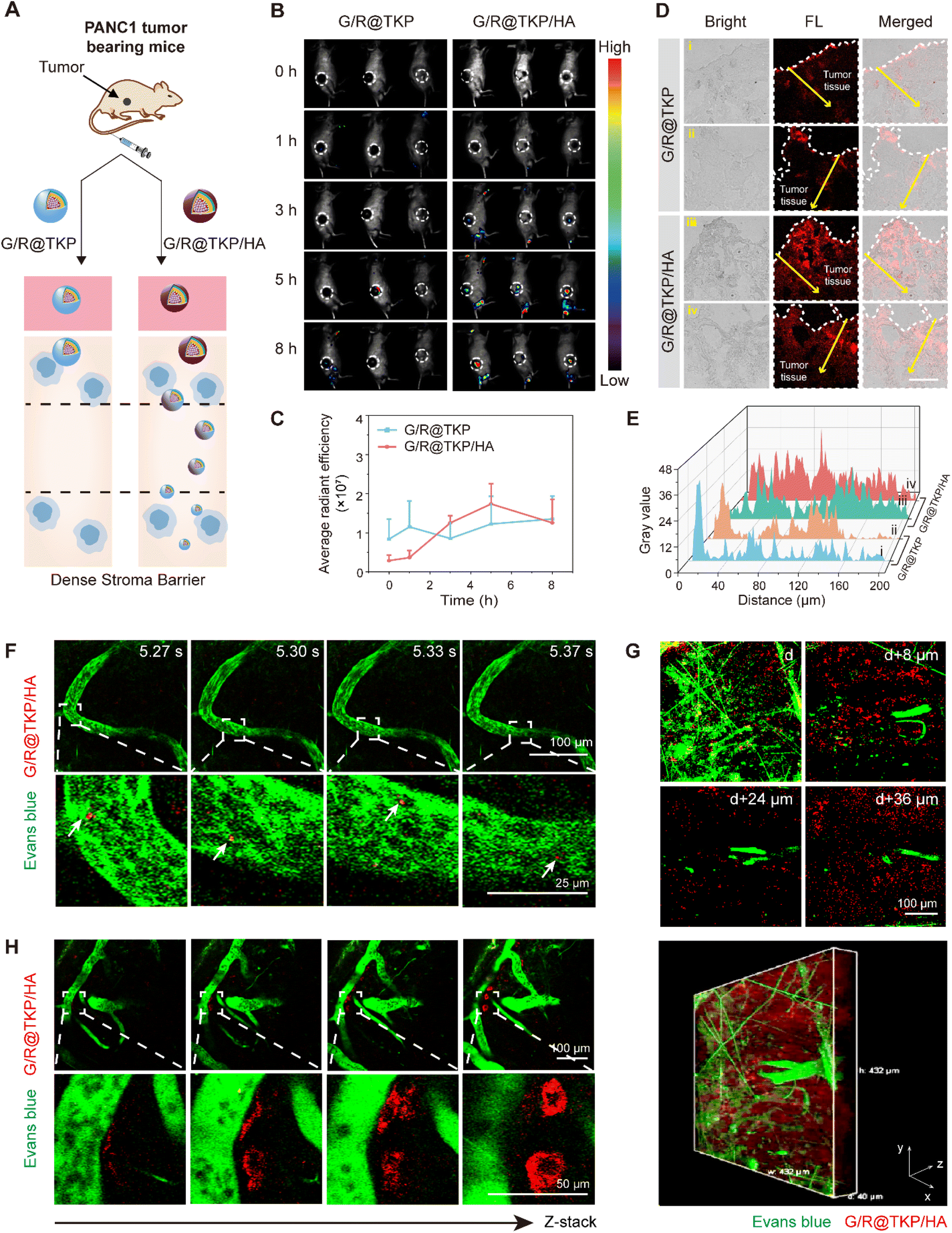 | ||
| Fig. 5 In vivo tumor targetability, penetrability and retention of the TME-cascade responsive btND with size/charge bidirectional-tunability. (A) Schematic illustration of the tumor stroma penetrability comparison between T@G/si and G/R@TKP/HA. (B) In vivo fluorescence images of PANC1 tumor bearing mice injected intravenously with G/R@TKP and G/R@TKP/HA at different time points (n = 3). (C) Quantitative analysis of fluorescence intensity in (B). (D) Tumor tissue penetration of G/R@TKP and G/R@TKP/HA. All images share the same scale bar of 100 μm. (E) Quantitative analysis of the fluorescence gray value in (D). (F) Confocal imaging of the time-resolved tumor region of PANC1 tumor bearing mice injected intravenously with G/R@TKP/HA. Vessels stained with Evans blue are shown in green (n = 1). (G) Confocal imaging of the tumor region along the z-axis at different depths of PANC1 tumor bearing mice injected intravenously with G/R@TKP/HA and its three-dimensional reconstruction. Vessels stained with Evans blue are shown in green (n = 1). (H) Confocal imaging of the tumor region along the z-axis at different depths of PANC1 tumor bearing mice injected intravenously with G/R@TKP/HA. Vessels stained with Evans blue are shown in green (n = 1). | ||
Following the initial assessment, we proceeded to conduct a comprehensive evaluation of the real-time in vivo tumor penetrability and retention of G/R@TKP/HA.31 This was achieved by tracking the AIE fluorescence of TCM (red fluorescence), while Evans blue was employed to stain blood vessels, providing visualization through green fluorescence signals. Real-time tracking enabled the observation of the flow of red fluorescent G/R@TKP/HA through vessels, offering clear visualization of its traversal (Fig. 5F and S18 Movie S3,†). Furthermore, to obtain a detailed understanding of G/R@TKP/HA distribution within the tumor microenvironment, fluorescence images of the tumor region were meticulously recorded by scanning along the z-axis. As depicted in Fig. 5G, G/R@TKP/HA demonstrated robust red fluorescence signals at the tumor site, which were discernible even in deeper layers (Fig. S19 and Movie S4†). This observation suggests the excellent tumor permeability of G/R@TKP/HA, which was facilitated by its size tuning in response to HAase, as demonstrated in previous experiments. Importantly, these findings were consistent with the results obtained from the aforementioned multi-section tumor tissue fluorescence analysis. Moreover, an intriguing observation was made regarding the accumulation pattern of G/R@TKP/HA relative to the distance from the vessel. It was noted that the farther G/R@TKP/HA was from the vessel, the greater its accumulation and the stronger the red AIE fluorescence signal. This phenomenon can be attributed to the size tuning mechanism triggered by the cascade ROS response at the tumor site, which enhances tumor retention (Fig. 4H and Movie S5†). Taken together, these results provide compelling evidence of the simultaneous penetration through dense pancreatic tumor stroma and abundant retention at tumor sites by G/R@TKP/HA. This comprehensive assessment underscores the promising potential of G/R@TKP/HA as an effective therapeutic agent for pancreatic cancer treatment.
In vivo antitumor efficacy and biosafety
The previous experiments unequivocally demonstrate the remarkable pancreatic tumor targetability, penetrability, and retention of the TME-cascade responsive G/R@TKP/HA in vivo, owing to its size and charge bidirectional-tunability. Additionally, this btND effectively executes three-in-one synergistic therapy, leading to the efficient eradication of PANC1 cells in vitro (Fig. 6A). Encouraged by these findings, we proceeded to evaluate the in vivo antitumor activity of G/R@TKP/HA using PANC1 tumor-bearing mice. The mice were randomly assigned to four groups: PBS, Free Gem and KRAS siRNAs (Free drugs), G/R@TKP/HA, and G/R@TKP/HA with white light (G/R@TKP/HA + L), as illustrated in Fig. 6B.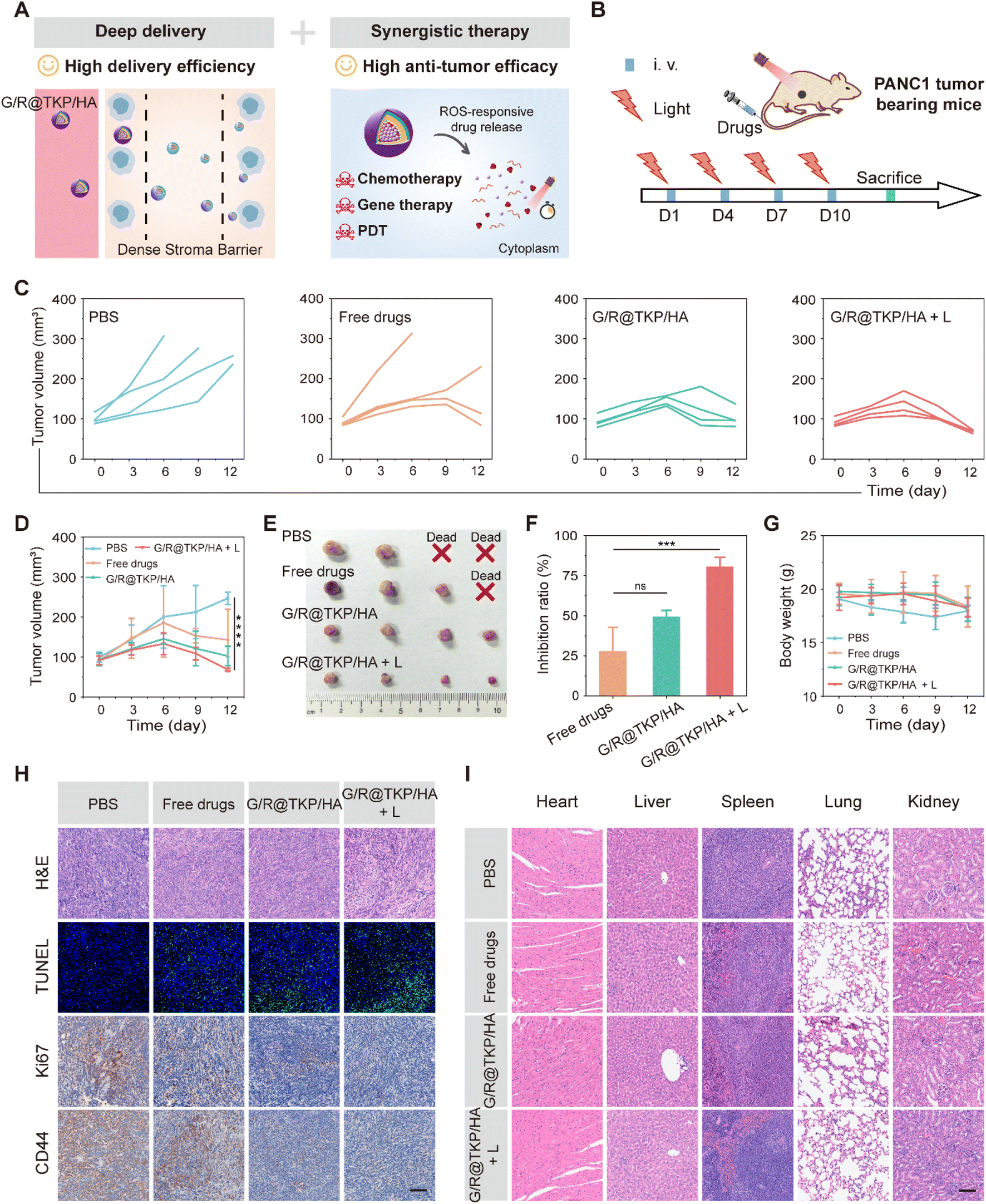 | ||
| Fig. 6 Excellent in vivo antitumor efficacy of the TME-cascade responsive btND with size/charge bidirectional-tunability. (A) Schematic illustration of two remarkable advantages of G/R@TKP/HA: high delivery efficiency and high antitumor efficacy. (B) Schematic illustration of the treatment procedure. (C and D) Tumor volume changes in PANC1 tumor-bearing mice treated with PBS, Free Gem and KRAS siRNAs (Free drugs), G/R@TKP/HA and G/R@TKP/HA with white light (G/R@TKP/HA + L), respectively by i.v. injection (n = 4). (E) Tumor images of different groups at the end of treatment (n = 4). (F) Tumor growth inhibition ratio of different treatment groups (n = 4). (G) Body weight changes of PANC1 tumor-bearing mice administered with different formulations during the treatment period (n = 4). (H) H&E, TUNEL, Ki67 and CD44 immunohistochemistry staining of harvested tumors after treatment. All images share the same scale bar of 100 μm. (I) H&E staining of major organs (heart, liver, spleen, lung, and kidney) after treatment. All images share the same scale bar of 100 μm. ns: P > 0.05, 0.01 < *P < 0.05, 0.001 < **P < 0.01, 0.0001 < ***P < 0.001, 0.00001 < ****P < 0.0001, calculated with the T-test. | ||
As depicted in Fig. 6C and D, PBS treated tumors grew rapidly and showed large individual differences, with two mice succumbing on the 6th and 9th day of treatment, respectively. In Free drugs treated tumors, tumor volume gradually increased over time, and the treatment had no therapeutic effect for some mice, leading to the death of one mouse on the 6th day of treatment and a significant increase in tumor volume for another mouse on the 9th day of treatment. Tumors treated with G/R@TKP/HA exhibited less individual variation, with controlled tumor volume growth. After 6 days of treatment, the overall trend of tumor volume showed a decrease, indicating that the HAase/ROS-responsive size/charge bidirectional-tunable btND facilitates drug delivery to deeper tumor sites, thereby enhancing therapeutic efficacy. Furthermore, ROS produced by G/R@TKP/HA upon white light irradiation can not only enhance the tumor retention but also enable PDT. Consequently, G/R@TKP/HA + L exhibited considerably better treatment outcomes compared to G/R@TKP/HA alone. At the end of treatment, each group of mice was sacrificed and tumors were collected. G/R@TKP/HA + L showed the smallest tumor size and the best tumor growth inhibition (∼80.57%) among the four groups (Free drugs: ∼27.81%, G/R@TKP/HA: ∼49.22%) (Fig. 6E, F and S20†). Additionally, unlike PBS and Free drugs treated groups, none of the mice in G/R@TKP/HA and G/R@TKP/HA + L died during the treatment, and the body weight of those in G/R@TKP/HA and G/R@TKP/HA + L remained stable, signifying the low toxic side effects of nanodelivery to tumor bearing mice (Fig. 6G).
After completing the treatment regimen, we conducted organ and tumor extraction from the mice for comprehensive analysis. The outcomes are delineated in Fig. 6H, wherein the H&E staining of tumor tissues exhibited a notable augmentation in the necrotic area of tumor cells across all groups, namely the Free drugs, G/R@TKP/HA, and G/R@TKP/HA + L groups, in comparison to the PBS group. Remarkably, G/R@TKP/HA + L demonstrated the most extensive necrotic area, indicative of its potent efficacy. In line with these observations, the results from terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays suggested that G/R@TKP/HA + L exerted the most robust effect in promoting tumor cell apoptosis. Furthermore, Ki67 immunohistochemistry revealed a substantial reduction in tumor cell proliferation within the G/R@TKP/HA + L group, thereby impeding tumor growth. Moreover, CD44 immunohistochemical staining exhibited a pronounced decrease in CD44 positivity in pancreatic cancer tissues treated with G/R@TKP/HA and G/R@TKP/HA + L, indicating the effective targeting of CD44 on the surface of pancreatic cancer cells by HA-modified G/R@TKP/HA, thereby facilitating their accumulation in tumor tissues. Notably, H&E staining of major organs demonstrated no significant differences among the G/R@TKP/HA, G/R@TKP/HA + L, and PBS groups, thus underscoring the superior biosafety profile of our btND (Fig. 6I). In summary, the TME-cascade responsive btND, G/R@TKP/HA, has shown remarkable tumor targeting, penetrability, and retention in vivo. Its significance lies in its ability to induce tumor cell apoptosis, suppress tumor cell proliferation, and enhance tumor targeting. Moreover, our btND has demonstrated excellent biosafety, bolstering its potential for clinical applications.
Conclusions
In summary, we proposed a HAase/ROS cascade-responsive nanodelivery (btND, G/R@TKP/HA) for excellent tumor barrier penetration via size and charge dual-bidirectional tunable transitions to augment treatment efficacy. Remarkably, the particle size of the btND exhibits a tuning process from large to small and then reverts to large upon TME-cascaded stimulation, accompanied by the surface charge reversal from negative to positive, followed by a decrease in positive charge. The smart bidirectional tunability of size and charge could break through almost all pancreatic tumor delivery obstacles simultaneously, encompassing blood circulation, tumor targetability, stroma penetration, tumor retention, cellular internalization, and endo/lysosomal escape, finally leading to controlled drug release within tumor cells. Furthermore, this btND containing chemotherapy, siRNA therapy and PDT could greatly suppress the growth of tumors and significantly extend the survival rate of pancreatic cancer mouse models. The design strategy of the size/charge dual-bidirectional tunable btND has the potential to engineer therapeutic delivery systems for malignant tumors in complex microenvironments.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Y. S. and J. L. contributed equally to this work.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA0910000), NSFC Excellent Young Scientist (EYS) Scheme (22222803), NSFC Science Center Program (21788102, 22404056, 22408105), Fellowship of China National Postdoctoral Program for Innovative Talents (BX20240114). All of the animal experimental procedures were performed according to the protocols approved by the Animal Care and Use Committee of the Shanghai Cancer Institute.Notes and references
- L. M. Liu, P. G. Kshirsagar, S. K. Gautam, M. Gulati, E. I. Wafa, J. C. Christiansen, B. M. White, S. K. Mallapragada, M. J. Wannemuehler, S. Kumar, J. C. Solheim, S. K. Batra, A. K. Salem, B. Narasimhan and M. Jain, Theranostics, 2022, 12, 1030–1060 CrossRef CAS PubMed.
- A. Forterre, H. Komuro, S. Aminova and M. Harada, Cancers, 2020, 12, 1852 CrossRef CAS PubMed.
- A. N. Hosein, R. A. Brekken and A. Maitra, Nat. Rev. Gastroenterol. Hepatol., 2020, 17, 487–505 CrossRef PubMed.
- A. Tiwari, R. Trivedi and S. Y. Lin, J. Biomed. Sci., 2022, 29, 83 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef CAS PubMed.
- M. J. Ernsting, M. Murakami, A. Roy and S. D. Li, J. Controlled Release, 2013, 172, 782–794 CrossRef CAS PubMed.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195–7206 CrossRef CAS PubMed.
- W. Q. Yu, R. Liu, Y. Zhou and H. L. Gao, ACS Cent. Sci., 2020, 6, 100–116 CrossRef CAS PubMed.
- S. Salatin, S. M. Dizaj and A. Y. Khosroushahi, Cell Biol. Int., 2015, 39, 881–890 CrossRef CAS PubMed.
- A. Kumari and S. K. Yadav, Expert Opin. Drug Delivery, 2011, 8, 141–151 CrossRef CAS PubMed.
- P. Zhang, D. Y. Chen, L. Li and K. X. Sun, J. Nanobiotechnol., 2022, 20, 31 CrossRef CAS PubMed.
- F. Veider, E. S. Armengol and A. Bernkop-Schnürch, Small, 2024, 20, 2304713 CrossRef CAS PubMed.
- L. D. Cavalcante and G. Monteiro, Eur. J. Pharmacol., 2014, 741, 8–16 CrossRef PubMed.
- S. Kamerkar, V. S. LeBleu, H. Sugimoto, S. J. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498–503 CrossRef CAS PubMed.
- Y. Takakura and Y. Takahashi, J. Controlled Release, 2022, 350, 486–493 CrossRef CAS PubMed.
- X. X. Kang, F. Q. Bu, W. L. Feng, F. Liu, X. K. Yang, H. F. Li, Y. J. Yu, G. F. Li, H. H. Xiao and X. Wang, Adv. Mater., 2022, 34, 2206765 CrossRef CAS PubMed.
- H. He, Y. Chen, Y. Li, Z. Song, Y. Zhong, R. Zhu, J. Cheng and L. Yin, Adv. Funct. Mater., 2018, 28, 1706710 CrossRef.
- A. Rinaldi, R. Caraffi, M. V. Grazioli, N. Oddone, L. Giardino, G. Tosi, M. A. Vandelli, L. Calzà, B. Ruozi and J. T. Duskey, Polymers, 2022, 14, 687 CrossRef CAS PubMed.
- G.-E. S. Chaudhry, A. Akim, M. Naveed Zafar, N. Safdar, Y. Y. Sung and T. S. T. Muhammad, Adv. Pharm. Bull., 2021, 11, 426–438 CrossRef CAS PubMed.
- E. Chiesa, A. Greco, F. Riva, R. Dorati, B. Conti, T. Modena and I. Genta, Pharmaceuticals, 2022, 15, 103 CrossRef CAS PubMed.
- G. David, L. Clima, M. Calin, C. A. Constantinescu, M. Balan-Porcarasu, C. M. Uritu and B. C. Simionescu, Polym. Chem., 2018, 9, 1072–1081 RSC.
- J. H. Wang, H. He, X. Xu, X. Wang, Y. B. Chen and L. C. Yin, Biomaterials, 2018, 171, 72–82 CrossRef CAS PubMed.
- J. X. Wang, X. Y. He, S. Shen, Z. Y. Cao and X. Z. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 1855–1863 CrossRef CAS PubMed.
- Y. Y. Yuan, C. J. Zhang and B. Liu, Angew. Chem., Int. Ed., 2015, 54, 11419–11423 CrossRef CAS PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed.
- Y. Wang, J. Liao, Y. Lyu, Q. Guo, Z. Zhu, X. Wu, J. Yu, Q. Wang and W.-H. Zhu, Adv. Funct. Mater., 2023, 33, 2301692 CrossRef CAS.
- Z. Liu, Q. Wang, W. Qiu, Y. Lyu, Z. Zhu, X. Zhao and W.-H. Zhu, Chem. Sci., 2022, 13, 3599–3608 RSC.
- F. Sivandzade, A. Bhalerao and L. Cucullo, Bio-Protoc., 2019, 9, e3128 Search PubMed.
- J. Xiang, Y. Shen, Y. Zhang, X. Liu, Q. Zhou, Z. Zhou, J. Tang, S. Shao and Y. Shen, Adv. Sci., 2022, 9, 2200173 CrossRef CAS PubMed.
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Nat. Nanotechnol., 2019, 14, 799–809 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04782f |
| This journal is © The Royal Society of Chemistry 2024 |