
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting type-I ROS production of molecular photosensitizers using bridge-assisted superexchange coupling†
Lei
Chen‡
,
Shirong
Yan‡
,
Wu-Jie
Guo
,
Lu
Qiao
,
Xinyue
Zhan
,
Bin
Liu
 * and
Hui-Qing
Peng
*
* and
Hui-Qing
Peng
*
State Key Laboratory of Chemical Resource Engineering, Beijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: binliu@buct.edu.cn; hqpeng@mail.buct.edu.cn
First published on 10th September 2024
Abstract
Bridge-assisted superexchange coupling capable of long-range electron transfer proves to be effective for charge separation. However, the exploitation of this photochemical process in engineering reactive oxygen species (ROS) production remains unexplored. Herein, piperazine serves as a bridging unit to facilitate a cascade electron transfer from the electron donor site (CO) to the acceptor site (CN) within the COCN molecule, ultimately boosting the generation of superoxide radicals (O2−˙) and hydroxyl radicals (˙OH). Experimental and theoretical studies elucidate that the long-range electron transfer is enabled by a superexchange interaction through the piperazine σ*-bridge, which leads to an effective generation of a radical ion pair CO+˙BCN−˙. The cationic radical CO+˙ can directly catalyze the oxidation of water, while the anionic radical CN−˙ transfers one electron to oxygen (O2). Additionally, COCN has an excited triplet state characterized by a 3(π–π*) electronic configuration, which further promotes sequential electron transfer to O2. These reactions enable the efficient production of ˙OH and O2−˙, respectively, thus completing a cascade electron cycling process. Based on these findings, nanoparticles of COCN exhibit satisfying O2−˙ and ˙OH production performance even under hypoxic environments and demonstrate potent photodynamic activity in addition to a notably high fluorescence quantum yield of 62.8%, rendering them promising candidates for cellular imaging and ablation assessments. This study contributes to the advancement of photosensitizers proficient in selectively generating ROS, offering valuable insights into the underlying mechanisms that govern ROS production.
Introduction
Photosensitizers (PSs) that exhibit high efficiency in generating strong oxidizing reactive oxygen species (ROS) upon light irradiation play crucial roles in the fields of photodynamic therapy (PDT).1,2 Under light irradiation, these PSs undergo a photophysical transition from the ground singlet state (S0) to the excited singlet state (Sn), followed by activation to the excited triplet state (Tn) through intersystem crossing (ISC).3–5 Ultimately, the triplet PSs interact with molecular oxygen (O2) via electron transfer processes, leading to the generation of type-I ROS such as hydroxyl radicals (˙OH) and superoxide radicals (O2−˙), and/or alternatively transfer energy to O2 resulting in the production of singlet oxygen (1O2) as a type-II ROS. By harnessing these ROS, PDT has been widely recognized as a promising clinical treatment modality, offering advantages of precise spatiotemporal control, minimal invasiveness, and non-drug resistance.6,7 Normally, conventional PSs, comprising methylene blue and Eosin Y, are capable of simultaneously releasing type-I and type-II ROS through these two photosensitization pathways.8 However, different ROS type exhibits distinct reactive and transient characteristics that can fulfill diverse biological demands.9,10 For this reason, the development of PSs possessing selective ROS-generating properties will offer significant advantages in optimizing ROS-mediated biological applications. Concurrently, it is believed that the ability to selectively produce specific ROS types is highly beneficial to simplifying the elucidation of underlying biological effects associated with ROS.Recently, PSs with efficient type-I ROS generation ability (type-I PSs) have attracted increasing attention due to its low dependence on O2, thereby overcoming the diminished therapeutic effect in tumor hypoxic microenvironments.11–15 Moreover, O2−˙, a highly cytotoxic ROS generated through the type-I mechanism, can react with proteins, DNA, and lipids, leading to irreversible cellular damage.16,17 And ˙OH, which can be generated through the cascade bioreaction of O2−˙ or direct oxidation of water, is considered the most reactive species due to its extremely high chemical reactivity towards nearly all biological molecules.18 The predominant strategy for facilitating charge separation by enhancing donor–acceptor interactions within a molecule has laid a solid foundation for the development of type-I PSs.19,20 By introducing light-induced intermolecular electron transfer between PSs and additional electron donors/acceptors can further boost the electron transfer pathway of PSs, which is able to overcome the rapid recombination of the photogenerated charges during their redox reactions.21,22 These previous studies have shown the viability of preparing type-I PSs using an electron transfer cascade strategy. Nevertheless, systems involving multiple components may be susceptible to phase separation and component leakage, potentially undermining the stability and efficacy of the PSs and thereby complicating their practical applications.
In the field of photochemistry, a long-range intramolecular electron transfer (>10 Å) can also occur from a specific electron donor site (D) to a well-defined acceptor site (A) through the intermediate linkage between D and A (Scheme 1a).23,24 For instance, the wave functions of the bridge (B) structures' σ* orbitals enable effective propagation of electronically coupled interactions between D and A fragments.25,26 Specifically, the LUMO of the D group overlaps with the LUMO of the σ* orbital located on the nearest bond within the bridge, promoting electron transfer to the LUMO of the A unit and further resulting in the formation of D+˙BA−˙ species. Such bridge-assisted superexchange coupling has been extensively investigated in quantum chemistry simulations due to its significance as a fundamental charge transfer process in chemical and biological systems, e.g., the primary charge separation in bacterial photosynthesis.27 However, compared to intermolecular electron transfer, the demonstration of long-range intramolecular electron transfer by superexchange coupling for facilitating the cascade electron transfer to O2 remains unexplored. The triplet state of O2 consists of two unpaired electrons occupying each of the two antibonding π orbitals (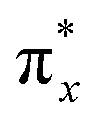 and
and  ) at the same energy level.28 Therefore, we hypothesized that O2−˙ could be generated by populating one of the π* orbitals with an electron from D+˙BA−˙. Simultaneously, the resulting cationic radical (D+˙) captures an electron from the surrounding water, thereby oxidizing the water to ˙OH. The utilization of bridge-assisted superexchange coupling is anticipated to offer a highly desirable alternative approach for boosting type-I ROS production, enabling the development of stable and efficient type-I PSs with well-defined molecular structures and selective ROS-generating abilities.
) at the same energy level.28 Therefore, we hypothesized that O2−˙ could be generated by populating one of the π* orbitals with an electron from D+˙BA−˙. Simultaneously, the resulting cationic radical (D+˙) captures an electron from the surrounding water, thereby oxidizing the water to ˙OH. The utilization of bridge-assisted superexchange coupling is anticipated to offer a highly desirable alternative approach for boosting type-I ROS production, enabling the development of stable and efficient type-I PSs with well-defined molecular structures and selective ROS-generating abilities.
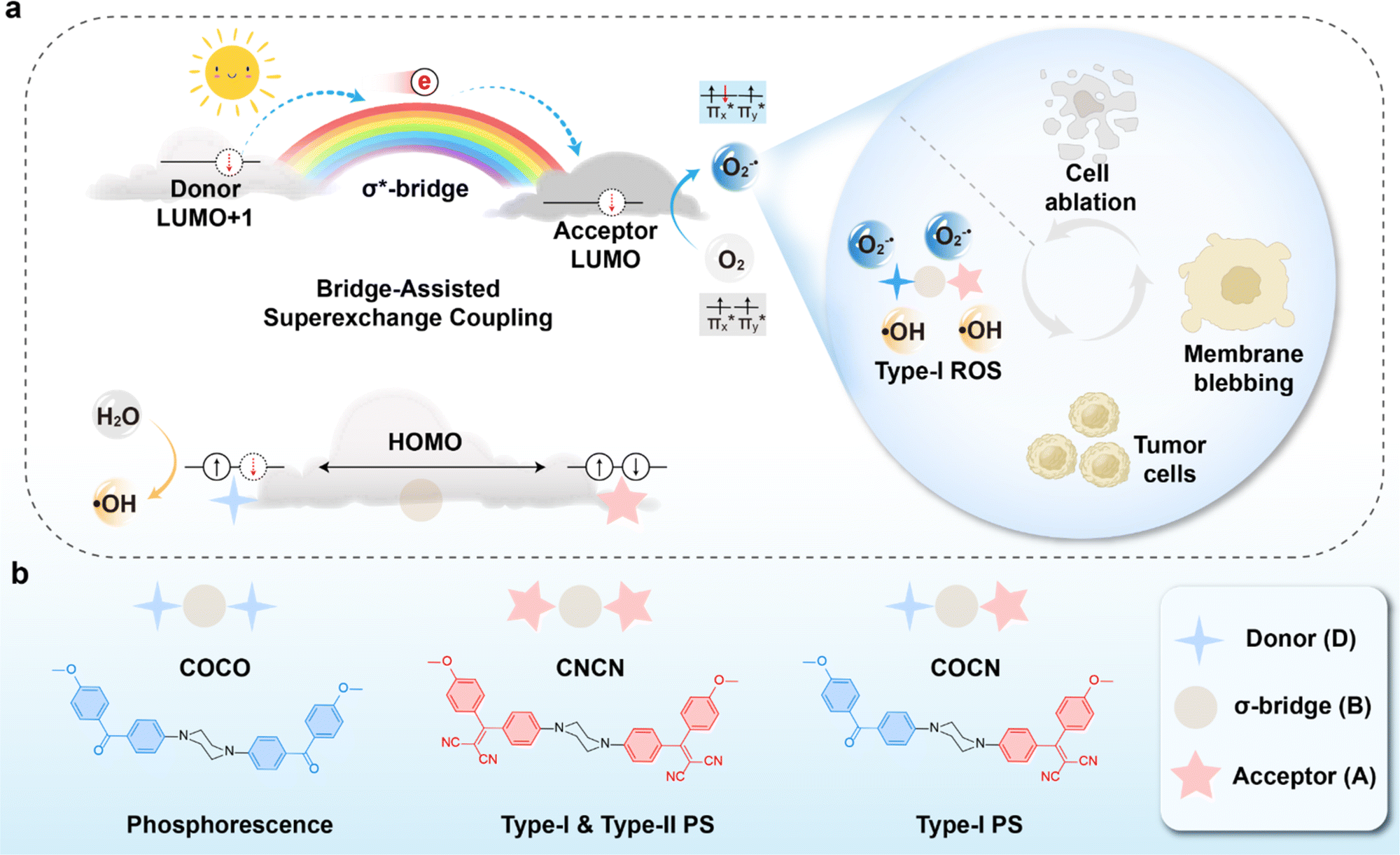 | ||
| Scheme 1 (a) Schematic illustration of bridge-assisted superexchange coupling mechanism for type-I ROS generation. (b) Molecular structures of COCO, CNCN, and COCN. | ||
Herein, we exploit the superexchange mechanism to facilitate the separation of electron–hole pairs in PSs for augmenting O2−˙ and ˙OH generation (Scheme 1). As a proof of concept, three homologous compounds COCO, COCN, and CNCN were rationally designed, utilizing piperazine as the bridging unit, benzophenone (CO) as the terminal D group, and 2-(diphenylmethylene)malononitrile (CN) as the A group (Scheme 1b). Both CO and CN are presumed to be capable of readily accessing Tn for reacting with O2 based on the design principle of organic phosphors.29 Among these compounds, COCO exhibits persistent phosphorescence and shows minimal production of ROS, whereas CNCN demonstrates a remarkable ability to generate type-I and type-II ROS through two photosensitization pathways. In stark contrast, the asymmetric molecule COCN manifests type-I ROS-generating property, resulting in significantly enhanced production of O2−˙ and ˙OH while the generation of 1O2 is negligible. Analysis of the phosphorescence lifetime and fluorescence spectra reveals that the excitation energy associated with the CO component within COCN has been substantially quenched by the CN group, implying an effective energy and/or electron transfer process from CO to CN. Theoretical studies verify that electron transfer occurs from the higher-energy LUMO+1 of CO to the lower-energy LUMO of CN via piperazine-assisted superexchange, resulting in the formation of a radical ion pair CO+˙BCN−˙. The cationic radical CO+˙ catalyze the oxidation of water, resulting in the generation of ˙OH. Concurrently, COCN has T1 state featuring a 3(π–π*) electronic configuration, which promotes cascade electron transfer from CN−˙ to O2 for efficient generation of O2−˙. Finally, nanoparticles of COCN were prepared with a remarkable fluorescence quantum yield of 62.8%, while maintaining efficient O2−˙ and ˙OH production ability in aqueous solution. They were successfully employed for cellular imaging and photodynamic cancer cell ablation. To our best knowledge, this is the first demonstration of a molecular photosensitizer design using bridge-assisted superexchange coupling to facilitate cascaded electron transfer and thus enable the engineering of type-I ROS generation.
Results and discussion
The primary purpose of this study is to design selective type-I PSs based on the electron transfer cascade strategy utilizing bridge-assisted superexchange coupling. For the CO group, benzophenone is a representative organic phosphor that can efficiently transition from Sn to Tn with nearly 100% ISC yield. For the CN fragment, the incorporation of a malononitrile group with strong electron-withdrawing ability not only promotes the charge transfer (CT) effect but also substantially reinforces the intramolecular steric hindrance, which results in a nonplanar molecular conformation. Such nonplanarity can destruct π–π stacking interactions and impart aggregation-induced emission (AIE) features to the compounds with CN moiety. The utilization of AIE-active PSs is expected to increase ROS generation by blocking nonradiative pathways through the restriction of molecular motions in aggregates, thereby realizing efficient fluorescence imaging-guided PDT.30 On the other hand, as per our previous findings, piperazine has a compact small molecular structure exhibiting a rigid chair conformation. It acts as the bridging unit not only fixes the D and A units at well-defined separations through the saturated hydrocarbon linkages but also plays a pivotal role in facilitating the electron transfer process by incorporating nitrogen atoms carrying lone pairs.31,32 Based on these reasons, the PSs in this study were designed and synthesized by involving benzophenone, malononitrile, and piperazine as the molecular components. COCO was readily prepared through a nucleophilic substitution reaction between (4-fluorophenyl)-(4-methoxyphenyl)methanone and piperazine. Subsequently, COCO can undergo Knoevenagel condensation with malononitrile to yield COCN and CNCN (Scheme S1†). The chemical structures of COCO, COCN, and CNCN were thoroughly characterized by 1H NMR, 13C NMR, and high-resolution mass spectroscopies with satisfactory outcomes (Fig. S1–S9†).The UV-vis and photoluminescence (PL) spectroscopies of COCO, COCN, and CNCN were systematically analyzed to gain a more comprehensive understanding of their photophysical properties. As illustrated in Fig. 1a, COCO in dimethyl sulfoxide (DMSO, 10 μM) exhibits maximum absorption and emission intensity at 350 nm and 500 nm, respectively. In contrast, the incorporation of the strong electron-withdrawing malononitrile group leads to a new absorption peak at 450 nm for both COCN and CNCN in DMSO (10 μM), while their emission intensity maximum peak at around 585 nm. Furthermore, the PL intensities of COCN and CNCN are observed to gradually increase in a mixture of tetrahydrofuran and water as the volume fraction of water increased, confirming their typical AIE characteristics (Fig. 1b and S10†). Nevertheless, the PL intensity of COCO exhibits a remarkable decrease as the fraction of the poor solvent increased due to the aggregation-caused quenching effect. Given the indispensability of accessing triplet states for ROS generation, we investigated the prompt and delayed emission spectra of COCO, COCN, and CNCN at the temperature of 77 K (Fig. S11†). With a delay time of 1 ms, the delayed spectra show a profile akin to their corresponding prompt spectra. This observation suggests that the emissions predominantly originate from the long-lived triplet levels, implying a high efficiency of these compounds in populating triplet excitons. The phosphorescence lifetime of COCO at 490 nm has been estimated to be 94.7 ms (Fig. 1c). However, the emission intensity of both COCN and CNCN is so faint that their emission lifetimes are hard to be detected. The comparative analysis of the results demonstrates that the presence of the CN group can block the relaxation of CO from the triplet state to the ground state via phosphorescence emission.
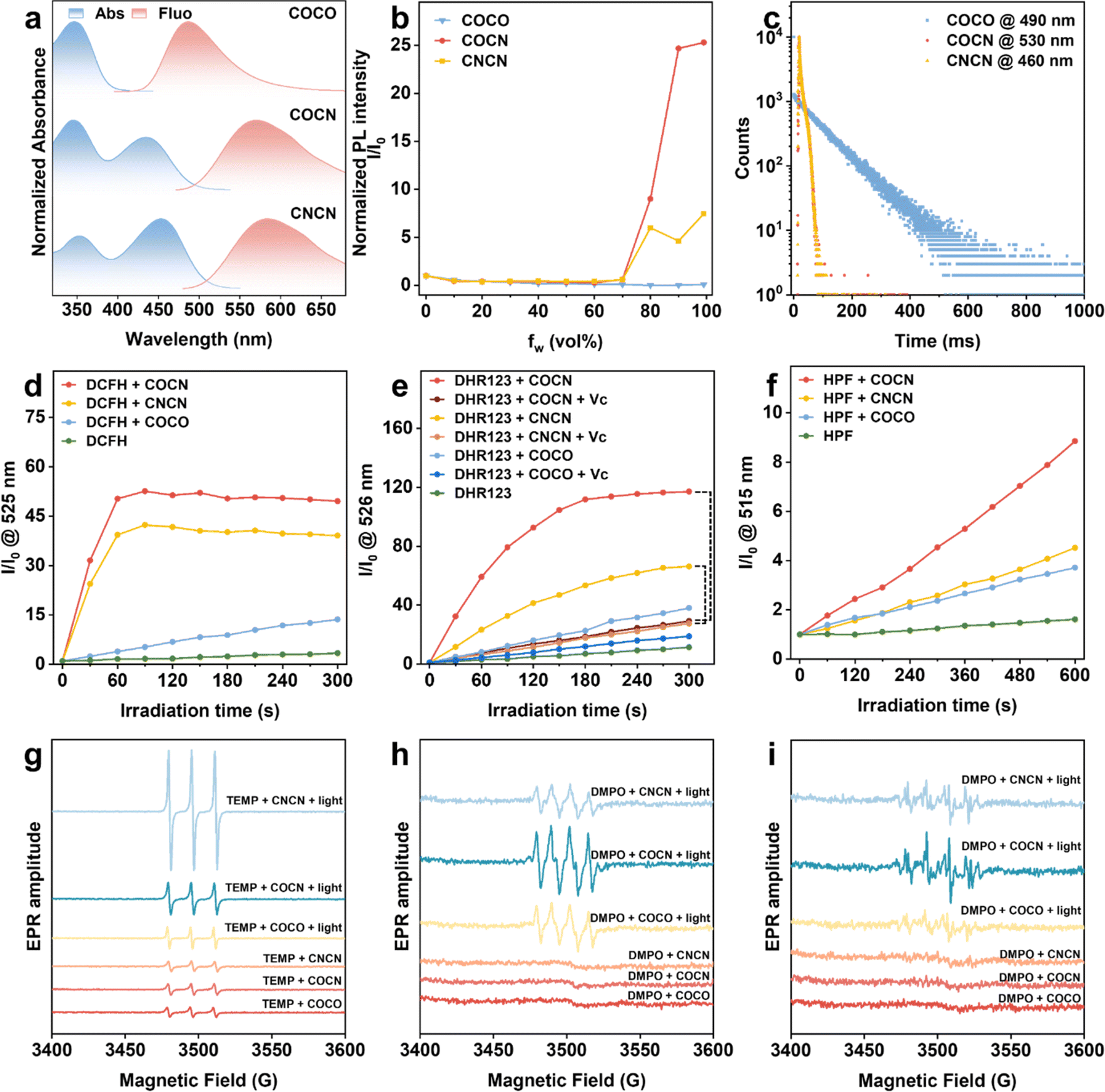 | ||
| Fig. 1 (a) Normalized absorption and PL spectra of COCO, COCN, and CNCN in DMSO. (b) Relative PL intensity (I/I0) versus H2O fraction. (c) Phosphorescent decay curves at 77 K for COCO (490 nm), COCN (530 nm), and CNCN (460 nm). (d) ROS generation using DCFH (1 μM) upon white light irradiation. (e) O2−˙ generation using DHR123 (10 μM) upon white light irradiation. (f) ˙OH generation using HPF (10 μM) upon white light irradiation. (g) EPR spectra for 1O2 detection using TEMP under irradiation. (h) EPR spectra for O2−˙ detection using DMPO in DMSO under irradiation. (i) EPR spectra for ˙OH detection using DMPO in water under irradiation. | ||
After confirming the excellent photostability of COCO, COCN, and CNCN (Fig. S12 and S13†), their overall ability to generate ROS was assessed using the commercially available 2,7-dichlorodihydrofluorescein (DCFH) as the indicator. As shown in Fig. 1d and S14,† under white light (35 mW cm−2) irradiation for 300 s, the PL intensities of DCFH solution significantly increase in the presence of COCN or CNCN, reaching a remarkable 49.5- and 39.1-fold enhancement, respectively. The results reveal the effective overall ROS generation by COCN and CNCN, with COCN possessing superior production efficacy. By comparison, the PL intensity of DCFH exhibits minimal changes when its solution with COCO is exposed to white light. To specifically identify the type of ROS, 9,10-anthracenediyl-bis(methylene)-dimalonic acid (ABDA) was utilized as a probe to selectively detect 1O2 (Fig. S15†). The experimental findings verify that the presence of either COCO or COCN cannot lead to significant attenuation in the ABDA absorbance after light exposure, suggesting their limited capacity for 1O2 production. Under identical conditions, an obvious reduction in absorbance is observed for the ABDA solution containing CNCN, indicative of type-II ROS generation by CNCN through the energy transfer pathway. Additionally, the generation of O2−˙ was evaluated using dihydrorhodamine 123 (DHR123) as an indicator, with vitamin C (Vc) serving as a reductant and free radical scavenger. The PL intensity of the DHR123 solution in the presence of COCN is apparently higher than that of observed in the presence of CNCN and COCO (Fig. 1e and S16†). Upon the addition of Vc, the fluorescence intensity of the DHR123 solution containing COCN is markedly reduced, with a significant 75% decrease from its maximum intensity after light irradiation for 300 s, whereas the presence of CNCN in DHR123 solution results in a modest reduction, retaining 42% of the peak fluorescence intensity. Afterwards, hydroxyphenyl fluorescein (HPF) was employed as an indicator to assess the production of ˙OH. Upon exposure to illumination, the PL intensity of HPF solution containing COCN is substantially higher than that of CNCN and COCO (Fig. 1f and S17†). These findings underscore stronger O2−˙ and ˙OH generation capacity of COCN. To further identify the ROS types, electron paramagnetic resonance (EPR) was conducted using 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the spin-trap agents for 1O2 and free radicals, respectively. Characteristic EPR signals indicative of 1O2, O2−˙, and ˙OH are detected in the presence of COCO, COCN, or CNCN under light irradiation (Fig. 1g–i). Among them, COCN elicits the most intense EPR signal for both O2−˙ and ˙OH, while CNCN produces the most pronounced signal corresponding to 1O2. Thus, we can conclude that an efficient type-I PS with free radical-generating ability has been developed.
The in-depth study of the photophysical and photochemical behaviors of COCO, COCN, and CNCN, as well as their different performances in the triplet states, is crucial for a fundamental understanding of the underlying mechanism associated with engineering O2−˙ and ˙OH production. We conducted systematic theoretical investigations using Gaussian 09, Multiwfn, and ORCA software packages.33–35 The transitions of the optimized states were analyzed based on the natural transition orbitals, taking into account the atomic contributions to both holes and electrons. The distribution of charge density suggests that the excitation of COCO arises from the local excitation of the n–π* transition, whereas the excitation of CNCN is attributable to the CT associated with the π–π* transition (Fig. S18†). In contrast to these symmetric structures, COCN displays distinct orbital occupancies for the CO and CN fragments, a disparity that can be ascribed to its asymmetrical nature at the piperazine bridge termini (Fig. 2a). In order to gain deeper insights into the distinctive properties of the σ*-bridge, the piperazine unit in COCN was replaced with a benzene ring, which serves as a π*-bridge and is referred to as COPhCN (Fig. S19†). Compared to COCN, the π*-bridge in COPhCN induces only a partial intramolecular charge transfer between the conjugated ends, making it challenging to completely separate the orbitals of the CO and CN fragments. Moreover, piperazine, acting as an electron donor, has a narrower HOMO–LUMO energy gap, thereby significantly facilitating electron transitions. Based on the isosurfaces, COCN can be split at the piperazine C–C σ-bond into two parts: the donor (CO) and the acceptor (CN), as shown in Fig. 2b. Considering a general electron transfer reaction (eqn (1)):
| D* + A → D+˙ + A−˙ | (1) |
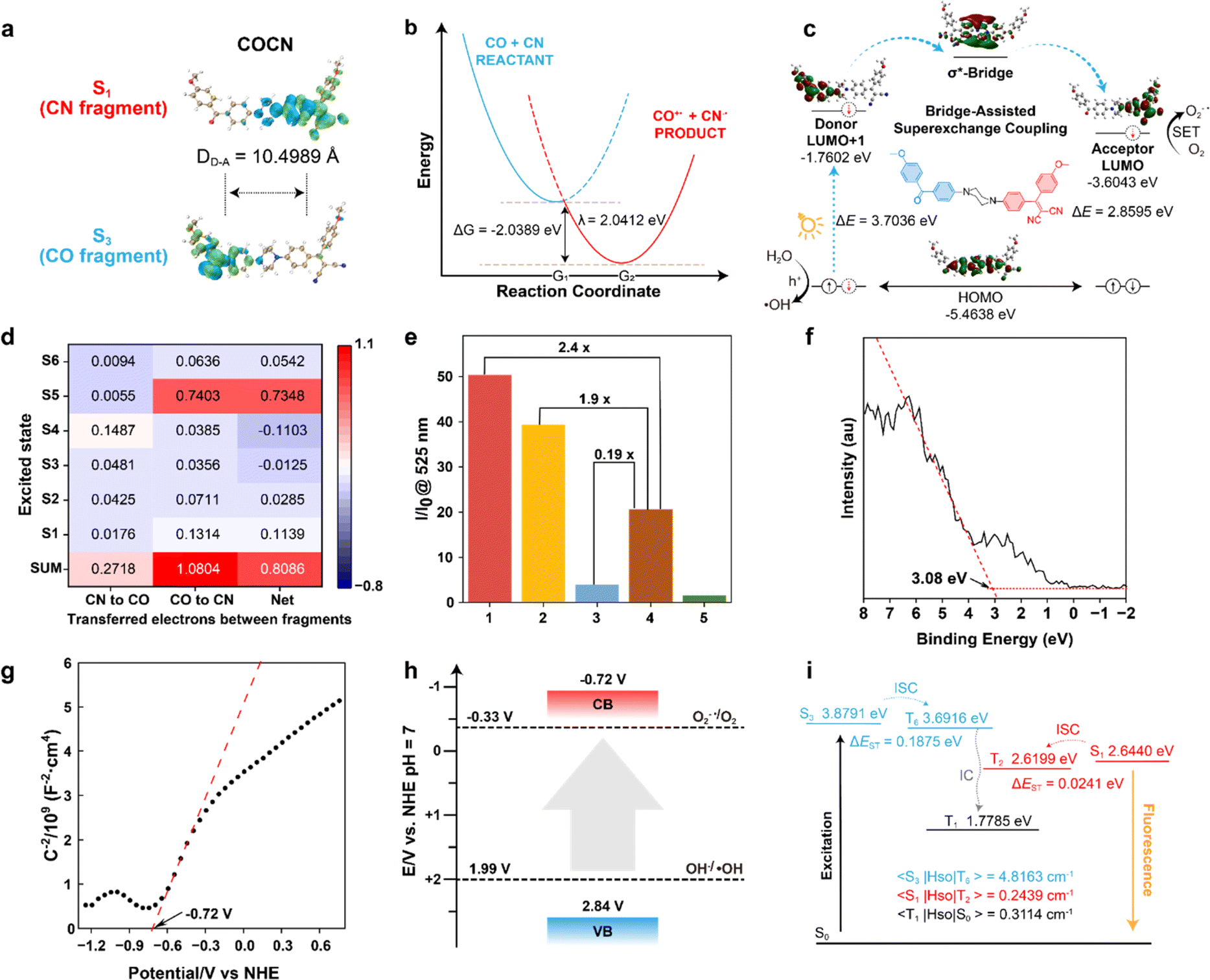 | ||
| Fig. 2 (a) Hole (blue)–electron (green) regions and distance of the D–A redox center (DD–A) of COCN at excited states. (b) Computation scheme for Marcus parameters. (c) The HOMO, LUMO, and LUMO+1 distribution of COCN and the proposed mechanism for electron transfer via bridge-assisted superexchange coupling. (d) Heat map of IFCT for COCN at excited states. (e) Enhancement of fluorescence intensity of DCFH at 525 nm after 60 s white light irradiation in absence or presence of PSs ((1) COCN + DCFH, (2) CNCN + DCFH, (3) COCO + DCFH, (4) COCO + CNCN + DCFH, and (5) DCFH). (f) XPS valence band spectrum of COCN. (g) Mott–Schottky plot of COCN at 2000 Hz, measured in the dark using a carbon paper working electrode, Hg/Hg2Cl2 reference electrode, platinum wire counter electrode, and 1.0 M Na2SO4 as the supporting electrolyte. (h) Energy level diagram of COCN. (i) Energy level diagram of singlet and triplet states for COCN (CO and CN fragments) with corresponding SOC values. | ||
Within the approximation of parabolic potential curves, the free energy of activation is calculated by Nelsen's four-point method (eqn (2)).
| ΔG0 = [ED+˙ + EA−˙ − (ED + EA)] | (2) |
The calculated free energy ΔG0 is −2.0389 eV, and the reorganization energy λ is 2.0412 eV (Tables S1–S3†). As shown in eqn (3), the associated rate constant kET exhibits a quadratic dependence on ΔG0 and λ.
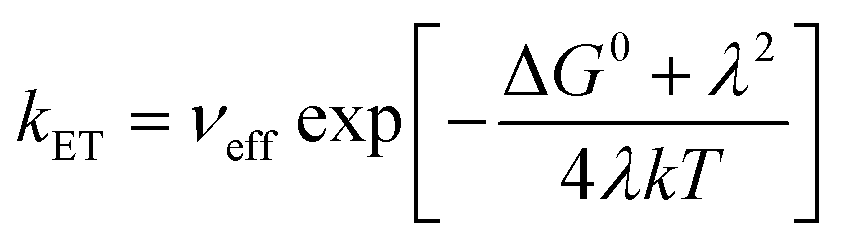 | (3) |
| A−˙ + O2 → A + O2−˙ | (4) |
The formed cationic radical (CO+˙) capture electrons from ambient water, oxidizing the water to ˙OH (eqn (5)).
| D+˙ + OH− → D + ˙OH | (5) |
Eventually, COCN returns to its ground state, enabling the completion of an electron cycling process. These findings support the adoption of a superexchange coupling mechanism for participating in the electron transfer cascade, thereby facilitating the type-I ROS generation.
To further disclose the underlying processes for O2−˙ production, a more comprehensive theoretical investigation was carried out. The electronic conformational information of COCN is provided in Tables S4–S6.† In COCN, the S1 excited states are dominated by the 1(π–π*) transitions of CN fragment (69.5%). Concurrently, its S3 states are mainly defined by the 3(n–π*) transitions of CO (65.2%). According to the EI-Sayed rule, the lowest-energy state proximal to S1 is T2 (with an energy gap ΔES1–T2 = 0.0241 eV), and the nearest 3(π–π*) to S3 is T6 (with a gap ΔES3–T6 = 0.1875 eV). These minimal energy gaps are conductive to high ISC efficiency (Fig. 2f). When contrasted with COCO (Table S7†), it is evident that the incorporation of the CN fragment in COCN significantly reduces the T1 energy level, while T1 characterized by a 3(π–π*) configuration exhibits a slow decay rate owing to the forbidden spin-flip transition from 3(π–π*) to 1π2, thus promoting the PS → O2 electron transfer pathway for producing O2−˙. Moreover, given that spin–orbit coupling (SOC) is a dominant driving force for ISC, the SOC values of COCN for S1 to T2 and S3 to T6 are calculated to be 0.2439 cm−1 and 4.8163 cm−1, respectively. In comparison to the photosensitizer CNCN, the inclusion of benzophenone carrying a lone pair of electrons in COCN greatly increases the likelihood of ISC (Table S8†), which ensures its high efficacy in generating ROS.
Recent studies have illustrated the benefits of type-I PSs, highlighting their capacity to maintain outstanding ROS generation performance under hypoxic microenvironments. Leveraging this advantage, we have preliminary validated the crucial potential of COCN for PDT. To enhance the compatibility of our PSs for biological applications, an amphiphilic copolymer, Pluronic F127, was utilized to encapsulate COCN or CNCN for preparing water-dispersible nanoparticles (COCN NPs and CNCN NPs, respectively). Their fundamental photophysical property assessments were conducted (Fig. 3 and Table S9†). Compared to the molecular counterparts in DMSO, the absorption and emission spectra of COCN NPs and CNCN NPs have negligible changes in aqueous solution (Fig. 3a). Remarkably, the fluorescence quantum yields (QY) of COCN NPs and CNCN NPs in aqueous media are measured at 62.8% and 22.53%, respectively. These values significant surpass those of the corresponding molecules in DMSO, corroborating the distinct AIE characteristics of COCN and CNCN. Dynamic light scattering (DLS) analysis discloses that COCN NPs and CNCN NPs are well-dispersed in water with average hydrodynamic diameters of 148.7 nm and 143.7 nm, respectively (Fig. 3b). Furthermore, transmission electron microscopy (TEM) images reveal uniform spherical morphology of these NPs, exhibiting sizes similar to those measured using DLS. Due to the demonstrated capabilities of COCN and CNCN for ROS generation, their corresponding nanoparticles were selected for further investigation. The presence of COCN NPs or CNCN NPs under continuous light irradiation markedly intensifies the PL signals of DCFH, while a negligible increase in PL intensity is observed DCFH alone, unequivocally confirming their exceptional ROS production capabilities (Fig. S20†). To simulate the biological environment, a specific redox indicator, dihydroethidium (DHE), was combined with RNA and applied to verify the generation of O2−˙. By comparison, the PL intensity of the DHE solution containing COCN NPs exhibited a substantially greater enhancement under white light irradiation than the solution with CNCN NPs (Fig. 3c and S21†). Then, HPF as a hydroxyl radical scavenger was used to detect ˙OH. The PL intensity of the HPF solution increases by a remarkable 32.2-fold in the presence of COCN NPs, while CNCN NPs enhance it by 17.1-fold after irradiation for 600 s (Fig. 3d and S22†). This provides convincing evidence that COCN NPs possess exceptional capacity to generate O2−˙ and ˙OH for potential applications.
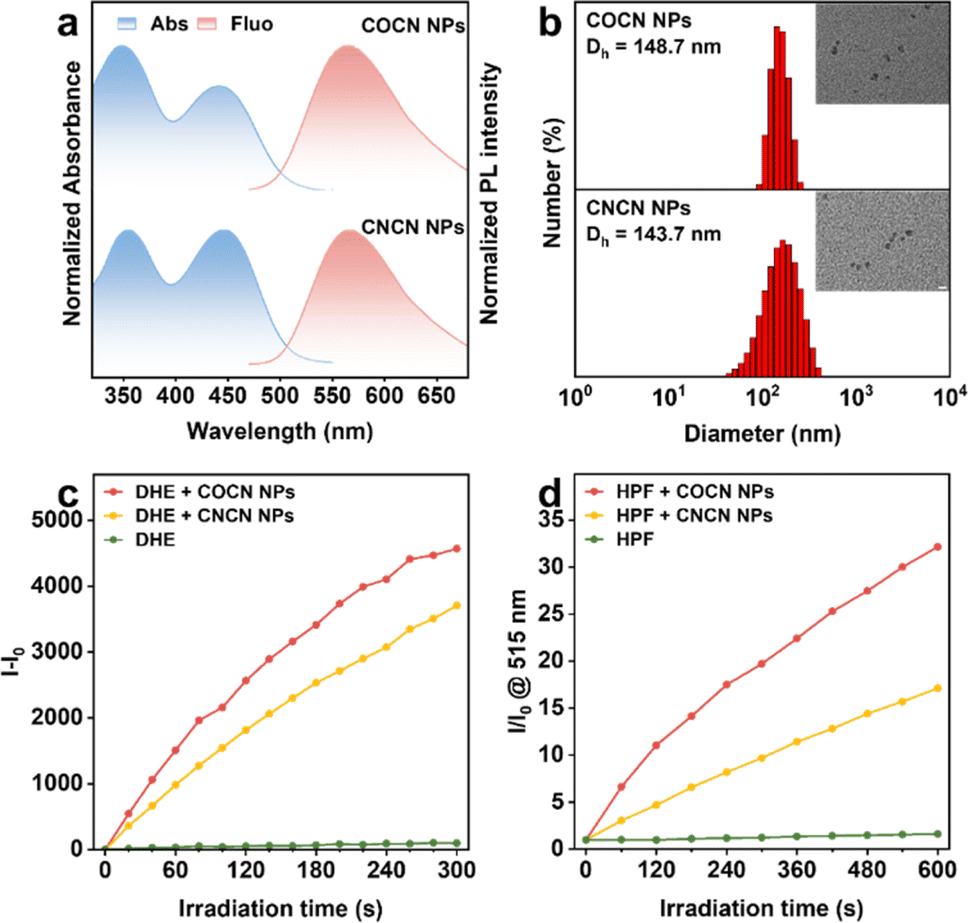 | ||
| Fig. 3 (a) Normalized absorption and PL spectra of COCN NPs and CNCN NPs in water. (b) Size distribution of COCN NPs and CNCN NPs. Inset: TEM image. Scale bar: 200 nm. (c) Plots of (I − I0) of DHE (40 μM) at 580 nm upon white light irradiation. (d) ˙OH generation of COCN NPs and CNCN NPs (5 μM) upon white light irradiation using HPF (10 μM) as an indicator. | ||
The high QYs of COCN NPs and CNCN NPs coupled with their robust ROS production highlight their potential for use in imaging-guided PDT. Prior to imaging, the cytotoxicity of these NPs toward 4T1 cancer cells were assessed using the CCK-8 assay. The 4T1 cell lines were purchased from the Cell Resource Center, Peking Union Medical College. The cell viability remains over 85% even after incubation with a 16 μM concentration of NPs for 24 h under dark conditions, indicative of outstanding biocompatibility and minimal dark toxicity of COCN NPs and CNCN NPs (Fig. S23†). On this basis, a concentration of 5 μM was chosen for subsequent cellular imaging studies. Live cell imaging experiments were performed on 4T1 cells incubated with the NPs, with evaluation via confocal laser scanning microscopy (CLSM). As shown in Fig. S24,† the fluorescence intensity within the 4T1 cells enhances over time, peaking at 4 h after NPs incubation, implying rapid cellular uptake of the NPs within this timeframe. Notably, cells treated with COCN NPs display the most intense fluorescence, which is consistent with their high fluorescence QY. In light of the significance of organelle-targeting specificity, co-localization experiments were performed (Fig. 4). 4T1 cells were treated with COCN NPs, followed by incubation with commercially available organelle-specific trackers. A significant overlap between the red signal from COCN NPs and the green fluorescence of the Lyso-Tracker Green is observed with a Pearson's correlation coefficient as high as 0.85. This indicates a predominant accumulation of COCN NPs within the lysosomes.
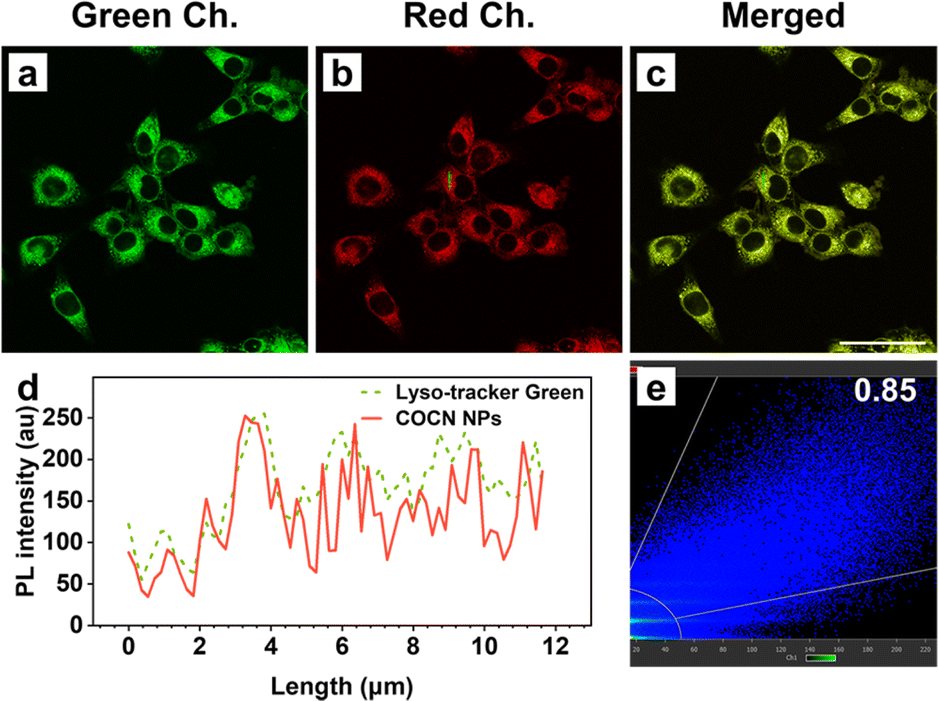 | ||
| Fig. 4 Confocal fluorescence images of 4T1 cells (a–c) stained with COCN NPs (5 μM) and Lyso-Tracker Green (50 nM). (d) Intensity profile of ROIs between COCN NPs (red line) and Lyso-Tracker Green (green line). (e) Fluorescence intensity correlation plot for COCN NPs and Lyso-Tracker Green. Scale bar: 50 μm. | ||
Motivated by the efficient O2−˙ and ˙OH generation and outstanding cellular imaging capabilities, the PDT performance of COCN NPs was studied under both normoxic and hypoxic conditions. Intracellular ROS generated by COCN NPs was detected using the general probe DCFH. Fig. 5a and b reveal that, following light irradiation, cells treated with COCN NPs exhibit bright green fluorescence, confirming the effective ROS generation. Satisfyingly, even in hypoxic conditions, notable green fluorescence is retaining 92% intensity compared to that observed under normoxic conditions, indicating that the efficacy of COCN NPs is not obviously diminished hypoxia. To specifically identify the type of the intracellular ROS, the O2−˙ indicator DHE and ˙OH indicator HPF were utilized. As anticipated, remarkable red and green fluorescence is observed in cells after exposure to light, both under normoxic and hypoxic conditions, which illustrates the potential ability of COCN NPs to yield O2−˙ and ˙OH even under the hypoxic microenvironment of tumor cells. Conversely, the detection of intracellular 1O2 generation using the singlet oxygen sensor green (SOSG) yields starkly different results from those of DHE. Minimal green fluorescence is observed in cells under hypoxic conditions, suggesting that the predominant cytotoxic mechanism of COCN NPs after light exposure is through O2−˙ and ˙OH rather than 1O2. Thereafter, the PDT effects of COCN NPs toward 4T1 cells under both normoxic and hypoxic conditions were evaluated using the standard CCK-8 assay. After exposure to white light for 15 min, a pronounced decline in cell viability was observed at concentrations of COCN NPs exceeding 4 μM in normoxic environments. Notably, similar outcomes were obtained even under hypoxic conditions. The reduction in O2 concentration has little impact on the cytotoxicity of COCN NPs, further highlighting their excellent PDT efficacy regardless of O2 levels (Fig. 5c). Further, live/dead cell staining assays with Calcein-AM (to indicate live cells with green fluorescence) and propidium iodide (PI, to indicate dead cells with red fluorescence) were performed to provide a clear visualization of the PDT efficacy. As shown in Fig. 5d and S25,† strong green fluorescence signals are detected in the three control groups, in stark contrast to the experimental group treated with COCN NPs and light irradiation, which exhibits predominantly red fluorescence, validating considerable cell mortality. To further elucidate the cancer-killing efficiency of COCN NPs at the cellular level, apoptosis/necrosis analysis was conducted employing flow cytometry with Annexin V-FITC and PI staining under normoxic conditions (Fig. 5e). It turns out that COCN NPs induce early apoptosis in 12.24% of cells and late apoptosis in 87.75% when exposed to white light. In contrast, either COCN NPs or white light irradiation alone doesn't inhibit cell growth. Collectively, these findings robustly support that COCN NPs possess potent therapeutic potential for cancer treatment.
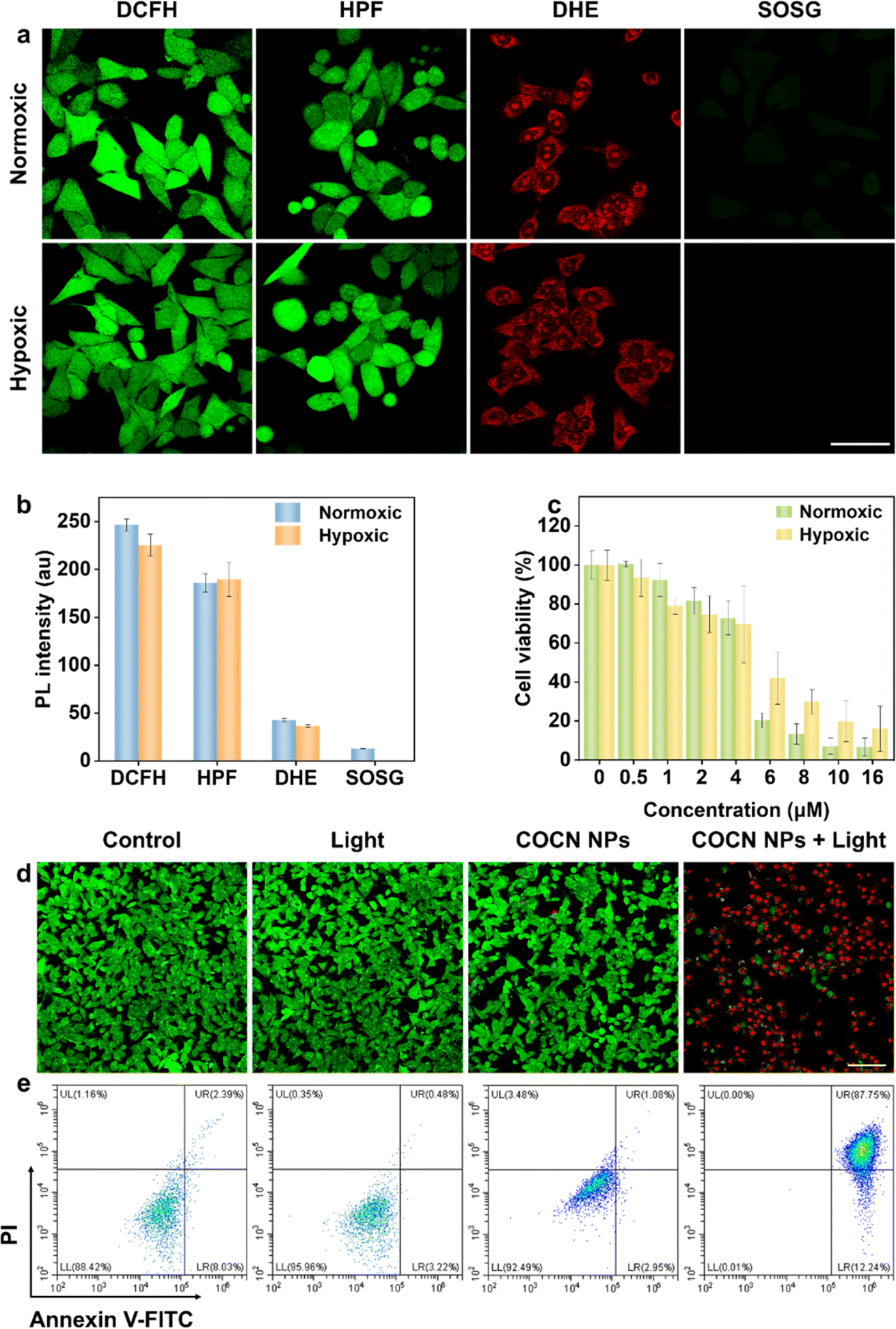 | ||
| Fig. 5 (a) ROS imaging and fluorescence intensity of 4T1 cells pretreated with COCN NPs (5 μM) and irradiated with white light (35 mW cm−2) under normoxic (21% O2) and hypoxic (2% O2) conditions. Scale bar: 50 μm. (b) Quantification analysis of panel (a). Data are mean ± SD (n = 3). (c) Viability of 4T1 cells subjected to various amounts of COCN NPs under normoxic and hypoxic conditions. Data are mean ± SD (n = 3). (d) Fluorescence images of 4T1 cells treated with COCN NPs (8 μM) and irradiated with white light (35 mW cm−2) under normoxic conditions. The live cells were stained with Calcein-AM, and the dead cells were stained with PI. Scale bar: 100 μm. (e) Apoptosis/necrosis analysis by flow cytometry using Annexin V-FITC and PI under normoxic conditions. | ||
Conclusions
In conclusion, we have reported the first demonstration of bridge-assisted superexchange coupling to facilitate an electron transfer cascade for engineering O2−˙ and ˙OH generation. By comparison, the asymmetric molecule COCN possesses the ability to produce O2−˙ and ˙OH, which is remarkably distinct from COCO with phosphorescence property and CNCN that generate both type-I and type-II ROS. Experimental and theoretical studies reveal that the amplified generation of free radicals from COCN is achieved by a long-range electron transfer, where the CO fragment donates one electron to the CN fragment through the piperazine σ*-bridge via superexchange coupling. Importantly, nanoparticles of COCN not only maintain their potent O2−˙ and ˙OH production capability but also exhibit a remarkable fluorescence high quantum yield of 62.8%. In vitro investigations confirm that COCN NPs can efficiently generate O2−˙ and ˙OH in cancer cells, regardless of the intracellular O2 level and thereby showcase excellent phototoxicity against cancer cells. Compared to intermolecular electron transfer, the utilization of bridge-assisted superexchange coupling to achieve long-range intramolecular electron transfer in a single molecule potentially offers the advantages of establishing a more explicit structure–activity relationship and enhancing stability. This approach is expected to be beneficial for the future advancement of PSs with selective ROS-generating properties for optimizing their biological effects and simplifying the underlying mechanisms. However, our photosensitizer's short emission wavelength limits its application in in vivo imaging and PDT. The development of red/NIR light-emitting PSs based on such an electron transfer cascade strategy for in vivo studies is currently ongoing in our laboratory.Data availability
Experimental procedures and the data supporting this article have been included as part of the ESI.†Author contributions
L. C. synthesized materials, performed all measurements, and wrote the manuscript. S. Y. designed and directed the investigations and composed the manuscript. W.-J. G., L. Q., and X. Z. revised the manuscript. B. L. and H.-Q. P. provided intellectual input and revised the manuscript. All the authors were involved the analysis of results and discussions of the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H.-Q. Peng thanks the National Natural Science Foundation of China (22105016) and the National Key R&D Program of China (2022YFA1505900).Notes and references
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- Z. Li, Z. Zhou, Y. Wang, J. Wang, L. Zhou, H.-B. Cheng and J. Yoon, Coord. Chem. Rev., 2023, 493, 215324 CrossRef CAS.
- J. Li, Z. Zhuang, Z. Zhao and B. Z. Tang, View, 2022, 3, 20200121 CrossRef CAS.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- Q. Sun, Q. Su, Y. Gao, K. Zhou, W. Song, P. Quan, X. Yang, Z. Ge, Y. Zhang and G. He, Aggregate, 2023, 4, e298 CrossRef CAS.
- S. Liu, Y. Pei, Y. Sun, Z. Wang, H. Chen, D. Zhu, M. R. Bryce, B. Z. Tang and Y. Chang, Aggregate, 2024, 5, e547 CrossRef CAS.
- H. Wang, W.-G. Li, K. Zeng, Y.-J. Wu, Y. Zhang, T.-L. Xu and Y. Chen, Angew. Chem., Int. Ed., 2019, 58, 561–565 CrossRef CAS.
- X. Wu, M. Yang, J. S. Kim, R. Wang, G. Kim, J. Ha, H. Kim, Y. Cho, K. T. Nam and J. Yoon, Angew. Chem., Int. Ed., 2022, 61, e202200808 CrossRef CAS.
- D. Chen, Q. Xu, W. Wang, J. Shao, W. Huang and X. Dong, Small, 2021, 17, 2006742 CrossRef CAS PubMed.
- K.-X. Teng, D. Zhang, B.-K. Liu, Z.-F. Liu, L.-Y. Niu and Q.-Z. Yang, Angew. Chem., Int. Ed., 2024, 63, e202318783 CrossRef CAS.
- K.-X. Teng, W.-K. Chen, L.-Y. Niu, W.-H. Fang, G. Cui and Q.-Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 19912–19920 CrossRef CAS.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, J. Am. Chem. Soc., 2023, 145, 8130–8140 CrossRef CAS.
- M. Li, J. Xia, R. Tian, J. Wang, J. Fan, J. Du, S. Long, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2018, 140, 14851–14859 CrossRef CAS PubMed.
- R. Lin, J. Liu, W. Xu, Z. Liu, X. He, C. Zheng, M. Kang, X. Li, Z. Zhang, H.-T. Feng, J. W. Y. Lam, D. Wang, M. Chen and B. Z. Tang, Adv. Mater., 2023, 35, 2303212 CrossRef CAS.
- J. An, S. Tang, G. Hong, W. Chen, M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, Nat. Commun., 2022, 13, 2225 CrossRef CAS.
- K. Zhang, Z. Yu, X. Meng, W. Zhao, Z. Shi, Z. Yang, H. Dong and X. Zhang, Adv. Sci., 2019, 6, 1900530 CrossRef.
- Y. Wang, Y. Li, Z. Zhang, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2021, 33, 2103748 CrossRef CAS.
- D. Li, X. Chen, D. Wang, H. Wu, H. Wen, L. Wang, Q. Jin, D. Wang, J. Ji and B. Z. Tang, Biomaterials, 2022, 283, 121476 CrossRef CAS.
- S. Ning, M. Lyu, D. Zhu, J. W. Y. Lam, Q. Huang, T. Zhang and B. Z. Tang, ACS Nano, 2023, 17, 10206–10217 CrossRef CAS PubMed.
- X. Hu, Z. Fang, F. Sun, C. Zhu, M. Jia, X. Miao, L. Huang, W. Hu, Q. Fan, Z. Yang and W. Huang, Angew. Chem., Int. Ed., 2024, 63, e202401036 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS.
- M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005–4018 RSC.
- M. U. Winters, K. Pettersson, J. Mårtensson and B. Albinsson, Chem.–Eur. J., 2005, 11, 562–573 CrossRef CAS PubMed.
- O. S. Wenger, Chem. Soc. Rev., 2011, 40, 3538–3550 RSC.
- C. Lambert, G. Nöll and J. Schelter, Nat. Mater., 2002, 1, 69–73 CrossRef CAS.
- M. Plato, K. Moebius, M. E. Michel-Beyerle, M. Bixon and J. Jortner, J. Am. Chem. Soc., 1988, 110, 7279–7285 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- W. Zhu, Z. Ding, S. Guo, W.-J. Guo, S. Yan, B. Liu, H. Li, Z. Liu, B. Z. Tang and H.-Q. Peng, Small, 2024, 2309424 CrossRef CAS PubMed.
- T. Peng and H.-Q. Peng, J. Mater. Chem. C, 2023, 11, 12894–12899 RSC.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Lu and F. Chen, Acta Chim. Sin., 2011, 69, 2393–2406 CAS.
- P. Gai, W. Yu, H. Zhao, R. Qi, F. Li, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 7224–7229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05345a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |