
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-driven asymmetric aldol reaction of ketones and glycinates via synergistic Lewis acid/photoredox catalysis†
Jiuqi
Tan
 ,
Longqing
Yang
,
Hanyu
Su
,
Yuntian
Yang
,
Ziwei
Zhong
,
Xiaoming
Feng
* and
Xiaohua
Liu
*
,
Longqing
Yang
,
Hanyu
Su
,
Yuntian
Yang
,
Ziwei
Zhong
,
Xiaoming
Feng
* and
Xiaohua
Liu
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: xmfeng@scu.edu.cn; liuxh@scu.edu.cn
First published on 16th September 2024
Abstract
Visible-light-driven direct asymmetric α-C(sp3)–H bond functionalization of glycinate provides a direct and efficient route for the synthesis of diverse optically enriched α-amino acid derivatives. However, asymmetric coupling between glycinate radical species and ketones faces significant challenges, including competitive pathways, mutable intermediates, as well as congested stereogenic centers. Herein, we disclose the first example for the asymmetric photocatalytic synthesis of a diverse array of β-diaryl-β-hydroxy-α-amino acetate derivatives from glycinates and heteroaryl ketones through the synergistic catalysis of achiral iridium photoredox catalyst and chiral lanthanide Lewis acid catalysts. The enantioselective radical addition pathway is supported by spectroscopic experiments, control experiments and DFT calculations.
Introduction
Glycine is the only achiral proteinogenic α-amino acid, but the α-functionalization of the C(sp3)–H bond of glycine derivatives provides a direct and efficient route for the synthesis of diverse natural or unnatural chiral α-amino acids and modified peptides.1 Visible-light-driven α-C(sp3)–H bond functionalization of glycinate has witnessed considerable developments benefiting from the harmonization of photoredox catalyst and/or metal complex/organocatalyst (Scheme 1a).2 The general mechanistic profile involves the oxidation of glycine ester into amino radical cation, which triggers rapid α-deprotonation to yield a carbon-centered α-amino radical (i), then further oxidation into cationic iminium (ii).3 Based on this strategy, a plethora of α-alkylation and α-arylation reactions of glycinate have been achieved using various alkyl sources and electron-rich aryls (Scheme 1a, path 1). Recently, the enantioselective variation was reported, which provides efficient access to optically enriched amino acid derivatives. Wang et al.3e,h reported enantioconvergent cross-coupling between racemic α-bromo ketones and N-substituted glycine esters by a cooperative photoredox/Brønsted acid catalysis (Scheme 1b). Xu et al. realized the enantioselective alkylation of quinolinyl-preinstalled glycinate derivative using N-hydroxyphthalimide (NHP) esters as the alkyl radical sources enabled by photoinduced chiral copper complexes.4 However, there are sporadic reports related to the addition or coupling with unsaturated compounds (Scheme 1a, path 2), such as carbonyl compounds reported by Xiao et al.,5a azo compounds by Fu et al.,5b or phenylpropyne by Breit et al.,5c which could serve as complementary approaches to the α-C(sp3)–H bond functionalization of glycinate. Moreover, König et al.6 utilized 3DPA2FBN as the photocatalyst to trigger the formation of carbanion intermediate (iii) bearing an exogenous alkylamine substitution, leading to the addition into carbonyl compounds.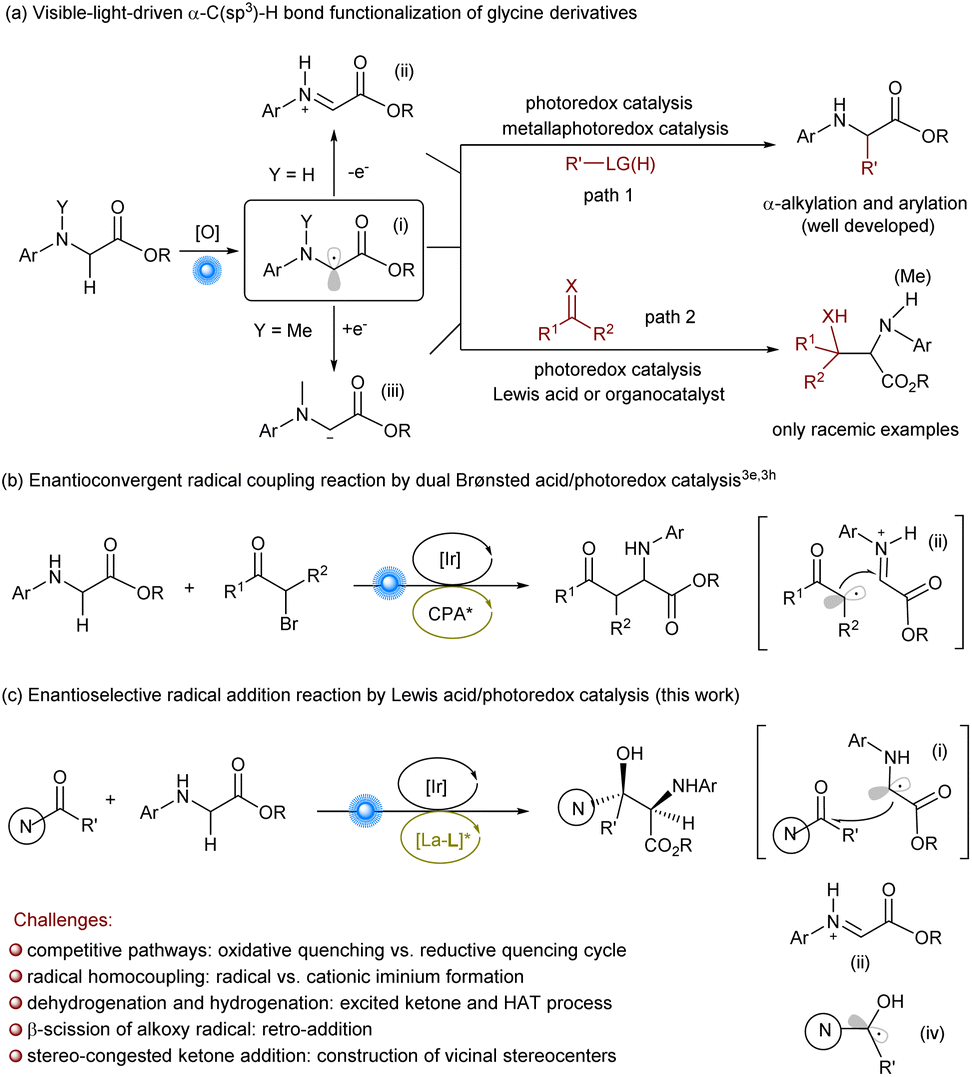 | ||
| Scheme 1 Visible light-driven α-C(sp3)–H bond functionalization of glycinates. | ||
β-Hydroxy-α-amino acids are a class of molecules commonly found in polypeptide natural products and pharmaceuticals.7 The direct aldol reaction of glycinates represents an intriguing and straightforward strategy.8,9 Zhao et al. realized biomimetic aldol reaction of trifluoromethyl ketones9f,g and aldehydes9h by carbonyl catalysis. The enantioselective α-C(sp3)–H functionalization of glycinate to general ketones remains elusive. Intrigued by visible-light-driven radical cross-coupling between glycinates and diaryl ketones,5a,6 we wish to achieve the more challenging enantioselective synthesis of β-diaryl-β-hydroxyl-α-amino acids via this approach.
There are several challenges for the target process (Scheme 1c). The use of photoredox catalysts is imperative but both glycinate and ketone can perform the SET process, which may cause competitive catalytic cycles: reductive quenching or oxidative quenching cycle.10 The excessive oxidation of glycinate into cationic iminium (ii) is disadvantageous to the coupling with ketone, enhancing the formation of the diamine byproduct. The conjugated system of diaryl ketones leads to the excited species via the direct absorption of visible light or energy transfer from photocatalysts,11–13 and the formed ketyl radical (iv) gives rise to the hydrogenation of ketone and dehydrogenation of glycinate. In addition, the addition to ketones results in the construction of vicinal stereo-congested centers, including a quaternary carbon center, which is one of the synthetic challenges in asymmetric catalysis. In addition, the alkoxy radical intermediate might undergo β-scission, leading to the decomposition of the product.14 The merging of a photoredox catalyst and a chiral catalyst for synergistic catalysis would provide an important platform, which could not only control the formation of the radical species to minimize undesired radical species but also guide the facial approach of the two reactive species. Herein, we disclosed a new synergistic catalysis by the combination of an Ir(III)-based photoredox catalyst and chiral lanthanide catalyst of N,N′-dioxide ligands developed by our group for the purpose.15,16 The first example for the visible light-driven asymmetric α-C(sp3)–H bond functionalization of glycinate for the synthesis of β-quaternary-stereocentric serine derivatives was achieved (Scheme 1c).
Results and discussion
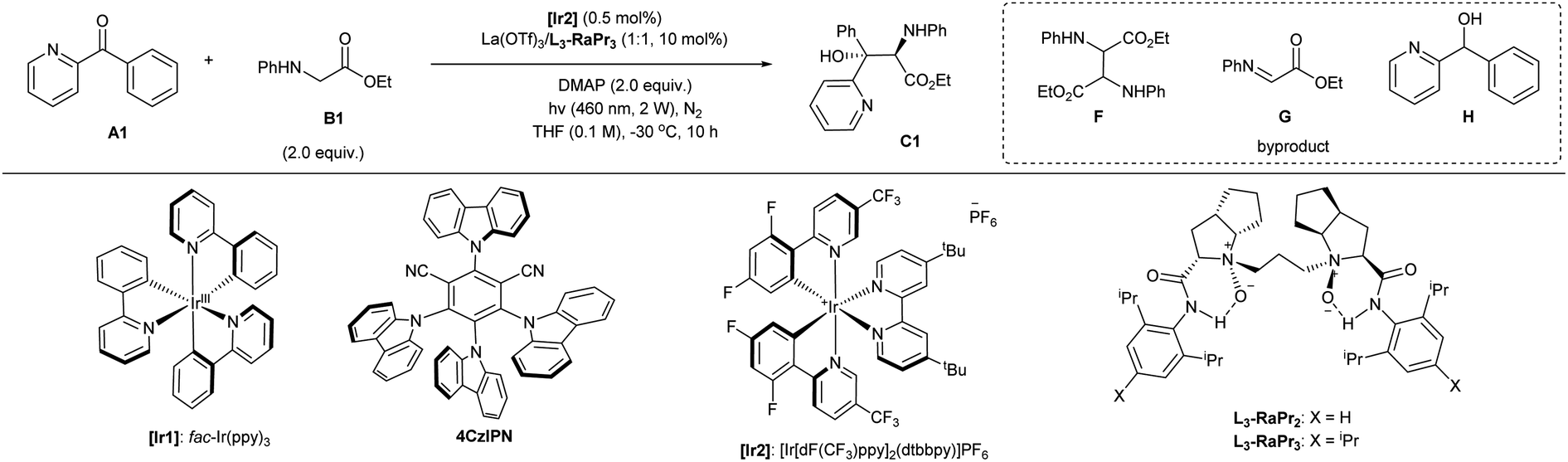
Initially, we chose phenyl(pyridin-2-yl)methanone A1 and ethyl phenylglycine B1 as the model substrates to identify the optimal catalytic system (Table 1 and see ESI† for details). The screening of a number of metal salts combined with chiral N,N′-dioxide L3-RaPr2 in the presence of photocatalyst [Ir1] and DMAP as the base at room temperature revealed that lanthanide salts with larger ion radii showed higher enantioselectivity. We found that La(OTf)3 could give product C1 excellent ee value for two diastereomers and moderate yield (Table 1, entry 1). The use of organic photocatalyst 4CzIPN (entry 2) or [Ir2] (entry 3) gave the product with slightly increased diastereoselectivity, although the yield dropped. Using the ligand L3-RaPr3 with increased steric hindrance at the amide subunits gave rise to the product with somewhat higher diastereoselectivity (entry 4). The yield of amino alcohol C1 is not good due to the strongly competitive reactions, which led to the generation of homo-coupling F and oxidated imine G from B1 and also the reduced byproduct H from A1. Later, when the transformation was carried out with weakened irradiation light (entry 5), decreased reaction temperature (entry 6), and lower amount of [Ir2] (entry 7) within less reaction time (10 hours; entry 8), the desired amino alcohol C1 could be isolated in a gradually increased yield as a result of minimizing the formation of byproducts and avoiding the decomposition of the product. In these conditions, product C1 could be isolated in 66% yield, with 98% and 92% ee for each diastereomers once the amount of glycinate increased to three equivalents (entry 9). Fortunately, the reaction can be further enhanced by the addition of 30 mol% of sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF4), resulting in a 93% NMR yield of the product and 84% isolated yield (entry 10). This result might contribute to the counteracting ion exchange with lanthanide catalyst and [Ir2] or the stabilization of the alkoxy radical intermediate (see ESI† for details). The concentration did not considerably affect the reaction results (entry 11). Considering environmental friendliness and economic efficiency, we chose a minimal quantity of the solvent. The homo-coupling product F was obtained in about 5% yield and the imine G was detected in 7% yield. In addition, the alcohol H was detected in less than 4% yield in this case. Finally, the optimal conditions (entry 11) giving the amino alcohol C1 in the best selectivity and isolated yield (84% yield, 68:32 dr, with 98% and 92% ee for each isomer) involved 0.5 mol% of [Ir2], chiral La(OTf)3/L3-RaPr3 catalyst, and DMAP in THF (0.2 M) under the irradiation of visible light (460 nm, 2 W) at −30 °C after 10 h. The system was sensitive to air but water and other protonic additives were tolerable (see ESI† for details). We also tested several other chiral ligands, such as bisoxazoline and phosphine ligands, as well as chiral phosphoric acid organocatalysts, but the stereoselectivity was poor (see ESI† for details). The comparison reaction in the presence of only [Ir2] and DMAP generated the racemic diastereomers in 19% yield, and ketone was recovered in 56% yield (entry 12 vs. entry 8), highlighting the acceleration of lanthanum catalyst. Note that the mixture of ketone A1 and glycinate B1 in the presence of chiral lanthanum catalyst could afford the product upon direct irradiation with 400 nm light without an additional photocatalyst, and the amino alcohol was isolated in 38% yield with maintained enantioselectivity (entry 13). It manifested that this addition reaction could be performed via different pathways depending on the photocatalyst, ketone, and light energy as well.|
|
||||
|---|---|---|---|---|
| Entry | Condition variation | Yieldb (%) | eec (%) | drc |
| a Unless otherwise noted, all reactions were carried out with A1 (0.1 mmol), B1 (2.0 equiv.), La(OTf)3/L3-RaPr3 (1:1, 10 mol%), DMAP (0.2 mmol), [Ir2] (0.5 mol%) in THF (1.0 mL) at −30 °C upon irradiation with LEDs (460 nm, 2 W) under nitrogen for 10 hours. b Total isolated yield of two diastereoisomers, and the data in the parentheses referred to the 1H NMR yield with dibromomethane as the internal standard. c ee and dr values were determined by HPLC analysis on a chiral stationary phase. DMAP = 4-dimethylaminopyridine. THF = tetrahydrofuran. r.t. = room temperature. | ||||
| 1 | L3-RaPr2/[Ir1] (2 mol%)/5 W/r.t./24 h | 35 | 99/91 | 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48 :48 |
| 2 | L3-RaPr2/4CzIPN (2 mol%)/5 W/r.t./24 h | 22 | 98/92 | 60:40 |
| 3 | L3-RaPr2/[Ir2] (2 mol%)/5 W/r.t./24 h | 10 | 99/91 | 60:40 |
| 4 | [Ir2] (2 mol%)/5 W/r.t./24 h | 16 | 93/90 | 66:34 |
| 5 | [Ir2] (2 mol%)/r.t./24 h | 30 | 95/93 | 66:34 |
| 6 | [Ir2] (2 mol%)/24 h | 36 | 95/91 | 72:28 |
| 7 | 24 h | 51 | 95/92 | 68:32 |
| 8 | — | 60 | 98/92 | 70:30 |
| 9 | B1 (3.0 equiv.) | 66 | 98/92 | 66:34 |
| 10 | B1 (3.0 equiv.)/NaBArF4 (30 mol%) | 85 (93) | 98/93 | 68:32 |
| 11 | B1 (3.0 equiv.)/NaBArF4 (30 mol%)/THF (0.5 mL) | 84 (93) | 98/92 | 68:32 |
| 12 | w/o L3-RaPr3/La(OTf)3 | 19 | —/— | 50:50 |
| 13 | w/o [Ir2]/400 nm/15 °C/24 h | 38 | 98/93 | 58:42 |
With the optimal reaction conditions (Table 1, entry 11) established, the substrate scope with respect to glycinates was investigated. As shown in Scheme 2, phenylglycinates with varied ester group regardless of the steric hindrance reacted with ketone A1 smoothly, providing the desired serine derivatives C1–C4 in good yield (77–84%) and excellent levels of enantioselectivity for each diastereomer (98–99% ee for major diastereomer, 91–98% ee for the minor one). The para-tertbutyl group introduced at the N-aryl substitution slightly raised the diastereoselectivity (C5–C6), and the absolute configuration of the major isomer of the product C5 was determined to be (2R,3R) by X-ray crystallography analysis.17 Ether glycinates bearing a series of substituted aryl groups as N-protection groups could be tolerated well. It revealed that ortho-substituents had a greater effect on both the yield and the stereoselectivity due to the steric hindrance (C7–C8); however, various substitutions at the meta- and para-position had no obvious influence on the enantioselectivity of each diastereomers (C9–C27; 91–99% ee for major diastereomer, and 82–93% ee for the minor one). The yields were satisfactory with para-cyano group (C21) as an exception, probably because its high oxidation potential is unfavorable to the photo-reductive quenching of [Ir2]. Notably, several functional groups, such as alkenyl (C22), alkynyl (C23), acyl (C24), ester (C25), boron ester group (C26), were compatible in the transformation. 1-Naphthyl (C28) and 2-naphthyl substitutions (C29) were competent in the reaction, but the former showed lower outcome as a result of steric hindrance. N-PMP-containing product C30 was isolated in 40% yield because of the thermodynamic instability in column chromatography purification. Moreover, this approach could be applied to late-stage modification (C31–C34), involving the introduction of alcohols to glycine as ester groups or peptide as the amide substitution.
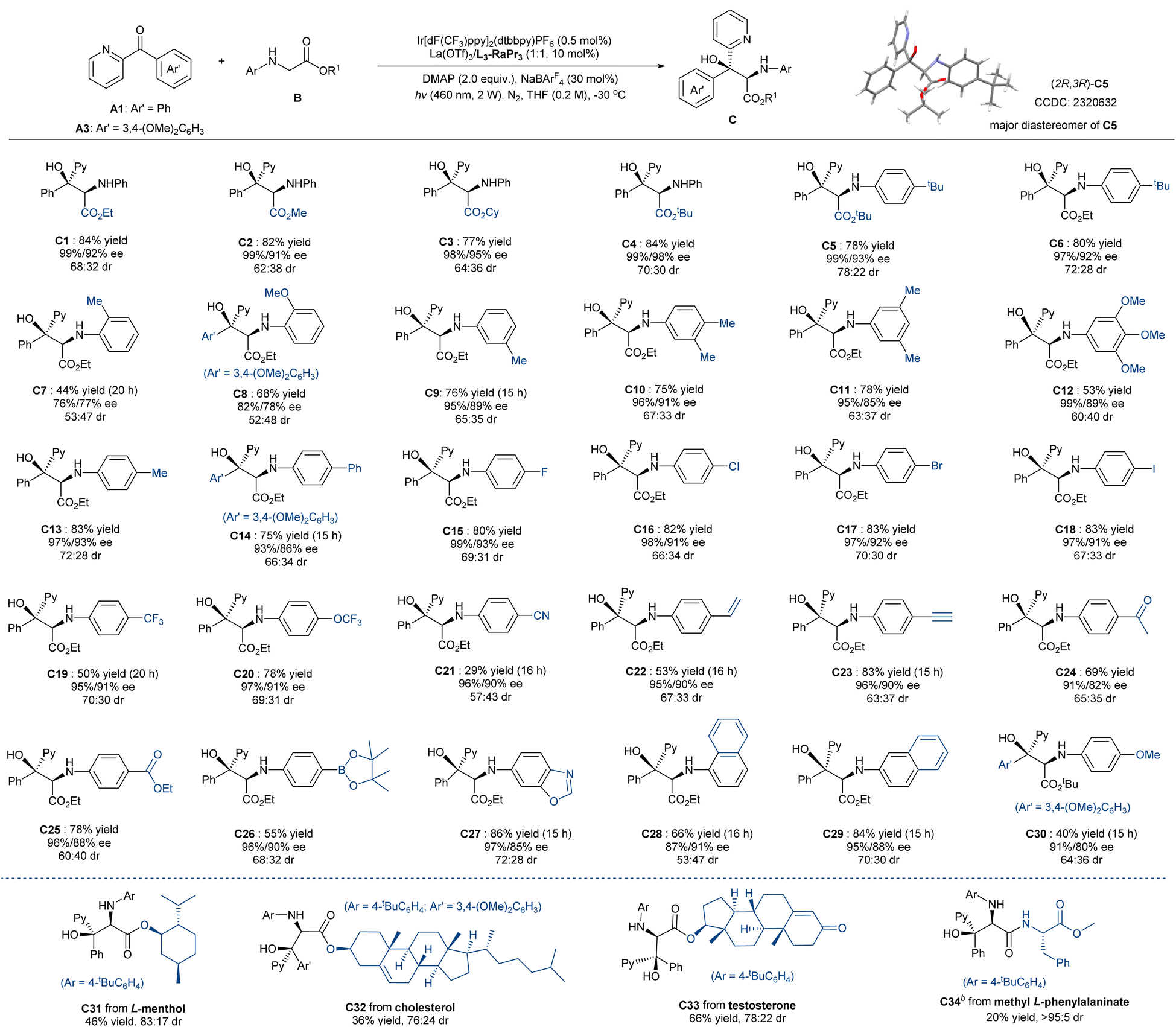 | ||
| Scheme 2 Substrate scope of N-aryl glycinate derivatives.a Unless otherwise noted, all reactions were carried out with A1 or A3 (0.1 mmol), B (3.0 equiv.), La(OTf)3/L3-RaPr3 (1:1, 10 mol%), DMAP (0.2 mmol), [Ir2] (0.5 mol%), NaBArF4 (30 mol%) in THF (0.2 M) at −30 °C upon irradiation with LEDs (460 nm, 2 W) under nitrogen for 10 hours. Total isolated yield of two diastereoisomers. ee values were determined by HPLC or UPC2 analysis on a chiral stationary phase. dr value was determined by 1H NMR.b4CzIPN (2 mol%), at 20 °C for 20 h. | ||
Subsequently, we turned our attention to the variation of ketones. As shown in Scheme 3, a series of 2-pyridine-based diaryl ketones bearing electron-deficient or -rich substitutions on the meta- or para-position of the phenyl ring performed well, and they delivered the corresponding adducts C35–C46 in moderate to good yields (66–81%) and excellent enantioselectivities (91–99% ee for the major diastereomer and 71–98% ee for the minor one), and the diastereoselectivity (up to 90:10 dr) was higher when there was a para-substituted phenyl group. Ortho-substitutes were not eligible to the addition, which might be due to the spatial congestion hindering the addition of radical species (see ESI† for details). Pyridin-2-yl(pyridin-4-yl)methanone delivered the related product C47 in 70% yield with good enantioselectivity (93%/87% ee) and 72:28 dr. Substitution at the C2 or C3-position of the pyridine subunit (C48–C51) could be compatible, but the C4-bromo substituted one led to the formation of C52 with decreased enantioselectivity, especially for the minor isomer. The 2-pyridinyl group of the ketone could be changed into the 3-isoquinolinyl (C53) or 2-pyrimidinyl group (C54) without diminishing the reactivity and enantioselectivity. In contrast, the addition of di(pyridin-2-yl)methanone yielded the product C55 with a poor ee value (20% ee), indicating that the diastereoselectivities of the unsymmetric ketones originate from the facially selective approach of glycinate and that the tertiary alcohol centers of the two isomers are likely to be identical in other cases. Additionally, 1-(pyridin-2-yl)ethan-1-one (C56) and benzothiazolphenone (C58) worked well despite there being a slight decrease in the reactivity (61–67% yield) and enantioselectivity. The reaction of cyclopropyl(pyridin-2-yl)methanone afforded the product C57 in moderate yield without ring-opening, manifesting that the addition process did not involve the ketyl radical intermediate.
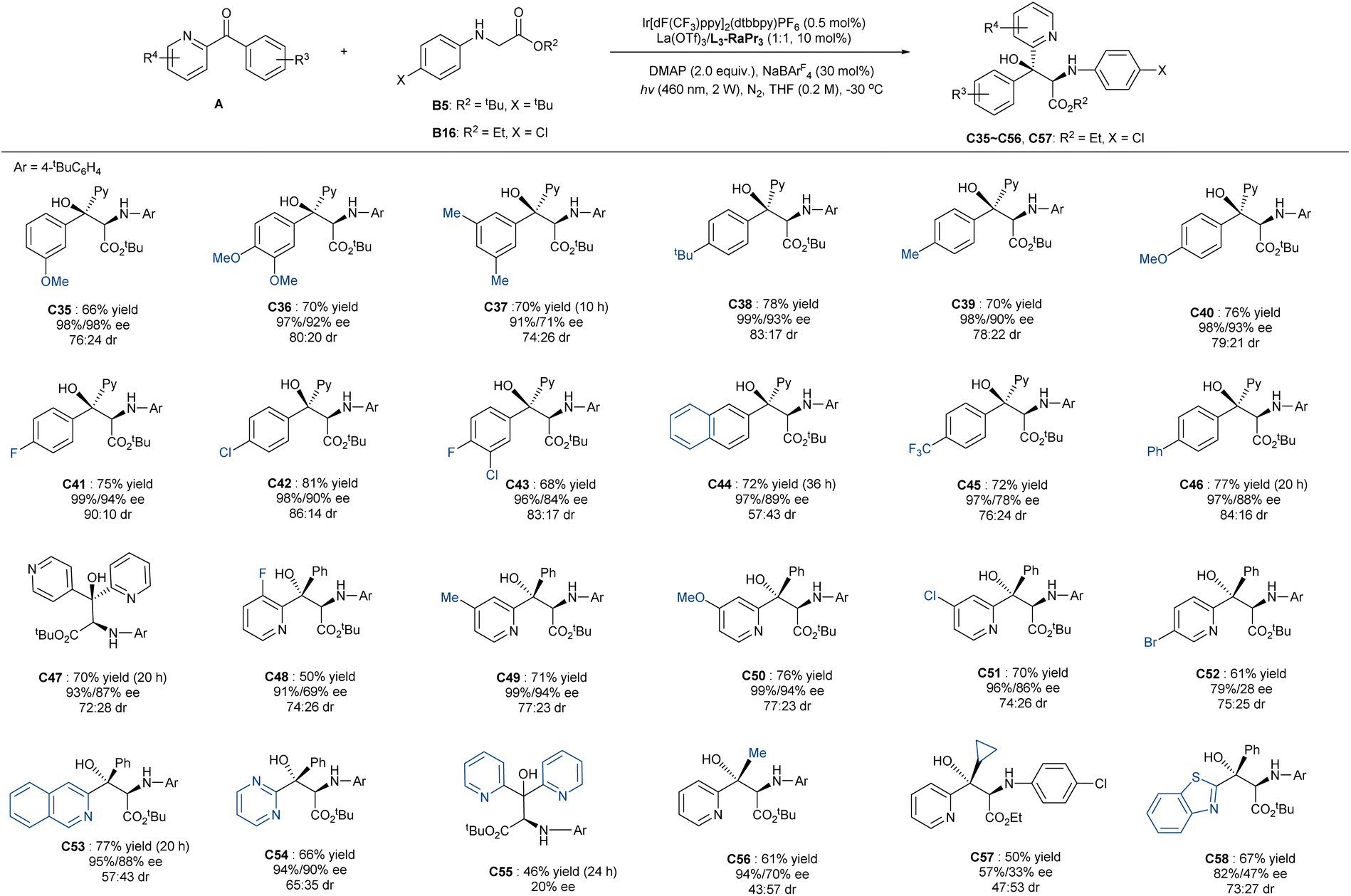 | ||
| Scheme 3 Substrate scope of ketones. a Unless otherwise noted, all reactions were carried out with A (0.1 mmol), B5 or B16 (3.0 equiv.), La(OTf)3/L3-RaPr3 (1:1, 10 mol%), DMAP (0.2 mmol), [Ir2] (0.5 mol%), NaBArF4 (30 mol%) in THF (0.2 M) at −30 °C upon irradiation with LEDs (460 nm, 2 W) under nitrogen for 15 hours. Total isolated yield of two diastereoisomers. ee values were determined by HPLC or UPC2 analysis on a chiral stationary phase. dr value was determined by 1H NMR. | ||
As shown in Scheme 4a, the reaction of ketone A1 at the 4 mmol scale with glycinate B5 performed well under the optimized reaction conditions, and the gram-scale synthesis of C5 gave 80% yield (1.42 g) with 99%/93% ee and 78:22 dr. Using the enantiomer of the ligand L3-RaPr3 instead, ent-C5 was delivered with comparable results (see ESI† for details). After recrystallization, nearly optically pure C5 (99% ee, >95:5 dr) was obtained, which could be readily reduced into the useful 2-amino-1,3-diol18D1 by diisobutylaluminium hydride (DIBAL-H) or be transformed into oxazolidinone D2 in moderate yield without the erosion of stereoselectivity after the treatment with triphosgene.
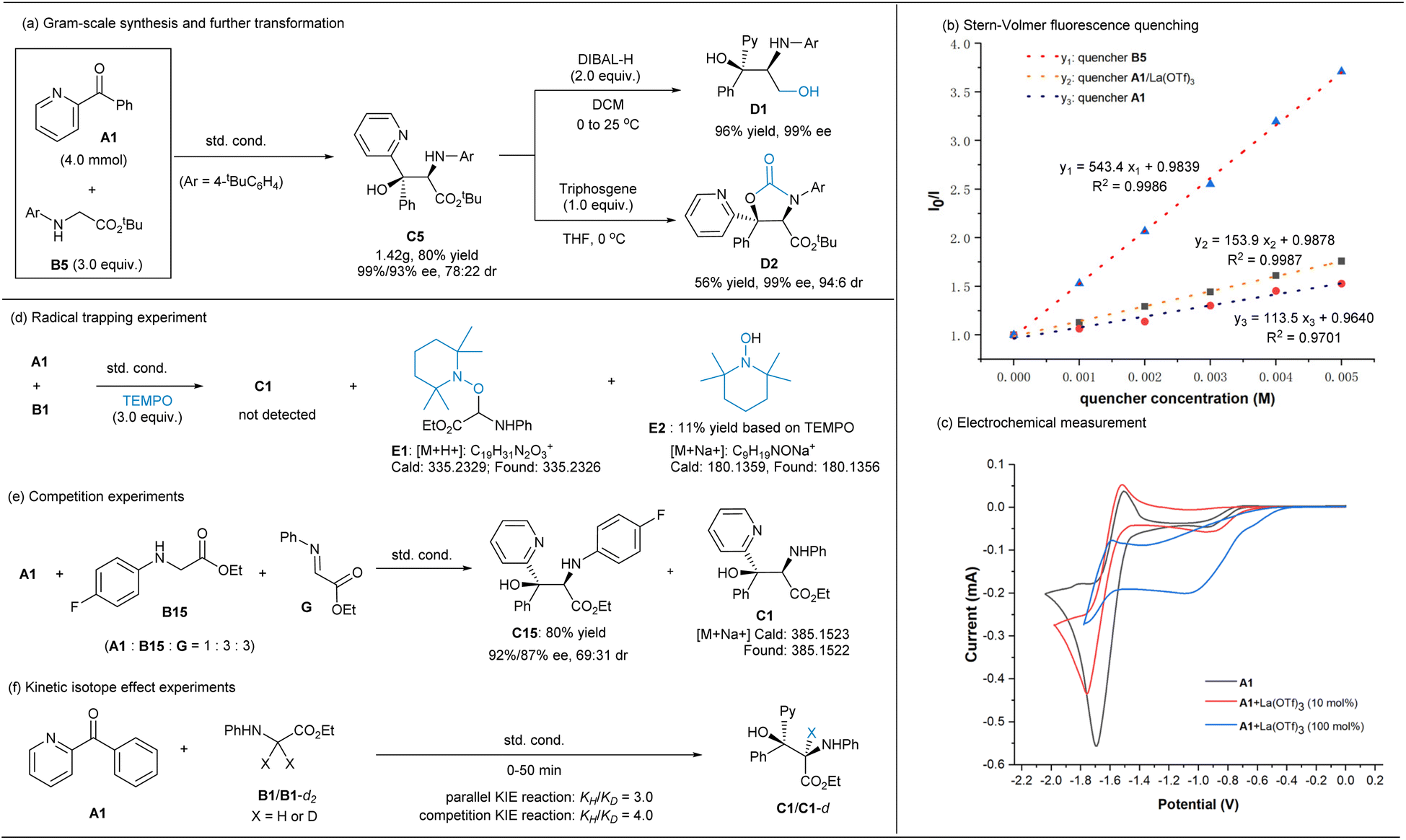 | ||
| Scheme 4 Synthetic application and mechanistic investigation. | ||
To glean insights into the overall mechanism, some spectroscopic studies and control experiments were conducted (Scheme 4 and ESI†). UV-vis absorption spectroscopy analysis suggested that the main light-absorbing species is the iridium photocatalyst rather than ketone A1 or glycine ester B5 under the irradiation at 460 nm (see ESI† for details). Stern–Volmer fluorescence quenching studies (Scheme 4b) showed that the excited photocatalyst [Ir2] (*Ered (*IrIII/IrII) = +1.21 V vs. SCE)10a could be quenched by both glycine ester B5 (Ep/2ox = +0.85 V vs. SCE) and A1 or La(III)-bonded A1, but the plot manifested that the quenching by the former is much stronger than that by the ketone, indicating a more favorable reductive quenching cycle. Furthermore, electrochemical measurement (Scheme 4c, all vs. SCE in MeCN) suggested that ketone A1 had lower reductive potential [Ep/2red (A1/A1˙−) = −1.6 V], which shifted to about −0.82 V in the presence of La(OTf)3, but the current intensity was low. Although the photoexcited state *Ir(III) [*Eox (IrIV/*IrIII) = −0.89 V] or Ir(II) [E1/2 = −1.37 V]10a might serve as a reductant to reduce Lewis acid-activated carbonyl A1 to afford the corresponding ketyl radical, this process will not dominate during the transformation in view of both the Stern–Volmer quenching experiments and radical clock experiments (see ESI† for details). This trend differed from the racemic reaction of benzophenone promoted by [Ir1]/LiBF4 cooperation catalysis in Xiao's report,5a in which the generation of the ketyl radical species of ketone via the reduction of Ir(III)* was proposed. The light on/off experiment between A1 and B5 indicated that continuous irradiation was essential for the formation of the product, and the quantum yield was found to be 0.022, excluding a radical chain-dominated process.
Next, radical trapping experiment was conducted (Scheme 4d) and it displayed that the addition process was completely inhibited by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), and the radical coupling adduct E1 was detected by high resolution mass spectrometry (HRMS), along with piperi-din-1-ol E2via the HAT process in 11% isolated yield (Scheme 4d). In connection with the detected byproducts F and G (Table 1), it is hypothesized that the reaction process involves reactive α-amino carbon radical species (see ESI† for details). Moreover, when imine G was added into the reaction system (Scheme 4e), the major coupling product is related to the glycinate B15, confirming that the formation of the ketyl radical is the least as stated above. A primary kinetic isotope effect (KIE) was obtained by parallel (KIE = 3.0) and competition reactions (KIE = 4.0), which indicated that the C–H cleavage of glycinate has a significant contribution to the rate-determining step (Scheme 4f).
In addition, a linear relationship between the enantiomeric excess of ligand L3-RaPr3 and the enantiomeric excess of product C50 was observed, indicating that the metal salt and the chiral N,N′-dioxide ligand were coordinated in a 1:1 ratio without catalyst aggregation in the active catalytic species (see the ESI† for details).
Based on the above analysis, a possible dominant mechanism is proposed in Fig. 1a to rationalize the synergistic catalysis. The reaction is initiated with excited *[IrIII] photocatalyst oxidation and DMAP deprotonation sequence of glycine ester B1 into α-amino radical Int I. Subsequently, the radical Int I rapidly adds to the chiral Lewis acid bidentate-bonded ketone complex (A1/[La]*), generating alkoxy radical species Int II bonded by the chiral Lewis acid catalyst (path a). Otherwise, the simple alkoxy radical would readily show β-scission14 to recover the ketone and Int I. However, DFT calculation revealed the stabilization of the alkoxy radical by the chiral catalyst or proton (see ESI† for details). We propose that after the release of the chiral Lewis acid catalyst, the protonated Int III of the alkoxy radical shows the reductive process with [IrII] species (Ered (IrIII/IrII) = −1.37 V vs. SCE)10a to form the product, along with the regeneration of the [IrIII] photocatalyst. During this process, the α-amino radical Int I may undergo further oxidation to yield the imine byproduct or the formal homocoupling diamine byproduct. On the other hand, in the absence of the photocatalyst, the chiral Lewis bonded ketone could be excited at 400 nm irradiation,16c,d which acts as a strong oxidant to transfer glycinate into the related radical species Int I or imine intermediate (path b). Following radical–radical cross-coupling or addition, the desired product could also be generated.
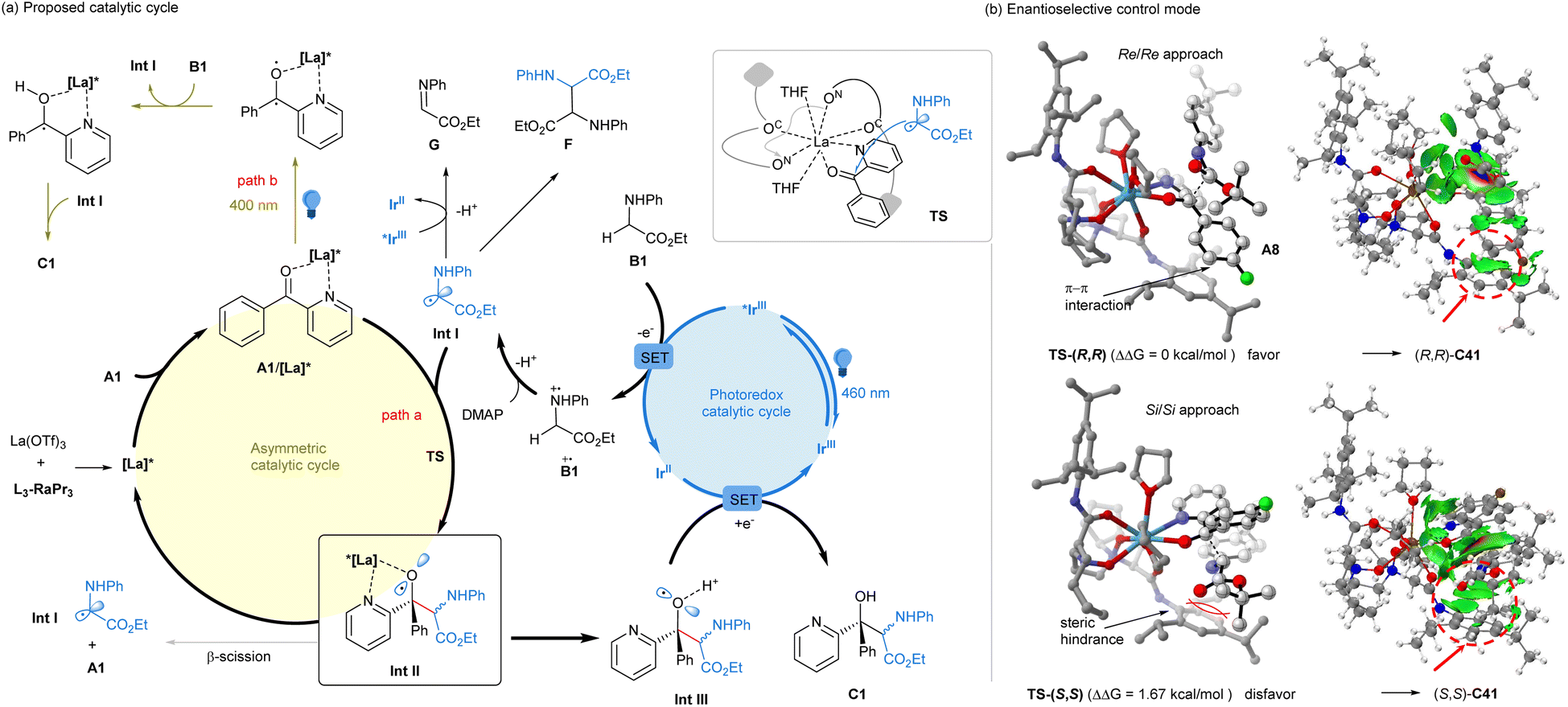 | ||
| Fig. 1 Proposed catalytic cycle. | ||
To elucidate the underlying cause of the enantioselectivity induced by the N,N′-dioxide ligand, we carried out DFT calculations employing the chiral La(OTf)3/L3-RaPr3 complex in the enantio-determining step of the intermediate Int II (see ESI† for details).19 As depicted in Fig. 1b, the eight coordination sites of La(III) were saturated by L3-RaPr3, THF and bidentate ketone A8. Two facial selective pathways during the radical addition process were to generate the major diastereomer Int II: (Re/Re) approach vs. the (Si/Si) approach between the ketone and radical Int I. The energy of the transition state TS-(R,R) in the (Re/Re) manner is 1.67 kcal lower than that of TS-(S,S). From the perspective of Boltzmann distribution, this energy difference corresponds to approximately 95% ee to generate C41, which is consistent with the experimental observations. The IGMH weak interaction analysis20 clearly reveals the reasons behind this difference. There is a weak π–π interaction between the aryl groups of ketone A8 and one aniline unit of the N,N′-dioxide ligand in TS-(R,R). However, there is steric hindrance between the ester group of radical Int I and the lower right aryl group of the ligand in TS-(S,S).
Conclusions
The synergistic catalysis of the iridium photoredox catalyst and the chiral lanthanide complex of N,N′-dioxide enables the direct asymmetric α-C(sp3)–H bond functionalization of glycinates with ketones. This effective protocol allows for the straightforward preparation of a wide range of valuable enantioenriched unnatural β-quaternary stereocentric serine derivatives with excellent enantioselectivities for both the diastereomers. The cooperation of the chiral Lewis acid catalyst not only harmonized the enantioselective radical addition step but also stabilized the alkoxy radical species before reduction. The multiplicative enhancement minimized the competitive byproduct formation and overwhelming racemic background reaction. This cooperative catalytic system holds promise for the development of a wide array of asymmetric catalytic direct α-C(sp3)–H bond functionalizations through visible light-driven pathways.Data availability
Further details of the experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, X-ray crystallographic data for C5 are available in the ESI.†Author contributions
J. Q. T. performed experiments and prepared the ESI† and paper. L. Q. Y. conducted the DFT calculation. Y. T. Y. repeated some experiments. H. Y. S. and Z. W. Z. participated in structure characterization and discussion. X. H. L. helped with modifying the paper and ESI.† X. H. L. and X. M. F. conceived and directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (22188101), the National Key Research and Development Program of China (2022YFA1504301), and Sichuan University (2020SCUNL204) for financial support. We thank Dr Yuqiao Zhou (Sichuan University) for the X-ray single crystal diffraction analysis.Notes and references
- (a) V. A. Soloshonok and K. Izawa, Asymmetric Synthesis and Application of a-Amino Acids, Oxford University Press, Washington, 2009 Search PubMed; (b) A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry, Analysis and Function of Amino Acids and Peptides, Wiley, Hoboken, 2013 Search PubMed.
- For selected reviews on α-C(sp3)–H bond functionalization of glycinate: (a) T. A. King, J. M. Kandemir, S. J. Walsh and D. R. Spring, Chem. Soc. Rev., 2021, 50, 39–57 RSC; (b) M. Hari Babu and J. Sim, Eur. J. Org Chem., 2022, e202200859 CrossRef CAS; (c) Y. Tian, X. B. Bu, Y. R. Chen, L. H. Wang, J. N. E, J. Zeng, H. Xu, A. H. Han, X. B. Yang and Z. Zhao, Catalysts, 2023, 13, 1502 CrossRef CAS; (d) C. Wang, R. P. Qi, R. Wang and Z. Q. Xu, Acc. Chem. Res., 2023, 56, 2110–2125 CrossRef CAS PubMed; (e) G. Q. Xu, W. D. Wang and P. F. Xu, J. Am. Chem. Soc., 2024, 146, 1209–1223 CrossRef CAS; (f) W. Wang, L. M. Xuan, Q. L. Chen, R. D. Fan, F. Zhao, J. H. Dong, H. F. Wang, Q. J. Yan, H. Zhou and F. E. Chen, J. Am. Chem. Soc., 2024, 146, 6307–6316 CrossRef CAS.
- For selected examples of cationic iminium: (a) X. W. Gao, Q. Y. Meng, J. X. Li, J. J. Zhong, T. Lei, X. B. Li, C. H. Tung and L. Z. Wu, ACS Catal., 2015, 5, 2391–2396 CrossRef CAS; (b) Y. Zhang, S. L. Li, Y. Zhu, X. R. Yang, H. Zhou and Y. Li, J. Org. Chem., 2020, 85, 6261–6270 CrossRef CAS PubMed; (c) Z. Q. Zhu, D. Guo, J. L. Ji, X. Zhu, J. Tang, Z. B. Xie and Z. G. Le, J. Org. Chem., 2020, 85, 15062–15071 CrossRef CAS PubMed; (d) H. Zhao and D. Leonori, Angew. Chem., Int. Ed., 2021, 60, 7669–7674 CrossRef CAS PubMed; (e) C. Che, Y. N. Li, X. Cheng, Y. N. Lu and C. J. Wang, Angew. Chem., Int. Ed., 2021, 60, 4698–4704 CrossRef CAS PubMed; (f) M. H. Babu and J. Sim, Eur. J. Org Chem., 2022, e2022008591 Search PubMed; (g) C. H. Jiang, X. F. Sha, C. Ni, W. Qin, X. J. Zhu, S. Wang, X. Li and H. F. Lu, J. Org. Chem., 2022, 87, 8744–8751 CrossRef CAS PubMed; (h) C. Che, Y. N. Lu and C. J. Wang, J. Am. Chem. Soc., 2023, 145, 2779–2786 CrossRef CAS; (i) S. T. Wang, Y. J. Ye, H. L. Shen, J. L. Liu, Z. Liu, Z. G. Jiang, J. Q. Lei and Y. Zhang, Org. Biomol. Chem., 2023, 21, 8364–8371 RSC; (j) C. Che, Y.-N. Lu, T. Fang, G.-J. Zhen, X. T. Qi and C.-J. Wang, ACS Cent. Sci., 2024 DOI:10.1021/acscentsci.4c00970.
- (a) R. P. Qi, C. Wang, Y. M. Huo, H. L. Chai, H. Y. Wang, Z. J. Ma, L. Y. Liu, R. Wang and Z. Q. Xu, J. Am. Chem. Soc., 2021, 143, 12777–12783 CrossRef CAS; (b) R. Qi, C. Wang, Z. Ma, H. Wang, Q. Chen, L. Liu, D. Pan, X. Ren, R. Wang and Z. Q. Xu, Angew. Chem., Int. Ed., 2022, 61, e202200822 CrossRef CAS PubMed.
- (a) W. Ding, L. Q. Lu, J. Liu, D. Liu, H. T. Song and W. J. Xiao, J. Org. Chem., 2016, 81, 7237–7243 CrossRef CAS; (b) G. Z. Wang, D. G. Liu, M. T. Liua and Y. Fu, Green Chem., 2021, 23, 5082–5087 RSC; (c) J. Zheng and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 3392–3397 CrossRef CAS PubMed.
- K. Murugesan, K. Donabauer, R. Narobe, V. Derdau, A. Bauer and B. König, ACS Catal., 2022, 12, 3974–3984 CrossRef CAS.
- For selected examples on bioactivitives of β-hydroxy-α-amino acids: (a) M. A. T. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed; (b) U. Galm, E. W. Pienkowski, L. Y. Wang, N. P. George, T. J. Oh, F. Yi, M. F. Tao, J. M. Coughlinc and B. Shen, Mol. BioSyst., 2009, 5, 77–90 RSC; (c) H. Hashizume, K. Iijima, K. Yamashita, T. Kimura, S. Wada, R. Sawa and M. Igarashi, J. Antibiot., 2018, 71, 129–134 CrossRef CAS; (d) C. J. Mathias, Clin. Auton. Res., 2008, 18, 25–29 CrossRef; (e) D. A. Wirtz, K. C. Ludwi, M. Arts, C. E. Marx, S. Krannich, P. Barac, S. Kehraus, M. Josten, B. Henrichfreise, A. Müller, G. M. König, A. J. Peoples, A. Nitti, A. L. Spoering, L. L. Ling, K. Lewis, M. Crüsemann and T. Schneider, Angew. Chem., Int. Ed., 2021, 60, 13579–13586 CrossRef CAS.
- For selected reviews of azomethine ylides: (a) L. Wei, L. Xiao, Y. Z. Hu, Z. F. Wang, H. Y. Tao and C. J. Wang, Chin. J. Org. Chem., 2019, 39, 2119–2130 CrossRef CAS; (b) L. Wei, X. Chang and C. J. Wang, Acc. Chem. Res., 2020, 53, 1084–1100 CrossRef CAS PubMed; (c) L. Wei, X. Chang, Z. P. Zhang, Z. F. Wang and C. J. Wang, Chin. Sci. Bull., 2023, 68, 3956–3968 Search PubMed.
- For selected reviews on carbonyl catalysis: (a) Y. C. Luo, H. H. Zhang, Y. Wang and P. F. Xu, Acc. Chem. Res., 2010, 43, 1317–1330 CrossRef CAS PubMed; (b) A. E. Sorochinsky, J. L. Aceña, H. Moriwaki, T. Sato and V. Soloshonok, Amino Acids, 2013, 45, 1017–1033 CrossRef CAS PubMed; (c) Q. Wang, Q. Gu and S. L. You, Angew. Chem., Int. Ed., 2019, 58, 6818–6825 CrossRef CAS; (d) W. Wen and Q. X. Guo, Synthesis, 2023, 55, 719–732 CrossRef CAS; (e) X. Xiao and B. G. Zhao, Acc. Chem. Res., 2023, 56, 1097–1117 CrossRef CAS For recent selected examples on asymmetric Aldol reaction:; (f) A. L. Cheng, L. L. Zhang, Q. H. Zhou, T. Liu, J. Cao, G. Q. Zhao, K. Zhang, G. S. Song and B. G. Zhao, Angew. Chem., Int. Ed., 2021, 60, 20166–20172 CrossRef CAS PubMed; (g) P. W. Ji, X. P. Liu, J. W. Xu, X. Zhang, J. H. Guo, W. W. Chen and B. G. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202206111 CrossRef CAS; (h) C. K. Hou, B. F. Peng, S. Ye, Z. Y. Yin, J. Cao, X. Xiao and B. G. Zhao, Nat. Catal., 2022, 5, 1061–1068 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) J. R. Chen, X. Q. Hu, L. Q. Lu and W. J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- For selected examples of excited ketones: (a) P. J. Wagner, Chemistry of Excited Triplet Organic Carbonyl Compounds. Triplet States III; Topics in Current Chemistry, Springer, vol. 66, 1976, pp. 1–52 Search PubMed; (b) P. J. Wagner and R. A. Leavitt, J. Am. Chem. Soc., 1973, 95, 3669–3677 CrossRef CAS; (c) P. J. Wagner, R. J. Truman, A. E. Puchalski and R. Wake, J. Am. Chem. Soc., 1986, 108, 7727–7738 CrossRef CAS; (d) C. Chen, Org. Biomol. Chem., 2016, 14, 8641–8647 RSC; (e) M. Fréneau and N. Hoffmann, J. Photochem. Photobiol., C, 2017, 33, 83–108 CrossRef; (f) Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC; (g) H.-M. Huang, P. Bellotti and F. Glorius, Acc. Chem. Res., 2022, 55, 1135–1147 CrossRef CAS; (h) X. B. Bai, J. L. Yao, W. X. Li, X. W. Zhao, Y. L. Yin, S. Y. Yu and Z. Y. Jiang, Org. Lett., 2024, 26, 5037–5042 CrossRef CAS PubMed.
- For selected reviews and examples on CLA–catalyzed photochemical reactions of aryl ketones: (a) L. L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS; (b) X. Q. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed; (c) L. Z. Hou, X. H. Liu, W. D. Cao and X. M. Feng, ChemCatChem, 2023, 15, e202300893 CrossRef CAS; (d) C. Y. Wang, J. Qin, X. D. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS; (e) J. J. Ma, A. R. Rosales, X. Q. Huang, K. Harms, R. Riedel, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 17245–17248 CrossRef CAS; (f) F. Y. Li, D. Tian, Y. F. Fan, R. Lee, G. Lu, Y. L. Yin, B. K. Qiao, X. W. Zhao, Z. W. Xiao and Z. Y. Jiang, Nat. Commun., 2019, 10, 1774 CrossRef; (g) Z. D. Tan, S. B. Zhu, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, 61, e202203374 CrossRef CAS PubMed; (h) J. B. Kidd, T. A. Fiala, W. B. Swords, Y. Park, K. A. Meyer, K. M. Sanders, L. A. Guzei, J. C. Wright and T. P. Yoon, J. Am. Chem. Soc., 2024, 146, 15293–15300 CrossRef CAS PubMed.
- (a) X.-K. He, L.-Q. Lu, B.-R. Yuan, J.-L. Luo, Y. Cheng and W.-J. Xiao, J. Am. Chem. Soc., 2024, 146, 18892–18898 CrossRef CAS PubMed; (b) Z. W. Zhong, H. D. Wu, X. F. Chen, Y. Luo, L. Q. Yang, X. M. Feng and X. H. Liu, J. Am. Chem. Soc., 2024, 146, 20401–20413 CrossRef CAS.
- L. Chang, Q. An, L. F. Duan, K. X. Feng and Z. W. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- For reviews on chiral N,N'-dioxides: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS; (e) Z. Wang, X. H. Liu and X. M. Feng, Aldrichim Acta, 2020, 53, 3–10 CAS; (f) L. L. Lin, Y. Q. Zhou, W. D. Cao, S. X. Dong, X. H. Liu and X. M. Feng, Sci. China: Chem., 2023, 53, 246–258 Search PubMed; (g) S. X. Dong, W. D. Cao, M. P. Pu, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2717–2735 CrossRef CAS; (h) G. H. Pan, C. L. He, M. Chen, Q. Xiong, W. D. Cao and X. M. Feng, CCS Chem., 2022, 4, 2000–2008 CrossRef CAS; (i) Z. W. Zhong, L. C. Ning, Y. C. Lu, J. Q. Tan, L. L. Lin and X. M. Feng, Sci. China: Chem., 2023, 66, 799–807 CrossRef CAS.
- For selected examples on photocatalytic reactions with chiral N,N'-dioxides: (a) H. Yu, S. X. Dong, Q. Yao, L. Chen, D. Zhang, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2018, 24, 19361–19367 CrossRef CAS PubMed; (b) L. K. Yang, W. Y. R. Li, H. L. Hou, T. Y. Zhan, W. D. Cao, X. H. Liu and X. M. Feng, Chem. Sci., 2022, 13, 8576–8582 RSC; (c) H. Yu, T. Y. Zhan, Y. Q. Zhou, L. Chen, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 5136–5144 CrossRef CAS; (d) L. Z. Hou, Y. Q. Zhou, H. Yu, T. Y. Zhan, W. D. Cao and X. M. Feng, J. Am. Chem. Soc., 2022, 144, 22140–22149 CrossRef CAS PubMed; (e) Y. Luo, Q. Wei, L. K. Yang, Y. Q. Zhou, W. D. Cao, Z. S. Su, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 12984–12992 CrossRef CAS; (f) L. L. Feng, X. F. Chen, N. Guo, Y. Q. Zhou, L. L. Lin, W. D. Cao and X. M. Feng, Chem. Sci., 2023, 14, 4516–4522 RSC; (g) T. Y. Zhan, L. K. Yang, Q. Y. Chen, R. Weng, X. H. Liu and X. M. Feng, CCS Chem., 2023, 5, 2101–2110 CrossRef CAS; (h) L. K. Yang, L. C. Ning, H. Yu, S. Y. Li, M. Yang, L. H. Yang, F. Wang, X. H. Liu, W. D. Cao and X. M. Feng, CCS Chem., 2024 DOI:10.31635/ccschem.024.202404257; (i) L. Z. Hou, L. Q. Yang, G. F. Yang, Z. Luo, W. L. Xiao, L. H. Yang, F. Wang, L.-Z. Gong, X. H. Liu, W. D. Cao and X. M. Feng, J. Am. Chem. Soc., 2024, 146, 23457–23466 CrossRef CAS PubMed; (j) Y. Luo, Y. Q. Zhou, F. N. Xiao, X. He, Z. W. Zhong, Q.-L. Zhou, W. D. Cao, X. H. Liu and X. M. Feng, ACS Catal., 2024, 14, 12031–12041 CrossRef CAS.
- CCDC 2320632 for C5 contains the supplentary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- For selected examples for application of 2-amino-1,3-diols: (a) M. P. Sibi, J. X. Chen and G. R. Cook, Tetrahedron Lett., 1999, 40, 3301–3304 CrossRef CAS; (b) J. V. Allen and J. M. J. Williams, Tetrahedron: Asymmetry, 1994, 5, 277–282 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. J. R. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu and Q. X. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC and UPC2 spectra. X-ray crystallographic data for C5. CCDC 2320632. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04900d |
| This journal is © The Royal Society of Chemistry 2024 |