
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDonor-modified asymmetric N/B/O multi-resonance TADF emitters for high-performance deep-blue OLEDs with the BT.2020 color gamut†
Jing
Jin‡
 a,
Zhaolong
He‡
a,
Di
Liu
*a,
Yongqiang
Mei
c,
Jiahui
Wang
c,
Huihui
Wan
d and
Jiuyan
Li
*bc
a,
Zhaolong
He‡
a,
Di
Liu
*a,
Yongqiang
Mei
c,
Jiahui
Wang
c,
Huihui
Wan
d and
Jiuyan
Li
*bc
aFrontier Science Center for Smart Materials, College of Chemistry, Dalian University of Technology, 2 Linggong Road, Dalian, 116024, China. E-mail: liudi@dlut.edu.cn
bShandong Laboratory of Yantai Advanced Materials and Green Manufacturing, Yantai Economic and Technological Development Zone, 300 Changjiang Road, Yantai, China. E-mail: greenjiuyanli@amgm.ac.cn
cFrontier Science Center for Smart Materials, College of Chemical Engineering, Dalian University of Technology, 2 Linggong Road, Dalian, 116024, China
dInstrumental Analysis Center, Dalian University of Technology, Dalian, 116024, China
First published on 7th October 2024
Abstract
Multi-resonance thermally activated delayed fluorescence (MR-TADF) materials of polycyclic heteroaromatics are attractive narrowband emitters in wide-color-gamut organic light-emitting diodes (OLEDs). However, deep-blue MR-TADF emitters with CIE coordinates fulfilling the BT.2020 standard and high efficiency still remain a significant challenge. Herein, two novel emitters NBO-mSAF and NBO-pSAF were developed by incorporating an electron donor, 10H-spiro[acridine-9,9′-fluorene] (SAF), at the para-position of the oxygen atom and the para-position of the boron atom in the nitrogen/boron/oxygen (N/B/O) ternary doped asymmetric MR skeleton. With appropriate electron-donating capacity and rigid spiro-structure, SAF was selected as the donor so that the long-range charge transfer triplet state (3LRCT) is induced to accelerate the reverse intersystem crossing (RISC) process, while the 1LRCT aligns higher than the short-range CT state (1SRCT) of the N/B/O core to retain the MR characters. As a result, these optimized emitters exhibit deep-blue TADF with narrow spectra and a high RISC rate constant of 3.4 × 105 s−1. In hyperfluorescence OLEDs with a TADF emitter DMAC-DPS as the sensitizer and PPF as the host, NBO-mSAF and NBO-pSAF achieved maximum external quantum efficiencies (EQEmax) of 26.7% and 25.2%. Interestingly, improved performance was realized in a traditional device configuration with a single bipolar host 26DCzPPy but without any sensitizer, where NBO-mSAF realized a higher EQEmax of 29.5% and CIE (0.128, 0.114), and NBO-pSAF exhibited an EQEmax of 20.5% and CIE of (0.147, 0.048). Narrow full width at half maximum (FWHM) values of 26–28 nm were achieved in both devices. Among all the deep-blue N/B/O type MR-TADF emitters with CIEx ≤ 0.15 and CIEy ≤ 0.12 ever reported so far, NBO-mSAF exhibited a highest EQEmax of 29.5%, which is even higher than those obtained with sensitizers, while the CIEy = 0.048 of the NBO-pSAF device is close to the standard blue (0.046) according to BT.2020, with a decent EQE of 20%.
Introduction
Organic light-emitting diodes (OLEDs) have garnered significant attention in display applications due to their multitude of advantages, encompassing lightweight design, superior color quality, expansive viewing angles, and ultra-fast response times.1–3 OLEDs are rapidly becoming the preferred display technology for a range of consumer electronics, from smartwatches, mobile phones, and laptops to large-screen televisions and displays used in the automotive industry. Ultrahigh-definition displays must meet ever more stringent industry color standards, the most recent of which being Broadcasting Television Services 2020 (BT.2020), which are defined according to the Commission International de l'Éclairage (CIE) 1931 as (0.131, 0.046), (0.170, 0.797) and (0.708, 0.292) for blue, green and red, respectively.4 The high performance of OLEDs relies on the efficient conversion of both electronically excited singlet (25%) and triplet (75%) excitons into photons, resulting in a high external quantum efficiency (EQE) and a small full width at half maximum (FWHM), ensuring extremely high emission color purity.5 Thermally activated delayed fluorescence (TADF) emitters have gained widespread recognition as the most promising category of luminescent materials in OLEDs due to their ability to overcome the limitations associated with the severe triplet exciton loss observed in traditional fluorescent materials, as well as the high production costs associated with noble metal based phosphorescent materials.6In comparison to the conventional donor–acceptor (D–A) materials, also termed as twisted intramolecular charge transfer (TICT) type TADF materials,7,8 that are typically characterized by broad emission spectra owing to the severe structural relaxation of the excited state and thus a wide FWHM, the newly developed multiple resonance TADF (MR-TADF) materials are absolutely advantageous in terms of narrow-band character and excellent color purity. The electron-poor boron (B) and electron-rich nitrogen (N) atoms are strategically incorporated into a rigid polycyclic aromatic hydrocarbon (PAH) framework,9,10 leveraging the anti-resonance effect between these atoms to achieve effective separation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) on adjacent atoms to form short-range charge transfer (SRCT) excited states. This innovative molecular design not only facilitates suitable overlap integral of the frontier molecular orbitals (FMO) and efficient radiative transition, but also crucially restricts structural relaxation and significantly narrows the FWHM of the S1 state (1SRCT). Accordingly, the MR-TADF emitters are generally characterized by a narrow-band, perfect color purity and high photoluminescence quantum yield (PLQY), and seem to be more suitable for ultrahigh-definition displays and have become the new favorite OLED emitters in both academic and industrial fields.1,3
Nevertheless, most of the reported MR-TADF emitters frequently suffer from the relatively large energy difference between the lowest singlet and triplet state (ΔEST) values compared to the well-elaborated D–A type TICT-TADF emitters, resulting in the typically lower reverse intersystem crossing rate (kRISC) (<105 s−1) and thus severe efficiency roll-off at high luminance intensities due to significant triplet exciton annihilation issues such as triplet–triplet annihilation (TTA) and triplet polaron quenching (TPQ).11 On the basis of the pioneering work of Hatakeyama and colleagues,9,10 enlarging the π-conjugated structure of the MR core has become one important modification strategy for novel MR emitters. Owing to the favorable properties of the famous DABNA-1 (Fig. 1),9 a number of MR-TADF materials were developed by constructing double or more N/B/N structures on a central benzene ring, which finally generated an enlarged π-conjugated MR core. Designed in such a way, ν-DABNA10 (Fig. 1) demonstrated even greater advantages in performance (e.g. FWHM = 18 nm, kRISC = 2.0 × 105 s−1, EQEmax = 34.4%) than the parent DABNA-1. However, the emission maximum of ν-DABNA (469 nm) was red-shifted by 10 nm from that of DABNA-1 because of the π-extension. As a result, the CIE coordinates (0.12, 0.11) deviated from the requirements defined by the National Television System Committee (0.14, 0.08).
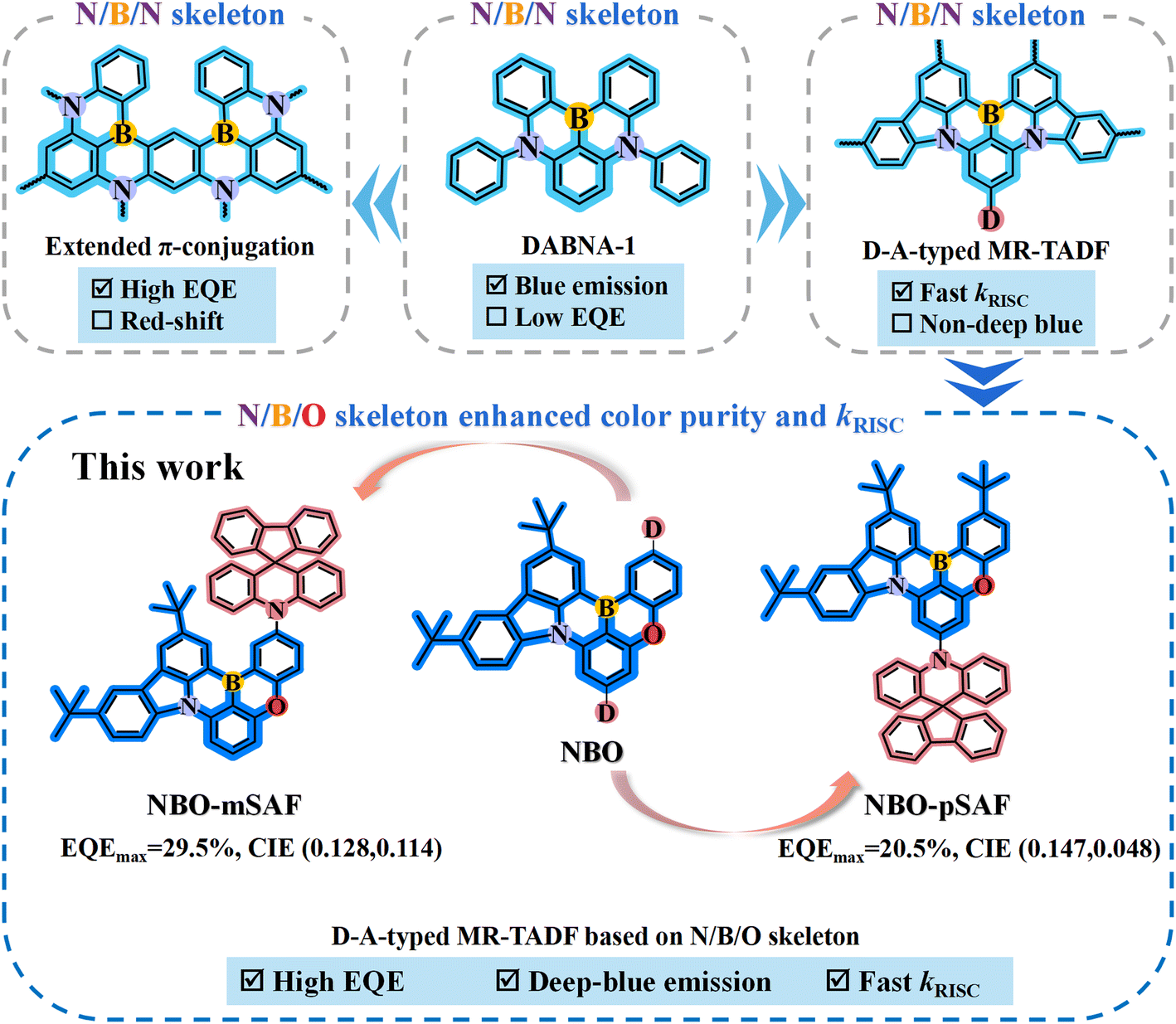 | ||
| Fig. 1 Molecular design strategies and the performance summary of NBO-mSAF and NBO-pSAF. | ||
Another typical structure modification method is to introduce an additional donor (Fig. 1) onto the N/B/N MR-skeleton to emulate the effect of traditional D–A-typed TADF emitters. By grafting a suitable electron donor, long-range charge transfer (LRCT) occurs between the MR core (acting as electron acceptor) and the donor, and the singlet LRCT excited state (1LRCT) aligns higher than the short-range CT state (1SRCT) of the MR core to guarantee the stability of the MR-TADF system and to ensure narrow-band characters and perfect emitting color purity, while the appearance of the triplet LRCT state (3LRCT) enriches the triplet states of the MR molecule and reduces the energy gaps between the adjacent triplet states (ΔETT) and dramatically facilitates the kRISC.12–14 Of course, the donor-modified MR-TADF derivatives definitely exhibit longer-wavelength fluorescence than the corresponding parent MR-core.15 However, the donor strength should be carefully controlled to avoid the case that the 1LRCT state falls below the 1SRCT state due to the too strong donor and the original characteristic narrow-band fluorescence of the MR core gives way to the broad spectra of the 1LRCT-TADF.12,13 It is clear that both the π-extension and donor incorporation strategies on N/B/N MR-skeletons are prone to cause a bathochromic shift of the emitting light, leading to further deviation from the pure- or deep-blue range. In the recent trend, continuous research has been focused on obtaining stable MR-TADF emitters having the compatibility of a narrower FWHM and large kRISC by engineering the MR effect to achieve highly stable and high color purity OLEDs.16,17 It is established that the N/B/O asymmetrically doped PAH (Fig. 1) usually has relatively shorter wavelength emission than the similar N/B/N skeletons, which may be better candidates if deep-blue MR-TADF is desired,18,19 especially when the rigorous BT. 2020 standard is employed.
Herein, we selected the N/B/O ternary asymmetrically doped PAH as the central MR skeleton to design two novel deep-blue MR-TADF materials, namely NBO-mSAF and NBO-pSAF (Fig. 1). 10H-Spiro[acridine-9,9′-fluorene] (SAF) was selected as the electron donor and was introduced for the first time at the para-position of the oxygen and boron atoms of the N/B/O MR-core. In comparison with the widely used acridine donor, SAF has weaker electron-donating ability. Simultaneous reduction of donor strength and acceptor strength is expected to generate relatively weak LRCT effect and make the 1LRCT state locate over the intrinsic 1SRCT state of the MR core to guarantee deep-blue emission.20 More importantly, its rigid spiro structure restrains excited-state structural relaxations and vibrations and thus benefits color purity and PLQY.20,21 Thanks to the appropriate electron-donating strength of the SAF group relative to the N/B/O MR-core and the suitably aligned 1LRCT state, both NBO-mSAF and NBO-pSAF not only have the same luminescence range relative to the corresponding donor-free N/B/O MR-core but also maintain narrower FWHMs (28 nm).22 The incorporation of the SAF donor at the periphery of the N/B/O MR nucleus played a key role in promoting the kRISC and triplet exciton utilization in comparison with the corresponding donor-free N/B/O MR-core due to the contribution from the additional 3LRCT state.
In the traditional structure OLEDs employing 2,6-bis(3-(9H-carbazol-9-yl)phenyl)pyridine (26DCzPPy) as a bipolar host, NBO-mSAF and NBO-pSAF exhibited the maximum device external quantum efficiency (EQEmax) of 29.5% and 20.5%, respectively, with the FWHM of 28 nm and 26 nm and CIE coordinates of (0.128, 0.114) and (0.147, 0.048), respectively. The EQEmax of 20.5% for NBO-pSAF is the highest efficiency so far for the N/B/O type deep-blue MR-TADF emitters under the BT.2020 standard (Fig. S1 and Table S1 in ESI†). Furthermore, enhanced efficiencies and dramatically suppressed efficiency roll-off were gained when a hyperfluorescence (HF) architecture with a D–A-type TADF emitter bis[4-(9,9-dimethyl-9,10-dihydroacridine)phenyl]sulfone (DMAC-DPS) as the sensitizer was utilized. The introduction of DMAC-DPS shortened the delayed fluorescence lifetime (τDF) without affecting the light color and narrow band character and significantly improved the PLQY to almost double. In addition to the remarkably improved EQEmax (25.2%) in the NBO-pSAF based HF device (vs. 26.7% for NBO-mSAF), the efficiency roll-off in both NBO-mSAF and NBO-pSAF HF devices was dramatically mitigated by means of the sensitized mechanism.
Results and discussion
Synthesis and theoretical calculation
NBO-mSAF and NBO-pSAF were synthesized through a palladium-catalyzed Buchwald cross-coupling reaction between the SAF fragment and the corresponding brominated MR core, NBO-mBr or NBO-pBr. The synthesis routes of intermediates and these two molecules are shown in Schemes S1 and S2,† and their structures were fully characterized by 1H NMR, 13C NMR and high-resolution mass spectrometry (Fig. S2–S9†).The molecular geometry and electronic properties of the studied emitters were investigated using Density Functional Theory (DFT) and Time-Dependent DFT (TD-DFT) calculations at the M062X/def2svp level.23 The optimized geometries of these molecules under vacuum conditions are shown in Fig. 2. The N/B/O plane and SAF donor present a highly distorted conformation, with high dihedral angles of 88.7° and 85.3° in NBO-mSAF and NBO-pSAF, respectively, similar to the general case of all acridine or acridine derivative based compounds due to the strong repulsion effect.24,25 The HOMOs of these emitters are mainly located on the SAF due to their relatively stronger electron-donating capability than the N/B/O core, while the LUMOs are mainly located on the boron atoms and their ortho/para-carbon atoms in the N/B/O skeleton, showing a similar FMO distribution to traditional D–A type TADF emitters. NBO-mSAF has a slightly deeper LUMO and a shallower HOMO than NBO-pSAF, and thus may be expected to show a slight red-shift in fluorescence. Simultaneously, the HOMO-1 orbitals are primarily located on the nitrogen/oxygen and carbon atoms in the ortho/para positions in the N/B/O skeleton (Fig. S10†).
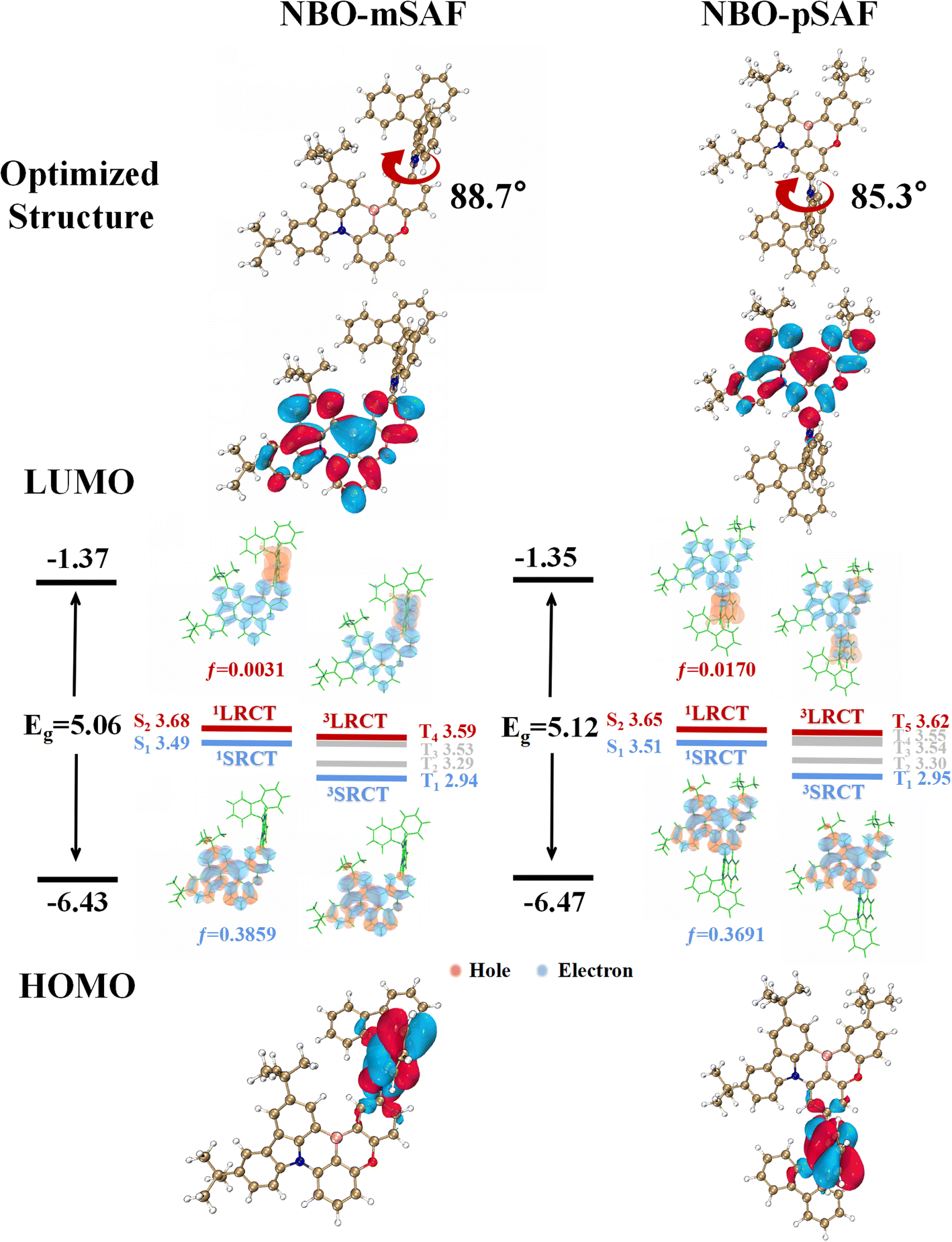 | ||
| Fig. 2 Optimized geometries, HOMO and LUMO distributions, simulated energy-level diagrams and natural transition orbital (NTO) distributions for NBO-mSAF and NBO-pSAF at the M062X/def2svp level. | ||
To clarify the emission properties, TD-DFT calculations were used to estimate the excited state energy level and analyze the natural transition orbital (NTO) (Fig. 2 and Table S2†). The results indicate that the S1 states of both NBO-mSAF and NBO-pSAF belong to the 1SRCT states, which ensures the MR characteristics of the whole molecule emission. NBO-mSAF and NBO-pSAF exhibit notable oscillator intensities (f) of 0.3859 and 0.3691, respectively, favoring the S1 → S0 radiation transition. The S2 states of NBO-mSAF and NBO-pSAF show an obvious 1LRCT state feature, in which holes and electrons are distributed on the SAF and N/B/O moieties, respectively. The 3LRCT is the T4 state of NBO-mSAF, and is the T5 state of NBO-pSAF, both of which are adjacent to their 1SRCT (S1) state. At the same time, the energy level difference between 1LRCT (S2) and 1SRCT (S1) is small for both emitters. This means that their 3LRCTs can probably act as the intermediary step to facilitate the T1 → S1 RISC conversion, which should be supported by the possibly favorable spin orbital coupling (SOC) between the 3LRCT and 1SRCT (S1) states (Table S3†). It is inferred that NBO-mSAF and NBO-pSAF may exhibit faster kRISC than the parent NBO molecule.
Electrochemical properties
The electrochemical properties of NBO-mSAF and NBO-pSAF were investigated by cyclic voltammetry (CV) measurements (Fig. S11†). Based on the almost identical profiles of the reduction and oxidation waves for these two compounds and the theoretically observed same HOMO and LUMO distribution (Fig. 2), it would be safe to deduce that the reduction should occur on the N/B/O skeleton moiety and the oxidation corresponds to the electron loss of the SAF donor in these two molecules. The HOMO energies were determined from the onset potential (Eonsetox) of the first oxidation wave according to the formula EHOMO = −(Eonsetox + 4.4). The LUMO energies of these compounds were determined from the onset potentials of the first reduction wave (Eonsetred) according to the formula ELUMO = –(Eonsetred + 4.4). The resultant EHOMO/ELUMO values were −5.37 eV/−2.90 eV and −5.41 eV/−2.91 eV for NBO-mSAF and NBO-pSAF, respectively, and the data are summarized in Table 1. It should be noted that these HOMO and LUMO levels match well with those widely used functional materials in OLEDs, indicating their high potential applications in optoelectronic devices.| Emitter | λ abs [nm] | λ em [nm] | FWHMa [nm] | E S [eV] | E T [eV] | ΔESTc [eV] | HOMO/LUMOd [eV] | E g [eV] |
|---|---|---|---|---|---|---|---|---|
| a Absorption and fluorescence peak wavelengths in dilute toluene solutions at room temperature. b Estimated from the wavelength onset of RT-FL in 2-MeTHF solution. c Estimated from the wavelength onset of LT-PH in 2-MeTHF solution at 77 K. ΔEST = ES − ET. d Determined from electrochemical measurements. | ||||||||
| NBO 22 | 434 | 448 | 25 | 2.88 | 2.77 | 0.11 | −5.40/−2.70 | 2.70 |
| NBO-mSAF | 440 | 458 | 28 | 2.78 | 2.56 | 0.22 | −5.37/−2.90 | 2.47 |
| NBO-pSAF | 432 | 446 | 28 | 2.83 | 2.64 | 0.19 | −5.41/−2.91 | 2.50 |
Photophysical properties
The fundamental photophysical properties of NBO-mSAF and NBO-pSAF were investigated by means of the ultraviolet-visible (UV-vis) absorption in dilute toluene solutions (10−5 mol L−1) and the photoluminescence (PL) spectra in dilute solutions in different polarity solvents (Fig. 3a and b). The essential photophysical parameters are compiled in Tables 1 and S4.† Absorption signals below 400 nm were attributed to the π–π* and n–π* transitions of arylamine and NBO segments, complemented by absorption bands at 440 and 432 nm for NBO-mSAF and NBO-pSAF that are characteristic of the SRCT transition within the MR segment. As predicted by the calculated results, NBO-mSAF exhibits a slight red-shift emission compared with NBO-pSAF. The fluorescence spectra of NBO-mSAF and NBO-pSAF revealed unique narrowband emissions with peaks at 458 and 446 nm and narrow FWHMs of 28 nm. Relative to the parent NBO that emits at 448 nm with a FWHM of 25 nm, the fluorescence of NBO-mSAF exhibits a 10 nm red-shift while NBO-pSAF has a 2 nm blue-shift, and both molecules exhibit a comparable FWHM, indicating that the introduction of the SAF donor has negligible influence on either the emitting color or the color purity. | ||
| Fig. 3 Normalized UV-vis absorption in dilute toluene (10−5 M) solution and solvatochromic effect on photoluminescence (PL) spectra of (a) NBO-mSAF and (b) NBO-pSAF, and the normalized PL spectra (c) and transient PL decay curves (d) of NBO-mSAF and NBO-pSAF in 6 wt% doped films of 26DCzPPy. | ||
In the solvatochromic study, the narrow-band fluorescence of both emitters exhibited tiny bathochromic effect in low polarity solvents, as illustrated in Fig. 3a, b and S12.† However, with solvent going to highly polar N,N-dimethylformamide (DMF) and acetonitrile (ACN), a new structureless and broad band appeared at near 560 nm for both emitters, which can be assigned to the fluorescence from the 1LRCT states. The 1LRCT states, i.e. the S2 states for these two emitters, were able to be detected in DMF and ACN because they originally have rather larger transition dipole moments than the 1SRCT states (S1) and are dramatically stabilized by strong polar solvents like DMF so as to approach the 1SRCT states or even drop below the 1SRCT states. As a result, both the narrow-band MR fluorescence from the 1SRCT state and the broad-band TICT emission from the 1LRCT state can be simultaneously detected in strong polar solvents. This is essential evidence to confirm the coexistence of 1SRCT and 1LRCT states by finely tuning the electronic properties of the MR-core and additional donor and selecting appropriate environmental conditions. The simultaneous detection of dual fluorescence in these two emitters is consistent with the NTO calculation observation that the 1SRCT and 1LRCT states are closely aligned in energy levels. According to the Lippert–Mataga equation (formula (S1)–(S4)),26 the excited dipole moments (μe) of the 1SRCT state are calculated from the relationship between the slope of the Stokes shift (νa − νf) and the orientation polarization (f) as 10.73 and 11.36 D for NBO-mSAF and NBO-pSAF, respectively (Fig. S12 and Table S5†), which are much smaller than those of the corresponding 1LRCT state (20.19 and 20.59 D). The onset points of the room temperature fluorescence (RT-FL) and low temperature (77 K) phosphorescence (LT-PH) spectra were utilized to calculate the ΔEST values, which are 0.22 eV and 0.19 eV for NBO-mSAF and NBO-pSAF, respectively (Fig. S13†). Given that the ΔESTs are sufficiently minimal, efficient RISC would be expected.
NBO-mSAF and NBO-pSAF were doped at 6 wt% in the 26DCzPPy host that has a large band gap and suitable T1 energy to make films for transient PL measurements to evaluate the TADF properties. As shown in Fig. 3c, the doped films exhibit similar PL spectra profiles to those solutions, but with red-shifts of 12 nm (from 458 nm to 470 nm) accompanied by an increase of 6 nm in the FWHM (from 28 to 34 nm) for NBO-mSAF due to intermolecular interactions in the solid film, while NBO-pSAF exhibits a smaller red-shift of 4 nm (from 448 nm to 452 nm) and the FWHM is broadened by 3 nm (28 to 31 nm), indicating relatively weak intermolecular interactions in the NBO-pSAF:26DCzPPy film.
Through the time-correlated single photon counting (TCSPC) technique, the prompt fluorescence lifetimes (τPF) (inset of Fig. 3d) of NBO-mSAF and NBO-pSAF were determined as 6.9 ns (91.7%) and 2.5 ns (86.4%), respectively. Through the multichannel scanning (MCS) method, in addition to the prompt fluorescence, a delayed component of each emitter was detected in the transient PL decay curve, which has an identical spectrum to the corresponding PF (Fig. S14†) and exhibits a lifetime of 17.1 μs (8.3%) for NBO-mSAF and 3.4 μs (13.6%) for NBO-pSAF (Fig. 3d and Table 2), confirming they are the delayed fluorescence (DF) of these emitters. The temperature dependent transient PL decay curves in Fig. S15† reveal that the PL intensity regularly increased with increasing temperature, especially in the short time range, confirming the TADF mechanism of the DF for these emitters. As anticipated, the introduction of donor SAF into the NBO-mSAF and NBO-pSAF resulted in a notable decrease of the τDF compared to the parent NBO (Table S6†), which should be favorable for effective up conversion of triplet excitons to singlet ones and inhibiting TTA and TPQ to a certain extent.
| Emitter | Host | Sensitizer | Φ PL [%] | Φ PF [%] | Φ DF [%] | τ PF [ns] | τ DF [μs] | k r [107 s−1] | k nr [107 s−1] | k ISC [107 s−1] | k RISC [104 s−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DMAC-DPS (x wt%) | |||||||||||
| a k r, knr, kISC, kRISC represent the rate constant of radiation, non-radiation, intersystem crossing, and reverse intersystem crossing, respectively; ΦPL, ΦPF, ΦDF, τPF and τDF represent total PLQY, quantum yield of PF, quantum yield of DF, and average lifetimes of PF and DF, respectively. | |||||||||||
| NBO-mSAF | 26DCzPPy | — | 60.0 | 55.0 | 5.0 | 6.9 | 17.1 | 8.0 | 5.3 | 1.2 | 6.4 |
| PPF | — | 55.0 | 42.3 | 12.7 | 11.1 | 22.8 | 3.8 | 3.1 | 2.1 | 5.7 | |
| 9 | 96.8 | 61.6 | 35.2 | 17.2 | 17.9 | 3.6 | 0.1 | 2.1 | 8.8 | ||
| 20 | 94.5 | 63.1 | 31.4 | 11.7 | 15.8 | 5.4 | 0.3 | 2.8 | 9.5 | ||
| NBO-pSAF | 26DCzPPy | — | 52.8 | 45.6 | 7.2 | 2.5 | 3.4 | 18.3 | 16.4 | 5.5 | 34.1 |
| PPF | — | 55.1 | 34.9 | 20.2 | 4.7 | 16.3 | 7.4 | 6.0 | 7.8 | 9.7 | |
| 9 | 84.8 | 42.1 | 42.7 | 9.9 | 18.3 | 4.2 | 0.8 | 5.1 | 11.0 | ||
| 20 | 90.4 | 39.2 | 51.2 | 11.3 | 15.5 | 3.5 | 0.4 | 5.0 | 14.8 | ||
The PLQYs (ΦPL) were measured as 60.0% and 52.8% for NBO-mSAF and NBO-pSAF in 26DCzPPy films (6 wt%), respectively, allowing determination of the quantum yields of PF and DF (ΦPF, ΦDF), and the rate constants of radiative decay (kr), of intersystem crossing (kISC), of RISC (kRISC) for each emitter (formula (S5)–(S11)). Relevant photophysical parameters are summarized in Table 2. As expected, NBO-mSAF and NBO-pSAF have larger kRISCs of 6.4 × 104 s−1 and 3.4 × 105 s−1 than the parent NBO (1.2 × 104 s−1, Table S6†). Evidently the enhanced kRISCs should be mainly attributed to the introduction of the SAF donor and the 3LRCT state (T4 or T5, Fig. 2) participates and facilitates the RISC process owing to both the tiny energy gap and the favorable SOC value for the 3LRCT → 1SRCT conversion. It is clear that NBO-mSAF and NBO-pSAF succeed in realizing an improved RISC process while preserving the 1SRCT as the emitting state. The optimal equilibrium between narrowband emission and fast spin-flip highlights the potential of these two materials as efficient deep-blue emitters in OLEDs.
Electroluminescence properties
To further understand the structure–performance relationship of these MR-TADF emitters, OLEDs were fabricated with the configuration of ITO/TAPC (40 nm)/TCTA (5 nm)/26DCzPPy:x wt% emitters (20 nm)/TmPyPb (40 nm)/LiF (1 nm)/Al (200 nm). In these devices, ITO (indium tin oxide) and LiF/Al acted as the anode and cathode, respectively; TAPC (1,1-bis((di-4-tolylamino)phenyl)-cyclohexane) and TmPyPb (1,3,5-tri(m-pyridin-3-ylphenyl)benzene) were used as hole-transporting and electron-transporting layers, respectively, TCTA (4,4,4-tris(N-carbazolyl)triphenylamine) as the exciton-blocking layer to confine the triplet excitons within the emitting layer (EML). The bipolar transporting 26DCzPPy was selected as the host material in the EML with the expectation that its appropriately high T1 energy (ET = 2.71 eV) would prevent reverse energy transfer from the dopant to the host without the need for a large energy gap for energy transfer. The chemical structures of all these functional materials are shown in Fig. S16.† The device energy level diagram, the electroluminescence (EL) spectra, current density–voltage–luminance (J–V–L) characteristics and efficiency curves are shown in Fig. 4. The key EL performance data are summarized in Table 3. | ||
| Fig. 4 Electroluminescence performance of the sensitizer-free OLEDs with 26DCzPPy as the host at a doping level of 6 wt%. (a) Device architecture and energy-level diagram, (b) J–V–L characteristics, (c) EQE-luminance curves (inset: EL spectra, CIE color coordinates and actual luminescence photography of NBO-mSAF (left) and NBO-pSAF (right)), and (d) comparison of the EQEmax and CIE coordinates of the N/B/O type blue MR-TADF emitters reported in the present study and in the literature. | ||
| Emitter | Host | Sensitizer | V on [V] | λ EL [nm] | FWHMc [nm] | L max [cd m−2] | CEmaxe [cd A−1] | PEmaxf [lm W−1] | EQEmax/100/1000g [%] | CIEh [x,y] |
|---|---|---|---|---|---|---|---|---|---|---|
| DMAC-DPS (x wt%) | ||||||||||
| a Turn-on voltage recorded at the luminance of 1 cd m−2. b Maximum EL wavelength. c FWHM of EL spectra. d Maximum luminance. e Maximum current efficiency. f Maximum power efficiency. g Efficiency at maximum and 100, 1000 cd m−2. h EL color coordinates in the CIE 1931 chromaticity diagram recorded. | ||||||||||
| NBO-mSAF | 26DCzPPy | — | 4.4 | 466 | 28 | 8201 | 27.7 | 19.3 | 29.5/14.2/6.0 | (0.128,0.114) |
| PPF | — | 3.4 | 472 | 34 | 2857 | 21.9 | 20.2 | 17.7/5.2/1.7 | (0.120,0.180) | |
| 9 | 3.2 | 462 | 34 | 3631 | 26.4 | 26.0 | 24.7/11.3/7.0 | (0.142,0.127) | ||
| 20 | 3.0 | 464 | 38 | 6408 | 32.4 | 34.0 | 26.7/16.4/11.7 | (0.137,0.154) | ||
| NBO-pSAF | 26DCzPPy | — | 4.5 | 451 | 26 | 4639 | 11.0 | 7.7 | 20.5/11.0/5.7 | (0.147,0.048) |
| PPF | — | 3.8 | 456 | 32 | 1029 | 13.0 | 10.7 | 16.1/3.9/1.2 | (0.142,0.087) | |
| 9 | 3.4 | 454 | 33 | 2671 | 18.7 | 16.3 | 23.4/10.4/5.5 | (0.146,0.089) | ||
| 20 | 3.2 | 451 | 38 | 3767 | 27.4 | 26.9 | 25.2/12.8/8.5 | (0.154,0.117) | ||
The doping concentration was first varied (3, 6, 10 wt%) in the 26DCzPPy host to optimize the EL performance. The results (Fig. S17 and Table S7†) indicate that a doping concentration of 6 wt% led to superior performance for both NBO-mSAF and NBO-pSAF.
As shown by the inset photographs in Fig. 4c, both NBO-mSAF and NBO-pSAF devices emit deep-blue fluorescence with similar EL spectral profiles to their PL. The NBO-mSAF device exhibited an emission peak at 466 nm, with a FWHM of 28 nm and CIE coordinates of (0.128, 0.114), while realizing a high EQEmax of 29.5%. The NBO-pSAF device showed an EL peak of 451 nm, with a FWHM of 26 nm and CIE coordinates of (0.147, 0.048), and achieved an EQEmax of 20.5%. Apparently both NBO-mSAF and NBO-pSAF devices exhibited typical device performances of the MR-TADF emitters while shifting the emission wavelength to the real deep-blue region, especially the CIEy = 0.048 for NBO-pSAF is almost identical to the standard blue value (0.046) under the rigorous BT.2020 standard. Furthermore, as far as we know, there have been only several examples18,19,27–29 whose CIEy can fulfil or is close to the BT.2020 standard among all the N/B/O type blue MR-TADF emitters ever reported so far,18,19,22,27–40 as shown in Fig. 4d and Table S1.† Evidently the EQEmax = 20.5% of NBO-pSAF is the highest efficiency so far for the N/B/O type deep-blue MR-TADF emitter with CIEy almost fulfilling the BT.2020 standard. Meanwhile, despite the slightly inferior color purity for the NBO-mSAF device, the EQEmax of 29.5% is also among the highest efficiencies for all the deep-blue N/B/O type MR-TADF emitters with CIEx ≤ 0.15 and CIEy ≤ 0.12 ever reported so far, which is even higher than those obtained with sensitizers (Table S1†).30 As illustrated in Table S1,† The FWHM values of NBO-mSAF and NBO-pSAF are narrower than most of the reported FWHM values for N/B/O cored MR-TADF emitters, and are also comparable to those of MR-TADF emitters based on N/B/N structures.12,14,27,35 Such improved color purity can be safely attributed to the high molecular rigidity induced molecular relaxation suppression in the excited state, as manifested in the photophysical behavior. It was observed that the turn on voltage (Von, the voltage to deliver a brightness of 1 cd m−2) was independent of the doping concentrations for both NBO-mSAF and NBO-pSAF devices, indicating the absence of a charge trapping effect in the emitting layer (Table S7†).41 It is evident that our strategy of introducing the SAF donor into the para-position of either the O or B atom of the N/B/O skeleton not only significantly improved the device efficiency but also maintained excellent deep-blue color purity. However, despite the high efficiency and perfect color purity, the NBO-mSAF and NBO-pSAF devices with this traditional configuration still suffer from severe efficiency decay, with efficiency roll-off at a high brightness of 1000 cd m−2 over 70% from the maximum efficiencies. Therefore, it is strongly desired to improve the efficiency stability through certain device techniques.
A special device technology called hyperfluorescence (HF), proposed by Adachi et al.42 and developed by Duan and co-workers,43,44 that generally uses a TADF molecule as the sensitizer for either a traditional fluorescence dye or a narrow-band MR-TADF emitter, is characterized by high efficiency, slow efficiency roll-off and high color purity. In this approach, a high triplet energy host material, a TADF sensitizer, and a final emitter with a narrow FWHM should be considered simultaneously. Enlightened by the HF strategy, the sensitized OLEDs were fabricated by selecting bis[4-(9,9-dimethyl-9,10-dihydroacridine)phenyl]sulfone (DMAC-DPS) as the sensitizer and dibenzo[b,d]furan-2, 8-diylbis (diphenyl phosphine oxide) (PPF) as the host based on the singlet and triplet state energy requirement (Fig. 5a). Significant overlap between the fluorescence of DMAC-DPS and the absorption of the two MR-TADF emitters (Fig. S18†) guarantees efficient and forward energy transfer from the sensitizer to the MR-TADF emitters. The HF OLEDs have a configuration of ITO/HAT-CN (5 nm)/TAPC (20 nm)/TCTA (5 nm)/mCP (5 nm)/PPF:x wt% DMAC-DPS:1 wt% emitters (20 nm)/PPF (5 nm)/TmPyPb (40 nm)/LiF (1 nm)/Al (200 nm). The electron-transporting PPF was selected as the host in the EML with the expectation that its higher T1 energy than the dopants is able to prevent reverse energy transfer from the dopant and TADF sensitizer to the host. TCTA, mCP (1,3-bis(N-carbazolyl)benzene) and PPF with sufficiently high triplet energies were used as exciton-blocking layers to confine the triplet excitons within the EML. The chemical structure of each functional layer material and the device energy level diagram are shown in Fig. S15† and 5b.
 | ||
| Fig. 5 Performance summary of the HF OLEDs with DMAC-DPS as the sensitizer. (a) Energy level diagram of each material in the EML layer and (b) the device structure and the energy level diagram of each functional material of the HF devices; (c) contrast of EL peaks and (d) EQEs of emitters in sensitizer-free devices of 26DCzPPy and PPF and in HF devices with DMAC-DPS (20 wt%). (e) Quantum yields and (f) the key rate constants of the sensitizer at different doping concentrations (sensitizer: DMAC-DPS) of NBO-mSAF and NBO-pSAF. | ||
In general, a fast Förster resonance energy transfer (FRET) from S1 of the TADF sensitizer to the S1 of the fluorescent emitter is crucial to activating the HF system. By deliberately employing a low dopant concentration of 1 wt% for the N/B/O terminal emitter, the Dexter energy transfer (DET) from the triplet excitons of the sensitizer to the MR emitter was intentionally suppressed, thereby favoring the dominance of FRET as the primary mechanism for the energy transfer from PPF to both the DMAC-DPS sensitizer and MR emitter. However, the doping content of the DMAC-DPS sensitizer was controlled relatively high at 9% or 20% in the EML to enable the Dexter energy transfer from the PPF host to the DMAC-DPS sensitizer. In such a way, the generated triplet excitons of DMAC-DPS can be converted, in the case of the forbidden Dexter energy from DMAC-DPS to the MR emitter, into its own singlet exciton through efficient RISC based on its intrinsic TADF nature (Fig. S19†). In a similar way, it was observed that a doping level of 6 wt% for NBO-mSAF and NBO-pSAF was the favorite concentration for optimized performance when they were doped into PPF in sensitizer-free OLEDs (Fig. S20 and Table S8†). Furthermore, in the HF OLEDs with the EML having the configuration of PPF:DMAC-DPS (x wt%):MR emitter (1 wt%), it was observed that a doping content of 20 wt% for the DMAC-DPS sensitizer in the EML led to better performance than 9 wt% (Fig. S21†). The key device parameters are summarized in Table 3.
As shown in Fig. 5c, the HF devices exhibited emission originating from the NBO-mSAF and NBO-pSAF emitters without the residual emission from the sensitizer or host, indicating effective energy transfer sensitizing processes. As illustrated in Fig. 5d, HF devices exhibit minimal efficiency roll-offs while maintaining high efficiency in comparison to those of unsensitized devices. The EQEmax of the NBO-mSAF-based HF device is 26.7%, which is slightly lower than that in the 26DCzPPy host. However, the efficiency roll-off was greatly reduced from 79.7% (in 26DCzPPy) to 56.2% (HF) at 1000 cd m−2. This improvement in efficiency stability is more pronounced in the PPF host, with a reduction from 90.4% (PPF) to 56.2% at 1000 cd m−2. Furthermore, NBO-pSAF realized a significant EQEmax increase to 25.2% in the HF device and a remarkably suppressed efficiency roll-off of 66.3% at 1000 cd m−2, in contrast to the values of 72.1% in 26DCzPPy and 92.5% in PPF.
The comparable or improved efficiencies and greatly suppressed efficiency roll-offs in the TADF-sensitized HF devices with the PPF:DMAC-DPS:MR emitter emitting layer configuration indicate the effective mitigation of triplet-related quenching facilitated by the sensitizer. This is evidenced by the change in the relevant photophysical parameters measured in the corresponding doping film (Table 2). In the two-component host-dopant films with 26DCzPPy or PPF as the host, both NBO-mSAF and NBO-pSAF exhibit almost comparable ΦPLs (60.0% vs. 55.0% for NBO-mSAF, 52.8% vs. 55.1% for NBO-pSAF), although both the kr and knr of 26DCzPPy are simultaneously superior to those of PPF (Table 2) for each TADF emitter. This is probably because the larger molecular dipole moment of the 26DCzPPy host (bipolar) than that of PPF (single polar) is more favorable for both the radiative and non-radiative transitions of the dopant molecules, and finally leads to comparable PLQYs in these different hosts, as illustrated by the formula (S8)–(S10) in the ESI.† Interestingly the introduction of the DMAC-DPS sensitizer greatly increases the ΦPL of the doped films. For NBO-mSAF and NBO-pSAF, the PLQYs increase from 55.0% to 94.5% and 90.4% respectively (Fig. 5e). As shown in Fig. 5f, the non-radiative rate constant (knr) reduced by an order of magnitude (from 3.1 × 107 s−1 and 6.0 × 107 s−1 to 3.0 × 106 s−1 and 4.0 × 106 s−1 for NBO-mSAF and NBO-pSAF, respectively), with the presence of the DMAC-DPS sensitizer, which well accounts for the substantial increase of PLQYs of the two MR-TADF emitters and thus the obvious increase of their EQEs. Furthermore, the kRISC has increased to a certain extent (from 5.7 × 104 s−1 and 9.7 × 104 s−1 to 9.5 × 104 s−1 and 1.5 × 105 s−1 for NBO-mSAF and NBO-pSAF, respectively). This is reflected in the correspondingly reduced τDF (from 22.8 μs and 16.3 μs to 15.8 μs and 15.5 μs for NBO-mSAF and NBO-pSAF, respectively) (Fig. S22† and Table 2). Such enhanced RISC in the HF OLEDs greatly improves the up-conversion and utilization rates of triplet excitons in the EML, and consequently improves the device efficiency and suppresses the efficiency roll-off.
Conclusion
Two novel MR-TADF emitters NBO-mSAF and NBO-pSAF were developed by decorating a N/B/O ternary doped asymmetrical MR skeleton with a highly rigid electron donor 10H-spiro[acridine-9,9′-fluorene] (SAF) at the para-position of the oxygen atom and the para-position of the boron atom. Due to the appropriate donor strength of the SAF group relative to the MR core as the acceptor, the long-range charge transfer singlet state (1LRCT) was located over the short-range charge transfer singlet state (1SRCT) of the N/B/O core, so that the intrinsic narrow-band emitting feature of MR-TADF was kept for both emitters. In addition, the presence of the 3LRCT state compressed the triplet states and thus facilitated the RISC process. As a result, the rate constants of RISC (kRISC) of NBO-mSAF and NBO-pSAF were increased by 5–10 times relative to the parent NBO molecule, e.g. to 3.4 × 105 s−1 for NBO-pSAF. In the traditional OLEDs with 26DCzPPy as the host, NBO-pSAF realized an EQEmax of 20.5% with a CIE of (0.147, 0.048) and FWHM of 26 nm, which is the highest efficiency for a N/B/O type deep-blue MR-TADF emitter with CIEy close to that of the BT.2020 standard (0.046). Meanwhile, despite the slightly inferior CIE coordinates of (0.128, 0.114), the EQEmax of 29.5% for the NBO-mSAF device is also among the highest efficiencies for all the deep-blue N/B/O type MR-TADF emitters with CIEx ≤ 0.15 and CIEy ≤ 0.12 ever reported so far, which is even higher than those obtained with sensitizers. In order to improve the efficiency stability, hyperfluorescence (HF) OLEDs were also fabricated with the emitting layer of the PPF:DMAC-DPS:MR emitter. Satisfactory EQEs of 26.7% and 25.2% were achieved for NBO-mSAF and NBO-pSAF, respectively. More importantly, the efficiency roll-off of these HF devices was dramatically suppressed owing to the reduced delay fluorescence lifetime and increased PLQY of the MR-TADF emitters with the presence of the sensitizer DMAC-DPS. As far as we know, among all the N/B/O type deep-blue MR emitters, NBO-mSAF is also the first example with an additional donor (SAF in the present case) incorporated into the para-site of the oxygen atom of the MR core, which in turn exhibited an excellent EQEmax of 29.5%. Furthermore, the EQEmax of 20.5% for NBO-pSAF is the highest efficiency recorded so far for N/B/O type deep-blue MR-TADF emitters with CIEy approaching that of the BT.2020 standard.Data availability
All the necessary data to support the findings of this study can be found within the main text and ESI.†Author contributions
Jing Jin: device preparation, characterization, data curation, investigation, writing – original draft. Zhaolong He: synthesis and methodology. Di Liu: investigation, project administration, resources, supervision. Yongqiang Mei: methodology and investigation. Jiahui Wang: software and formal analysis. Huihui Wang: methodology and data curation. Jiuyan Li: funding acquisition, investigation, supervision, writing – review and editing.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22078051 and 22478063), the Fundamental Research Funds for the Central Universities (DUT22LAB610) and the Open Fund of the Key Laboratory of Advanced Display and System Applications, Ministry of Education, Shanghai University (OF202401) for financial support of this work.References
- Y. X. Hu, J. Miao, T. Hua, Z. Huang, Y. Qi, Y. Zou, Y. Qiu, H. Xia, H. Liu, X. Cao and C. Yang, Nat. Photonics, 2022, 16, 803–810 CrossRef CAS.
- T. Huang, Q. Wang, H. Zhang, Y. Zhang, G. Zhan, D. Zhang and L. Duan, Nat. Photonics, 2024, 18, 516–523 CrossRef CAS.
- M. Mamada, A. Aoyama, R. Uchida, J. Ochi, S. Oda, Y. Kondo, M. Kondo and T. Hatakeyama, Adv. Mater., 2024, 2402905 CrossRef CAS PubMed.
- R. Zhu, Z. Luo, H. Chen, Y. Dong and S.-T. Wu, Opt. Express, 2015, 23, 23680 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Cai and S. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- D. Karthik, Y. H. Jung, H. Lee, S. Hwang, B. Seo, J. Kim, C. W. Han and J. H. Kwon, Adv. Mater., 2021, 33, 2007724 CrossRef CAS PubMed.
- Y. Shi, H. Ma, Z. Sun, W. Zhao, G. Sun and Q. Peng, Angew. Chem., 2022, 134, e202213463 CrossRef.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678–682 CrossRef CAS.
- H. Lim, S. Woo, Y. H. Ha, Y. Kim and J. Kim, Adv. Mater., 2022, 34, 2100161 CrossRef CAS PubMed.
- Z. Huang, H. Xie, J. Miao, Y. Wei, Y. Zou, T. Hua, X. Cao and C. Yang, J. Am. Chem. Soc., 2023, 145, 12550–12560 CrossRef CAS PubMed.
- Q. Wu, J. Li, D. Liu, Y. Mei, B. Liu, J. Wang, M. Xu and Y. Li, Dyes Pigm., 2023, 217, 111421 CrossRef CAS.
- S. Chen, Y. Wang, J. Lin, R. Tian, S. Li, Y. Man, S. Chen, J. Zhang, C. Duan, C. Han and H. Xu, Chem. Eng. J., 2024, 489, 151517 CrossRef CAS.
- F. Liu, Z. Cheng, L. Wan, Z. Feng, H. Liu, H. Jin, L. Gao, P. Lu and W. Yang, Small, 2022, 18, 2106462 CrossRef CAS PubMed.
- Z. Ye, H. Wu, Y. Xu, T. Hua, G. Chen, Z. Chen, X. Yin, M. Huang, K. Xu, X. Song, Z. Huang, X. Lv, J. Miao, X. Cao and C. Yang, Adv. Mater., 2024, 36, 2308314 CrossRef CAS PubMed.
- L. Wu, X. Mu, D. Liu, W. Li, D. Li, J. Zhang, C. Liu, T. Feng, Y. Wu, J. Li, S.-J. Su and Z. Ge, Angew. Chem., Int. Ed., 2024, e202409580 CAS.
- I. S. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2022, 34, 2107951 CrossRef CAS PubMed.
- I. S. Park, H. Min and T. Yasuda, Angew. Chem., Int. Ed., 2022, 61, e202205684 CrossRef CAS PubMed.
- H. Lim, H. J. Cheon, S. Woo, S. Kwon, Y. Kim and J. Kim, Adv. Mater., 2020, 32, 2004083 CrossRef CAS PubMed.
- Y. H. Lee, W. Lee, T. Lee, J. Jung, S. Yoo and M. H. Lee, Chem. Eng. J., 2023, 452, 139387 CrossRef CAS.
- G. Liu, H. Sasabe, K. Kumada, H. Arai and J. Kido, Chem.–Eur. J., 2022, 28, e202201605 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- B. Chen, H. Liu, J. Yang, M. Ahmadi, Q. Chen, N. Yin, S. Zhang, M. Xiao, H. Zhang, L. Xu and P. Chen, Adv. Funct. Mater., 2024, 2402522 CrossRef CAS.
- H. Liu, Y. Fu, J. Chen, B. Z. Tang and Z. Zhao, Adv. Mater., 2023, 35, 2212237 CrossRef CAS PubMed.
- Y. Yin, X. Lai, Q. Ma, H. Ma, W. Zhu, J. Y. Lee and Y. Wang, Adv. Mater., 2024, 36, 2313656 CrossRef CAS PubMed.
- J. Park, J. Lim, J. H. Lee, B. Jang, J. H. Han, S. S. Yoon and J. Y. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 45798–45805 CrossRef CAS PubMed.
- C.-Y. Chan, S. Madayanad Suresh, Y.-T. Lee, Y. Tsuchiya, T. Matulaitis, D. Hall, A. M. Z. Slawin, S. Warriner, D. Beljonne, Y. Olivier, C. Adachi and E. Zysman-Colman, Chem. Commun., 2022, 58, 9377–9380 RSC.
- J. Jin, M. Chen, H. Jiang, B. Zhang, Z. Xie and W.-Y. Wong, ACS Mater. Lett., 2024, 3246–3253 CrossRef CAS.
- X. Huang, Y. Xu, J. Miao, Y.-Y. Jing, S. Wang, Z. Ye, Z. Huang, X. Cao and C. Yang, J. Mater. Chem. C, 2023, 11, 11885–11894 RSC.
- Y. Xu, J. Han, N. Li, Z. Huang, J. Miao and C. Yang, J. Mater. Chem. C, 2023, 11, 13733–13739 RSC.
- H. Tanaka, S. Oda, G. Ricci, H. Gotoh, K. Tabata, R. Kawasumi, D. Beljonne, Y. Olivier and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 17910–17914 CrossRef CAS PubMed.
- X. Luo, H. Ni, A. Lv, X. Yao, H. Ma and Y. Zheng, Adv. Opt. Mater., 2022, 10, 2200504 CrossRef CAS.
- J. Liu, L. Chen, X. Wang, Q. Yang, L. Zhao, C. Tong, S. Wang, S. Shao and L. Wang, Macromol. Rapid Commun., 2022, 43, 2200079 CrossRef CAS PubMed.
- J. Han, Z. Huang, X. Lv, J. Miao, Y. Qiu, X. Cao and C. Yang, Adv. Opt. Mater., 2022, 10, 2102092 CrossRef CAS.
- Z. Yan, L. Yuan, Y. Zhang, M. Mao, X. Liao, H. Ni, Z. Wang, Z. An, Y. Zheng and J. Zuo, Adv. Mater., 2022, 34, 2204253 CrossRef CAS PubMed.
- J. Jin, C. Duan, H. Jiang, P. Tao, H. Xu and W. Wong, Angew. Chem., Int. Ed., 2023, 62, e202218947 CrossRef CAS PubMed.
- K. R. Naveen, J. H. Oh, H. S. Lee and J. H. Kwon, Angew. Chem., Int. Ed., 2023, 62, e202306768 CrossRef CAS PubMed.
- X. He, J. Lou, B. Li, X. Dong, F. Zhong, W. Liu, X. Feng, D. Yang, D. Ma, Z. Zhao, Z. Wang and B. Z. Tang, Adv. Mater., 2024, 36, 2310417 CrossRef CAS PubMed.
- J. Hu, Y. Wei, X. Wang, X. Liang, X. Liao, L. Yuan, H. Ni and Y. Zheng, Adv. Opt. Mater., 2024, 12, 2302987 CrossRef CAS.
- M. Tanaka, C.-Y. Chan, H. Nakanotani and C. Adachi, Nat. Commun., 2024, 15, 5950 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- X. Song, D. Zhang, Y. Lu, C. Yin and L. Duan, Adv. Mater., 2019, 31, 1901923 CrossRef PubMed.
- C. Yin, Y. Zhang, T. Huang, Z. Liu, L. Duan and D. Zhang, Sci. Adv., 2022, 8, eabp9203 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04896b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |