
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing hydrogen evolution reaction activity through defects and strain engineering in monolayer MoS2†
Renjith
Nadarajan
 ,
Sraboni
Dey
,
Arijit
Kayal
,
Joy
Mitra
and
Manikoth M.
Shaijumon
*
,
Sraboni
Dey
,
Arijit
Kayal
,
Joy
Mitra
and
Manikoth M.
Shaijumon
*
School of Physics, Indian Institute of Science Education and Research Thiruvananthapuram, Maruthamala PO, Thiruvananthapuram, Kerala 695551, India. E-mail: shaiju@iisertvm.ac.in
First published on 14th October 2024
Abstract
Molybdenum disulfide (MoS2) has recently emerged as a promising electrocatalyst for the hydrogen evolution reaction (HER). However, the poor in-plane electrical conductivity and inert basal plane activity pose major challenges in realizing its practical application. Herein, we demonstrate a new approach to induce biaxial strain into CVD-grown MoS2 monolayers by draping it over an array of patterned gold nanopillar arrays (AuNAs) as an efficient strategy to enhance its HER activity. We vary the magnitude of applied strain by changing the inter-pillar spacing, and its effect on the HER activity is investigated. To capitalize on the synergistic effect of improved ΔGHvia strain engineering and leverage basal plane activation by introduction of sulphur vacancies, we further exposed the strained MoS2 monolayers to oxygen plasma treatment to create S-vacancies. The strained MoS2 on AuNAs with optimal inter-pillar spacing is exposed to oxygen plasma treatment for different durations, and we study its electrocatalytic activity towards the HER using on-chip microcell devices. The strained and vacancy-rich monolayer MoS2 draped on AuNAs with a 0.5 μm inter-pillar spacing and exposed to plasma for 50 s (S0.5μmV50-MoS2) is shown to exhibit remarkable improvement in HER activity, with an overpotential of 53 mV in 0.5 M H2SO4. Thus, the synergistic creation of additional vacancy defects, along with strain-induced active sites, results in enhancement in HER performance of CVD-grown monolayer MoS2. The present study provides a highly promising route for engineering 2D electrocatalysts towards efficient hydrogen evolution.
1. Introduction
2-Dimensional (2D) transition metal dichalcogenides (TMDs) have been widely explored as promising electrocatalysts for the hydrogen evolution reaction (HER).1,2 In particular, MoS2 has shown huge potential as a HER electrocatalyst owing to its favorable characteristics such as earth abundance, low cost, high catalytic activity, good stability, etc. though the poor in-plane electrical conductivity and inert basal plane activity of MoS2 pose major challenges in realizing its practical application.3 Over the last decade, research has been focused on exploring strategies to optimize active edge sites in MoS2 and, more importantly, to fully utilize the basal plane activity.4,5 For instance, tailoring MoS2 nanostructures to expose active edge sites,6,7 making nanocomposites with a conducting matrix,8,9 and forming metallic 1T forms10 have all shown significant enhancement in HER activity. Recently, there has been a lot of interest in exploring strategies such as strain and defect engineering for increasing the number of active sites, conductivity, and surface adsorption ability of 2H-MoS2.11,12 According to the d-band theory, the optimization of hydrogen adsorption energy is ascribed to the movement of the d-band center, where the coordination environment can influence the d-band structure.13,14 Strain can change the metal–ligand bond lengths, resulting in strong interactions when the d-band center moves toward the Fermi level, making the Gibbs free energy (ΔGH*) close to zero. Then, strain can improve the electron transfer ability between catalytically active sites and adsorbed hydrogen.13,15–17 Different strategies have been explored to introduce strain in 2D materials, including physical (material deformation under external forces) and chemical (structural defects and atomic doping) pathways.18–20 Introducing sulphur vacancies in MoS2 causes strain in the lattice and acts as catalytically active sites that could enhance the basal plane activity.21–23 Epitaxial growth is also shown to cause strain because of lattice mismatch at the interface. For instance, epitaxy growth of WS2 nanosheets on mesoporous graphene showed inherent strain and S-vacancies, resulting in enhanced HER activity.24 Coexistence of strain and S-vacancies could synergistically contribute towards basal plane activation, leading to an overall enhancement in the HER activity in MoS2.Transferring thin 2D materials onto a patterned substrate induces non-uniform stresses, causing the material to wrinkle. Since the strong covalent bonds in the 2D material allow for a significant elastic strain along the surface, this approach primarily works with their atomic thickness, which is flexible and prone to deformation.25 Using nanostructured substrates to constrain atomically thin 2D materials seems like a promising approach.26 Importantly, the strain distribution is controlled by the substrate's pattern and quality, necessitating optimal substrate patterning and superior processing.27
Herein, we demonstrate a new approach to induce non-uniform strain into CVD-grown MoS2 monolayers by draping it over an array of patterned gold nanopillars (AuNAs), which effectively enhances the HER activity. We vary the magnitude of applied non-uniform strain by changing the inter-pillar spacings, and its effect on the HER activity is investigated. To capitalize on the synergistic effect of improved ΔGHvia strain engineering and leverage basal plane activation through the introduction of sulphur vacancies, we subjected the strained monolayers to plasma treatment to create S-vacancies (Fig. 1). The strained MoS2 on AuNAs with optimal inter-pillar spacing, in terms of HER performance, is plasma-treated for different durations, and we study its electrocatalytic activity towards the HER by using on-chip microcell devices. Strained and vacancy-rich monolayer MoS2 draped on AuNAs with a 0.5 μm inter-pillar spacing and exposed to plasma for 50 s (S0.5μmV50-MoS2) is shown to exhibit the best electrochemical characteristics for the HER, with an overpotential of 53 mV in 0.5 M H2SO4. Thus, the synergistic creation of additional vacancy defects, along with strain-induced active sites, could potentially result in enhancement in HER performance of CVD-grown MoS2. The results are promising, and we believe that this could lead to significant advancements in the field of 2D materials for electrocatalysis.
 | ||
| Fig. 1 Schematic representation of (a) gold nanopillar arrays on a Si/SiO2 substrate, (b) CVD-grown MoS2 monolayer transferred over the AuNAs (S-MoS2), and (c) vacancy-rich strained MoS2 (SV-MoS2). | ||
2. Experimental
2.1 Growth of monolayer MoS2
Monolayer MoS2 was synthesized on a Si/SiO2 substrate by standard chemical vapor deposition. The Na2MoO4 powder (99.9%, Sigma-Aldrich) was drop-cast on a cleaned Si/SiO2 substrate which is placed in the central hot zone (T ≈ 750 °C) of a quartz tube (single zone Thermo Fisher Lindberg Blue M split tube furnace), and 500 mg of sulphur powder (99%, Sigma Aldrich) was kept at the upstream end. The deposition was carried out under an argon atmosphere at 750 °C for 10 minutes and then naturally cooled down to room temperature.2.2 Fabrication of Au NAs and SV-MoS2
300 nm diameter circular patterns were fabricated over the Si/SiO2 substrate using e-beam lithography (arrays of 300 × 300 μm area) with PMMA as the resist using a Raith Pioneer 2 electron beam system. Metal deposition of Cr/Au (≈50 nm) was done using a thermal evaporator, followed by standard lift-off using acetone. The diameter and the height of the gold nanopillars were fixed to ≈300 nm and ≈50 nm. The overall strain can be tuned by the nanopillar separation (2 μm, 1 μm, and 0.5 μm). The maximum strain is induced at the pillar edges, and the entire flake is non-uniformly strained via the wrinkles, which crucially modifies the local electron and phonon band structures.26 The transfer of MoS2 flakes was done using the polymethyl methacrylate (PMMA) assisted wet transfer method. The PMMA layer is spin-coated over the as-grown substrate and kept floating in a 3 M KOH solution until the PMMA layer is lifted from the Si/SiO2 substrate. These PMMA layers containing MoS2 flakes were washed with DI water and then scooped onto the Au NA pre-patterned substrate, and the PMMA layer was then removed using acetone. Depending on the pillar distance, the strained MoS2 samples were given the names S2μm-MoS2, S1μm-MoS2, and S0.5μm-MoS2. Later, the strained MoS2 domains (S-MoS2) were subjected to plasma treatment for several seconds. S-MoS2 was placed in a plasma chamber for 10–50 seconds for introducing vacancies (SV-MoS2).2.3 Materials characterization
The MoS2 flakes and the Au NA's morphology were identified using optical microscopy (Olympus BX41). The Raman and photoluminescence spectra were measured with a 532 nm laser excitation source using a Horiba Raman Xplora Plus confocal Raman spectroscope. All the spectra and mappings were collected at room temperature. The height profile and atomic force microscopy (AFM) images were obtained using a JPK BIOAFM Nano Wizard. An X-ray photoelectron spectrometer (XPS) (Scienta omicron 540, using a monochromatic Mg Kα source; 1200 eV excitation) is used to identify chemical composition. Transmission electron microscopy was done with an FEI Tecnai F30 (300 keV), and the samples were prepared on a TEM grid by the PMMA-assisted transfer method.2.4 Device fabrication and electrochemical measurements for the HER
Electrochemical measurements were performed on MoS2, S-MoS2, and SV-MoS2 domains using microcell devices fabricated by using standard lithography techniques. S-MoS2 was identified for device fabrication using an optical microscope. The contact pads are defined using standard photolithography (ML3, DMO), and the microelectrodes from the contact pad to the specific domains are patterned using e-beam lithography, followed by Cr/Au metallization (50 nm) via thermal evaporation followed by lift-off. Later, the device is spin-coated with the PMMA polymer, and a reaction window is unwrapped on top of S-MoS2 through e-beam lithography. The electrochemical HER measurements are done using a standard three-electrode setup. For microreactor measurements, S-MoS2 – gold electrodes were used as the working electrode. The counter and reference electrodes were a sharp platinum rod and an Ag/AgCl reference microelectrode. The electrochemical measurements were measured in a small drop of 0.5 M H2SO4 solution. The linear sweep voltammetry was used to measure polarization curves from 0 to −400 mV vs. the reversible hydrogen electrode (RHE) at a scan rate of 5 mV s−1 using an Origa Flex-OGF500 potentiostat–galvanostat workstation. The current densities were calculated by normalizing the measured currents by the window surface area exposed to the electrolyte solution.3. Results and discussion
3.1 Synthesis and characterization of strained and vacancy-rich MoS2
We synthesized MoS2 domains on Si/SiO2 substrates via the chemical vapor deposition method (Fig. S1a and b†), as detailed in the Experimental methods section. The optical image (Fig. 2a) and atomic force microscopy (AFM) image (Fig. 2b) clearly show the presence of typical triangular domains with lateral size greater than 50 μm. The AFM height profile (inset of Fig. 2b) reveals a thickness of ∼0.71 nm that corresponds to monolayer flakes. The MoS2 domains on the Si/SiO2 substrate are transferred onto copper grids via the PMMA-assisted wet transfer method28 and are further characterized by transmission electron microscopy (TEM) to reveal the surface morphology (Fig. S1c†). The selected area electron diffraction (SAED) pattern (inset of Fig. S1c†) reveals the hexagonal lattice structure of MoS2 flakes. The lattice fringes with 0.26 nm separation marked in the high-resolution TEM (HRTEM) image correspond to the (110) planes of MoS2 (Fig. S1d†). MoS2 domains were further characterized using Raman and photoluminescence (PL) spectroscopy. The Raman spectrum showed characteristic peaks at 385.3 cm−1 (E2g1) and 403.5 cm−1 (A1g), with a peak separation of about 18.2 cm−1, corresponding to monolayer domains (Fig. S1e†). The PL spectrum shows photoluminescence peaks at 1.82 eV and 1.97 eV, corresponding to the A1 and B1 direct excitonic transitions, respectively, for the MoS2 monolayer (Fig. S1f†). The Raman and PL intensity mapping was performed for the A1g mode at 403 cm−1 and the A1 peak at 1.82 eV, respectively, which showed uniform intensity (Fig. S1g and h†), suggesting the uniform thickness of MoS2 crystals.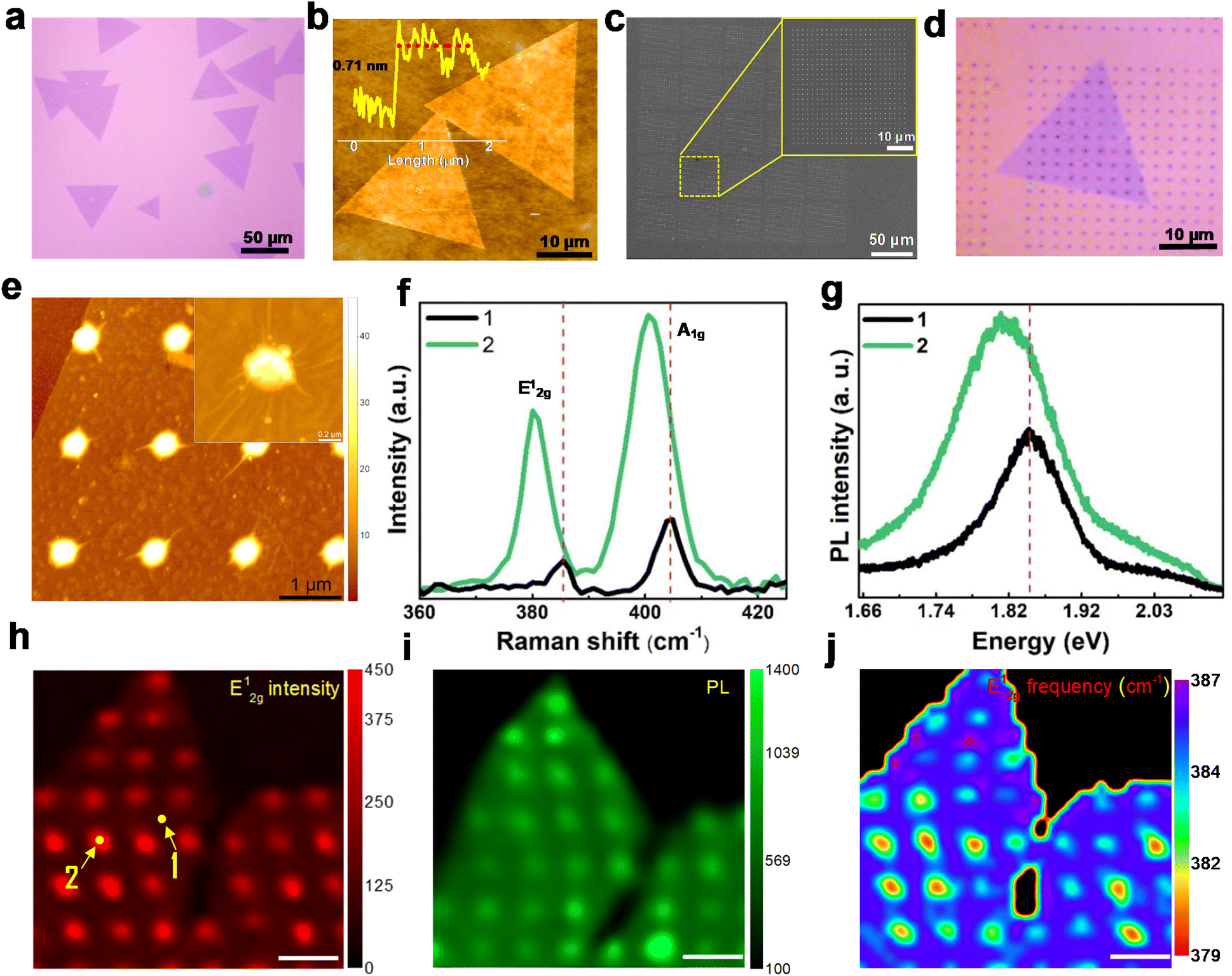 | ||
| Fig. 2 (a) Optical microscopy image and (b) AFM image of CVD-grown MoS2 monolayer domains; the inset shows the height profile in the marked region. (c) SEM image of AuNAs with a separation of 2 μm, and the inset shows the magnified image. (d) Optical image of S2μm-MoS2. (e) AFM image of S2μm-MoS2 clearly shows wrinkles on the layers. (f) Raman and (g) PL spectra of S2μm-MoS2 recorded from spots corresponding to the locations 1 and 2 marked on the Raman map image (E2g1 intensity) shown in (h). The clear shift in the spectra shows the action of biaxial strain. (i) PL map image at 1.82 eV of S2μm-MoS2, and (j) Raman map image (E2g1 peak position) (scale bars of (h–j) are 2 μm). | ||
To introduce non-uniform strain in the 2D MoS2 structure, the CVD-grown flakes are transferred via the PMMA-assisted wet chemical transfer method onto Au nanopillar arrays that are pre-patterned on Si/SiO2 substrates using e-beam lithography (see experimental methods). Subsequently, the monolayer MoS2 domain is subjected to a biaxial strain, whose magnitude is further modulated by varying the periodicity of the nanopillars. Keeping the diameter (∼300 nm) and height (∼50 nm) of the pillars fixed, the inter-pillar separation (center to center) is varied at 2 μm, 1 μm, and 0.5 μm, thereby altering the average strain experienced by the MoS2 domain. The strained-MoS2 (S-MoS2) domains with varying magnitudes of non-uniform strain are hereafter named S2μm-MoS2, S1μm-MoS2, and S0.5μm-MoS2. The wrinkles formed on the monolayer MoS2 domains, nucleated during their draping over the AuNAs, are critical to realizing the overall strain distribution in the flakes. The strain induced by draping MoS2 flakes over periodic Au nanopillars modifies their local electronic properties26 and could enhance the HER activity of monolayer domains.
Fig. 2c shows the scanning electron microscopy (SEM) image of Au nanopillar arrays (AuNAs) that reveal uniformly patterned pillars on Si/SiO2 substrates. The inset shows the magnified image of the AuNAs with 2 μm spacing. MoS2 monolayer domains are randomly placed on the AuNAs with 2 μm, 1 μm, and 0.5 μm spacing (Fig. 2d and S2†). This facilitates wrinkle formation on the strained domain, as clearly revealed in the AFM images given in Fig. 2e for S2μm-MoS2. With the decrease in the separation of AuNAs, the overall biaxial strain exerted on the monolayer MoS2 domains increases, resulting in more wrinkles, as clearly observed from the SEM images of S0.5μm-MoS2 (Fig. S3a and b†).
The strain-induced changes in local optical and electronic properties of strained monolayer MoS2 flakes are investigated with Raman and PL spectroscopy. On average, the 2D flake (S-MoS2) experiences higher strain at the AuNAs edges than in the region between pillars. This has been verified from the redshift in the Raman and PL spectra, as shown in Fig. 2f and g, respectively, which are recorded from the two regions marked in the Raman map of the E2g1 peak, displayed in Fig. 2h. The region between pillars and that atop the gold pillar are represented by the numbers 1 and 2, respectively, in the Raman map (Fig. 2h). Due to the funneling effect, the PL peak intensity enhancement at the strained area is higher than that in the unstrained regions.29,30 We further quantified the strain to be in the range of 0.2–1.3%, based on the redshift (1–3.5 cm−1) observed in the E2g1 Raman peak,31 indicative of increased yet non-uniform local strain across the flakes. The redshift in the PL spectrum has been reported to be ∼45 meV/% strain for uniaxial tensile strain and ∼−99 meV/% under biaxial strain (Fig. 2i). Consequently, S2μm-MoS2 showed a shift of ∼70 meV, corresponding to 0.7 to 1.5% strain, depending on the uniaxial or biaxial nature of the local strain.31–33 The local strain variation across the MoS2 flake can be spatially resolved from the Raman and PL intensity maps. The variation in the E2g1 mode intensity (Fig. 2h) clearly shows that the Raman signal is more intense on top of the AuNAs, accompanied by mode softening and redshift. Similarly, the Raman A1g intensity map and PL intensity map at 1.82 eV for S2μm-MoS2 are shown in Fig. S4 and 2i,† respectively. Spatial variations in the wavenumber of the E2g1 mode are depicted in Fig. 2j, which clearly distinguishes the strained and unstrained regions.
Creating sulphur vacancy defects is another effective approach for activating the inert basal plane of MoS2, by which new interstitial states are generated close to the Fermi level, thereby enhancing the HER performance of MoS2.19,34 We employed oxygen plasma treatment to introduce sulphur vacancy defects in MoS2. Samples exposed to oxygen plasma for 10, 20, 30, and 50 s, are named V10-MoS2, V20-MoS2, V30-MoS2, and V50-MoS2, respectively, and the corresponding domains clearly reveal a gradual reduction in the optical contrast (Fig. S5a–e†). Sulphur vacancies are created in MoS2 (Fig. S6a†) following oxygen plasma treatment, which is characterized by using various techniques, including Raman and PL measurements. A marked difference in SEM image contrast is observed between the as-grown MoS2 and V30-MoS2, as shown in Fig. S6b and c,† respectively. Isolated cracks are observed in the AFM image of V30-MoS2 (Fig. S6d†). We found that plasma exposure beyond 50 s resulted in several cracks in the sample, and assessing the electrochemical performance thus becomes very challenging. Raman and photoluminescence (PL) spectra further illustrate the structural changes of CVD-grown MoS2 upon plasma exposure. As shown in Fig. S6e,† Raman spectra of V-MoS2 show a significant decrease in the intensity of both A1g and E2g1 modes compared to its pristine counterpart. The separation between the two characteristic peaks (∼18.2 cm−1) confirms the monolayer nature of MoS2. After subjecting the CVD-grown MoS2 monolayer domains to 10 seconds of plasma exposure, the peak position of the A1g mode is found to shift from 403.5 cm−1 to 405.0 cm−1, and that of E2g1 mode from 385.3 cm−1 to 382.5 cm−1, respectively. The separation between the two characteristic peaks for MoS2 and V10-MoS2 is recorded at 18.2 cm−1 and 22.5 cm−1, respectively, and the peak shift can be attributed to the surface defects.35,36 Furthermore, the decrease in intensity of the PL peak (1.82 eV) observed for the samples subjected to increasing plasma exposure time (Fig. S6f†) suggests that more defects and cracks are formed.37 Thus, the observed PL quenching implies that oxygen bombardment has altered the allowed recombination mechanisms of pristine MoS2 due to lattice distortion.35
We further adopted another interesting approach, wherein the strained MoS2 (S-MoS2) domains are subjected to plasma treatment, that would ensure synergistic creation of additional vacancy defects along with strain-induced active sites, that could potentially result in further enhancement in HER performance of CVD-grown MoS2. The SEM and AFM images of S0.5μm-MoS2 subjected to 30 s of plasma treatment, which is named S0.5μmV30-MoS2, clearly reveal the presence of wrinkles (Fig. 3a and b). X-ray photoelectron spectroscopy (XPS) was used to examine the surface elemental composition of MoS2 samples. The high-resolution Mo 3d and S 2p peaks provide essential information about the stoichiometry and the valence state of the MoS2 flakes (Fig. 3c, d). The Mo 3d5/2 and 3d3/2 peaks are observed at 229.5 and 232.7 eV, respectively, while the S 2p3/2 and S 2p1/2 peaks are seen at 162.5 and 163.8 eV, respectively. These peak positions are consistent with the reported values for 2H-MoS2 flakes.34 The XPS spectra of Mo 3d and S 2p peaks for V30-MoS2 show a shift in the binding energy towards lower energy, as shown in Fig. S7.†38 The decrease in the ratio of intensities of S 2p to Mo 3d, indicates the formation of sulphur vacancies. For the S2μm-MoS2 samples, the Mo 3d5/2 and Mo 3d3/2 peaks are observed at 228.4 and 231.5 eV, respectively, while the S 2p3/2 and S 2p1/2 peaks are seen at 161.4 and 162.5 eV, respectively. The S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mo ratio was calculated to be 1.99, based on the peak area ratio of Mo 3d5/2 and S 2p3/2 peaks. The S:Mo ratio of as-grown MoS2 is normalized to 2.39 Finally, the high-resolution XPS spectra of Mo and S for the S2μmV30-MoS2 samples showed a significant shift toward lower binding energy as the Mo 3d5/2 and Mo 3d3/2 peaks are observed at 228.3 and 231.5 eV, respectively, while the S 2p3/2 and S 2p1/2 peaks are seen at 161.3 and 162.3 eV, respectively. The S:Mo peak area ratio for S2μmV30-MoS2 (1.74) is about 13% lower than that of as-grown MoS2, indicative of 13% S-vacancies in the sample.
:Mo ratio was calculated to be 1.99, based on the peak area ratio of Mo 3d5/2 and S 2p3/2 peaks. The S:Mo ratio of as-grown MoS2 is normalized to 2.39 Finally, the high-resolution XPS spectra of Mo and S for the S2μmV30-MoS2 samples showed a significant shift toward lower binding energy as the Mo 3d5/2 and Mo 3d3/2 peaks are observed at 228.3 and 231.5 eV, respectively, while the S 2p3/2 and S 2p1/2 peaks are seen at 161.3 and 162.3 eV, respectively. The S:Mo peak area ratio for S2μmV30-MoS2 (1.74) is about 13% lower than that of as-grown MoS2, indicative of 13% S-vacancies in the sample.
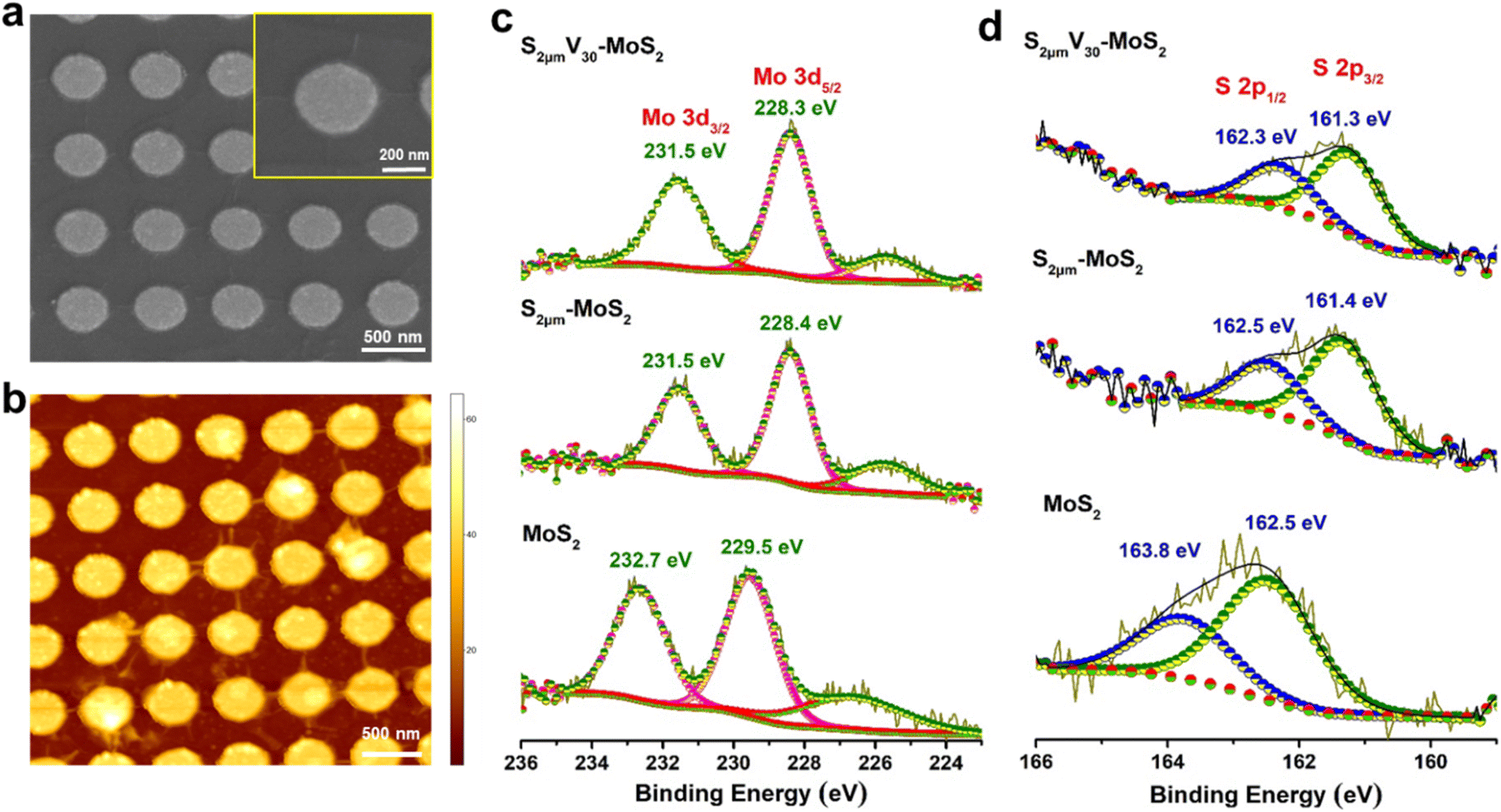 | ||
| Fig. 3 (a) SEM and (b) AFM images of S0.5μmV30-MoS2. XPS spectra of (c) Mo 3d and (d) S 2p of MoS2, S2μm-MoS2 and S2μmV30-MoS2. | ||
3.2 Electrochemical properties of the strained MoS2 microcell assembly towards the HER
Electrochemical performance of strained MoS2 domains is studied by fabricating on-chip microcell devices (Fig. 4a). After making necessary contacts from the desired domains of interest, the device is coated with PMMA, exposing a rectangular area of the respective domain (Fig. 4b and S8†). This would ensure accurate quantification of the obtained electrochemical data corresponding to the sample area under the exposed window of a single MoS2 domain (Fig. 4b). The detailed fabrication procedure involving a three-step lithography process, deposition, and lift-off is detailed in the Experimental methods (Section 2.4). A three-electrode cell configuration is employed using the strained MoS2 domain on AuNAs in contact with the Cr/Au pad as the working electrode, covered with a drop of 0.5 M H2SO4 electrolyte, Ag/AgCl as the reference electrode, and a Pt wire as the counter electrode. To study the effect of strain on the HER performance of MoS2 domains, we recorded linear sweep voltammetry (LSV) polarization curves and obtained Tafel plots for all the on-chip microcell devices fabricated with monolayer MoS2, S2μm-MoS2, S1μm-MoS2, and S0.5μm-MoS2 (Fig. 4c). The low overpotential at a constant current density (10 mA cm−2) and low Tafel slopes are indicative key parameters for good electrocatalytic performances. The overpotential (at 10 mA cm−2) of the pristine MoS2 single domain on a planar substrate is measured to be 282 mV. The strained domains, S2μm-MoS2, S1μm-MoS2, and S0.5μm-MoS2, exhibited lower overpotentials of 153 mV, 128 mV, and 107 mV, respectively (Fig. 4c). The as-grown MoS2 base plane exhibits relatively poor HER activity because of its dangling bond-free inert basal plane.40 However, all the strained domains are shown to exhibit lower overpotentials, clearly indicating improved HER performance with an increase in overall strain. The draping of MoS2 flakes over the nanopillar arrays generates non-uniform strain on the domains, which increases with a decrease in the spacing between Au nanopillars, as reflected in the formation of wrinkles and bubbles on the domains. On comparison of the Tafel slopes derived for all the samples from the respective LSVs, we further observe that S0.5μm-MoS2 with the highest strain (least spacing between the Au nanopillars) shows the lowest value of 79 mV dec−1, compared to pristine MoS2 (188 mV dec−1), S2μm-MoS2 (155 mV dec−1), and S1μm-MoS2 (154 mV dec−1), as depicted in Fig. 4d. This clearly indicates that increasing non-uniform strain enhances the HER performance, which is consistent with the results reported in the literature.17,41 The overpotential and the Tafel slope measured for different MoS2 samples under strain are graphically depicted in Fig. S9.† | ||
| Fig. 4 (a) Schematic illustration of SV-MoS2 microcell device employed for the electrocatalytic HER studies. (b) Optical microscope image of the fabricated microcell device. (c) Linear sweep voltammetry (LSV) curves recorded for pristine MoS2 and S-MoS2 with varying inter-pillar spacings, and (d) the corresponding Tafel plots. (e) LSV curves and (f) the corresponding Tafel plots recorded for S0.5μmV-MoS2 with varying plasma exposure durations. | ||
We further studied the effect of sulphur vacancy/defects in improving the basal plane activity of CVD-grown monolayer MoS2 single domains towards enhanced HER performance. For instance, V10-MoS2 exhibited considerably higher current density compared to pristine MoS2 (Fig. S10†). Such improvements in HER activity come from the nonhomogeneous distribution of surface atoms that generate intrinsic catalytic activity at its basal plane.39,42 To further comprehend the synergistic impact of vacancy/defects and strain on the HER performance of MoS2, our highly strained MoS2 (S0.5μm-MoS2) is subjected to plasma treatments under varying exposure times. Thus, obtained samples are labeled as S0.5μmV10-MoS2, S0.5μmV20-MoS2, S0.5μmV30-MoS2, and S0.5μmV50-MoS2, corresponding to a plasma exposure time of 10 s, 20 s, 30 s, and 50 s, respectively. The overpotential and Tafel slope values are estimated from the respective LSV curves (Fig. 4e), and their comparison bar plots for all the studied SV-MoS2 samples are represented in Fig. S11.† Upon generation of vacancies in the strained MoS2 domain (S0.5μm-MoS2), the overpotential of S0.5μmV30-MoS2 is reduced further to 53 mV and exhibited a much lower Tafel slope of 36.1 mV dec−1 (Fig. 4e and f). We found that the overall HER performance of the MoS2 single domain, following strain engineering and vacancy generation, improved dramatically compared to the pristine MoS2 domain, and the obtained overpotential and Tafel slope for our best sample (S0.5μmV50-MoS2) are among the best reported in the literature (Table S1†). The non-uniform strain exerted on the MoS2 domain would result in the variation of the coordination environment of Mo atoms in the lattice, leading to the modulation of its band gap. This would in turn change the density of states (DoS) near the Fermi energy level (Ef), resulting in an upshift of the d-band centre.11 The d-band center being closer to the Ef indicates stronger adsorption of hydrogen with the catalyst surface, as per the d-band theory, leading to enhanced HER performance.17,43 A similar upshift in the d-band center is realized with the creation of vacancies in the TMD lattice, which alters the coordination environment of Mo atoms in the lattice. Coincidence of both strain and vacancies in the lattice is expected to act synergistically towards increasing the DoS near the Ef, resulting in enhanced electrocatalytic activity towards the HER.
4. Conclusion
In summary, we fabricated highly strained and vacancy-rich monolayer MoS2 catalysts for efficient electrocatalytic hydrogen evolution. CVD-grown monolayer MoS2 domains are transferred onto gold nanopillar arrays pre-patterned on Si/SiO2 substrates and subsequently exposed to oxygen plasma treatment for inducing S-vacancies. The overall strain was varied by adjusting the inter-pillar spacing from 2 μm to 0.5 μm, keeping the height and diameter of the pillars constant. Furthermore, the duration of oxygen plasma treatment was varied, and optimized parameters were established to achieve high performance HER activity. The HER studies are performed in a microcell assembly, and the S0.5μmV50-MoS2 device showed exceptional electrocatalytic properties with a low overpotential of 53 mV. The synergistic effect of strain and vacancy-induced basal plane activation has resulted in improved electrocatalytic activity of MoS2 towards the HER.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
RN, AK, and MMS conceived the idea and designed the experiments. RN performed materials synthesis and electrochemical measurements. SD performed the microcell fabrication. RN, SD, and AK performed the data analysis. RN and MMS wrote the manuscript with inputs from SD, AK, and JM. MMS supervised the work.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
MMS acknowledges the Science and Engineering Research Board, Department of Science and Technology, India (CRG/2021/006246) for the financial support. JM acknowledges financial support from the MOE-STARS (STARS-2/2023-1012) and SPARC-UKIERI (Project No. 3086). R. N. is grateful to the University Grants Commission (UGC), Government of India, A. K. acknowledges the PhD fellowship from IISER Thiruvananthapuram, and SD acknowledges DST INSPIRE for the financial support.References
- A. Mondal and A. Vomiero, Adv. Funct. Mater., 2022, 32, 2208994 CrossRef CAS.
- H. Liu, R. Xie, Y. Luo, Z. Cui, Q. Yu, Z. Gao, Z. Zhang, F. Yang, X. Kang, S. Ge, S. Li, X. Gao, G. Chai, L. Liu and B. Liu, Nat. Commun., 2022, 13, 6382 CrossRef CAS PubMed.
- J. Zhang, J. Wu, H. Guo, W. Chen, J. Yuan, U. Martinez, G. Gupta, A. Mohite, P. M. Ajayan and J. Lou, Adv. Mater., 2017, 29, 1701995 Search PubMed.
- C. Zhang, Y. Luo, J. Tan, Q. Yu, F. Yang, Z. Zhang, L. Yang, H.-M. Cheng and B. Liu, Nat. Commun., 2020, 11, 3724 CrossRef CAS PubMed.
- Y. Luo, L. Tang, U. Khan, Q. Yu, H.-M. Cheng, X. Zou and B. Liu, Nat. Commun., 2019, 10, 269 CrossRef PubMed.
- D. Voiry, R. Fullon, J. Yang, C. De Carvalho Castro E Silva, R. Kappera, I. Bozkurt, D. Kaplan, M. J. Lagos, P. E. Batson, G. Gupta, A. D. Mohite, L. Dong, D. Er, V. B. Shenoy, T. Asefa and M. Chhowalla, Nat. Mater., 2016, 15, 1003–1009 CrossRef CAS PubMed.
- D. Gopalakrishnan, D. Damien and M. M. Shaijumon, ACS Nano, 2014, 8, 5297–5303 CrossRef CAS PubMed.
- Z. Zheng, L. Yu, M. Gao, X. Chen, W. Zhou, C. Ma, L. Wu, J. Zhu, X. Meng, J. Hu, Y. Tu, S. Wu, J. Mao, Z. Tian and D. Deng, Nat. Commun., 2020, 11, 3315 CrossRef CAS PubMed.
- X. Han, X. Tong, X. Liu, A. Chen, X. Wen, N. Yang and X.-Y. Guo, ACS Catal., 2018, 8, 1828–1836 CrossRef CAS.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- X. Mao, Z. Qin, S. Ge, C. Rong, B. Zhang and F. Xuan, Mater. Horiz., 2022, 10, 340–360 RSC.
- S. Ozden, S. Bawari, S. Vinod, U. Martinez, S. Susarla, C. Narvaez, J. Joyner, C. S. Tiwary, T. N. Narayanan and P. M. Ajayan, Nanoscale, 2019, 11, 12489–12496 RSC.
- S. Jiao, X. Fu and H. Huang, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- M. Luo and S. Guo, Nat. Rev. Mater., 2017, 2, 17059 CrossRef CAS.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- G. Wu, X. Han, J. Cai, P. Yin, P. Cui, X. Zheng, H. Li, C. Chen, G. Wang and X. Hong, Nat. Commun., 2022, 13, 4200 CrossRef CAS PubMed.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1–28 CrossRef.
- D. Pierucci, H. Henck, Z. Ben Aziza, C. H. Naylor, A. Balan, J. E. Rault, M. G. Silly, Y. J. Dappe, F. Bertran, P. Le Fèvre, F. Sirotti, A. T. C. Johnson and A. Ouerghi, ACS Nano, 2017, 11, 1755–1761 CrossRef CAS PubMed.
- D. Lloyd, X. Liu, N. Boddeti, L. Cantley, R. Long, M. L. Dunn and J. S. Bunch, Nano Lett., 2017, 17, 5329–5334 CrossRef CAS PubMed.
- J. Wang, M. Han, Q. Wang, Y. Ji, X. Zhang, R. Shi, Z. Wu, L. Zhang, A. Amini, L. Guo, N. Wang, J. Lin and C. Cheng, ACS Nano, 2021, 15, 6633–6644 CrossRef CAS PubMed.
- L. Li, Z. Qin, L. Ries, S. Hong, T. Michel, J. Yang, C. Salameh, M. Bechelany, P. Miele, D. Kaplan, M. Chhowalla and D. Voiry, ACS Nano, 2019, 13, 6824–6834 CrossRef CAS PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- G. Li, D. Zhang, Q. Qiao, Y. Yu, D. Peterson, A. Zafar, R. Kumar, S. Curtarolo, F. Hunte, S. Shannon, Y. Zhu, W. Yang and L. Cao, J. Am. Chem. Soc., 2016, 138, 16632–16638 CrossRef CAS PubMed.
- W. Han, Z. Liu, Y. Pan, G. Guo, J. Zou, Y. Xia, Z. Peng, W. Li and A. Dong, Adv. Mater., 2020, 32, 1–9 Search PubMed.
- A. Castellanos-Gomez, R. Roldán, E. Cappelluti, M. Buscema, F. Guinea, H. S. J. van der Zant and G. A. Steele, Nano Lett., 2013, 13, 5361–5366 CrossRef CAS PubMed.
- A. Kayal, S. Dey, H. Gopalakrishnan, R. Nadarajan, S. Chattopadhyay and J. Mitra, Nano Lett., 2023, 23, 6629–6636 CrossRef CAS PubMed.
- H. Li, A. W. Contryman, X. Qian, S. M. Ardakani, Y. Gong, X. Wang, J. M. Weisse, C. H. Lee, J. Zhao, P. M. Ajayan, J. Li, H. C. Manoharan and X. Zheng, Nat. Commun., 2015, 6, 7381 CrossRef CAS PubMed.
- P. V. Sarma, A. Kayal, C. H. Sharma, M. Thalakulam, J. Mitra and M. M. Shaijumon, ACS Nano, 2019, 13, 10448–10455 CrossRef CAS PubMed.
- J. Lee, S. J. Yun, C. Seo, K. Cho, T. S. Kim, G. H. An, K. Kang, H. S. Lee and J. Kim, Nano Lett., 2021, 21, 43–50 CrossRef CAS PubMed.
- A. Castellanos-Gomez, R. Roldán, E. Cappelluti, M. Buscema, F. Guinea, H. S. J. van der Zant and G. A. Steele, Nano Lett., 2013, 13, 5361–5366 CrossRef CAS PubMed.
- S. Manzeli, A. Allain, A. Ghadimi and A. Kis, Nano Lett., 2015, 15, 5330–5335 CrossRef CAS PubMed.
- H. J. Conley, B. Wang, J. I. Ziegler, R. F. Haglund, S. T. Pantelides and K. I. Bolotin, Nano Lett., 2013, 13, 3626–3630 CrossRef CAS PubMed.
- G. Plechinger, A. Castellanos-Gomez, M. Buscema, H. S. J. van der Zant, G. A. Steele, A. Kuc, T. Heine, C. Schüller and T. Korn, 2D Mater., 2015, 2, 015006 CrossRef.
- J. Chen, W. Tang, B. Tian, B. Liu, X. Zhao, Y. Liu, T. Ren, W. Liu, D. Geng, H. Y. Jeong, H. S. Shin, W. Zhou and K. P. Loh, Adv. Sci., 2016, 3, 1600033 Search PubMed.
- N. Kang, H. P. Paudel, M. N. Leuenberger, L. Tetard and S. I. Khondaker, J. Phys. Chem. C, 2014, 118, 21258–21263 CrossRef CAS.
- L. Gao, Q. Liao, X. Zhang, X. Liu, L. Gu, B. Liu, J. Du, Y. Ou, J. Xiao, Z. Kang, Z. Zhang and Y. Zhang, Adv. Mater., 2020, 32, 1906646 CrossRef PubMed.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16, 1097–1103 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed.
- C. Tsai, H. Li, S. Park, J. Park, H. S. Han, J. K. Nørskov, X. Zheng and F. Abild-Pedersen, Nat. Commun., 2017, 8, 15113 CrossRef PubMed.
- H. Li, C. Tsai, A. L. Koh, L. Cai, A. W. Contryman, A. H. Fragapane, J. Zhao, H. S. Han, H. C. Manoharan, F. Abild-Pedersen, J. K. Nørskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- X. Liu, Z. Li, H. Jiang, X. Wang, P. Xia, Z. Duan, Y. Ren, H. Xiang, H. Li, J. Zeng, Y. Zhou and S. Liu, Small, 2024, 20, 2307293 CrossRef CAS PubMed.
- J. Seok, J.-H. Lee, S. Cho, B. Ji, H. W. Kim, M. Kwon, D. Kim, Y.-M. Kim, S. H. Oh, S. W. Kim, Y. H. Lee, Y.-W. Son and H. Yang, 2D Mater., 2017, 4, 025061 CrossRef.
- K. Yan, T. A. Maark, A. Khorshidi, V. A. Sethuraman, A. A. Peterson and P. R. Guduru, Angew. Chem. Int. Ed., 2016, 55, 6175–6181 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04874a |
| This journal is © The Royal Society of Chemistry 2024 |