DOI:
10.1039/D4SC04843A
(Edge Article)
Chem. Sci., 2024,
15, 18067-18075
A homoleptic AgIII complex stabilized by succinimidate ligands†
Received
21st July 2024
, Accepted 16th September 2024
First published on 30th September 2024
Abstract
Herein, the first example of a homoleptic AgIII complex stabilized by a monodentate N-donor ligand is presented. Na2[S2O8] oxidizes the linear AgI complex, Na[Ag(succ)2] (1Na), to form a square planar argentate(III) ion, [Ag(succ)4]−, which crystallizes with a polymeric chain-structure, M[Ag(succ)4] (2M), when treated with alkali metal sulfate M2SO4 (M = K, Rb, Cs). A mixed-valent Robin-Day class I system, [(H2O)Ag][Ag(succ)4] (2Ag), forms in the absence of K+/Rb+/Cs+ ion. Diamagnetic 2Cs displays a succinimide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency at higher energy than does the isoelectronic PdII complex, [(H2O)Na]2[Pd(succ)4] (3Na). Moreover, 2Cs displays UV-vis absorptions that are more intense and occur at lower energy than those in 3Na. The electron-deficient nature of 2Cs is further evident from its ability to oxidize water to O2. From 109Ag magic-angle spinning NMR studies, a highly deshielded AgIII environment in 2Cs (2080 ppm) relative to the AgI starting material 1Na (492 ppm) is observed.
O stretching frequency at higher energy than does the isoelectronic PdII complex, [(H2O)Na]2[Pd(succ)4] (3Na). Moreover, 2Cs displays UV-vis absorptions that are more intense and occur at lower energy than those in 3Na. The electron-deficient nature of 2Cs is further evident from its ability to oxidize water to O2. From 109Ag magic-angle spinning NMR studies, a highly deshielded AgIII environment in 2Cs (2080 ppm) relative to the AgI starting material 1Na (492 ppm) is observed.
Introduction
AgIII complexes remain elusive due to the limited selection of ligands that can coordinate to this high-valent metal center while resisting oxidation.1 Accordingly, AgIII complexes show utility as oxidizing agents,2 and their electronic structure opens to applications in both superconducting3 and semiconducting4 materials. More recently, reductive elimination from the strongly oxidizing AgIII ion has gained traction in C(sp3)–C(sp3) coupling reactions5 as well as cross-coupling of arenes, leading to heteroatom functionalization,6 trifluoromethylation,7 and alkylation.8 The earliest examples of AgIII complexes were bis(orthoperiodate)9 and bis(orthotellurate)10 complexes reported by Malaprade9d and by Malatesta10, as well as an ethylenebis(biguanide) complex reported by Rây in 1943 (Chart 1A).11 The stability of these classic AgIII chelate complexes reflects both the ability of the ligands to resist oxidation as well as their ability to enforce square-planar coordination around the d8 AgIII ion. Along these lines, considerable development of AgIII chemistry has revolved around chelating12 and macrocyclic13 ligands, in particular porphyrinoid systems14 (Chart 1B). By contrast, homoleptic AgIII systems with monodentate ligands, are confined to an exceedingly narrow range of oxidation-resistant donors: in 1957, Hoppe isolated the square planar fluoro complexes, K[AgF4] and Cs[AgF4],15 (Chart 1C) and subsequently octahedral Cs2K[AgF6] in 1966.16 In 1968, Cohen and Atkinson generated [Ag(OH)4]− in solution by anodic oxidation of silver;17 this hydroxo complex could be trapped using periodate or tellurate as chelates (Chart 1A).18 The reducing nature of nitrogen ligands has, heretofore, precluded unequivocal identification of any homoleptic AgIII complex.19 On the other hand, in 1986, Dukat and Naumann demonstrated how the [Ag(CF3)2]− ion disproportionates to metallic silver and the mixed-valent salt, [AgI][AgIII(CF3)4].20 The remarkable stability of trifluoromethyl silver complexes21 has since opened to isolation of numerous heteroleptic [Ag(CF3)4−n(X)n]− argentate complexes (n = 1–3, X = halide, pseudohalide, hydrocarbyl)7b,21a,22 as well as neutral [Ag(CF3)3(L)] complexes (L = CH3CN, PPh3, AsPh3, pyridine, s-2,4,6-trimethyltriazine, N-heterocyclic carbene).23 More recently, Menjón and co-workers isolated a unique D3h-symmetric (Ph4P)2[Ag(CF3)2Br3] complex,24 ushering in five-coordinate geometries in AgIII chemistry.7,23b
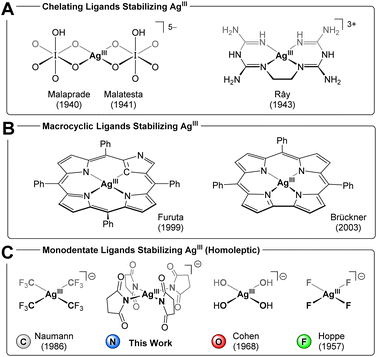 |
| | Chart 1 AgIII complexes isolated using (A) chelating ligands, (B) macrocyclic ligands, (C) monodentate ligands. | |
Akin to AgIII, a square planar (Ph4P)[Cu(CF3)4] motif exists for the CuIII ion.25 Despite being d8 CuIII by customary electron counting rules,26 this complex may be physically better described as a d10 CuI system bearing partially oxidized ligands, i.e. having an inverted ligand field (coined by Hoffmann).27 This proposition was originally promulgated in a theoretical study by Snyder28 and recently affirmed experimentally in X-ray spectroscopic studies by Lancaster29 and high-resolution crystallographic studies by Overgaard.30 Snyder extended the inverted ligand field description to square planar silver,31 and against this background, it is interesting to consider electronic structure studies of AgIII complexes. Grochala and Hoffmann studied binary and ternary silver fluorides by X-ray photoelectron spectroscopy and calculated density of states; for K[AgF4], the Ag 4d states contribute more to the ligand band than do the F 2p states.32 In a similar fashion, Ghosh established how variously substituted [Ag(corrole)] complexes fall into both redox innocent, [Ag[III](corrole[3−])], and redox non-innocent, [Ag[II](corrole[˙2−])], regimes.14k,m More recently, Menjón identified inverted ligand fields in a range of AgIII complexes, including [Ag(CF3)4]−, through experimental and computational studies.22b,24 From an electronic structure standpoint, the d8 AgIIIversus d10 AgI dichotomy represents two idealized points, [Ag[+III](CF3[−I])4]− and [Ag[+I](CF3[−½])4]−, along a continuum of covalent interactions describing the Ag–CF3 bond. The true electronic structure falls somewhere along this continuum, but, as the degree of covalency increases for higher oxidation states, the notion of electrons as belonging exclusively to any one metal- or ligand fragment inevitably breaks down. In a recent analysis of bonding in organometallic systems, Deeth pointed out that the oxidation state in [Cu(CF3)4]− reflects which bonding model is employed. While MO theory suggests an inverted LF with d10 CuI, the aiLFT subspace modeling implemented in the ORCA program package suggests a normal LF with a square planar d8 CuIII, leading to the salient conclusion that these are very covalent systems no matter the modeling approach.33 Inspired by the elusive nature and enticing electronic structure of AgIII complexes, we sought strongly donating ligands to stabilize the high oxidation state. Considering Collins's seminal work on high-valent amide complexes (including CuIII),34 coupled with the ability of halosuccinimides to act as synthons for oxidized species such as Cl+ and Br+, we inquired whether the succinimide anion (succ−) could stabilize a homoleptic AgIII complex. Herein, we present the synthesis of a square-planar argentate(III) ion, [Ag(succ)4]−, isolated as polymeric Cs+, Rb+, and K+ salts. We also present the isolation of a mixed-valent [(H2O)AgI][AgIII(succ)4] salt. We probe the electronic structure of the highly oxidized AgIII complex alongside the isoelectronic PdII analog, [Pd(succ)4]2−, combining vibrational, UV-visible, and solid-state 109Ag NMR spectral data.
Results and discussion
Synthesis of a succinimide-stabilized AgIII complex (2Cs)
Succinimide buffers have emerged as non-toxic alternatives to cyanide baths for electrodeposition of silver,35 and accordingly, AgI/succ− systems have been subject to a wide range of electrochemical studies. In one study, Lessard observed that [M][Ag(succ)2] complexes (M+ = Ag+, Et4N+) undergo an oxidation process in acetonitrile, whose limiting current increases when excess succinimide anion is present.36 In a subsequent study, the oxidation product was proposed to be succinimidyl radical, succ˙, based on the isolation of free succinimide formed upon H-atom abstraction from the solvent.37 In a separate study, Masaki reported that silver anodes develop a crystalline covering in aqueous succinimide buffers, assigned to a rare AgII species, such as [Ag(succ)2].38 Given that Na2[S2O8] oxidizes AgNO3 in pyridine to afford the orange and paramagnetic AgII complex, [Ag(pyridine)4][S2O8],39 we conducted a chemical oxidation of AgNO3 in aqueous succinimide buffer, using Na2[S2O8]. Heating the solution converted the in situ generated AgI complex, Na[Ag(succ)2] (1Na), to a new yellow-colored species, which could be isolated in good yield as large, bright yellow crystals by addition of Cs2SO4 (54%) or Rb2SO4 (45%), and low yield (11%) from solutions containing only K(succ) (Scheme 1). The new species could also be prepared by electrocrystallization. In a two-chamber system with a silver cathode in aqueous AgNO3 and a platinum anode in a succinimide buffer containing Cs2SO4 and AgNO3, the new species deposits on the anode when the potential difference exceeds 0.80 V. The surface area of the anode limits the yield from electrocrystallization (∼1 mg cm−2), and thus, chemical oxidation is most suitable for generating larger quantities of the new species.
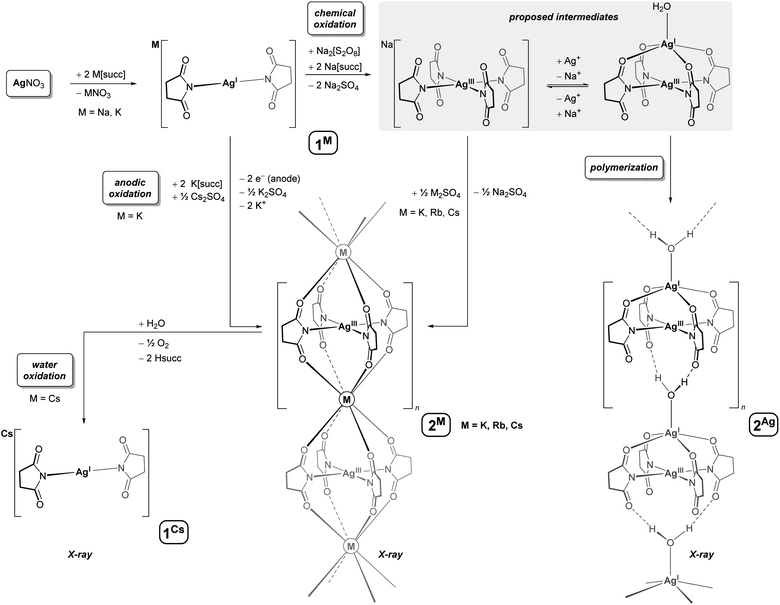 |
| | Scheme 1 Chemical or anodic oxidation converts [Ag(succ)2]− to a square-planar argentate(III) ion, [Ag(succ)4]−; complexation with Cs+, Rb+, K+, or Ag+ yields polymeric salts, 2Cs, 2Rb, 2K, and 2Ag. Complex 2Cs oxidizes water to form 1Cs and O2. | |
The color of the new species isolated in our studies (yellow) differs strikingly from Masaki's putative [Ag(succ)2] complex (white), which called for further characterization. Interestingly, 1H and 13C NMR spectra of solutions (D2O) of the new species showed sharp singlets in the normal chemical shift range for succinimide complexes, and solid-state DC SQUID magnetometry studies (χM = −1.2 × 10−4 cm3 mol−1, 70–300 K) affirmed a diamagnetic system, which would be inconsistent with a magnetically uncoupled AgII complex. Electrospray ionization mass spectrometry (ESI-MS) in negative mode revealed a molecular ion with m/z corresponding to [Ag(succ)4]−, as well as heavier aggregates, {Cs[Ag(succ)4]2}− and {Cs2[Ag(succ)4]3}−. In line with the other characterization data, X-ray crystallographic studies revealed the new species to be a salt of composition Cs[Ag(succ)4]·4H2O (2Cs, spacegroup I4/m, Fig. 1A). The silver ion resides at the intersection between a mirror-plane and a fourfold axis, with the nitrogen-bound succinimide ligands oriented perpendicular to the square planar silver ion. The carbonyl oxygens from two [Ag(succ)4]− ions coordinate to the cesium ion, generating an elongated square prismatic motif (O⋯O edges: 2 × 3.275(3), 1 × 4.206(2) Å), while four molecules of water engage in hydrogen bonding with the same carbonyl oxygens, resulting in a polymeric chain-structure (Fig. 1B). Bond distances in the isostructural rubidium derivative, Rb[Ag(succ)4]·4H2O (2Rb), reflect the smaller ionic radius of Rb+versus Cs+ (O⋯O edges: 2 × 3.253(2), 1 × 3.967(2) Å), whereas metrics within the [Ag(succ)4]− ion are essentially unperturbed. A similar situation is observed for the potassium derivative, K[Ag(succ)4]·4H2O (2K), given the even smaller ionic radius for K+ (O⋯O edges: 2 × 3.242(2), 1 × 3.880(2) Å). The Ag–N bonds in 2Cs (2.009(3) Å) are short compared to the range known for [AgI(succ)2]− complexes (2.067(5)–2.088(2) Å),40 and in fact, the Ag–N bond distance in 2Cs falls among the shortest 0.5% Ag–N bond distances in the Cambridge Structural Database, in line with the presence of a contracted AgIII ion in 2Cs.
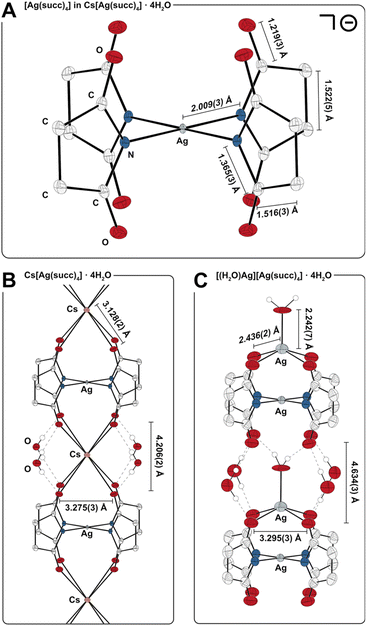 |
| | Fig. 1 X-ray crystallographic studies (H atoms omitted, except on H2O). (A) Geometry of the [Ag(succ)4]− ion in 2Cs (50% probability, 122(2) K). (B) Bonding interactions between CO groups, Cs+, and H2O in the polymeric chain-structure of 2Cs. (C) Bonding interactions between CO groups, Ag+, and H2O in the polymeric structure of 2Ag (30% probability, 293(2) K); orientational disorder of the [H2O–Ag]+ unit is omitted. | |
DFT computational assignment of the silver oxidation state in 2Rb
The assignment of a high formal oxidation state of +III for silver bound to mono-anionic succinimidate ligands appears reasonable based on the structural data. However, for high-valent, late transition metals, the question of ligand non-innocence leading to internal redox and concomitant inverted ligand fields has received increasing focus as described above.22b,24,27–33 In order to gauge the level of covalency and the appropriateness of the AgIII formulation, we performed a single point DFT calculation employing the B3LYP functional and the experimental geometry for 2Rb with two Rb+ cations terminating the silver complex and yielding a mono cationic fragment for the computation. As the DFT is known to be biased towards covalent description of the M–L bonding,41 the results are expected to represent an upper estimate on the covalency. The system is very covalent as indicated by a low Mulliken charge on the Ag (+0.22) and only a fractional hole (0.77 e) in the silver d-shell. The σ-interaction between Ag and the succinimidate ligands has an anti-bonding part, which in the normal LF ordering should be a pure, unoccupied 4dx2−y2(Ag) orbital. The KS-LUMO is indeed the one with most 4dx2−y2(Ag) character among all of the orbitals, but the Ag-dx2−y2 contribution totals only 35%. A plot of the LUMO is given in Fig. S35† illustrating the delocalization throughout the complex. The bonding counterpart has only ca. 12% of Ag-dx2−y2 contribution (Fig. S35,† bottom), which aligns with a non-inverted LF, but the remaining ca. 45% of Ag-dx2−y2 is shared between many, mainly bonding, KS-orbitals. In summary, the DFT data suggest that the complex is borderline between having a normal and an inverted ligand field. Considering the covalency bias of the DFT calculations on transition metal complexes, we employ the classical LF picture with an empty 4dx2−y2(Ag) orbital for the remaining discussions. Similar modeling of the AgI salt of [Ag(succ)4]− (vide infra) yielded a clear distinction between the two silver centers with the AgIII having the lower Mulliken charge (+0.11 versus +0.49 for AgI), but with Mayer total valencies of 2.52 versus 0.98 for the high and low valent Ag-centers.
Isolation of a mixed-valent AgI–AgIII complex (2Ag)
Complex 2Cs was reproducibly isolated in 50–55% yield when AgNO3 was used as the limiting reagent, and modification of the synthetic procedure did not increase the yield. When left to crystallize, yellow reaction mixtures containing [Ag(succ)4]− and Cs2SO4 produce very little crystalline material over an initial period of two days, whereas the solutions typically decolorize and deposit large amounts of yellow crystals of 2Cs after three days. Addition of a solution of NaCl to the isolated mother liquor resulted in precipitation of AgCl within seconds, demonstrating the presence of an unreacted AgI species, after crystallization of 2Cs is complete. In view of these observations, we further examined reaction mixtures containing the [Ag(succ)4]− ion. By heating a concentrated solution containing Na(succ), AgNO3, and Na2[S2O8] in a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, we isolated dark yellow crystals within few minutes. The resulting tetragonal crystalline phase has crystallographic a- and b-axes similar to 2Cs/2Rb/2K, but a longer c-axis, indicative of a [Ag(succ)4]− system with a larger spacer between the AgIII centers. Solving the crystal structure revealed a polymeric, mixed-valent salt of composition [(H2O)Ag][Ag(succ)4]·4H2O (2Ag, spacegroup I4/m, Fig. 1C). The [(H2O)Ag]+ fragment is located between four of the carbonyl oxygens from a [Ag(succ)4]− unit, forming a square pyramidal motif with water at the apical position (τ5 = 0.00).42 The apical water molecule forms hydrogen bonds to the free set of carbonyl oxygens from a neighboring [(H2O)Ag][Ag(succ)4] fragment, generating a polymeric structure. Given that the [Ag(succ)4]− fragment resides at the intersection between a crystallographic four-fold rotation axis and a mirror plane, the [(H2O)Ag]+ fragments are disordered over two orientations, pointing in either direction of the polymeric chain structure. The [(H2O)Ag]+ fragments separate neighboring [Ag(succ)4]− units in 2Ag more effectively than Cs+, Rb+, and K+ do in 2Cs/2Rb/2K, resulting in a significantly elongated square prismatic motif in 2Ag (O⋯O edges: 2 × 3.295(3), 1 × 4.634(3) Å). Overall, 2Ag conforms to a Robin-Day class I description,43 given its structure and spectroscopic traits: The [Ag(succ)4]− fragments in 2Cs, 2Rb, 2K, and 2Ag display the same gross geometry. On the other hand, a structurally distinct [(H2O)Ag]+ site is present in 2Ag, suggesting distinctly different oxidation states in the mixed-valent salt. Moreover, the UV-vis spectrum of 2Ag is devoid of intervalence charge-transfer bands and is essentially superimposable with that of 2Cs (see discussion below as well as ESI, Fig. S12†). Returning to the observation that isolated yields of 2Cs are consistently close to 50%, we propose that a monomeric species akin to 2Ag forms in equilibrium in the reaction mixtures. The Ag+ ion would prevent Cs+ from coordinating to the argentate(III) ion, [Ag(succ)4]−, to form polymeric 2Cs. Once the substitution of Ag+ for Cs+ eventually succeeds, most of the oxidant, Na2[S2O8] may have then been consumed in other oxidation processes, leaving close to half of the AgI ions untouched.
:2:1, we isolated dark yellow crystals within few minutes. The resulting tetragonal crystalline phase has crystallographic a- and b-axes similar to 2Cs/2Rb/2K, but a longer c-axis, indicative of a [Ag(succ)4]− system with a larger spacer between the AgIII centers. Solving the crystal structure revealed a polymeric, mixed-valent salt of composition [(H2O)Ag][Ag(succ)4]·4H2O (2Ag, spacegroup I4/m, Fig. 1C). The [(H2O)Ag]+ fragment is located between four of the carbonyl oxygens from a [Ag(succ)4]− unit, forming a square pyramidal motif with water at the apical position (τ5 = 0.00).42 The apical water molecule forms hydrogen bonds to the free set of carbonyl oxygens from a neighboring [(H2O)Ag][Ag(succ)4] fragment, generating a polymeric structure. Given that the [Ag(succ)4]− fragment resides at the intersection between a crystallographic four-fold rotation axis and a mirror plane, the [(H2O)Ag]+ fragments are disordered over two orientations, pointing in either direction of the polymeric chain structure. The [(H2O)Ag]+ fragments separate neighboring [Ag(succ)4]− units in 2Ag more effectively than Cs+, Rb+, and K+ do in 2Cs/2Rb/2K, resulting in a significantly elongated square prismatic motif in 2Ag (O⋯O edges: 2 × 3.295(3), 1 × 4.634(3) Å). Overall, 2Ag conforms to a Robin-Day class I description,43 given its structure and spectroscopic traits: The [Ag(succ)4]− fragments in 2Cs, 2Rb, 2K, and 2Ag display the same gross geometry. On the other hand, a structurally distinct [(H2O)Ag]+ site is present in 2Ag, suggesting distinctly different oxidation states in the mixed-valent salt. Moreover, the UV-vis spectrum of 2Ag is devoid of intervalence charge-transfer bands and is essentially superimposable with that of 2Cs (see discussion below as well as ESI, Fig. S12†). Returning to the observation that isolated yields of 2Cs are consistently close to 50%, we propose that a monomeric species akin to 2Ag forms in equilibrium in the reaction mixtures. The Ag+ ion would prevent Cs+ from coordinating to the argentate(III) ion, [Ag(succ)4]−, to form polymeric 2Cs. Once the substitution of Ag+ for Cs+ eventually succeeds, most of the oxidant, Na2[S2O8] may have then been consumed in other oxidation processes, leaving close to half of the AgI ions untouched.
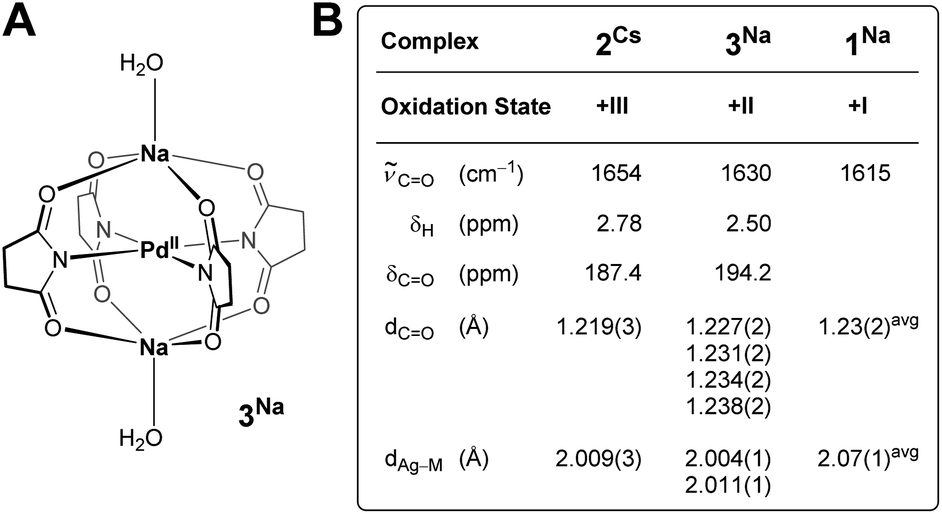
Isolation of a PdII complex (3Na) and comparison to the isoelectronic AgIII complex (2Cs)
To assess how the trivalent silver ion affects the molecular and electronic structure of the [Ag(succ)4]− anion, we prepared the isoelectronic palladium anion, [Pd(succ)4]2−, by dissolving [PdCl2] in succinimide buffer. From the reaction mixture, colorless crystals of [(H2O)Na]2[Pd(succ)4]·2H2O (3Na) were isolated in 77% yield and characterized by elemental analysis, 1H and 13C NMR spectroscopies, ESI-MS, and X-ray crystallography (Chart 2A). The [Pd(succ)4]2− fragment adopts a gross geometry akin to the AgIII analog, with Pd–N bond distances that are statistically indistinguishable from Ag–N bond distances in 2Cs (Chart 2B). Given the larger ionic radius of PdII as compared to the more contracted AgIII ion, these metrics indicate that metal-succinimide bonding in 2Cs and 3Na might display variable degrees of covalency. In complex 3Na, the two sets of carbonyl oxygens from the [Pd(succ)4]2− ion coordinate to one Na+ ion above – and one below – the plane of the PdII center, and the coordination sphere of each Na+ ion is completed by an axial water molecule to form a square pyramidal motif (τ5 = 0.02). In complex 3Na, the relatively small Na+ ion adopts a buried position between the four carbonyl oxygens, similar to the Ag+ ion in 2Ag (3Na: PdII⋯Na = 3.0321(5) Å, 2Ag: AgIII⋯AgI = 2.972(1) Å). In contrast, the Cs+, Rb+, and K+ ions in 2Cs, 2Rb, and 2K show a more pronounced departure from the AgIII centers (AgIII⋯Cs = 4.3722(2) Å, AgIII⋯Rb = 4.2535(3) Å, AgIII⋯K = 4.2066(2) Å). This highlights the role of the counterion, as well as hydrogen bonding interactions, in the isolation of the polymeric AgIII salts, 2Cs, 2Rb, 2K, and 2Ag.
 |
| | Chart 2 (A) Molecular structure of 3Na. (B) Spectroscopic and metric data for succinimide complexes 2Cs (AgIII), 3Na (PdII), and 1Na (AgI). | |
We then turned to spectroscopic methods to glean more information about the electronic structure of the AgIII, PdII, and AgI systems, 2Cs, 3Na, and 1Na (Chart 2B). The IR spectrum of crystalline 2Cs is dominated by a strong out-of-phase44 CO stretching frequency at 1654 cm−1 (this is also the case for 2Rb and 2K). In contrast, PdII complex 3Na displays a CO vibration at 1630 cm−1, and AgI precursor 1Na displays a CO vibration at 1615 cm−1. In this series (AgIII > PdII > AgI), the shift of the CO vibrations toward lower frequencies illustrates how the succinimide anion is sensitive to the oxidation state of the metals. This reflects a balance between the succinimide ligand, on the one hand, being engaged in donor interactions to stabilize a high-valent metal ion such as AgIII, or, on the other hand, forming a delocalized π-system involving the carbonyl and imide nitrogen groups. Accordingly, CO bond distances in complex 3Na are elongated by ∼0.01 Å (avg. 1.232(2) Å) as compared to complex 2Cs. From NMR measurements, the 1H resonance of 2Cs in D2O displays a singlet at 2.78 ppm, whereas 3Na displays a more shielded proton environment (2.50 ppm), in accord with the lower negative charge and higher oxidation state of the [Ag(succ)4]− anion versus [Pd(succ)4]2−. By contrast, the carbonyl 13C NMR resonance in 2Cs is more shielded than that in 3Na, in line with the shorter CO bond in the AgIII complex leading to a more efficient orbital overlap within the carbonyl group, and hence a more shielded carbon nucleus.
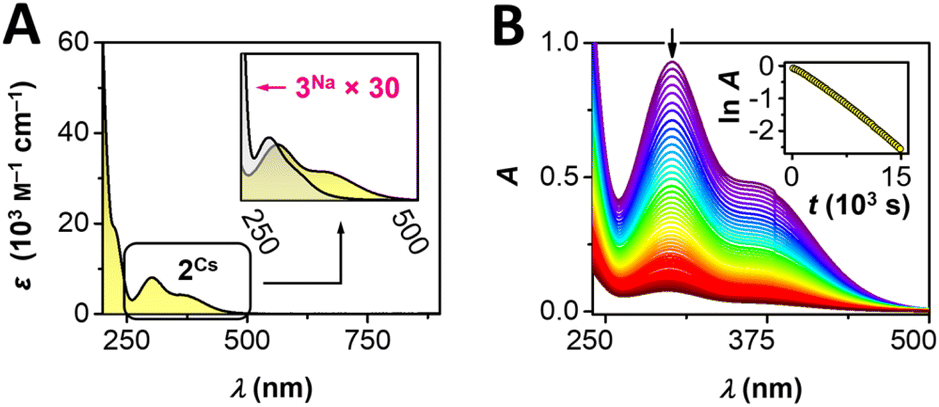
UV-visible spectroscopic studies of a AgIII complex (2Cs)
Given the bright yellow color of complex 2Cs, we subjected an aqueous sample to UV-visible spectroscopy (Fig. 2). The AgIII complex displays a very strong UV absorption at 222 nm (ε = 19400 M−1 cm−1), with energy and intensity falling close to 4 eq. of free succinimidate (215 nm, ε = 5900 M−1 cm−1, see ESI Fig. S15†). Moreover, 2Cs displays two intense bands in the near-UV range at 302 and 361 nm (ε = 8100, 4300 M−1 cm−1, Fig. 2A); the lowest-energy band extends into the visible range and vanishes around 500 nm, accounting for the yellow color of the complex. In a similar fashion, aqueous solutions of 2Ag display absorptions at energies essentially identical to those of 2Cs but with ∼50% higher intensity, in line with the darker yellow color of the mixed-valent silver complex.
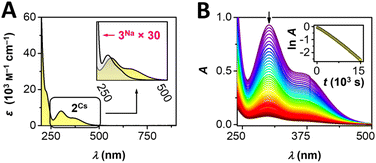 |
| | Fig. 2 (A) UV-vis spectroscopic studies of 2Cs and 3Na. (B) Evolution of UV-vis spectra of 2Cs in neutral solution; inset: time dependence of lnA at λmax = 302 nm. | |
Comparison of 2Cs and 2Ag in the solid state (KBr pellet) reveals very similar UV-vis features, in accord with the solution data (ESI, Fig. S12 and S13†). We also recorded UV-visible spectral data for the isoelectronic [Pd(succ)4]2− anion in 3Na. Akin to 2Cs, the palladium complex exhibits a very strong UV-absorption at 216 nm (ε = 49100 M−1 cm−1) as well as two bands in the near-UV range (289, 336 nm, ε = 290, 120 M−1 cm−1), which exhibit a similar band shape to 2Cs. However, PdII complex 3Na displays markedly lower absorption intensity than the trivalent silver system 2Cs (×1/30, Fig. 2A, inset). The qualitatively similar appearance of electronic transitions observed for [Ag(succ)4]− and [Pd(succ)4]2− is indicative of these isoelectronic complexes possessing similar electronic structures. In view of the more oxidizing nature of the AgIII ion as compared to PdII, complex 2Cs displays electronic transitions with more ligand-to-metal charge-transfer character, resulting in lower transition energies and higher molar absorptivity. TDDFT calculation on 2Rb yielded three states with significant intensities with wavelengths of 372, 346 and 247 nm, of which the two lowest energy transitions involved excitations from bonding orbitals of mainly ligand character to the heavily admixed LUMO having the most 4dx2−y2 (Ag) character of any of the KS-orbitals. The transition at 346 nm is calculated as the most intense by a factor of 10. The change in electron density for this transition shows a shift of electron density from the succ-π⊥ orbitals involving the nitrogen and carbonyl groups towards the σ-orbitals on silver and nitrogen (cf. Fig. S36†). This corroborates the description of the transitions as having significant LMCT character. The high-valent nature of 2Cs can be further ascertained from its ability to oxidize water (Scheme 1). In aqueous solution, electronic transitions between 250–500 nm decrease in a first-order process with a half-life of 70 minutes (293 K, Fig. 2B). The process proceeds with formation of O2, succinimide, and a new AgI complex, Cs[Ag(succ)2]·4H2O (1Cs), identified by a combination of NMR spectroscopy and X-ray crystallography. These findings are interesting given Lessard's suggestion that electrochemical oxidation of [Ag(succ)2]− affords a succinimidyl radical, which acts as a hydrogen atom abstractor to form succinimide;37 our studies, on the other hand, implicate a AgIII complex.
Solid-state 109Ag NMR spectroscopic studies of a AgIII complex (2Cs)
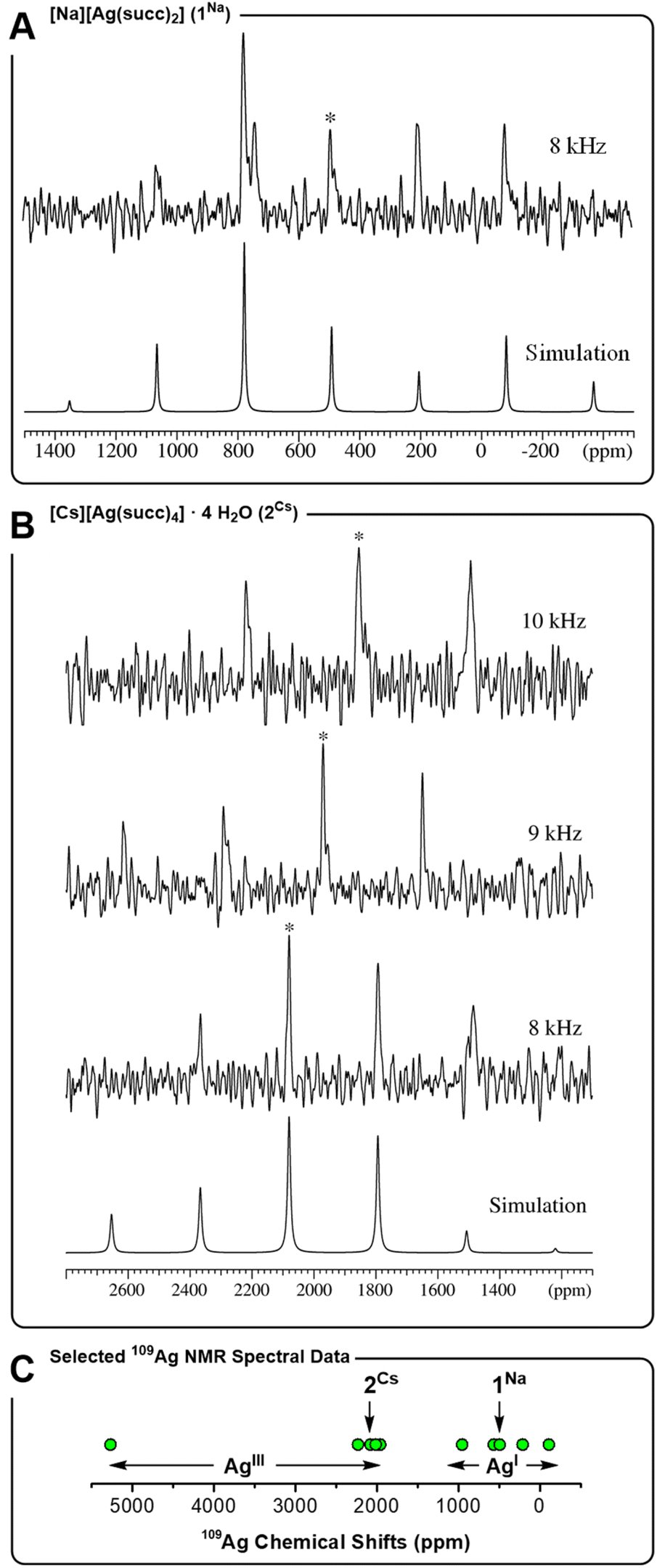
Given the tendency for 2Cs to decay in aqueous solution, clean NMR spectral data can be recorded within a timeframe of ∼5 minutes after a sample of 2Cs has been prepared. This permits characterization of reasonably sensitive nuclei, such as 1H and 13C. In view of the lower sensitivity of a nucleus such as 109Ag, we conducted a solid-state NMR spectroscopic study of 2Cs to gain more information about the trivalent silver ion. 109Ag magic-angle spinning (MAS) NMR spectra (14.09 T) were recorded for monovalent complex 1Na and trivalent complex 2Cs (Fig. 3). The acquisition of these solid-state NMR spectra is complicated by the low gyromagnetic ratio for 109Ag, as well as long spin-lattice relaxation times (T1's), which may be on the order of several hundred seconds for diamagnetic silver compounds.45 Accordingly, single-pulse 109Ag NMR spectra were recorded with relaxation delays of 180–600 s (not corresponding to full relaxation), resulting in rather low signal-to-noise ratios even at acquisition times spanning several days. Both of the complexes, 1Na and 2Cs, displays a single isotropic peak, which in each case is flanked by one or two rotational spinning sidebands on each side, due to 109Ag chemical shift anisotropy (CSA). The intensities of the centerband and spinning sidebands allow estimation of the 109Ag CSA parameters from spectral simulations for 1Na and 2Cs (Table 1).
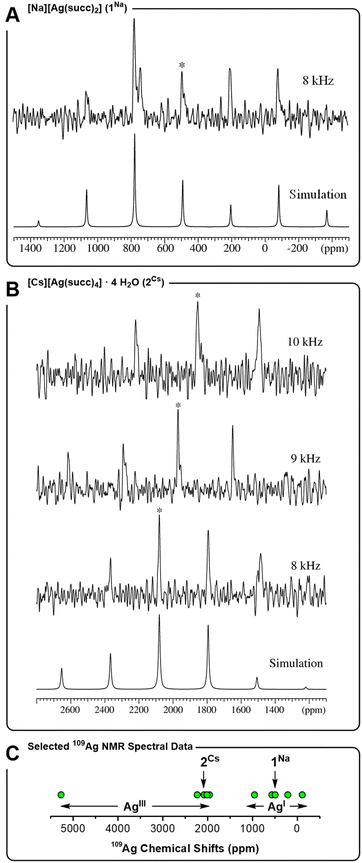 |
| | Fig. 3
109Ag MAS NMR spectra 14.09 T (27.9 MHz). (A) 1Na measured at a spinning frequency of υR = 8.0 kHz (T = 36 °C, relaxation delay d1 = 180 s, 2304 scans). (B) 2Cs measured at spinning frequencies of υR = 10.0 kHz (T = 43 °C, d1 = 600 s, 542 scans), 9.0 kHz (T = 40 °C, d1 = 300 s, and 1356 scans) and 8.0 kHz (T = 36 °C, d1 = 600 s, and 1004 scans). Isotropic peaks indicated by *. (C) Representative 109Ag chemical shifts (ppm) for AgIII and AgI complexes: [AgIIIF4]− (5270),46 [AgIII(CF3)4]− (2233),22a2Cs (2080), AgIII2O3 (2015),45b AgIAgIIIO2 (1960),45b AgIAlO2 (953),45b [AgI(CF3)2]− (566),22a,471Na (492), [AgIN(SO2Me)2] (210),45d AgIF (−110).45d | |
Table 1
109Ag chemical shift parameters for 1Na and 2Cs
|
|
υ
R (kHz) |
T (°C) |
δ
iso
(ppm) |
δ
σ
(ppm) |
η
σ
|
|
109Ag isotropic chemical shift referenced to a 1.0 M AgNO3 solution (T = 21 °C). At these conditions, δiso = −118 ± 1 ppm is observed for solid AgNO3 at T = 43 °C.
109Ag chemical shift anisotropy parameters, defined as δiso = ⅓(δxx + δyy + δzz), δσ = (δiso − δzz), and ησ = (δxx − δyy)/δzz, where δxx, δyy, and δzz are the principal elements of the chemical shift tensor defined as |δzz − δiso| ≥ |δxx − δiso| ≥ |δyy − δiso|.
|
|
1Na
|
8.0 |
36 |
492 ± 2 |
1100 ± 100 |
0.2 ± 0.2 |
|
2Cs
|
8.0 |
36 |
2080 ± 2 |
−750 ± 100 |
0.5 ± 0.2 |
| 9.0 |
40 |
1971 ± 2 |
n.d. |
n.d. |
| 10.0 |
43 |
1855 ± 2 |
n.d. |
n.d. |
A clear distinction in isotropic chemical shifts is apparent for the two complexes, with 1Na displaying an isotropic shift at 492 ppm, whereas trivalent 2Cs gives rise to a much more deshielded isotropic shift at 2080 ppm at the same spinning frequency and sample temperature. In this regard, 1Na and 2Cs show a similar correlation between oxidation state and chemical shift as observed between a AgI complex such as [Ag(CF3)2]− (566 ppm) and a covalent AgIII complex such as [Ag(CF3)4]− (2233 ppm).22a Generally speaking, monovalent AgI compounds, including oxides45b–d,48 and nitrogen-coordinated complexes,45a,49 exhibit 109Ag chemical shifts in the range −100 to +1000 ppm. Trivalent silver complexes, on the other hand, display strongly deshielded resonances, often in the range 1900 to 2500 ppm, and even as high as 5270 ppm for K[AgF4] (Fig. 3C). Thereby, the 109Ag MAS NMR spectra support the presence of a AgIII center in complex 2Cs, and the observation of a unique AgIII site by NMR is in accord with the crystallographic symmetry. Furthermore, 109Ag CSA parameters (δσ and ησ) were determined, albeit with rather low precision, reflecting the low S/N ratio of the acquired spectra. Notably, the shielding anisotropy (δσ) changes sign between 1Na and 2Cs. This is in line with the change in geometry about the silver ion, going from axial donation in linear 1Na to equatorial donation in square planar 2Cs. Finally, we note that δiso(109Ag) is highly temperature dependent for 2Cs, as apparent from the spectra obtained at different spinning frequencies, causing different degrees of frictional heating of the sample. The chemical shifts observed at three different rotation frequencies (Table 1), indicate δiso to have a temperature dependence of −32 ppm/°C, leading to a variation of several hundred ppm within a few °C (in contrast, 1Na displays no temperature dependence of δiso over the small studied temperature range of ±5 °C). Although not record-setting, the temperature dependence of the chemical shift for 2Cs is quite large for a d-block metal.50 Moreover, it is clear that comparisons of the numerical value of 109Ag NMR chemical shifts for AgIII complexes (such as those in Fig. 3C), should be made with care, given the effect of temperature.
Conclusions
In summary, we have isolated the first example of a homoleptic and square planar AgIII complex stabilized by a monodentate N-donor ligand, namely the Cs+, Rb+, K+, and Ag+ salts of the argentate(III) ion [Ag(succ)4]− (2Cs, 2Rb, 2K, and 2Ag). Dissolving AgNO3 in succinimide buffer affords AgI precursor 1Na, which is oxidized chemically by Na2[S2O8] or electrochemically by anodic oxidation to form a square planar [Ag(succ)4]− motif. Complexation with Cs+, Rb+, K+, or Ag+ leads to polymeric chain-structures, which in the case of 2Ag encompasses a mixed-valent Robin-Day class I system. 1H and 13C NMR spectral studies combined with SQUID magnetometry attest to the diamagnetic nature of 2Cs. IR spectroscopy reveals a high-frequency CO stretching mode for the succinimide ligands in 2Cs as compared to the isoelectronic PdII complex, 3Na. DFT modeling of the ground state electronic structure suggests the systems to be placed at the border between having a normal LF (AgIII) and an inverted LF (AgI). In addition, UV-vis spectroscopy reveals electronic transitions at lower energy and with higher molar absorption for 2Cs than for 3Na. These spectroscopic results point to the oxidizing nature of the [Ag(succ)4]− ion, which is further manifested in the ability of 2Cs to oxidize water, forming succinimide, O2, and a AgI complex 1Cs. TDDFT computation of the lowest excitations in 2Rb reveal LMCT character (ligand-to-Ag(dx2−y2)) to dominate in the observed spectral range. Solid-state 109Ag magic-angle spinning NMR studies reveal a resonance at 492 ppm for AgI precursor 1Na, whereas AgIII complex 2Cs exhibits a highly deshielded resonance at 2080 ppm, in the range expected for AgIII, but towards the low end due to the covalent nature of the bonding in the trivalent silver complex with nitrogen donors. The NMR studies directly map the change in geometry between 1Na (linear) and 2Cs (square planar) through a change of sign for the shielding anisotropy (δσ). Moreover, the 109Ag chemical shift of 2Cs is highly temperature-dependent (−32 ppm/°C), leading to variations of several hundred ppm within a temperature range of few °C. The possibility of generating 2Cs by chemical as well as electrochemical oxidation is of interest in both coordination chemistry and materials science, given that the identification of argentate(III) systems such as [Ag(succ)4]− illustrates one possible fate of silver anodes used for electroplating. We are presently examining the new family of succinimide complexes as ditopic metalloligands and seeking entries to other AgIII complexes to probe their reactivity and electronic structure.
Data availability
All experimental data are available in the ESI;† synthetic procedures; NMR, IR, UV-vis, X-ray diffraction, and magnetic data. CCDC entries 2365916–2365920, and 2372376 contain full crystallographic data (.cif) reported herein. The data supporting this article have been included as part of the ESI.†
Author contributions
The synthetic work was carried out by E. M. H. L., A. R. and J. B. All authors contributed to data collection and to writing the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
JB acknowledges support from and the Independent Research Fund Denmark (1026-00082B).
Notes and references
-
(a) W. Levason and M. D. Spicer, Coord. Chem. Rev., 1987, 76, 45–120 CrossRef;
(b) M. A. García-Monforte, S. Martínez-Salvador and B. Menjón, Eur. J. Inorg. Chem., 2012, 2012, 4945–4966 CrossRef.
-
(a) B. Zemva, K. Lutar, A. Jesih, W. J. Casteel, A. P. Wilkinson, D. E. Cox, R. B. Von Dreele, H. Borrmann and N. Bartlett, J. Am. Chem. Soc., 1991, 113, 4192–4198 CrossRef;
(b) N. Bartlett, G. Lucier, C. Shen, W. J. Casteel, L. Chacon, J. Munzenberg and B. Žemva, J. Fluorine Chem., 1995, 71, 163–164 CrossRef;
(c) J. Zhang, S. Huo, H. Shi, S. Shen and Y. Shang, Transition Met. Chem., 2013, 38, 15–20 CrossRef.
-
(a) J. A. Schlueter, K. D. Carlson, U. Geiser, H. H. Wang, J. M. Williams, W. K. Kwok, J. A. Fendrich, U. Welp, P. M. Keane, J. D. Dudek, A. S. Komosa, D. Naumann, T. Roy, J. E. Schirber, W. R. Bayless and B. Dodrill, Phys. C: Supercond. Appl., 1994, 233, 379–386 CrossRef;
(b) J. A. Schlueter, L. Wiehl, H. Park, M. de Souza, M. Lang, H.-J. Koo and M.-H. Whangbo, J. Am. Chem. Soc., 2010, 132, 16308–16310 CrossRef CAS PubMed;
(c) D. Guterding, M. Altmeyer, H. O. Jeschke and R. Valentí, Phys. Rev. B, 2016, 94, 024515 CrossRef.
- U. Geiser, J. A. Schlueter, J. D. Dudek, J. M. Williams, D. Naumann and T. Roy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 1779–1782 CrossRef.
- S. Weske, R. A. Hardin, T. Auth, R. A. J. O'Hair, K. Koszinowski and C. A. Ogle, Chem. Commun., 2018, 54, 5086–5089 RSC.
-
(a) M. Font, F. Acuña-Parés, T. Parella, J. Serra, J. M. Luis, J. Lloret-Fillol, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 4373 CrossRef PubMed;
(b) L. Capdevila, E. Andris, A. Briš, M. Tarrés, S. Roldán-Gómez, J. Roithová and X. Ribas, ACS Catal., 2018, 8, 10430–10436 CrossRef.
-
(a) L. Demonti, N. Saffon-Merceron, N. Mézailles and N. Nebra, Chem.–Eur. J., 2021, 27, 15396–15405 CrossRef;
(b) Z. Lu, S. Liu, Y. Lan, X. Leng and Q. Shen, Organometallics, 2021, 40, 1713–1718 CrossRef.
- M. Deuker, Y. Yang, R. A. J. O'Hair and K. Koszinowski, Organometallics, 2021, 40, 2354–2363 CrossRef.
-
(a) L. Malatesta, Gazz. Chim. Ital., 1941, 71, 467–474 CAS;
(b) R. Masse and A. Simon, J. Solid State Chem., 1982, 44, 201–207 CrossRef CAS;
(c) C. J. Spina, J. E. Notarandrea-Alfonzo, E. D. Guerra, C. Goodall, D. S. Bohle and R. Precht, ACS Omega, 2021, 6, 27017–27025 CrossRef CAS PubMed;
(d) L. Malaprade, C. R. Acad. Sci., 1940, 210, 504–505 CAS.
- L. Malatesta, Gazz. Chim. Ital., 1941, 71, 580–584 Search PubMed.
-
(a) P. Rây, Nature, 1943, 151, 643 CrossRef;
(b) P. Rây and K. Chakravarty, J. Indian Chem. Soc., 1944, 21, 47–50 Search PubMed;
(c) N. R. Kunchur, Nature, 1968, 217, 539 CrossRef.
-
(a) L. F. Warren and M. A. Bennett, Inorg. Chem., 1976, 15, 3126–3140 CrossRef;
(b) L. J. Kirschenbaum and J. D. Rush, J. Am. Chem. Soc., 1984, 106, 1003–1010 CrossRef;
(c) L. J. Kirschenbaum, R. K. Panda, E. T. Borish and E. Mentasti, Inorg. Chem., 1989, 28, 3623–3628 CrossRef.
- E. K. Barefield and M. T. Mocella, Inorg. Chem., 1973, 12, 2829–2832 CrossRef CAS.
-
(a) H. Furuta, T. Ogawa, Y. Uwatoko and K. Araki, Inorg. Chem., 1999, 38, 2676–2682 CrossRef CAS;
(b) H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2000, 122, 803–807 CrossRef;
(c) M. A. Muckey, L. F. Szczepura, G. M. Ferrence and T. D. Lash, Inorg. Chem., 2002, 41, 4840–4842 CrossRef PubMed;
(d) C. Brückner, C. A. Barta, R. P. Briñas and J. A. Krause Bauer, Inorg. Chem., 2003, 42, 1673–1680 CrossRef;
(e) H. Furuta, T. Morimoto and A. Osuka, Org. Lett., 2003, 5, 1427–1430 CrossRef;
(f) H. Maeda, A. Osuka, Y. Ishikawa, I. Aritome, Y. Hisaeda and H. Furuta, Org. Lett., 2003, 5, 1293–1296 CrossRef;
(g) M. Pawlicki and L. Latos-Grażyński, Chem.–Eur. J., 2003, 9, 4650–4660 CrossRef PubMed;
(h) K. M. Bergman, G. M. Ferrence and T. D. Lash, J. Org. Chem., 2004, 69, 7888–7897 CrossRef PubMed;
(i) M. Pawlicki and L. Latos-Grażyński, J. Org. Chem., 2005, 70, 9123–9130 CrossRef;
(j) M. Stefanelli, M. Mastroianni, S. Nardis, S. Licoccia, F. R. Fronczek, K. M. Smith, W. Zhu, Z. Ou, K. M. Kadish and R. Paolesse, Inorg. Chem., 2007, 46, 10791–10799 CrossRef;
(k) K. E. Thomas, A. B. Alemayehu, J. Conradie, C. Beavers and A. Ghosh, Inorg. Chem., 2011, 50, 12844–12851 CrossRef;
(l) W. Sinha, M. G. Sommer, N. Deibel, F. Ehret, B. Sarkar and S. Kar, Chem.–Eur. J., 2014, 20, 15920–15932 CrossRef;
(m) K. E. Thomas, H. Vazquez-Lima, Y. Fang, Y. Song, K. J. Gagnon, C. M. Beavers, K. M. Kadish and A. Ghosh, Chem.–Eur. J., 2015, 21, 16839–16847 CrossRef;
(n) M. Soll, K. Sudhakar, N. Fridman, A. Müller, B. Röder and Z. Gross, Org. Lett., 2016, 18, 5840–5843 CrossRef;
(o) J. Yan, Y. Yang, M. Ishida, S. Mori, B. Zhang, Y. Feng and H. Furuta, Chem.–Eur. J., 2017, 23, 11375–11384 CrossRef;
(p) C. M. Lemon, D. C. Powers, M. Huynh, A. G. Maher, A. A. Phillips, B. P. Tripet and D. G. Nocera, Inorg. Chem., 2023, 62, 3–17 CrossRef.
- R. Hoppe, Z. Anorg. Allg. Chem., 1957, 292, 28–33 CrossRef.
- R. Hoppe and R. Homann, Naturwissenschaften, 1966, 53, 501 CrossRef.
- G. L. Cohen and G. Atkinson, J. Electrochem. Soc., 1968, 115, 1236–1242 CrossRef.
-
(a) G. L. Cohen and G. Atkinson, Inorg. Chem., 1964, 3, 1741–1743 CrossRef;
(b) L. J. Kirschenbaum, J. H. Ambrus and G. Atkinson, Inorg. Chem., 1973, 12, 2832–2837 CrossRef CAS.
- J. Iqbal, D. W. A. Sharp and J. M. Winfield, J. Chem. Soc., Dalton Trans., 1989, 461–464 RSC.
-
(a) W. Dukat and D. Naumann, Rev. Chim. Miner., 1986, 23, 589–603 CAS;
(b) L. Demonti, H. Tabikh, N. Saffon-Merceron and N. Nebra, Eur. J. Inorg. Chem., 2023, 26, e202300042 CrossRef CAS.
-
(a) R. Eujen, B. Hoge and D. J. Brauer, Inorg. Chem., 1997, 36, 3160–3166 CrossRef CAS;
(b) D. Joven-Sancho, M. Baya, A. Martín and B. Menjón, Chem.–Eur. J., 2018, 24, 13098–13101 CrossRef CAS PubMed;
(c) M. Baya, D. Joven-Sancho, P. J. Alonso, J. Orduna and B. Menjón, Angew. Chem., Int. Ed., 2019, 58, 9954–9958 CrossRef CAS.
-
(a) R. Eujen, B. Hoge and D. J. Brauer, Inorg. Chem., 1997, 36, 1464–1475 CrossRef CAS PubMed;
(b) D. Joven-Sancho, M. Baya, L. R. Falvello, A. Martín, J. Orduna and B. Menjón, Chem.–Eur. J., 2021, 27, 12796–12806 CrossRef CAS PubMed;
(c) D. Joven-Sancho, M. Baya, A. Martín, J. Orduna and B. Menjón, Chem.–Eur. J., 2020, 26, 4471–4475 CrossRef CAS PubMed.
-
(a) D. Naumann, W. Tyrra, F. Trinius, W. Wessel and T. Roy, J. Fluorine Chem., 2000, 101, 131–135 CrossRef CAS;
(b) D. Joven-Sancho, L. Demonti, A. Martín, N. Saffon-Merceron, N. Nebra, M. Baya and B. Menjón, Chem. Commun., 2023, 59, 4166–4168 RSC;
(c) J. Pueyo, D. Joven-Sancho, A. Martín, B. Menjón and M. Baya, Chem.–Eur. J., 2024, 30, e202303937 CrossRef CAS;
(d) L. Demonti, D. Joven-Sancho, N. Saffon-Merceron, M. Baya and N. Nebra, Chem.–Eur. J., 2024, 30, e202400881 CrossRef CAS.
- D. Joven-Sancho, M. Baya, A. Martín, J. Orduna and B. Menjón, Angew. Chem., Int. Ed., 2021, 60, 26545–26549 CrossRef CAS.
-
(a) D. Naumann, T. Roy, K.-F. Tebbe and W. Crump, Angew. Chem., Int. Ed. Engl., 1993, 32, 1482–1483 CrossRef;
(b) N. Nebra and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 16998–17001 CrossRef CAS;
(c) A. M. Romine, N. Nebra, A. I. Konovalov, E. Martin, J. Benet-Buchholz and V. V. Grushin, Angew. Chem., Int. Ed., 2015, 54, 2745–2749 CrossRef CAS.
- M. Kaupp and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1995, 34, 986 CrossRef CAS.
- R. Hoffmann, S. Alvarez, C. Mealli, A. Falceto, T. J. Cahill, T. Zeng and G. Manca, Chem. Rev., 2016, 116, 8173–8192 CrossRef CAS.
- J. P. Snyder, Angew. Chem., Int. Ed. Engl., 1995, 34, 80–81 CrossRef CAS.
-
(a) R. C. Walroth, J. T. Lukens, S. N. MacMillan, K. D. Finkelstein and K. M. Lancaster, J. Am. Chem. Soc., 2016, 138, 1922–1931 CrossRef CAS PubMed;
(b) I. M. DiMucci, J. T. Lukens, S. Chatterjee, K. M. Carsch, C. J. Titus, S. J. Lee, D. Nordlund, T. A. Betley, S. N. MacMillan and K. M. Lancaster, J. Am. Chem. Soc., 2019, 141, 18508–18520 CrossRef CAS PubMed.
- C. Gao, G. Macetti and J. Overgaard, Inorg. Chem., 2019, 58, 2133–2139 CrossRef CAS PubMed.
- J. P. Snyder, Angew. Chem., Int. Ed. Engl., 1995, 34, 986–987 CrossRef CAS.
-
(a) W. Grochala and R. Hoffmann, Angew. Chem., Int. Ed., 2001, 40, 2742–2781 CrossRef CAS;
(b) W. Grochala, R. G. Egdell, P. P. Edwards, Z. Mazej and B. Žemva, ChemPhysChem, 2003, 4, 997–1001 CrossRef CAS;
(c) W. Grochala and Z. Mazej, Philos. Trans. R. Soc., A, 2015, 373, 20140179 CrossRef.
- R. J. Deeth, Phys. Chem. Chem. Phys., 2024, 26, 18138–18148 RSC.
-
(a) T. J. Collins, Acc. Chem. Res., 1994, 27, 279–285 CrossRef CAS;
(b) T. J. Collins, T. R. Nichols and E. S. Uffelman, J. Am. Chem. Soc., 1991, 113, 4708–4709 CrossRef CAS;
(c) F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835–838 CrossRef;
(d) M. R. Mills, A. C. Weitz, M. P. Hendrich, A. D. Ryabov and T. J. Collins, J. Am. Chem. Soc., 2016, 138, 13866–13869 CrossRef CAS PubMed;
(e) F. C. Anson, T. J. Collins, T. G. Richmond, B. D. Santarsiero, J. E. Toth and B. G. R. T. Treco, J. Am. Chem. Soc., 1987, 109, 2974–2979 CrossRef CAS.
-
(a) S. Jayakrishnan, S. R. Natarajan and K. I. Vasu, Met. Finish., 1996, 94, 12–15 CrossRef CAS;
(b) S. Masaki, H. Inoue and K. Yamakawa, J. Surf. Finish. Soc. Jpn., 1997, 48, 559–564 CrossRef CAS;
(c) S. Masaki, H. Inoue and K. Yamakawa, J. Surf. Finish. Soc. Jpn., 1997, 48, 643–646 CrossRef CAS.
- J.-Y. Huot, D. Serve and J. Lessard, Can. J. Chem., 1983, 61, 1890–1898 CrossRef CAS.
- J.-Y. Huot, D. Serve, S. Desjardins and J. Lessard, Can. J. Chem., 1988, 66, 35–44 CrossRef CAS.
- S. Masaki, H. Inoue and H. Honma, Met. Finish., 1998, 96, 52–54 CrossRef CAS.
-
(a) G. A. Barbieri, Gazz. Chim. Ital., 1912, 42, 7–14 Search PubMed;
(b) G. B. Kauffman, R. A. Houghten, R. E. Likins, P. L. Posson, R. K. Ray, J. P. Fackler Jr and R. Theron Stubbs, Inorg. Synth., 1998, 177–181 CrossRef CAS.
-
(a) W. Khayata, D. Baylocq, F. Pellerin and N. Rodier, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 765–767 CrossRef;
(b) J. Perron and A. L. Beauchamp, Inorg. Chem., 1984, 23, 2853–2859 CrossRef CAS;
(c) X. Tao, K.-C. Shen, Y.-L. Wang and Y.-Z. Shen, Z. Anorg. Allg. Chem., 2011, 637, 1394–1400 CrossRef CAS.
-
(a) E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS;
(b) M. Atanasov, C. A. Daul and C. Rauzy, Chem. Phys. Lett., 2003, 367, 737–746 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
M. B. Robin and P. Day, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- T. Woldbæk, P. Klæboe and D. H. Christensen, Acta Chem. Scand., 1976, 30A, 531–539 CrossRef.
-
(a) L. H. Merwin and A. Sebald, J. Magn. Reson., 1992, 97, 628–631 CAS;
(b) L. van Wüllen, S. Vensky, W. Hoffbauer and M. Jansen, Solid State Sci., 2005, 7, 920–924 CrossRef;
(c) G. H. Penner and X. Liu, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 151–167 CrossRef CAS;
(d) G. H. Penner and W. Li, Inorg. Chem., 2004, 43, 5588–5597 CrossRef CAS.
- R. Eujen and B. Zemva, J. Fluorine Chem., 1999, 99, 139–140 CrossRef CAS.
-
(a) P. J. Leszczynski, M. J. Jadwiszczak and W. Grochala, ChemistrySelect, 2023, 8, e202301775 CrossRef CAS;
(b) L. Demonti, D. Joven-Sancho and N. Nebra, Chem. Rec., 2023, 23, e202300143 CrossRef CAS PubMed.
- M. Weil, M. Puchberger, E. Füglein, E. J. Baran, J. Vannahme, H. J. Jakobsen and J. Skibsted, Inorg. Chem., 2007, 46, 801–808 CrossRef CAS.
- G. A. Bowmaker, R. K. Harris, B. Assadollahzadeh, D. C. Apperley, P. Hodgkinson and P. Amornsakchai, Magn. Reson. Chem., 2004, 42, 819–826 CrossRef CAS.
- Ö. Üngör, T. M. Ozvat, Z. Ni and J. M. Zadrozny, J. Am. Chem. Soc., 2022, 144, 9132–9137 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures; NMR, IR, UV-vis, X-ray diffraction, and magnetic data. CCDC 2365916–2365920 and 2372376. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04843a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Theis
Brock-Nannestad
a,
Theis
Brock-Nannestad